

Abstract
Background
Mosses are the largest of the three extant clades of gametophyte-dominant land plants and remain poorly studied using comparative genomic methods. Major monophyletic moss lineages are characterised by different types of a spore dehiscence apparatus called the peristome, and the most important unsolved problem in higher-level moss systematics is the branching order of these peristomate clades. Organellar genome sequencing offers the potential to resolve this issue through the provision of both genomic structural characters and a greatly increased quantity of nucleotide substitution characters, as well as to elucidate organellar evolution in mosses. We publish and describe the chloroplast and mitochondrial genomes of Tetraphis pellucida, representative of the most phylogenetically intractable and morphologically isolated peristomate lineage.
Results
Assembly of reads from Illumina SBS and Pacific Biosciences RS sequencing reveals that the Tetraphis chloroplast genome comprises 127,489 bp and the mitochondrial genome 107,730 bp. Although genomic structures are similar to those of the small number of other known moss organellar genomes, the chloroplast lacks the petN gene (in common with Tortula ruralis) and the mitochondrion has only a non-functional pseudogenised remnant of nad7 (uniquely amongst known moss chondromes).
Conclusions
Structural genomic features exist with the potential to be informative for phylogenetic relationships amongst the peristomate moss lineages, and thus organellar genome sequences are urgently required for exemplars from other clades. The unique genomic and morphological features of Tetraphis confirm its importance for resolving one of the major questions in land plant phylogeny and for understanding the evolution of the peristome, a likely key innovation underlying the diversity of mosses. The functional loss of nad7 from the chondrome is now shown to have occurred independently in all three bryophyte clades as well as in the early-diverging tracheophyte Huperzia squarrosa.
Keywords: Moss, Tetraphis pellucida, Peristome, nad7, petN, Phylogeny, Organellar genomes, Sequencing
Background
Land plants (embryophytes) comprise three extant gametophyte-dominant clades (mosses, liverworts and hornworts) and one extant sporophyte-dominant clade (tracheophytes). The former comprise a paraphyletic grade known as the “bryophytes”, a morphological grouping of convenience that includes all lineages in which the diploid generation (the sporophyte) is unbranched, has only a single sporangium, and remains attached to a generally more complex, well-developed, and persistent haploid generation (the gametophyte).
Compared to the tracheophytes, there has been very little study of the organellar genomes of bryophytes. More genomic data are needed from representatives of each major clade, to solidify phylogenetic understanding as well as to investigate organellar evolution within the group. A key phylogenetic question that genomic data may be able to contribute towards solving is the branching order of the major peristomate clades (the peristome is the collective term for the elaborate teeth around the mouth of the capsule that provide a mechanism to control spore release). Sampling from the more isolated and morphologically distinct lineages surviving from this relatively early diversification event may also be an efficient strategy for uncovering genomic diversity of relevance to organellar evolution in land plants.
In contrast to liverworts, moss sporophytes exhibit considerable structural diversity, particularly in the morphology and function of the peristome. Although peristomes are lacking in the earliest-diverging moss lineages (Takakiopsida, Sphagnopsida, Andreaeopsida, and Andreaeobryopsida), they characterise all other major groups of mosses, representing about 97% of species diversity [1], except for the phylogenetically enigmatic Oedipodiopsida. Results from molecular systematic studies have corroborated the conclusion of early investigations (e.g., [2, 3]) that distinct peristome types define the primary phylogenetic divisions among peristomate mosses (e.g.; [4–8]).
Although each of these peristomate clades is clearly monophyletic (the Polytrichopsida, the Tetraphidopsida, the Buxbaumiales, and the arthrodontous clade, in which peristomes consist entirely of cell wall remnants), their relationships to each other and to the non-peristomate Oedipodiopsida is perhaps the most significant unsolved problem remaining in higher-level moss systematics (e.g., [4, 5, 9–13]). It has profound implications for hypotheses of homology among peristome types and whether peristomes have evolved more than once (Figure 1). Central to this problem is the position of the Tetraphidopsida, a small group of two genera and five species with a seemingly paradoxical combination of morphological and molecular traits.
Figure 1.
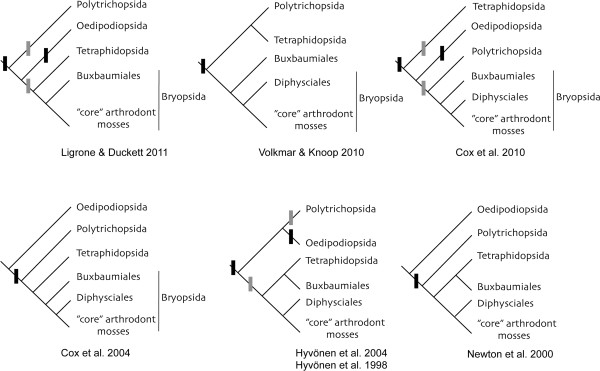
Various topologies proposed for peristomate mosses in recent phylogenetic analyses. Possible character state changes for gain and/or loss of peristomes with teeth are marked on trees with bars. In some cases alternative optimisations are indicated with lighter coloured bars.
Members of the Tetraphidopsida have peristomes unlike those in any other group of mosses, with the entire apical, peristome-forming portion of the sporophyte divided into four massive teeth. Based solely on the observation that these teeth are comprised of entire, elongated, thick-walled cells rather than cell wall remnants as in the arthrodontous mosses, the “nematodontous” Tetraphidopsida have traditionally been considered to be related to the Polytrichopsida, in which the very different peristomes also have these features. Developmentally, however, Shaw & Anderson [14] demonstrated that unlike in the Polytrichopsida, the peristome of Tetraphis pellucida is identical to that of arthrodontous mosses until a fairly late stage. Furthermore, the mature peristome in the Polytrichopsida is structurally very different from that of Tetraphis, and recent studies of relationships within the group strongly suggest that it is a derived feature, since the earliest diverging lineages lack peristome teeth altogether [13, 15]. All of these observations cast doubt on the idea that a generalized nematodontous peristome, homologous between Tetraphidopsida and Polytrichopsida, is plesiomorphic to the arthodontous peristome.
It seems plausible that peristomes (at least as structures composed of multiple linear processes remaining after dehiscence of the operculum, or capsule lid) might have arisen independently in the Polytrichopsida and in the common ancestor of the Tetraphidopsida and arthrodontous mosses (Figure 1), or even separately in all three lineages, perhaps from a primitive “pre-peristomate” structure. In this case it would be most parsimonious to interpret Oedipodium as also primitively non-peristomate, even if it is sister to the Tetraphidopsida, Buxbaumia, and the arthrodontous mosses [9] rather than to all of the peristomate mosses [10]. Clearly, however, the nearly identical and highly regular development of the peristome-forming cell layers in Tetraphis, Buxbaumia and the arthrodontous mosses, and to a lesser extent the Polytrichopsida [16, 17], is likely to be homologous, even if it did not originally gave rise to what we would now call a peristome.
In order to address these questions it is necessary to expand the quantity and quality of the phylogenetic characters available to inform relationships among the major peristomate groups, and in particular to clarify the position of the Tetraphidopsida. Sequencing of complete organellar (plastid and mitochondrial) genomes offers the potential to considerably increase the number of nucleotide characters and to provide new genomic-level characters, as well as to elucidate organellar evolution. Potential genomic characters include gene presence/absence, gene inversions, intron insertion and deletion, pseudogenisation processes, and changes in gene order, and can be viewed as morphological characters at the genetic level [18–22]. Currently there are still only two complete chloroplast genomes fully published for mosses, for Physcomitrella patens[23] and Tortula ruralis[24], although plastid genomes have been sequenced for a number of other species including Takakia lepidozioides, Sphagnum palustre, and Andreaea nivalis, with genomic level characters (mainly gene losses and pseudogenisation) being found that are congruent with currently accepted relationships [25]. Similarly, there are only two complete mitochondrial genomes published for mosses at time of writing, for P. patens[26] and Anomodon rugelii[27], although genomes from a range of mosses have been assembled, with the composition reported as highly conserved [28]. As a step towards providing genomic level data to address the question of relationships among the peristomate moss groups, we sequenced and fully assembled the chloroplast and mitochondrial genomes of Tetraphis pellucida using a mixture of Illumina SBS and Pacific Biosciences (PacBio) RS sequencing. The results provide preliminary insights into phylogeny and organellar evolution, which we anticipate will be developed further when similar data becomes available for other key lineages.
Results
Chloroplast genome
The chloroplast genome of Tetraphis pellucida has a length of 127,489 bp and retains the general structure common to most land plants, with two inverted repeat (IR) regions of 9,564 bp separated by a small single copy region (SSC) of 18,927 bp and a large single copy region (LSC) of 89,434 bp. Overall GC content is 29.4%, similar to other known bryophyte chloroplast genomes (28-33% [23]) and significantly less than the 34-40% found in seed plants [29]. The IR gene content is identical to that of the mosses Tortula ruralis and Physcomitrella patens, with the trnV-GAC and trnN-GUU transfer RNA genes terminating the IRs. Figure 2 illustrates the structure of the genome and the relative positions of all genes.
Figure 2.
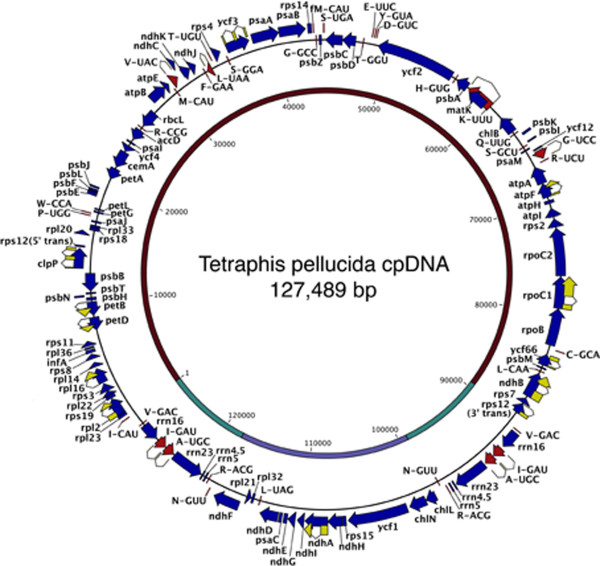
Gene map of the Tetraphis pellucida chloroplast genome. Protein-encoding and rRNA-encoding genes are in blue and tRNA-encoding genes are in red. All genes reading clockwise are shown, with their names, outside of the circle. All genes reading counter-clockwise are shown on the circle with their names inside. All genes with multiple exons have these shown in yellow flanking the gene names. Single letters designate tRNA genes according to the one-letter code for the corresponding amino acid with “fM” indicating the tRNA expected to be charged with formyl-methionine. tRNA-encoding genes are further differentiated with the codon expected to be recognized in cases where there are more than one tRNA for the same amino acid. As is common, rps12 occurs in two non-contiguous regions that are spliced in trans to form a complete transcript, so these are shown with the words “5′ trans” and “3′ trans” to indicate this. The inner circle shows the large single-copy (LSC) region and small single-copy (SSC) region in brown and blue, respectively, and the inverted repeat regions in green, with nucleotides numbered starting at the beginning of the LSC.
In common with Tortula ruralis, Tetraphis pellucida lacks the sizeable inversion of around 71 kb in the LSC that characterizes P. patens and other Funariales [30]. It also shares with Tortula ruralis the absence of petN, these two species being the only land plants currently known to lack this gene in the chloroplast. Otherwise, functional gene content and order are identical in all three of these mosses. The assembly also corroborates the absence of the rpoA gene, previously reported to have been absent in Tetraphis as well as in all arthrodontous groups (including Diphyscium), although present in all other major moss lineages, including Buxbaumia[31].
In Tortula ruralis a pseudogenised copy of the trnP-GGG gene was reported, but a nucleotide BLAST (BLASTn) of the corresponding area in the Tetraphis pellucida assembly matched only a small part of the spacer region adjacent to this pseudogene in Tortula ruralis. Similarly, while a tufA pseudogene is present in the Takakia chloroplast genome (accession number AB367138, incomplete assembly), a BLASTn of the corresponding Tetraphis pellucida DNA did not match this.
Despite identical or near identical gene content, the Tetraphis pellucida chloroplast genome is approximately 5 kb longer than those of Tortula ruralis (122,530) and P. patens (122,890). This difference is nearly entirely accounted for by an increased total length of intragenic spacer regions in the LSC (see Table 1 for basic compositional statistics).
Table 1.
Selected statistics for chloroplast and mitochondrial genomes of Tetraphis pellucida
| Chloroplast | Mitochondrion | |
|---|---|---|
| Total length (bp) | 127,489 | 107,730 |
| Protein coding (genes/ORFs [exons bp total]) | 82/83 [69,897] | 41/41 [36,285]* |
| Ribosomal (genes [exons bp total]) | 8 [4,532] | 3 [4,822] |
| tRNA (genes [exons bp total]) | 36 [2,705] | 24 [1,795] |
| Other (introns, spacers, pseudogenes etc., bp total) | 50,355 | 64,828 |
| Adenine, A (bp [%]) | 45,073 [35.4%] | 31,162 [28.9%] |
| Cytosine, C (bp [%]) | 18,834 [14.8%] | 22,098 [20.5%] |
| Guanine, G (bp [%]) | 18,635 [14.6%] | 23,720 [22%] |
| Thymine, T (bp [%]) | 44,941 [35.3%] | 30,750 [28.5%] |
| Weight (single-stranded) | 39.371 MDa | 33.313 MDa |
| Weight (double-stranded) | 78.749 MDa | 66.558 MDa |
*All counts include the two unidentified ORFs (see Figure 3).
A scan for candidate RNA editing sites (C => U) using the protein sequence BLAST prediction method (BLASTx) implemented in PREPACT 2.0 [32] found 15 potential sites predicted by 100% of references and 25 potential sites predicted by 75% of references. Only one of the sites predicted by 75% or 100% of references corresponded to a previously identified RNA editing event in a reference (in psbB in Anthoceros formosae, label psbBeU38PL). A scan of the published Tortula ruralis chloroplast genome for comparative purposes yielded only three potential sites predicted by 100% of references and 10 predicted by 75% of references, with none of these corresponding to previously identified editing events.
Mitochondrial genome
The Tetraphis pellucida mitochondrial genome (structure illustrated in Figure 3) has a total length of 107,730 bp and an overall GC content of 42.5% (see Table 1 for statistics). Both of these figures are similar to, if slightly higher than, those for P. patens (105,340 bp, 40.6% GC; [26]) and Anomodon rugelii (104,239 bp, 41.2% GC; [27]). Gene order is identical to that of the other two known moss chondromes, but the nad7 gene, which is present as a functional gene in the published gene maps and GenBank annotations for Physcomitrella patens and A. rugelii, is present only as a partial pseudogene in T. pellucida. The region between the trnT and rpl2 genes in T. pellucida consists of only 1,329 bp, with BLASTn results for this area indicating 81% and 86% alignment to sections of the nad7 genes of A. rugelii and P. patens respectively and 90% sequence identity in both cases. However, in both A. rugelii and P. patens the nad7 gene occurs in three exons of 140 bp, 69 bp and 973 bp separated by two large introns, while in T. pellucida the largest downstream exon and most of the preceding intron are entirely absent. Also absent in T. pellucida is the pseudogenised rps10 gene, present in A. rugelii and P. patens downstream of nad7 and preceding the start of the rpl2 gene. Nonetheless the two shorter nad7 exons and intervening intron are present and alignable with those of A. rugelii and P. patens, the exons apparently with uninterrupted reading frames.
Figure 3.
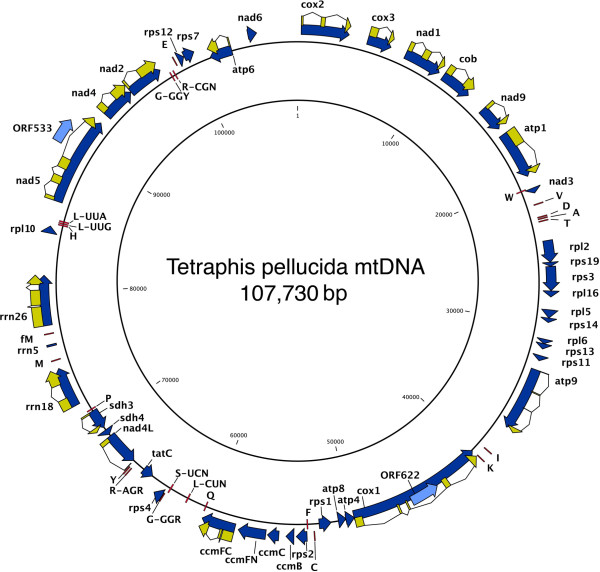
Gene map of the Tetraphis pellucida mitochondrial genome. Genes are shown as explained for Figure 2.
In T. pellucida there is only a short region of 283 bp between the second 69 bp exon of the pseudogenised nad7 and the rpl2 gene. Most of the first 161 bp is alignable with the start of the downstream intron in the nad7 genes of A. rugelii and P. patens, but the final 122 bp return only a single match in a BLASTn search (87% identity), to the same region immediately upstream of the rpl2 gene in the lycopod Huperzia squarrosa, a species in which the nad7 gene is also functionally absent [33].
The pseudogenised copy of rps8 occurring next to the rpl6 gene in A. rugelii and P. patens is identifiable also in T. pellucida.
Scanning for RNA editing sites (C => U) using the BLASTx prediction method in PREPACT 2.0 found 62 potential sites predicted by 100% of references and 89 potential sites predicted by 75% of references. Of these, 45 and 54 respectively corresponded to previously identified RNA editing events in at least one reference.
Discussion
The chloroplast genome of Tetraphis pellucida is identical in overall structure, gene content, and gene order to that of Tortula ruralis and highly similar to that of Physcomitrella patens, while the mitochondrial genome is identical in gene order to those of Anomodon rugelii and P. patens, except differing from both in the absence of a functional nad7 gene. The latter is due to the absence of a region that includes the largest exon of nad7 as well as the pseudogenised rps10 gene. Although the full value of the data for phylogenetic reconstruction and the study of organellar evolution within the mosses will not become apparent until genomes from other major moss lineages are fully assembled and published, a number of observations can be made at this stage based on comparison with data that are already available.
Predicted levels of organellar RNA editing in Tetraphis pellucida are consistent with a hypothesis of a general trend towards reduction in numbers of edited sites in more derived (or at least later diverging) clades within the mosses, with numbers of both chloroplast and mitochondrial sites being somewhat higher than those found in arthrodontous mosses and numbers of chloroplast sites very much lower than those found in the early-diverging moss lineage Takakia as well as in hornworts [34–37]. Yura et al. [34] found 302 putative RNA editing sites in the chloroplast genome of Takakia lepidozioides, while Miyata et al. [36] found only two in P. patens and our own results predict three in Tortula ruralis (an arthrodont, like P. patens). This compares with our predicted figure of 15 sites for Tetraphis pellucida. Similarly, our predicted figure of 62 mitochondrial RNA editing sites for Tetraphis pellucida is considerably higher than the 11 sites present in P. patens[37] and the 39 predicted for Anomodon rugelii by Lenz & Knoop [32].
The pseudogenisation of nad7 in the Tetraphis pellucida chondrome is significant, as while it is also absent or existing only as a non-functional pseudogene in hornworts and in most liverworts [38–40] as well as in the lycopod Huperzia squarrosa[33], it appears to be functionally present in all other known tracheophyte and moss chondromes investigated, including representatives of Takakia, Ulota, Leucobryum and Dichodontium[41] as well as P. patens and A. rugelii. In liverworts, both Haplomitrium[38] and Treubia[27] retain a functional mitochondrial nad7 gene, while all other species investigated have pseudogenised copies, varying greatly in their degrees of degeneration but remarkable for having persisted throughout the long evolutionary history of the group [38]. This is consistent with current hypotheses of Haplomitrium and Treubia forming the sister clade (Haplomitriopsida) to the rest of the liverworts (Marchantiopsida and Jungermanniopsida, e.g. [42, 43]) and the psuedogenisation of nad7 having occurred in a common ancestor of the extant Marchantiopsida and Jungermanniopsida. In Marchantia polymorpha at least, there is clear evidence for endosymbiotic gene transfer to the nucleus and a functional copy of nad7 in the nuclear genome [44], and it can reasonably be assumed that this is the case for all other non-Haplomitriopsid liverworts. Interestingly, the functional nad7 gene found in other mosses shares its two introns with the version found in vascular plants [41], while in the liverwort nad7 gene these are lacking, and two other non-homologous introns occur instead [41, 38]. The current finding that nad7 is present only as a pseudogenised remnant in the T. pellucida chondrome demonstrates that loss of a functional mitochondrial copy of this gene has occurred independently in all three bryophyte clades as well as in at least one early-diverging tracheophyte (Huperzia). However, while this appears to have been a very early and defining event in the evolution of both hornworts and liverworts, it is a derived condition in mosses and conceivably even unique to Tetraphis (as it may be unique to Huperzia within the tracheophytes). Wahrmund et al. [45] showed that the entire trnA-trnT-nad7 gene cluster has been subject to extensive recombination during the evolutionary history of liverworts. However, it is striking that a BLASTn search for the region of 122 bp adjacent to the rpl2 gene in T. pellucida matches only the same region in H. squarrosa, suggesting the possibility of a shared mechanism for the independent loss of nad7 in Tetraphis and Huperzia. Unlike T. pellucida however, Huperzia appears to lack any traces of the two smaller nad7 exons (which are present with uninterrupted reading frames in Tetraphis), suggesting that pseudogenisation following the loss of the large exon is considerably more advanced in Huperzia.
Although the role of nad7 in encoding a subunit of respiratory complex I (RC 1, NADH-ubiquinone oxidoreductase) might imply that it is indispensable, in plants the existence of alternative NAD(P)H-ubiquinone oxidoreductases means that mutants with a deficient RC I are potentially viable [46]. In the tobacco mutant CMSII, which lacks RC I activity, this has been shown to be due to deletion of the nad7 gene from the mitochondrion [46]. Although these plants exhibit slow development and reduced vegetative and floral organs they are apparently viable in cultivation. Although it is much more probable that in T. pellucida the nad7 gene has been transferred to the nuclear genome (as in Marchantia) than that nad7 and/or RC I activity is lacking, perhaps the relative simplicity and slow growth rates of bryophytes make endosymbiotic transfer of this gene more likely to occur, because temporary loss of RC I functionality might have less chance of being immediately fatal.
Of further note is the absence of petN in the plastome of Tetraphis pellucida, while this gene is present in Physcomitrella patens[23], as well as in all other known embryophytes except Tortula ruralis. Unpublished data additionally suggest that petN is present in at least Sphagnum palustre[25]. On noting the absence of petN in Tortula ruralis, Oliver et al. [24] considered it most probable that it has been transferred to the nuclear genome, or else that another nuclear-encoded gene product performs the same function as a subunit of the photosynthetic cytochrome b6f complex. The shared absence of petN in Tortula and Tetraphis and its presence in Physcomitrella and other Funariales [47] requires either that a loss has occurred more than once, that petN has been regained in the Funariales (which seems improbable), or that Tetraphis (or the Tetraphis chloroplast) is more closely related to Tortula than Tortula is to Physcomitrella. As both Physcomitrella and Tortula are unambiguously arthrodontous, highly supported by phylogenetic analyses of nucleotide data, the latter also seems unlikely and is at odds with all recent phylogenetic analyses (Figure 1). Both the unique structure of the tetraphidopsidan peristome (see Background) and existing molecular phylogenetic studies strongly suggest that the Tetraphidopsida are outside of the major clade of arthrodontous mosses, while generally also implying that Buxbaumia is more closely related to the arthrodonts than Tetraphis is. Further studies of presence/absence of petN in moss plastomes are needed.
The rps16 gene appears to be plesiomorphically present in the plastomes of exemplars of non-peristomate moss lineages [25], including Andreaea nivalis, representative of the Andreaeopsida (the sister lineage to the peristomate mosses + Oedipodium), although it is absent in Physcomitrella and Tortula. The current results show that it is also absent in Tetraphis pellucida, which is consistent with a loss in a common ancestor of the Tetraphidopsida and the arthrodontous mosses, originating after divergence with the Andreaeopsida. Thus the presence of this gene in the plastome has the potential to be phylogenetically informative for relationships amongst the major peristomate lineages, depending on which other groups (Oedipodopsida, Polytrichopsida, and Buxbaumiales) it occurs in. Further adding to the pattern of gene presence/absence, it is known that the rpoA gene is present in all earlier diverging moss lineages (including Buxbaumia and the Polytrichopsida) but absent in all lineages of arthrodontous mosses as well as in Tetraphis[31].
Despite consensus on the monophyly of the arthrodontous mosses, molecular sequence comparisons have failed to consistently support any hypothesis of a relationship between the Tetraphidopsida and any other major group e.g. [4, 5, 10], (Figure 1). Thus after failing to corroborate the results of Cox et. al [10], in which Tetraphis pellucida was placed as sister to Buxbaumia plus the arthrodontous mosses, Cox et al. [4] concluded that (in relation to nematodontous peristomes) “the origin of the arthrodontous peristome”… “remains obscure”. Ligrone & Duckett [9] instead stressed putatively conserved morphological and molecular characters to support a phylogenetic hypothesis in which the Tetraphidopsida is sister to a clade comprising Buxbaumia and the arthrodontous mosses including Diphyscium (as in [10]), but with Oedipodium sister to this group rather than to a larger peristomate clade including the Polytrichopsida. The Polytrichopsida lack a distinctive placental morphology in the gametophyte/sporophyte junction that is found in all other peristomate mosses as well as in Oedipodium. The authors suggested that a nematodontous peristome arose once in the ancestor of all peristomate mosses and was lost in Oedipodium, while being retained in the Tetraphidopsida and also giving rise to the arthrodontous peristome. As discussed above, however, all hypotheses implying that the tetraphidopsidan peristome inherits features from a common ancestor shared with the Polytrichopsida are questionable, given that the Polytrichopsida appear to be primitively non-peristomate [13, 15].
It is likely that the evolutionary innovation represented by the development of peristomes in mosses was associated with a period of rapid lineage diversification, and that the time period separating the origin of the most recent common ancestor of all extant peristomate lineages from that of each individual lineage was short relative to the age of these events. Based on a phylogenetic reconstruction in which Oedipodium was resolved as sister to the peristomate mosses, Newton et al. [1] estimated the age of the node representing this initial split at 291 MYA (late Carboniferous), with a final split between Buxbaumia and the arthrodontous mosses estimated to have occurred in the mid Permian (approximately 275 MYA). The inability of phylogenetic analyses using nucleotide substitution data to satisfactorily resolve the sequence of branching events among the peristomate lineages may be due to multiple changes in most nucleotide characters evolving rapidly enough to potentially have been informative within this narrow hypothesised 15 MY window. In such cases, morphological characters representing significant structural innovations may be particularly useful because their rate of evolution may increase during periods of rapid diversification and decrease during periods of evolutionary stasis [48]. Similarly, it is possible that genomic rearrangements may be more frequent during periods of increased speciation, as suggested by accelerated rates of chondrome evolution in parasitic arthropods (e.g., [49–51]). Certainly, as relatively rare events they should be less subject to homoplasy than point mutations of nucleotides and thus particularly useful for phylogeny reconstruction [19, 20].
Unfortunately, organellar gene losses (if assumed to be associated with transference of function to the nucleus) may be the least reliable of such characters due to a relatively high potential for convergent evolution. We would expect transfer of an indispensable gene to the nucleus to occur prior to loss from the organellar genome, followed by the acquisition of regulatory signals for the gene and plastid import signals for the protein. If the initial transfer occurred in the common ancestor of a large clade, subsequent loss from the plastid would be significantly favoured in descendent lineages, but with a large element of chance governing when and in which lineages it occurred. Although such considerations caution against placing undue emphasis on gene loss events in isolation, such data nonetheless provide valuable characters that must be interpreted in the context of any given phylogenetic hypothesis as well as all other characters.
The functional absence of nad7 from the chondrome of Tetraphis pellucida is currently uninformative phylogenetically, as no other mosses are known to share this feature. However, if a similar loss was found in representatives of one or more of the other lineages that have not yet been investigated for this gene, such as Buxbaumia or Oedipodium, the character could be highly informative for branching order of the major peristomate groups. Otherwise, the structure of the mitochondrial genome appears to be highly conserved amongst peristomate mosses, based on the three (relatively phylogenetically distant) exemplars for which fully assembled chondromes are available. It is possible that the phylogenetic utility of mitochondrial genome sequences in mosses may lie principally in nucleotide-level data. Conserved protein coding genes from the mitochondrion, such as nad5, have proven to be highly useful for phylogeny reconstruction at relatively deep nodes in mosses, such as at the ordinal level within subclass Bryidae [52–54].
The distribution of the chloroplast rpoA gene [31] suggests an unconventional phylogeny if a single state change is assumed, although not an entirely incredible one. Although Goffinet et al. [31] assumed a topology in which Buxbaumia is sister to Diphyscium and the other arthrodontous mosses and then reconstructed an independent loss of rpoA in Tetraphis and in the arthrodonts using likelihood (consistent with DELTRAN optimisation under parsimony), if Tetraphis is sister to the arthrodonts and Buxbaumia sister to that clade in turn, it would be necessary to assume only a single loss. Although the peristomes of Buxbaumia and Diphyscium are superficially highly similar and Shaw et al. [17] demonstrated that Diphyscium is effectively entirely arthrodontous in its peristomial structure, no such detailed developmental study exists for Buxbaumia, which has a considerably more developed “parastome”, apparently with some teeth comprised of entire cells (nematodontous) and involving exothecial layers outside of the OPL [55]. Buxbaumia aphylla also has more columns of cells in its peristomal layers (Diphyscium has 16 PPL cells and 32 OPL cells at maturity, as in most arthrodonts [17]) and a more irregular peristomal structure, further distancing it from the arthrodontous mosses. Assuming that Tetraphis is highly autapomorphic it is conceivable that it could be derived from an ancestor having the shared peristomal features of Buxbaumia and Diphyscium. Nonetheless, a significant number of characters link Buxbaumia to Diphyscium and the arthrodonts (including a characteristic deletion in the rps4-trnA intergenic spacer [10, 56]), and Goffinet at al.’s [31] proposed scenario currently seems most credible.
Conclusions
Both the chloroplast and mitochondrial genomes of Tetraphis pellucida exhibit structural features that have the potential to be informative for reconstructing the branching order of the peristomate moss lineages. In order to exploit these data, fully assembled organellar genomes are urgently required from representatives of the other major groups (Buxbaumia, Oedipodium, and the Polytrichopsida). In particular, presence/absence of the mitochondrial nad7 gene and the chloroplast petN gene (and potentially also the chloroplast rps16) will be of paramount importance when considered together with that of the rpoA chloroplast gene, which is better known at present. Although homoplasy (probably in the form of multiple losses of individual genes rather than reversals) is quite possible, when all of these characters are available for all lineages and can be analysed together, a single scenario, or a much smaller set of scenarios, may emerge as uniquely credibly supported. Furthermore, simultaneous phylogenetic analysis of the full complement of alignable nucleotide data from both organellar genomes may retrieve sufficient signal to resolve the ambiguities in the relatively small volume of such data that currently exists. Whether Tetraphis eventually is revealed to be a unique experiment in moss sporophyte dehiscence or as providing clues to stages in the evolutionary development of the arthrodontous peristome, understanding the precise relationships of this enigmatic moss to other extant taxa will be a significant piece in the puzzle of land plant evolutionary history.
Methods
DNA isolation, sequencing, and assembly
Material of Tetraphis pellucida was collected from two wild populations situated within 15 m of each other, both on rotten wood in dense, mixed coniferous-deciduous forest in Espoo, Finland (approximately 60°19′10″N, 24°30′00″E). Voucher specimens (Bell 03.03.10.001, Bell 03.03.10.002) are held in the herbarium of the Botanical Museum, Finnish Museum of Natural History (H). Two DNA extractions were made using different methods, both including material from both collections due to the large volume of material required (relative to the size of the plants) by one of the extraction methods. Plants of T. pellucida are small and sometimes grow mixed with other organisms, thus single shoots were separated under the dissecting microscope over several days with each shoot checked individually for identity and for contaminants visible at 40× magnification.
For Illumina SBS sequencing, a modified CTAB extraction protocol was used [57], incorporating an initial differential centrifugation step in an attempt to concentrate chloroplast DNA in the final preparation. A volume of shoots weighing 0.4 g was ground in liquid nitrogen and mixed by inversion in 20 ml of a buffer containing 50 mM Tris–HCl pH 8.0, 10 mM EDTA, 20% sucrose, 5 mM 2-mercaptoethanol and 0.1% BSA (from [58]). This suspension was passed through five layers of fine mesh cloth and then centrifuged for 30 minutes in a 50 ml centrifuge tube at 1000 rpm to remove unbroken cells and any remaining leaf and shoot fragments. The supernatant was centrifuged at 5000 rpm for 30 minutes to produce a (theoretically) chloroplast-rich pellet, this subsequently being used for CTAB extraction. However, later analysis of sequencing reads suggested that chloroplast DNA was not significantly increased in the final extraction by this method relative to DNA from other genomic compartments. For PacBio RS sequencing, a standard genomic extraction was prepared with a much smaller starting volume (approximately 10 mg) of material from both collections using the Invisorb spin plant mini kit (Invitek, Berlin, Germany).
This DNA was sheared into fragments averaging approximately 250 bp and then processed for sequencing on an Illumina GAIIx instrument in paired-end format using 100 cycles. The resulting reads were processed using Illumina’s Cassava v. 1.8 software (http://www.illumina.com), then trimmed to a quality standard approximating PHRED Q20 in a sliding window, then all reads shorter than 30 nts or containing any ambiguous nucleotides (e.g., “N”) were eliminated. This retained 108,620,955 reads with an average length of 91.9 nts. These were assembled using a deBruijn graph method within the CLC Genomics Workbench (http://www.clcbio.com) to produce 607 contigs at least 200 nts in length. These were screened separately with tBLASTx [59] with the entire Physcomitrella patens mtDNA and cpDNA to identify 15 and 11 candidate contigs for these organelles, respectively, with at least some BLAST E-values of 0. Careful manual examination of these candidates based on length, consistency of matching throughout, and depth of coverage reduced these to six contigs of mtDNA and five contigs of cpDNA in which we had high confidence and which summed, separately, to a rough expectation of each genome size.
To order and orient these contigs and to close any gaps between them, we then produced 40,925 long-read (up to ~8,000 nts) sequences from these same DNA sources on a PacBio RSII instrument using the nanopore-based technology from Pacific Biosciences (http://www.pacificbiosciences.com). These were filtered and broken into subreads by splitting at adaptor sequences using PacBio’s “SMRT” software. We searched these reads using BLASTn with the terminal 300 nts of each of these organelle scaffolds to identify reads that may span junctions between pairs of contigs, and then manually used these reads to order and orient these scaffolds and, in some cases, to supply short stretches of intervening sequences. In the latter case, we then searched back to Illumina reads to identify those that “walk” through the PacBio-only portions to ensure accuracy.
Gene identification and annotation
Gene and inferred protein sequences of other plant organelle genomes were searched to the Tetraphis pellucida mtDNA and cpDNA to identify homologs, first using MAKER [60], then manually for correction and additions. All open reading frames were collected from unannotated regions and searched against GenBank to check for whether any novel genes are present. Protein sequences were inferred assuming the universal genetic code. Careful manual examination was made specifically for the small number of genes inferred here to be missing, but are otherwise present in other bryophyte organelle genomes.
Prediction of RNA editing sites
The online tool PREPACT 2.0 [32] was used to search for candidate RNA editing sites (C => U) in both the mtDNA and cpDNA assemblies of Tetraphis pellucida as well as the published cpDNA of Tortula ruralis. The BLASTx prediction method was used with the “commons” option and the default settings. For the mtDNA search the CDS/protein databases used were those for Chaetospaeridium globosum, Chara vulgaris, Isoetes engelmannii, Lotus japonicas, Marchantia polymorpha, Physcomitrella patens, Selaginella moellendorfii and Silene latifolia (the same set used for prediction of sites in the Anomodon rugelii mtDNA by Lenz & Knoop [32]). For the cpDNA searches the databases were for Adiantum capillus-veneris, Anthoceros formosae, Chaetospaeridium globosum, Chara vulgaris, Gossypium hirsutum, Marchantia polymorpha, Oryza sativa, Pellia endiviifolia and Physcomitrella patens.
Availability of supporting data
The data sets supporting the results of this article are available in the NCBI GenBank repository, accession numbers KJ817845 (cpDNA) and KJ817846 (mtDNA).
Acknowledgements
This research was mostly funded by Academy of Finland project no. 1128112, awarded to JH and BDM. NEB additionally acknowledges support from Academy of Finland research fellowship no. 258554 and thanks Gunilla Stahls and Yu Sun for assistance in connection with work in the DNA Laboratory of the Finnish Museum of Natural History and the Molecular Ecology and Systematics Laboratory in the Department of Biosciences at the University of Helsinki. The authors thank Heather Koshinsky and Tudor Constantin at Eureka Genomics, Hercules, CA for work on library preparation and the first round of Illumina sequencing, Sonia Nosratina and Minyong Chung at the University of California, Berkeley for organising the second round of Illumina sequencing at the QB3 Vincent J. Coates Genomics Sequencing Laboratory, Petri Auvinen for implementing PacBio sequencing at the DNA Sequencing and Genomics Laboratory of the Institute of Biotechnology at the University of Helsinki, and Susan Fuerstenberg for initial work on genome assembly.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NEB undertook fieldwork, planned and undertook genomic DNA extractions, assisted in planning the sequencing work and in interpretation of final genomic assemblies, and drafted the manuscript. JLB assisted in planning the study, performed quality assessment, genome assembly, and gene annotation for the organellar genomes, and participated in interpretations and drafting of the manuscript. JH and BDM jointly planned the study and obtained funding. They also participated in preparing the manuscript at various stages. All authors have read and approved the final manuscript.
Contributor Information
Neil E Bell, Email: neil.bell@helsinki.fi.
Jeffrey L Boore, Email: jlboore@berkeley.edu.
Brent D Mishler, Email: bmishler@berkeley.edu.
Jaakko Hyvönen, Email: jaakko.hyvonen@helsinki.fi.
References
- 1.Newton AE, Wikström N, Bell NE, Forrest LL, Ignatov MS. Dating the diversification of the pleurocarpous mosses. In: Newton AE, Tangney RS, editors. Pleurocarpous Mosses: Systematics and Evolution. Boca Raton: CRC Press; 2007. pp. 337–366. [Google Scholar]
- 2.Philibert H. De l’importance du péristome pour les affinities naturelles des mousses. Rev Bryologique. 1884;11:49–52. [Google Scholar]
- 3.Fleischer M. Die Musci der Flora Buitenzorg. Leiden: EJ Brill; 1923. [Google Scholar]
- 4.Cox CJ, Goffinet B, Wickett NJ, Boles SB, Shaw AJ. Moss diversity: a molecular phylogenetic analysis of genera. Phytotaxa. 2010;9:175–195. [Google Scholar]
- 5.Volkmar U, Knoop V. Introducing intron locus cox1i624 for phylogenetic analyses in bryophytes: on the issue of Takakia as sister genus to all other extant mosses. J Mol Evol. 2010;70:506–518. doi: 10.1007/s00239-010-9348-9. [DOI] [PubMed] [Google Scholar]
- 6.Newton AE, Cox CJ, Duckett JG, Wheeler JA, Goffinet B, Hedderson AJ, Mishler BD. Evolution of the major moss lineages: phylogenetic analysis based on multiple gene sequences and morphology. Bryologist. 2000;103:187–211. doi: 10.1639/0007-2745(2000)103[0187:EOTMML]2.0.CO;2. [DOI] [Google Scholar]
- 7.Magombo ZLK. The phylogeny of basal peristomate mosses: evidence from cpDNA, and implications for peristome evolution. Syst Bot. 2003;28:24–38. [Google Scholar]
- 8.Cox CJ, Hedderson TA. Phylogenetic relationships among the ciliate arthrodontous mosses: evidence from chloroplast and nuclear DNA sequences. Plant Syst Evol. 1999;215:119–139. doi: 10.1007/BF00984651. [DOI] [Google Scholar]
- 9.Ligrone R, Duckett JG. Morphology versus molecules in moss phylogeny: new insights (or controversies) from placental and vascular anatomy in Oedipodium griffithianum. Plant Syst Evol. 2011;296:275–282. doi: 10.1007/s00606-011-0496-1. [DOI] [Google Scholar]
- 10.Cox CJ, Goffinet B, Shaw AJ, Boles SB. Phylogenetic relationships among the mosses based on heterogeneous Bayesian analysis of multiple genes from multiple genomic compartments. Syst Bot. 2004;29:234–250. doi: 10.1600/036364404774195458. [DOI] [Google Scholar]
- 11.Hyvönen J, Hedderson TA, Smith Merrill GL, Gibbings JG, Koskinen S. On phylogeny of the Polytrichales. Bryologist. 1998;101:489–504. doi: 10.1639/0007-2745(1998)101[489:OPOTP]2.0.CO;2. [DOI] [Google Scholar]
- 12.Hyvönen J, Koskinen S, Smith Merrill GL, Hedderson TA, Stenroos S. Phylogeny of the Polytrichales (Bryophyta) based on simultaneous analysis of molecular and morphological data. Mol Phylogenet Evol. 2004;31:915–928. doi: 10.1016/j.ympev.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Bell NE, Hyvönen J. Rooting the Polytrichopsida: the Phylogenetic Position of Atrichopsis and the Independent Origin of the Polytrichopsid Peristome. In: Mohamed H, Baki BB, Nasrulhaq-Boyce A, Lee PKY, editors. Bryology in the New Millennium. Kuala Lumpur: University of Malaya; 2008. pp. 227–239. [Google Scholar]
- 14.Shaw AJ, Anderson LE. Peristome development in mosses in relation to systematics and evolution. II. Tetraphis pellucida (Tetraphidaceae) Am J Bot. 1988;75:1019–1032. doi: 10.2307/2443770. [DOI] [Google Scholar]
- 15.Bell NE, Hyvönen J. Phylogeny of the moss class Polytrichopsida (BRYOPHYTA): generic-level structure and incongruent gene trees. Mol Phylogenet Evol. 2010;55:381–398. doi: 10.1016/j.ympev.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Shaw AJ, Renzaglia K. Phylogeny and diversification of bryophytes. Am J Bot. 2004;91:1557–1581. doi: 10.3732/ajb.91.10.1557. [DOI] [PubMed] [Google Scholar]
- 17.Shaw AJ, Anderson LE, Mishler BD. Peristome development in mosses in relation to systematics and evolution. I. Diphyscium foliosum (Buxbaumiaceae) Mem NY Bot Gard. 1987;45:55–70. [Google Scholar]
- 18.de Pinna MCC. Concepts and tests of homology in the cladistics paradigm. Cladistics. 1991;7:367–394. doi: 10.1111/j.1096-0031.1991.tb00045.x. [DOI] [Google Scholar]
- 19.Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/S0959-437X(98)80035-X. [DOI] [PubMed] [Google Scholar]
- 20.Kelch DG, Driskell A, Mishler BD. Inferring phylogeny using genomic characters: a case study using land plant plastomes. In: Goffinet B, Hollowell V, Magill R, editors. Molecular Systematics of Bryophytes [Monographs in Systematic Botany 98] St. Louis: Missouri Botanical Garden Press; 2004. pp. 3–12. [Google Scholar]
- 21.Mishler BD. The logic of the data matrix in phylogenetic analysis. In: Albert VA, editor. Parsimony, Phylogeny, and Genomics. Oxford New York: University Press; 2005. pp. 57–70. [Google Scholar]
- 22.Mishler BD, Kelch DG. Phylogenomics and early land plant evolution. In: Shaw AJ, Goffinet B, editors. Bryophyte Biology. 2. New York: Cambridge University Press; 2009. pp. 173–197. [Google Scholar]
- 23.Sugiura C, Kobayashi Y, Aoki S, Sugita C, Sugita M. Complete chloroplast DNA sequence of the moss Physcomitrella patens: evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucl Acids Res. 2003;31:5324–5331. doi: 10.1093/nar/gkg726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliver MJ, Murdock AG, Mishler BD, Kuehl JV, Boore JL, Mandoli DF, Everett KDE, Wolf PG, Duffy AM, Karol KG. Chloroplast genome sequence of the moss Tortula ruralis: gene content, polymorphism, and structural arrangement relative to other green plant chloroplast genomes. BMC Genomics. 2010;11:143. doi: 10.1186/1471-2164-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimamura M, Sadamitsu A, Yamaguchi T, Deguchi H. XVIII International Botanical Congress, Melbourne, Australia, 23–30 July 2011, Abstracts. 2011. Chloroplast genome structure of a primitive moss,Sphagnum palustre(Bryophyta): DNA sequencing and direct observation of single molecules [abstract] pp. 641–642. [Google Scholar]
- 26.Terasawa K, Odahara M, Kabeya Y, Kikugawa T, Sekine Y, Fujiwara M, Sato N. The mitochondrial genome of the moss Physcomitrella patens sheds new light on mitochondrial evolution in land plants. Mol Biol Evol. 2007;24:699–709. doi: 10.1093/molbev/msl198. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Xue J-Y, Wang B, Li L, Qiu Y-L. The mitochondrial genomes of the early land plants Treubia lacunosa and Anomodon rugelii: dynamic and conservative evolution. PLoS ONE. 2011;6:e25836. doi: 10.1371/journal.pone.0025836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Medina R, Goffinet B. The evolution of mitochondrial genomes in mosses. [abstract] New Orleans, LA, USA: Botany; 2013. [Google Scholar]
- 29.Jansen RK, Ruhlman TA. Plastid genomes of seed plants. In: Bock R, Knoop V, editors. Genomics of Chloroplasts and Mitochondria, Advances in Photosynthesis and Respiration 35. Dordrecht: Springer; 2012. pp. 103–126. [Google Scholar]
- 30.Goffinet B, Wickett NJ, Werner O, Ros RM, Shaw AJ, Cox CJ. Distribution and phylogenetic significance of the 71-kb inversion in the plastid genome in Funariidae (Bryophyta) Ann Bot - London. 2007;99:747–753. doi: 10.1093/aob/mcm010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goffinet B, Wickett NJ, Shaw AJ, Cox CJ. Phylogenetic significance of the rpoA loss in the chloroplast genome of mosses. Taxon. 2005;54:353–360. doi: 10.2307/25065363. [DOI] [Google Scholar]
- 32.Lenz H, Knoop V. PREPACT 2.0: predicting C-to-U and U-to-C RNA editing in organelle genome sequences with multiple references and curated RNA editing annotation. Bioinform Biol Insights. 2013;7:1–19. doi: 10.4137/BBI.S11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Wang B, Cui P, Li L, Xue J-Y, Yu J, Qiu Y-L. The mitochondrial genome of the lycophyte Huperzia squarrosa: the most archaic form in vascular plants. PLoS ONE. 2012;4:e35168. doi: 10.1371/journal.pone.0035168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yura K, Miyata Y, Arikawa T, Higuchi M, Sugita M. Characteristics and prediction of RNA editing sites in transcripts of the moss Takakia lepidozioides chloroplast. DNA Res. 2008;15:309–321. doi: 10.1093/dnares/dsn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K. RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res. 2003;31:2417–2423. doi: 10.1093/nar/gkg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyata Y, Sugiura C, Kobayashi Y, Hagiwara M, Sugita M. Chloroplast ribosomal S14 protein transcript is edited to create a translation initiation codon in the moss Physcomitrella patens. Biochim Biophys Acta. 2002;1576:346–349. doi: 10.1016/S0167-4781(02)00346-9. [DOI] [PubMed] [Google Scholar]
- 37.Rüdinger M, Funk HT, Rensing SA, Maier UG, Knoop V. RNA editing: only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol Genet Genomics. 2009;281:473–481. doi: 10.1007/s00438-009-0424-z. [DOI] [PubMed] [Google Scholar]
- 38.Groth-Malonek M, Wahrmund U, Polsakiewicz M, Knoop V. Evolution of a pseudogene: exclusive survival of a functional mitochondrial nad7 gene supports Haplomitrium as the earliest liverwort lineage and proposes a secondary loss of RNA editing in Marchantiidae. Mol Biol Evol. 2007;24:1068–1074. doi: 10.1093/molbev/msm026. [DOI] [PubMed] [Google Scholar]
- 39.Xue J-Y, Liu Y, Li L, Wang B, Qiu Y-L. The complete mitochondrial genome sequence of the hornwort Phaeoceros laevis: retention of many ancient pseudogenes and conservative evolution of mitochondrial genomes in hornworts. Curr Genet. 2010;56:53–61. doi: 10.1007/s00294-009-0279-1. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Wang B, Liu Y, Qiu Y-L. The Complete mitochondrial genome sequence of the hornwort Megaceros aenigmaticus shows a mixed mode of conservative yet dynamic evolution in early land plant mitochondrial genomes. J Mol Evol. 2009;68:665–678. doi: 10.1007/s00239-009-9240-7. [DOI] [PubMed] [Google Scholar]
- 41.Pruchner D, Nassal B, Schindler M, Knoop V. Mosses share mitochondrial group II introns with flowering plants, not with liverworts. Mol Genet Genomics. 2001;266:608–613. doi: 10.1007/s004380100577. [DOI] [PubMed] [Google Scholar]
- 42.He-Nygrén X, Juslén A, Ahonen I, Glenny D, Piippo S. Illuminating the evolutionary history of liverworts (Marchantiophyta) – towards a natural classification. Cladistics. 2006;22:1–31. doi: 10.1111/j.1096-0031.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 43.Crandall-Stotler B, Stotler RE, Long DG. Phylogeny and classification of the Marchantiophyta. Edinb J Bot. 2009;66:155–198. doi: 10.1017/S0960428609005393. [DOI] [Google Scholar]
- 44.Kobayashi Y, Knoop V, Fukuzawa H, Brennicke A, Ohyama K. Interorganellar gene transfer in bryophytes: the functional nad7 gene is nuclear encoded in Marchantia polymorpha. Mol Gen Genet. 1997;256:589–592. doi: 10.1007/pl00008616. [DOI] [PubMed] [Google Scholar]
- 45.Wahrmund U, Groth-Malonek M, Knoop V. Tracing plant Mitochondrial DNA evolution: rearrangements of the ancient mitochondrial gene cluster trnA-trnT-nad7 in liverwort phylogeny. J Mol Evol. 2008;66:621–629. doi: 10.1007/s00239-008-9114-4. [DOI] [PubMed] [Google Scholar]
- 46.Pineau B, Mathieu C, Gérard-Hirne C, De Paepe R, Chétrit P. Targeting the NAD7 subunit to mitochondria restores a functional complex I and a wild type phenotype in the Nicotiana sylvestris CMS II mutant lacking nad7. J Biol Chem. 2005;280:25994–26001. doi: 10.1074/jbc.M500508200. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Budke J, Goffinet B. Phylogenetic inference rejects sporophyte based classification of the Funariaceae (Bryophyta): rapid radiation suggests rampant homoplasy in sporophyte evolution. Mol Phylogenet Evol. 2012;62:130–145. doi: 10.1016/j.ympev.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Eldredge N, Gould SJ. Punctuated equilibria: an alternative to phyletic gradualism. In: Schopf TJM, editor. Models in Paleobiology. San Francisco: Freeman Cooper; 1972. pp. 82–115. [Google Scholar]
- 49.Oliveira DCSG, Raychoudhury R, Lavrov DV, Werren JH. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae) Mol Biol Evol. 2008;25:2167–2180. doi: 10.1093/molbev/msn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shao R, Campbell NJH, Barker SC. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera) Mol Biol Evol. 2001;18:858–865. doi: 10.1093/oxfordjournals.molbev.a003867. [DOI] [PubMed] [Google Scholar]
- 51.Xiao J-H, Jia J-G, Murphy RW, Huang D-W. Rapid evolution of the mitochondrial genome in chalcidoid wasps (Hymenoptera: Chalcidoidea) driven by parasitic lifestyles. PLoS ONE. 2011;6:e 26645. doi: 10.1371/journal.pone.0026645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beckert S, Steinhauser S, Muhle H, Knoop V. A molecular phylogeny of bryophytes based on nucleotide sequences of the mitochondrial nad5 gene. Plant Syst Evol. 1999;218:179–192. doi: 10.1007/BF01089226. [DOI] [Google Scholar]
- 53.Quandt D, Bell NE, Stech M. Unravelling the knot: the Pulchrinodaceae fam. nov. (Bryales) Beih Nova Hedwigia. 2007;131:21–39. [Google Scholar]
- 54.Bell NE, Quandt D, O’Brien TJ, Newton AE. Taxonomy and phylogeny in the earliest diverging pleurocarps: square holes and bifurcating pegs. Bryologist. 2007;110:533–560. doi: 10.1639/0007-2745(2007)110[533:TAPITE]2.0.CO;2. [DOI] [Google Scholar]
- 55.Edwards S. Homologies and Inter-relationships of Moss Peristomes. In: Schuster RM, editor. New Manual of Bryology Vol. 2. Nichinan: The Hattori Botanical Laboratory; 1984. pp. 658–695. [Google Scholar]
- 56.Goffinet B, Buck WR. Systematics of the Bryophyta (mosses): from molecules to a revised classification. In: Goffinet B, Hollowell V, Magill R, editors. Molecular Systematics of Bryophytes [Monographs in Systematic Botany 98] St. Louis: Missouri Botanical Garden Press; 2004. pp. 205–239. [Google Scholar]
- 57.Rogers SO, Bendich AJ. Extraction of total cellular DNA from plants, algae and fungi. In: Gelvin SB, Schilperoot RA, editors. Plant Molecular Biology Manual. London: Kluwer Academic Publishers; 2004. p. D1,1-8. [Google Scholar]
- 58.Kugita AK, Yamamoto Y, Takeya Y, Matsumoto T, Yoshinaga K. The complete nucleotide sequence of the hornwort (Anthoceros formosae) chloroplast genome: insight into the earliest land plants. Nuc Acids Res. 2003;31:716–721. doi: 10.1093/nar/gkg155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altschul S, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 60.Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]