

Abstract
Metabotropic transmission typically occurs through the spillover activation of extrasynaptic receptors. This study examined the mechanisms underlying somatodendritic dopamine and noradrenaline transmission and found that the extent of spillover and pooling varied dramatically between these two transmitters. In the mouse ventral tegmental area, the time course of D2-receptor-mediated IPSCs (D2-IPSCs) was consistent between cells and was unaffected by altering stimulation intensity, probability of release, or the extent of diffusion. Blocking dopamine reuptake with cocaine extended the time course of D2-IPSCs and suggested that transporters strongly limited spillover. As a result, individual release sites contributed independently to the duration of D2-IPSCs. In contrast, increasing the release of noradrenaline in the rat locus ceruleus prolonged the duration of α2-receptor-mediated IPSCs even when reuptake was intact. Spillover and subsequent pooling of noradrenaline activated distal α2-receptors, which prolonged the duration of α2-IPSCs when multiple release sites were activated synchronously. By using the rapid application of agonists onto large macropatches, we determined the concentration profile of agonists underlying the two IPSCs. Incorporating the results into a model simulating extracellular diffusion predicted that the functional range of noradrenaline diffusion was nearly fivefold greater in the locus ceruleus than dopamine in the midbrain. This study demonstrates that catecholamine synapses differentially regulate the extent of spillover and pooling to control the timing of local inhibition and suggests diversity in the roles of uptake and diffusion in governing metabotropic transmission.
Keywords: addiction, integration, monoamine, psychostimulant
Introduction
Synaptic potentials mediated by G-protein-coupled receptors (GPCRs) typically occur through the activation of extrasynaptic receptors by transmitter spillover from nearby synapses (Otis and Mody, 1992; Isaacson et al., 1993; Scanziani, 2000; Kulik et al., 2002; Beenhakker and Huguenard, 2010). These slow events differ from fast ligand-gated ion channel mediated events, in which a high concentration of transmitter rapidly rises and falls within the synapse (Clements et al., 1992; Barbour et al., 1994; Beato, 2008). Because GPCRs are primarily located outside of the synapse (Sesack et al., 1994), reuptake by transporters tightly regulates the spatial range of receptor activation (Isaacson et al., 1993; Scanziani, 2000; Beenhakker and Huguenard, 2010). Bursts of stimulation are therefore often required to overcome uptake to facilitate transmitter pooling and to evoke GPCR-mediated synaptic events (Otis and Mody, 1992; Isaacson et al., 1993; Scanziani, 2000).
Transmission mediated by monoamines such as dopamine and noradrenaline is classically thought to occur via an extended form of spillover known as volume transmission (Zoli et al., 1998; Agnati et al., 2010). Catecholamine transients measured electrochemically in the extracellular space can be detected for several seconds after release (Kawagoe et al., 1992; Garris et al., 1994; Palij and Stamford, 1994; Cragg and Rice, 2004; Rice and Cragg, 2008). Modeling studies have predicted that these low concentration transients may diffuse many micrometers away from the site of release before activating distant extrasynaptic receptors (Cragg and Rice, 2004; Rice and Cragg, 2008; Agnati et al., 2010; Dreyer et al., 2010). However, functional studies in the ventral tegmental area (VTA) have found that, at somatodendritic synapses, saturating concentrations of dopamine are required to evoke D2-receptor-mediated IPSCs (Ford et al., 2009). It therefore remains unclear to what extent the prolonged diffusion of catecholamines within the extracellular space contributes to the timing of transmission.
Within the locus ceruleus (LC), somatodendritic noradrenaline release evokes α2-receptor-mediated IPSCs through an analogous G-protein inwardly rectifying potassium (GIRK) conductance (Egan et al., 1983) to that which mediates somatodendritic dopamine D2-IPSCs in the VTA (Geffen et al., 1976; Cheramy et al., 1981; Lacey et al., 1987; Palij and Stamford, 1994; Rice et al., 1994; Jaffe et al., 1998; Beckstead et al., 2004). The present work compares dopamine and noradrenaline transmission mediated by autoreceptors in the VTA and LC in brain slices. The results show that diffusion and reuptake of catecholamines play different roles at these two synapses. Whereas the kinetics of transmission mediated by noradrenaline is dependent on both diffusion and reuptake, dopamine-dependent transmission is regulated primarily by reuptake. This difference maintains independence between dopamine synapses in the VTA, yet allows for pooling of noradrenaline in the extracellular space of the LC that prolongs transmission when multiple terminals are active synchronously. These results show that, by regulating the extent of spillover, transporters control the timing and the extent of cross talk that occurs between dopamine synapses.
Materials and Methods
Slice preparation and visualization.
All procedures were approved and performed in accordance with Case Western Reserve University Institutional Animal Care and Use Committee guidelines. Horizontal brain slices were obtained from deeply anesthetized 3- to 6-week-old male and female Sprague Dawley rats (Charles River Laboratories) containing the LC (240 μm) or 3- to 6-week-old male and female C57BL/6 mice (Jackson Laboratories) containing the VTA (220 μm). Brain slices were cut using a vibratome (Leica) in ice-cold cutting solution containing the following (in mm): 75 NaCl, 2.5 KCl, 6 MgCl2, 0.1 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, 2.5 d-glucose, and 50 sucrose and continuously bubbled with 95% O2 and 5% CO2. Slices were transferred to artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 d-glucose bubbled with 95% O2 and 5% CO2 and incubated at 35°C for at least 45 min before use. MK-801 (10 μm) was included during the incubation to block NMDA receptors. After incubation, slices were transferred to a recording chamber and constantly perfused at 2 ml/min with oxygenated ACSF warmed to 34 ± 2°C. Slices were visualized with a BX51WI microscope (Olympus) with custom-built infrared gradient contrast optics.
Electrophysiology.
Whole-cell voltage-clamp recordings were made using an Axopatch 200B amplifier (Molecular Devices). Patch pipettes (2.0–2.5 MΩ) were pulled from borosilicate glass (World Precision Instruments). The pipette internal solution contained the following (in mm): 115 K-methylsulphate, 20 NaCl, 1.5 MgCl2, 10 K-HEPES, 10 BAPTA-tetrapotassium, 2 ATP, 0.3 GTP, and 6 sodium phosphocreatine, pH 7.4, 275 mOsm. Data were acquired using an ITC-18 interface (Instrutech) and Axograph X (Axograph Scientific) at 10 kHz and filtered to 2 kHz. Series resistance was not compensated and recordings were discarded if the access resistance rose above 15 MΩ. In the case of experiments with reduced extracellular calcium, MgCl2 was substituted to maintain a constant divalent ion concentration. All drugs, except where otherwise noted, were applied by bath perfusion. The VTA was defined as the area within and medial to the fibers of the medial lemniscus (Ford et al., 2006). Dopamine neurons within the VTA were identified by the presence of a dopamine D2-receptor-mediated dopamine conductance, a capacitance of 25–40 pF, an input resistance of <300 MΩ, an h-current, and a tonic firing rate of 0.5–2 Hz, as described previously (Ford et al., 2006). The LC was identified by its location between the corner of the fourth ventricle and the mesencephalic tract motor neurons. Noradrenergic neurons within the LC were identified by having an α2-sensitive noradrenaline conductance, a capacitance of >40 pF, an input resistance of <100 MΩ, and a tonic firing rate of 0.5–2 Hz.
Catecholamine release was elicited with an extracellular, ACSF-filled monopolar glass electrode (∼5 MΩ). In slices containing the VTA, the stimulating electrode was placed 50–100 μm away from the neuron being recorded (or the tip of the carbon fiber where applicable). In slices containing the LC, the stimulating electrode was placed into the caudal side of the LC. Either single pulses (0.5 ms) or trains of stimuli (0.5 ms at 40 Hz) were used to drive catecholamine release (∼40–50 μA unless otherwise noted). D2- and α2-receptors were pharmacologically isolated with DNQX (10 μm), MK801 (10 μm), picrotoxin (100 μm), and CGP55845 (200 nm) to block AMPA, NMDA, GABAA, and GABAB receptors, respectively. BAPTA (10 mm) in the internal pipette solution was used to block intracellular Ca2+ signaling and associated mGluR responses. For experiments in the LC in which the rising kinetics were analyzed, the stimulation artifact was removed by antagonist subtraction (idazoxan, 2 μm). The effect of cocaine on transmission was verified to be due to inhibition of reuptake by comparisons to a DAT-specific antagonist (GBR-12909, 1 μm) and a NET-specific antagonist (nisoxetine, 1 μm).
Electrochemistry.
Fast-scan cyclic voltammetry (FSCV) recordings were made using glass-encased carbon fiber electrodes (34–700; Goodfellow) with an exposed diameter of 7 μm and an exposed length of 50–100 μm. Before use, the exposed carbon fiber tip was soaked in isopropanol purified with activated carbon for at least 20 min. The tip of the fiber was placed directly into either the LC or VTA ∼50 μm below the surface of the slice. While holding the fiber at −0.4 V, either triangular waveforms (−0.4 to 1.3 V vs Ag/AgCl at 400 V/S) or sinusoidal waveforms (−0.4 to 1.0 V vs Ag/AgCl over 10 ms) were applied at 10 Hz. Background-subtracted cyclic voltammogram currents were obtained by subtracting the average of 10 voltammograms obtained before stimulation from each voltammogram obtained after stimulation. The time course of dopamine and noradrenaline were determined by plotting their peak oxidation potentials versus time. After experimentation, each electrode was calibrated against known concentrations of either dopamine or noradrenaline.
To confirm the chemical identify of the signal, we compared the voltammogram of evoked transmitter release with voltammograms obtained by applying serotonin, dopamine, and noradrenaline directly onto the carbon fiber via iontophoresis. The single-stimulation-evoked signal had a peak reduction at −257 or −223 mV in the LC or VTA, respectively, which was consistent with the voltammograms of noradrenaline and dopamine. Serotonin did not contribute to either evoked signal as evidenced by the lack of characteristic twin reduction peaks (Rice et al., 1997). Inhibiting dopamine reuptake with the DAT inhibitor GBR12909 (1 μm) had no effect on evoked catecholamine signal in the LC (p > 0.4, data not shown, n = 5, Student's paired t test), confirming that dopamine likely does not contribute to the evoked signal in the LC (Palij and Stamford, 1994).
Isolated macropatches.
Large, nucleated, outside-out macropatches were obtained from either dopamine cells from the VTA or noradrenaline cells from the LC, as described previously (Ford et al., 2009). Patches were pulled out of the slice and into the free solution above the tissue. In some experiments, the patches were lowered onto the surface of the slice to attempt to detect the evoked release of noradrenaline or dopamine. For these experiments, monopolar electrode stimulation was used, similar to the method used for evoking IPSCs. Other experiments used fast-flowing solutions through a theta tube flow pipe to rapidly apply known concentrations of dopamine or noradrenaline. Macropatches were raised ∼500 μm above the surface of the slice and placed in front of the theta tube. Once in place, solution was allowed to flow through both barrels of the flow pipette at a rate of ∼20 μl per minute. The bath volume was adjusted such that 1 cm of the flow pipette was immersed in the bath, allowing the temperature of the flow solution to reach equilibrium with the temperature of the bath (34 ± 2°C). One barrel of the flow pipette perfused a control solution containing the following (in mm): 121 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.4 NaH2PO4, 0.6 ascorbate-NA, and 5 HEPES-Na, pH 7.3, while a similar solution with the addition of a known concentration of dopamine or noradrenaline was perfused through the other barrel. The flow pipette was attached to a piezoelectric bimorph. Passing 25 V across the bimorph changed the location of the flow pipette tip such that the solution bathing the macropatch was rapidly exchanged. To determine the kinetics of this exchange, open tip junction potentials were measured with the flow pipette perfusing solutions with different concentrations of KCl. The average 10–90% exchange time, as measured by changes in junction potential, was 2 ± 1 ms.
Point-release diffusion modeling.
The spatiotemporal profile of transmitter diffusing away from a single vesicle of release was approximated using a point diffusion model (Cragg et al., 2001; Cragg and Rice, 2004; Rice and Cragg, 2008). In brief, we solved for the concentration (C) of dopamine or noradrenaline as a function of radius (r) from the release site and time after release (t) using the equation:
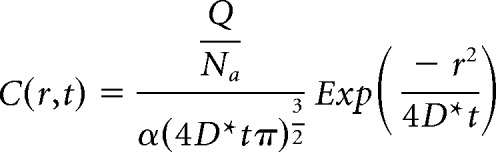 |
Where Q is the quantal content of a vesicle in molecules, Na is Avogadro's number, α is the volume fraction of the tissue, and D* is the tissue corrected diffusion constant equal to D/λ2 where D is the diffusion constant of the neurotransmitter and λ is the tortuosity of the tissue. For the diffusion of dopamine in the VTA, we used the following values obtained from the literature (Cragg et al., 2001): α = 0.30, λ = 1.62, Q = 14,000 (Jaffe et al., 1998), and D = 7.63 × 10−6 cm2 sec−1. For the diffusion of noradrenaline in the LC, we used: Q = 37,000 (Huang et al., 2007), D = 7.63 × 10−6 cm2 sec−1 (adapted from Bennett et al., 2004). The values of α = 0.30 and λ = 1.58 were used in the LC.
Statistics and data analysis.
All data are shown as mean ± SEM. Statistical significance (p < 0.05) was assessed by Student's paired t test, Student's unpaired t test, ANOVA, repeated-measures ANOVA, or Pearson's correlation where applicable (InStat 3.0). Decay kinetics of D2-IPSCs and α2-IPSCs and extracellular catecholamine transients were fit with a single exponential using a Simplex algorithm optimized by the sum of squared errors in Axograph X (Axograph Scientific). Single exponential decays provided accurate fits to the decay kinetics and were used to indicate the duration of the IPSC decay phase. Multiple exponential fits were not considered during analysis (*p < 0.05, **p < 0.01, ***p < 0.001, and n.s., p ≥ 0.05 for all figures).
Materials.
Picrotoxin, DNQX, CGP 55845, and MK-801 were from Ascent Scientific. S-(−)-sulpiride, idazoxan hydrochloride, and pirlindole were from Tocris Bioscience. Dextran (35,000–50,000 kDa) was from MP Biomedical. K-Methylsulphate was from Acros Organics. BAPTA was from Invitrogen. Cocaine hydrochloride was obtained from the National Institute of Drug Abuse, National Institutes of Health. Dopamine hydrochloride, noradrenaline hydrochloride, d-glucose, sodium l-ascorbate, and HEPES sodium salt were from Sigma-Aldrich. All other chemicals were from Fisher Scientific.
Results
Time courses of dopamine and noradrenaline transmission are different
To determine the mechanisms regulating catecholamine synapses, voltage-clamp recordings were made from either noradrenaline neurons in the LC or dopamine neurons in the VTA. A single stimulation in the LC from rat brain slices evoked a slow IPSC (67.3 ± 6.9 pA, n = 26) that was blocked by the noradrenaline α2-receptor antagonist idazoxan (2 μm; Fig. 1A). Bath application of noradrenaline (30 μm) also evoked large outward currents in these LC neurons (298 ± 34 pA, n = 3). Because the amplitude of α2-receptor-mediated outward currents in LC neurons was significantly smaller in slices from the mouse (bath application 30 μm: 83 ± 14 pA, n = 3; p = 0.001, Student's t test), all noradrenergic α2-receptor-mediated transmission was subsequently examined in LC slices from the rat.
Figure 1.

Dopamine and noradrenaline IPSCs have different time courses. A, α2-IPSC from a LC neuron under control conditions and in the presence of idazoxan (2 μm). Ticks illustrate time of stimulation. The current artifact remaining in idazoxan was subtracted to allow for analysis of rise kinetics. Left, Schematic of whole-cell voltage-clamp recordings of noradrenaline neurons in the LC. B, Averaged D2-IPSCs from a VTA neuron under control conditions and in the presence of sulpiride (200 nm). C, Summary of the 10% onset and time to peak of α2-IPSCs (n = 26) or D2-IPSCs (n = 17; Student's t test). Artifacts remaining in the presence of antagonists (idazoxan in the LC or sulpiride in the VTA) were subtracted from control α2-IPSCs or D2-IPSCs when analyzing each cell. D, Representative averages of α2-IPSCs grouped by amplitude. Traces in the presence of idazoxan were subtracted to remove stimulation artifact. Bottom, Traces from top scaled to their peak amplitude. E, Decay time correlates with amplitude for α2-IPSCs. Circles represent IPSCs recorded from individual cells (Pearson's correlation). F, Summary data of the half-width and τdecay of α2-IPSCs (n = 26) and D2-IPSCs (n = 17; Student's t test). G, Representative averages of D2-IPSCs grouped by amplitude. Stimulation artifacts blanked for clarity. H, Decay time does not correlate with amplitude for D2-IPSCs (Pearson's correlation). Circles represent IPSCs recorded from individual cells. I, Lack of correlation of rise time (10–90%) with amplitude of IPSCs (Pearson's correlation). Circles represent IPSCs recorded from individual cells. ***p < 0.001; error bars indicate ± SEM.
Previous work has shown that D2-IPSCs recorded from the VTA of rats and mice have similar kinetics, although the amplitudes are significant larger in mice (Courtney et al., 2012). Because bath application of dopamine (30 μm) evoked significantly larger outward currents in VTA neurons from brain slices from mice than from rats (mouse: 223 ± 8 pA, n = 3, rat: 126 ± 22 pA, n = 4, p = 0.014, Student's t test), dopamine D2-receptor-mediated transmission was examined in mouse VTA brain slices. A single stimulation in the VTA evoked a dopamine-mediated IPSC (29.9 ± 3.5 pA, n = 14) that was abolished by the D2-receptor antagonist sulpiride (200 nm; Fig. 1B). Both IPSCs reversed near the potassium equilibrium potential and were inhibited by BaCl2 (300 μm), suggesting that both currents resulted from GIRK channel activation (Egan et al., 1983; Lacey et al., 1987). A comparison of the time courses of D2-IPSCs in the VTA with those of α2-IPSCs in the LC revealed that D2-IPSCs had markedly faster kinetics. When evoked with similar stimulation intensity, dopamine D2-IPSCs occurred after a latency of 64 ± 3 ms (10% onset) and had 10–90% rise times of 163 ± 6 ms (Fig. 1C). This led to D2-IPSCs reaching their peak in <300 ms (n = 17; Fig. 1C). In contrast, the kinetics of noradrenaline α2-IPSCs from the LC were slower (latency to 10% onset: 154 ± 6 ms; 10–90% rise time: 388 ± 11 ms; n = 26, p < 0.001 vs D2-IPSC, Student's t test). Therefore, α2-IPSCs in the LC did not peak until nearly 900 ms (Fig. 1C). Because the rate of GIRK current activation is determined by the concentration of agonist at the receptor (Sodickson and Bean, 1996; Ingram et al., 1997; Ford et al., 2009), the faster activation of D2-IPSCs suggests that a higher effective concentration of dopamine may underlie transmission at D2-receptors in the VTA than noradrenaline at α2-receptors in the LC.
In addition to activating more slowly, α2-IPSCs were longer than D2-IPSCs and slower to decay (τdecay α2-IPSC: 1.1 ± 0.1 s, n = 26; τdecay D2-IPSC: 0.4 ± 0.1 s, n = 17; p < 0.001; half-width α2-IPSC: 1.2 ± 0.1 s, n = 26; half-width D2-IPSC: 0.5 ± 0.02 s, n = 17; p < 0.001; Fig. 1D,F,G). Noradrenaline-mediated α2-IPSCs also varied widely in decay time (τDecay SD: 0.56 s, n = 26; Fig. 1D) and the amplitude of larger events correlated with longer decay times (R2 = 0.176, n = 26, p = 0.016, Pearson's correlation; Fig. 1E). Surprisingly, the fast-decay kinetics of dopamine IPSCs were remarkably similar among neurons (τDecay SD: 0.09 s, n = 17; Fig. 1G) such that there was no correlation with amplitude (Fig. 1H). Because the rise time (10–90%) of neither α2-IPSCs nor D2-IPSCs correlated with the amplitude of each event (Pearson's correlation; α2-IPSCs: R2 = −0.1, p = 0.8; n = 26; D2-IPSCs: R2 = 0.04, p = 0.4; n = 17; Fig. 1I), the slow time courses of α2-IPSCs are unlikely to result from the asynchronous release of noradrenaline in the LC. These results are consistent with the idea that an increase in the amount of noradrenaline released leads to increased exposure of α2-receptors. The consistent time course of D2-IPSCs, in contrast, suggests that dopamine is regulated such that the duration of transmission remains independent of the number of active release sites.
Pooling of transmitter prolongs the duration of noradrenaline transmission
Spillover of transmitter from within the synapse to distal extrasynaptic sites can prolong of the duration of synaptic events (Isaacson et al., 1993; Carter and Regehr, 2000; Scanziani, 2000; DiGregorio et al., 2002; Cathala et al., 2005; Szabadics et al., 2007; Olah et al., 2009; Beenhakker and Huguenard, 2010). In several cases, spillover can allow for pooling in the extracellular space of transmitter that originated from multiple presynaptic release sites (Barbour et al., 1994; Otis et al., 1996; Silver et al., 1996; Overstreet and Westbrook, 2003; Balakrishnan et al., 2009). To determine whether pooling contributed to the time course of transmission at these two synapses, we next varied the release of dopamine and noradrenaline to determine whether increased transmitter release could result in longer duration IPSCs.
The stimulus of a single pulse was varied to determine the intensity that produced ∼85% of the maximal IPSC amplitude (high stimulation). The stimulus intensity was then lowered to reduce the IPSC amplitude by 62 ± 4% for D2-IPSCs (n = 8) and 59 ± 2% for α2-IPSCs (n = 9, p = 0.5 vs D2-IPSCs, Student's t test; Fig. 2A). Despite the reduction in amplitude, the decay kinetics of dopamine D2-IPSCs remained constant (0.4 ± 0.1 to 0.4 ± 0.1 s, high- to low-intensity stimulation, p = 0.9, Student's paired t test; Fig. 2A). In contrast, reducing the stimulation intensity significantly accelerated the decay kinetics of α2-IPSCs (0.8 ± 0.1 s to 0.4 ± 0.1 s, high- to low-intensity stimulation, p < 0.001, Student's paired t test; Fig. 2A).
Figure 2.
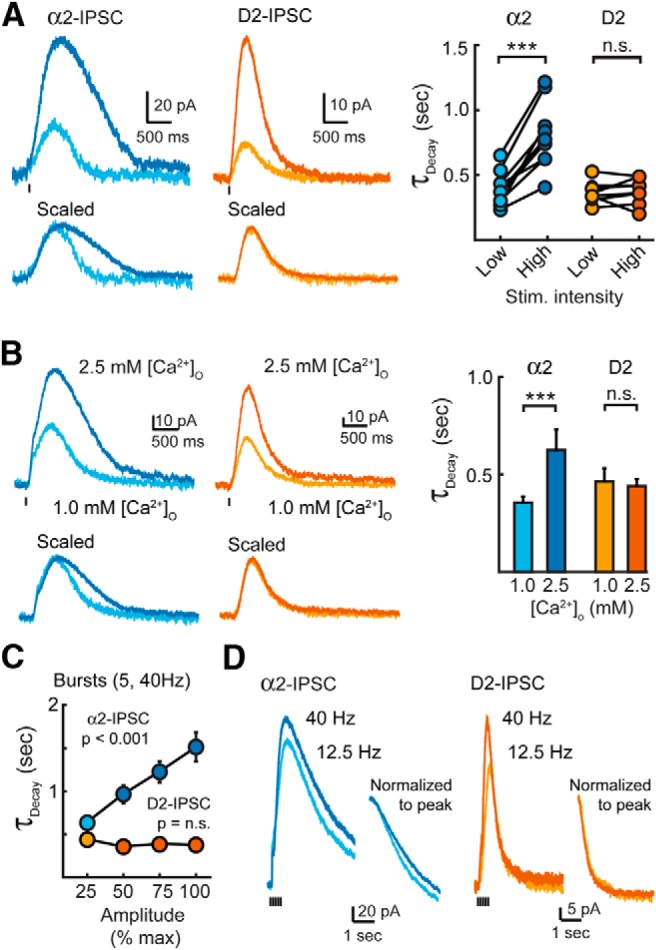
Increasing noradrenaline release prolongs the decay time of α2-IPSCs. A, Averaged α2-IPSCs (n = 9) and D2-IPSCs (n = 8) evoked by high- and low-intensity stimulation. Right, Quantification of the decay times of individual IPSCs. Changing the stimulation intensity had no effect on the decay time of D2-IPSCs (Student's paired t test). B, Averaged α2-IPSCs (n = 8) and D2-IPSCs (n = 5) evoked with a single stimulation and recorded in 1.0 and 2.5 mm extracellular Ca2+ ([Ca]o). Right, Summary data of decay times in different extracellular [Ca2+] (Student's paired t test). C, Summary quantification of the decay time of IPSCs evoked by bursts when divided into quartiles based upon their maximum amplitude (α2-IPSCs: n = 15, D2-IPSCs: n = 12). For α2-IPSCs, longer decay times were significantly correlated with larger amplitude quartiles (Pearson's correlation). D, Averaged noradrenaline α2-IPSCs (n = 5) and D2-IPSCs (n = 5) evoked by a burst of stimuli at 12.5 and 40 Hz. Inset, Decay phase of IPSCs normalized and aligned to their peak when evoked by a burst of stimuli at 12.5 or 40 Hz. Stimulation artifacts were blanked for clarity. n.s., p > 0.05; **p < 0.01; error bars indicate ± SEM.
Next, we lowered the release probability (Pr) by decreasing the extracellular concentration of calcium. The amplitude of D2-IPSCs and α2-IPSCs were reduced to 48 ± 3% and 53 ± 3% of control levels when Ca2+ was reduced from 2.5 to 1.0 mm. Lowering extracellular Ca2+ had no effect on the decay time of D2-IPSCs in the VTA (τdecay: 106 ± 8% of 2.5 mm Ca, n = 5, p = 0.54, Student's paired t test), but it accelerated the decay of α2-IPSCs in the LC by 48 ± 5% (n = 8, p < 0.001, Student's paired t test; Fig. 2B).
Finally, the intensity (ranging from 10 to 80 μA) of bursts of stimuli (5 pulses at 40 Hz) was varied. Despite increasing the amplitude fourfold over the range of stimulation, the kinetics of D2-IPSC decay remained constant (0.4 ± 0.1 to 0.4 ± 0.1 s, lowest versus highest, n = 12, p = 0.18, Pearson's correlation; Beckstead et al., 2004). Increasing the intensity of stimulation in the LC, however, resulted in larger amplitude α2-IPSCs that also had longer decay times (0.7 ± 0.1 s to 1.5 ± 0.2 s, lowest vs highest; n = 15 cells; p < 0.001, Pearson's correlation; Fig. 2C). Similar results were obtained when the frequency of stimulations during the train was varied (Fig. 2D). IPSCs evoked at low frequency (12.5 Hz) in both the VTA and LC had smaller amplitudes (Fig. 2D). This reduced the decay time of noradrenaline IPSCs in the LC by 23% (n = 5, p = 0.014, Student's paired t test), but did not alter the rate of decay of the D2-IPSC (n = 5, p = 0.13, Student's paired t test; Fig. 2D). Therefore, in all cases, the time course of dopamine transmission remained constant regardless of the size of D2-IPSC.
Because spillover of transmitter from neighboring sites is limited when the probability of release is lowered (Hartzell et al., 1975; Trussell et al., 1993; Silver et al., 1996; DiGregorio et al., 2002; Balakrishnan et al., 2009), the acceleration in the decay of α2-IPSCs under conditions of low release suggests that pooling of noradrenaline from adjacent release sites contributed to the extended time course of the α2-IPSC. In contrast, the consistent duration of dopamine D2-IPSCs suggests that limited dopamine spillover occurs during phasic transmission.
Differences in receptor signaling and dendritic filtering cannot account for the variation in kinetics of catecholamine transmission
We next investigated whether differences in receptor signaling or the rate of release could alternatively explain the different time courses between the two IPSCs. First, the efficiency of α2-/D2-receptor signaling to GIRK channels was investigated to determine whether the kinetics of dopamine IPSCs could result from faster coupling of D2-receptors to potassium channels. Diffusion and reuptake severely slow the local application of exogenous agonists (Barbour and Häusser, 1997). To avoid this, outside-out macropatches were pulled from either noradrenergic neurons of the LC or dopaminergic neurons of the VTA and moved out of the slice into free solution (Ford et al., 2009). Currents were evoked from macropatches by rapid-flow application of either noradrenaline or dopamine via a two-barrel theta tube flow pipe. One barrel of the flow pipette contained a control solution and the second barrel contained a known concentration of dopamine or noradrenaline. The flow pipe was attached to a piezoelectric bimorph to allow for rapid exchange between the two solutions (Fig. 3A). The application of noradrenaline (100 μm, 100 ms) onto LC macropatches resulted in idazoxan-sensitive outward currents that initiated within 58 ± 8 ms (10% onset, n = 6) and had a time to peak of 202 ± 14 ms (n = 6; Fig. 3A,B). The kinetics of these currents were indistinguishable from the kinetics of sulpiride-sensitive currents evoked from patches from VTA neurons when dopamine was similarly applied (100 μm, 100 ms; 10% onset: 66 ± 4 ms; time to peak: 217 ± 22; n = 13, p = 0.33 compared with NA onto LC patches, Student's t test; Fig. 3A,B). This indicates that the kinetics of both the α2- and D2-receptor-mediated currents are similar and that coupling efficiency cannot explain the difference in time course between D2- and α2-IPSCs.
Figure 3.
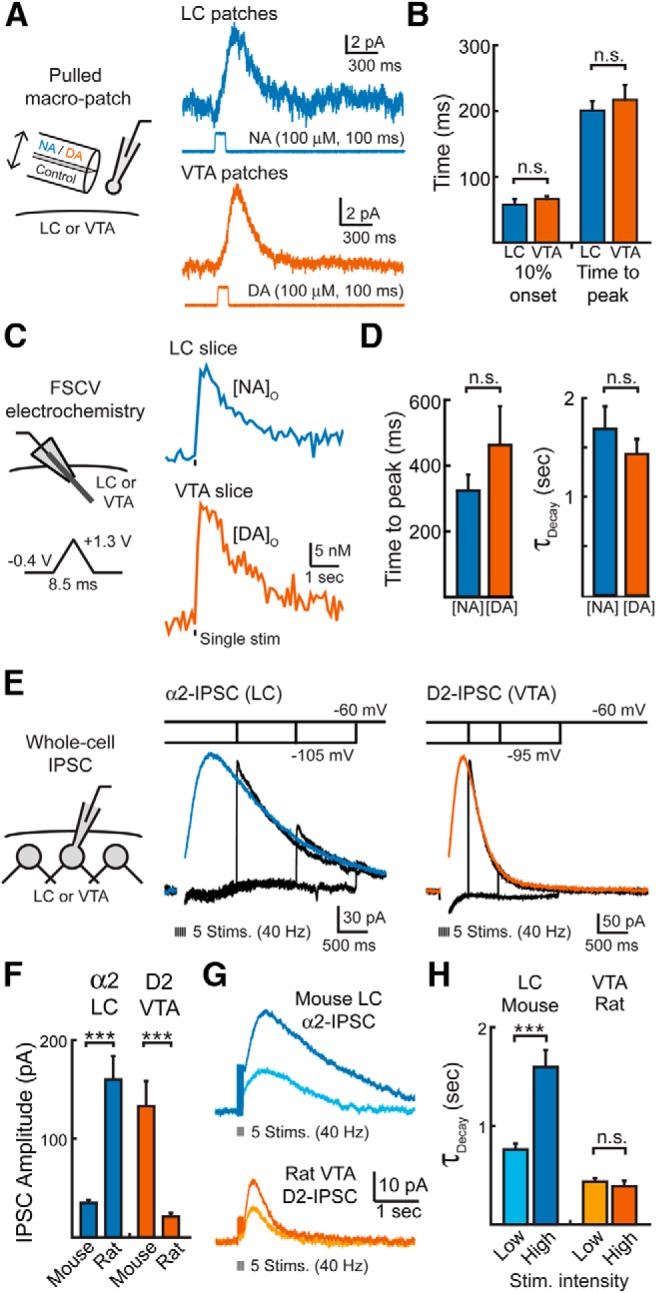
The duration of GIRK signaling, transmitter release and the synaptic conductance are similar for dopamine and noradrenaline. A, Left, schematic of experimental condition for catecholamine rapid-flow application onto nucleated patches. Right, Representative traces of currents evoked after the rapid-flow application of noradrenaline (100 μm; 100 ms) onto nucleated patches of LC cells or dopamine application (100 μm; 100 ms) onto nucleated patches of VTA cells. B, Summary data illustrating the time until 10% onset and the time to peak of currents evoked in patches (Student's t test). C, Left, Schematic of the FSCV experimental condition. Right, Averaged FSCV traces measuring the extracellular concentration of noradrenaline [NA]o in the LC (n = 13) or the extracellular concentration of dopamine [DA]o in the VTA (n = 9). Release was evoked by a single pulse. D, Summary data of the time to peak and decay kinetics of [NA]o and [DA]o (Student's t test). E, Left, Schematic of whole-cell voltage-clamp recordings. Right, Current responses to a series of voltage jumps after synaptic stimulation. Voltage jumps were given from near the potassium equilibrium potential (between −95 and −105 mV) to the test potential of −60 mV at 500, 1000, and 1500 ms after stimulation while recording D2-IPSCs and 1000, 2000, and 3000 ms after stimulation while recording α2-IPSCs (black traces). IPSCs were evoked by a burst of stimuli. Each trace is the average of three to five events from three VTA dopamine cells and six LC noradrenaline cells. IPSCs after the jump were separated from the capacitance transients recorded with no extracellular stimulation. After the voltage jump, the instantaneous IPSCs (black) rapidly approached control IPSCs (color) recorded at −60 mV, indicating that the synaptic conductance was still active. The stimulation artifact was blanked for clarity. F, Summary data of the peak amplitude of D2-IPSCs or α2-IPSCs evoked by five stimulations (40 Hz) in either the mouse or rat (Student's t test). G, Average traces illustrating the D2-IPSCs and α2-IPSCs in the mouse and rat. Top, α2-IPSCs evoked in the mouse LC have different kinetics when evoked by high- or low-intensity stimulation. Bottom, D2-IPSCs evoked in the rat VTA have the same kinetics independent of stimulation intensity. H, Quantification of the decay kinetics shown in G (Student's paired t test). n.s., p > 0.05; error bars indicate ± SEM.
Next, the rise and fall of the concentration of dopamine and noradrenaline in the extracellular space was measured with FSCV. A carbon-fiber electrode was placed into either the LC or VTA and cycled through a triangular waveform to detect either noradrenaline or dopamine (10 Hz, Fig. 3C,D). The rise and duration of catecholamines in the extracellular space was similar between the VTA and LC (Fig. 3C,D). Using a triangular voltage protocol of −0.4 to +1.3 V at 400 V/s to maximize sensitivity (Heien et al., 2003), both dopamine and noradrenaline release peaked in <500 ms after a single stimulation (0.5 ± 0.1 s, n = 9, VTA; 0.3 ± 0.1 s, n = 13, LC; p = 0.24) and decayed with time constants of ∼1.5 s (1.4 ± 0.2 s, n = 9, VTA; 1.7 ± 0.2 s, n = 13, LC, p = 0.41). Similarly, the kinetics of both release and decay were comparable for both dopamine and noradrenaline if a sine-wave voltage protocol of −0.4 to +1.0 V at 300 V/s was used to more accurately measure the kinetics of release (Heien et al., 2003). Therefore, the release of catecholamines into the extracellular space had similar time courses in both regions.
Because the dendritic location of inputs could differ between dopamine and noradrenaline neurons, the slow decay of α2-IPSCs may result from filtering of faster events at distal dendritic sites within the LC. To determine whether the noradrenaline IPSC extended beyond the time when synaptic receptor signaling was active, we applied a voltage jump during the IPSC (Pearce, 1993; Barbour et al., 1994; Häusser and Roth, 1997). Voltage steps from near the potassium equilibrium potential to depolarized potentials increased the synaptic current to levels equivalent to that under control conditions for both D2-IPSCs and α2-IPSCs when given after the peak of the IPSC (Fig. 3E). As measured by the average current for 500 ms after the depolarizing voltage step, there was no difference between IPSCs with or without the voltage steps for either D2-IPSCs (without voltage vs with voltage step; 500 ms poststimulation step: 161 vs 162 pA, p = 0.99; 1000 ms poststimulation step: 28 vs 28 pA, p = 0.95; 2000 ms poststimulation step: 6 vs 7 pA, p = 0.94; n = 6; Student's paired t test) or α2-IPSCs (without voltage vs with voltage step; 1000 ms poststimulation step: 109 vs 113 pA, p = 0.72; 2000 ms poststimulation step: 50 vs 56 pA, p = 0.64; 3000 ms poststimulation step: 24 vs 24 pA, p = 0.95; n = 3; Student's paired t test). This indicates that synaptic conductances were active even at late times and that the slow decay of α2-IPSCs in the LC is due to the intrinsic slow kinetics of a receptor-mediated event.
Finally, we investigated whether the differences in kinetics could be explained by differences in transmission between the two species examined. First, we compared D2-IPSCs evoked from slices from the rat with those from the mouse. Due the small amplitude of D2-receptor-mediated GIRK currents in the rat VTA, trains of five stimulations (40 Hz) were used to evoke dopamine release. The amplitude of D2-IPSCs from brain slices from the mouse was ∼sixfold greater than D2-IPSCs from brain slices from the rat (mouse: 133 ± 21 pA, n = 11; rat: 21 ± 4 pA, n = 7; p < 0.001, Student's t test; Fig. 3F). However, despite the difference in amplitude, D2-IPSCs in both species had similar decay kinetics (τdecay; mice: 0.34 ± 0.02 s, n = 11; rats: 0.39 ± 0.05 s, n = 7; p = 0.37, Student's t test). Similar to brain slices from the mouse, lowering the stimulation intensity had no effect on the kinetics of rat D2-IPSCs despite a 47 ± 4% reduction in amplitude (τdecay: 0.44 ± 0.04 s, n = 7, p = 0.48 maximal stimulation, Student's paired t test; Fig. 3G,H). We next investigated whether the kinetics of α2-IPSCs recorded in the LC of mice were similar to those recorded in the LC of rats. Again, due the small amplitude of α2-receptor-mediated GIRK currents in mouse LC slices, noradrenaline was evoked by a burst of five stimulations (40 Hz). The amplitude of α2-IPSCs recorded in the rat LC were >sixfold greater than α2-receptor-mediated IPSCs recorded in the mouse LC (rats: 226 ± 30 pA, n = 10; mice: 35 ± 3 pA, n = 9; p < 0.001, Student's t test; Fig. 3F). The decay kinetics of α2-IPSCs, however, were indistinguishable in slices obtained from the mouse and the rat (τdecay; rats: 1.3 ± 0.2 s, n = 10; mice: 1.6 ± 0.2 s, n = 9; p = 0.35, Student's t test). As in the rat, lowering the stimulation intensity reduced both the amplitude (58 ± 7% reduction; Fig. 3G) and time course of decay of α2-IPSCs in the LC of slices from the mouse (50 ± 5% reduction, n = 9, p < 0.001, Student's paired t test; Fig. 3H). Therefore, in both rats and mice, different mechanisms regulate the timing of dopamine and noradrenaline transmission. Together, these findings suggest that the difference in synaptic timing between dopamine and noradrenaline is unlikely to be explained by differences in dendritic filtering, receptor signaling kinetics, the kinetics of release, or a species-specific difference.
Reuptake transporters maintain the temporal fidelity of dopamine transmission in the VTA
Because the diffusion of transmitter is regulated by neurotransmitter transporters (Isaacson et al., 1993; Barbour et al., 1994; Brasnjo and Otis, 2001; Overstreet and Westbrook, 2003; Beenhakker and Huguenard, 2010), we next investigated how D2-IPSCs and α2-IPSCs were affected after blocking dopamine and noradrenaline uptake transporters (DAT, NET) with cocaine (2 μm). Cocaine potentiated both the amplitude and duration of D2-IPSCs and α2-IPSCs (Fig. 4A) and prolonged their rising phases, as indicated by a delayed time to peak. Cocaine blocks DAT and NET with similar potency (Han and Gu, 2006), yet consistently had a greater effect on the amplitude of D2-IPSCs than that of α2-IPSCs. Cocaine also prolonged the rising phase of D2-IPSCs more than α2-IPSCs (time to peak; D2-IPSC: 76 ± 6% increase in cocaine, n = 11; α2-IPSC: 36 ± 8% increase in cocaine, n = 10; p < 0.001, Student's t test). A similar effect of cocaine was observed between recordings from the rat and mouse LC and the rat and mouse VTA (D2-IPSCs, rat VTA: 360 + 44% increase in amplitude, n = 4; α2-IPSCs, mouse LC: 114 + 11% increase in amplitude, n = 4; p = 0.002 for change in D2-IPSCs vs α2-IPSCs; Student's t test). These results suggest that reuptake plays a more dominant role in regulating dopamine transmission in the VTA than noradrenaline transmission in the LC.
Figure 4.

Transporters limit dopamine spillover. A, Left, Representative traces of single-stimulation IPSCs in control conditions and the increase in amplitude and duration in the presence of cocaine (2 μm, gray). Right, Quantification showing the increase in amplitude of IPSCs recorded in the presence of cocaine. Cocaine potentiated the amplitude of D2-IPSCs more than α2-IPSCs (Student's t test). B, Left, Averaged α2-IPSCs (n = 11) and D2-IPSCs (n = 11) evoked by single stimulation or a train of five stimuli. Insets, Average IPSCs normalized to peak amplitude. C, Averaged α2-IPSCs and D2-IPSCs evoked by either a single stimulation or a train of five stimuli in the presence of cocaine (2 μm). Inset, Averaged IPSCs in the presence of cocaine normalized to peak amplitude. D, Quantification of IPSC decay times when evoked by a single stimulation or bursts of 5 or 10 stimulations in the absence or presence of cocaine (2 μm, gray). Trains of stimuli failed to potentiate the decay time of D2-IPSCs under control conditions. Blocking reuptake with cocaine allowed trains of stimuli to evoke longer duration dopamine IPSCs (repeated-measures ANOVA, Bonferroni post hoc test). n.s., p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; error bars indicate ± SEM.
We next applied brief bursts of stimuli to recruit multiple release sites. Again, bursts facilitated the amplitude of D2-IPSCs without altering their decay times, but increased both the amplitude and duration of α2-IPSCs (D2-IPSCs: single stimulus: 32 ± 5 pA, 0.4 ± 0.1 s; 5 stimuli: 133 ± 21 pA, 0.3 ± 0.1 s; n = 11; p < 0.001 for amplitude, p = 0.16 for decay time. α2-IPSCs: single stimulus: 64 ± 10 pA, 0.8 ± 0.1 s; 5 stimuli: 160 ± 22 pA, 1.3 ± 0.2 s; n = 11; p < 0.001 for amplitude, p = 0.002 for decay time; Fig. 4B). In the presence of cocaine, however, the decay times of D2-IPSCs were increased as a result of increased stimulation (Fig. 4C,D). Therefore, when scaled to peak amplitude, the D2-IPSC evoked by a burst was no longer superimposable on D2-IPSCs evoked by a single stimulus (Fig. 4C, inset). A similar effect was also observed in the VTA of the rat in cocaine (n = 6, p = 0.03, Student's paired t test). Therefore, blocking reuptake transporters in the VTA extended the diffusion of dopamine such that the duration of the D2-IPSC was now dependent on the amount of transmitter released. These results suggest that, in the VTA, DATs limit spillover to prevent dopamine pooling and activation of distal extrasynaptic D2-receptors.
Dopamine transmission in the VTA does not occur via volume transmission
Because both reuptake and diffusion govern the spatial limit over which spillover activates postsynaptic receptors (Barbour and Häusser, 1997; Rusakov and Kullmann, 1998), the time course of IPSCs were investigated after slowing diffusion through the addition of dextran to the extracellular solution (5%, MW = 35,000–50,000; Min et al., 1998; Ford et al., 2010). Slowing diffusion with dextran alters the amplitude and time course of spillover events (Min et al., 1998; Nielsen et al., 2004; Szabadics et al., 2007; Markwardt et al., 2009; Ford et al., 2010), so dextran was predicted to have a greater effect on α2-IPSCs than on D2-IPSCs. After application of dextran, the amplitude of noradrenaline α2-IPSCs increased by 137 ± 16% (n = 10, p = 0.047, Student's paired t test; Fig. 5A,D), suggesting that noradrenaline does not saturate receptors mediating the IPSC. Dextran also increased the decay time of α2-IPSCs by 157 ± 16% (n = 10, p = 0.006, Student's paired t test; Fig. 5A,E). Because dextran altered the kinetics of the α2-IPSC, diffusion is likely a key clearance mechanism regulating noradrenaline transmission in the LC. In contrast, dextran had no effect on the kinetics or amplitude of dopamine IPSCs from the VTA (amplitude: 88 ± 7%, n = 9, p = 0.12; τDecay: 112 ± 9%, n = 9, p = 0.23; Fig. 5B,D,E). The lack of an effect of dextran on D2-IPSCs implies that, under control conditions, extended diffusion plays little role in determining the kinetics of the D2-IPSC. However, in the presence of a subsaturating concentration of cocaine (500 nm), dextran slowed the kinetics of D2-IPSCs (amplitude: 48 ± 5%, n = 6, p < 0.001; τDecay: 148 ± 16%, n = 6, p = 0.03, Student's paired t test; Fig. 5C,E). Because hindering diffusion reduced the amplitude of the D2-IPSC in the presence of cocaine, dextran likely prevented the activation of receptors away from the release site. Therefore, diffusion of dopamine only contributes to D2-IPSCs when reuptake is blocked, confirming that transporters prevent the long-range spillover activation of D2-receptors.
Figure 5.
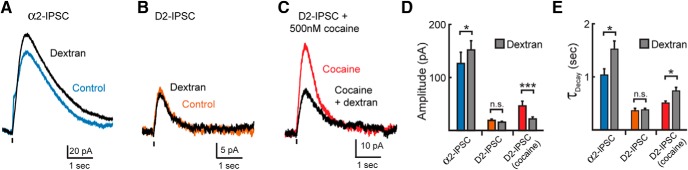
Slowing diffusion alters α2-IPSCs, but not D2-IPSCs, unless reuptake is inhibited. A, Averaged α2-IPSC (n = 10) illustrating the effect of dextran (5% w/v). B, Averaged D2-IPSC (n = 9) illustrating the lack of effect of dextran. C, Averaged D2-IPSC in the presence of cocaine (500 nm; n = 6) illustrating the effect of dextran. D, E, Quantification of the amplitude (D) and decay time (E) of IPSCs recorded in control solution (colored bars) and dextran (gray bars; Student's paired t test). n.s., p > 0.05; *p < 0.05; ***p < 0.001; error bars indicate ± SEM.
We also examined the effects of enzymatic degradation on transmitter clearance using the monoamine oxidase inhibitor (MAO-I) pirlindole (10 μm). Acute application (10 min) of pirlindole, however, had no effect on the amplitude or decay kinetics of either α2-IPSCs (n = 3, p = 0.79 for amplitude and p = 0.77 for τDecay, Student's paired t test) or D2-IPSCs (n = 3, p = 0.98 for amplitude and p = 0.75 for τDecay, Student's paired t test). Therefore, enzymatic degradation by MAO is unlikely to acutely influence the time course of dopamine- or noradrenaline-mediated IPSCs.
To further investigate how signaling at a distance may lead to activation of catecholamine receptors in the VTA and LC, we pulled macropatches from either dopamine or noradrenaline neurons out from the slice and placed them back onto the surface of the brain slice (Fig. 6A). Using these patches as a sensor for evoked release, bursts of stimuli within the LC evoked outward currents in the majority of LC-noradrenaline sensor patches (16 ± 3 pA; n = 12; Fig. 6B). Currents were abolished by the α2-receptor antagonist idazoxan (2 μm; 95 ± 2% reduction, n = 3, p < 0.001, Student's paired t test). The kinetics and time course of these events were similar to those of whole-cell α2-IPSCs recorded separately from LC neurons in other control experiments. When scaled to the peak of their amplitude, the evoked α2-currents from patches were indistinguishable from an average α2-IPSC (Fig. 6C). Therefore, despite patches being located at a distance from the site of release, the concentration profile of noradrenaline was similar to the α2-receptors that mediate the IPSC within the slice.
Figure 6.
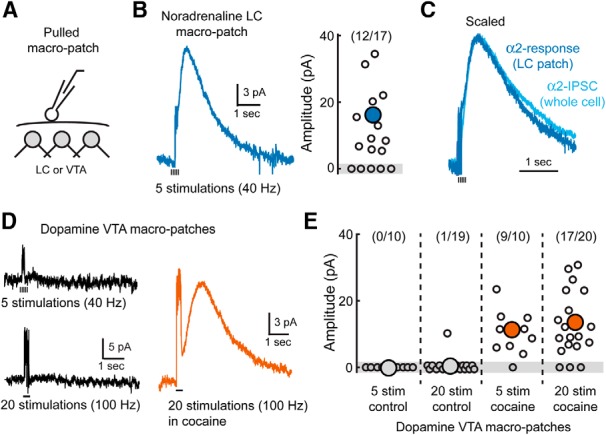
Spillover activates D2 receptors when uptake is inhibited. A, Schematic of experimental condition. Large sensor patches from the LC or VTA were pulled out from the slice and repositioned at the surface. B, Left, Averaged α2-receptor-mediated current from noradrenaline neuron sensor patches (n = 17). Noradrenaline release was evoked from within the LC by a train of stimuli. The stimulating electrode was positioned in the LC in a similar location used to evoke α2-IPSCs. Right, Distribution of current amplitudes recorded in all sensor patches from the LC. Outward currents could be detected in the majority of LC patches. C, Normalized, averaged whole-cell α2-IPSC recorded within the LC (n = 15) and α2-receptor-mediated current from a sensor patch recorded at the surface of the LC (n = 12). D, Averaged D2-receptor-mediated currents from dopamine neuron sensor patches. Left, Control recordings from VTA sensor patches did not respond to dopamine release evoked by a burst of stimuli (five pulses, 40 Hz; or 20 pulses, 100 Hz). Right, Averaged D2-receptor current in the presence of cocaine (5 μm; 20 pulses, 100 Hz). E, Distribution of amplitudes of dopamine sensor patch currents in all conditions.
In contrast, when sensor patches were pulled from dopamine cells in the VTA or SNc and placed on the surface of the slice, bursts of stimuli failed to evoke D2-receptor-mediated outward currents (Fig. 6D). Increasing the intensity of stimulation (20 stimulations, 100 Hz) also failed to produce measurable outward currents (Fig. 6D,E). This suggests that, under control conditions, the concentration of dopamine that escapes from the slice is too low to activate D2-receptors. In the presence of cocaine (5 μm), however, bursts of stimuli were able to evoke outward currents in the majority of patches (Fig. 6D,E). Currents were abolished by sulpiride (200 nm; 93 ± 5% reduction, n = 4, p < 0.001, Student's paired t test), confirming that they were mediated by D2-receptor activation. Therefore, dopamine transporters prevented dopamine from diffusing out of the slice at a high enough concentration to activate D2-receptors. This observation is consistent with the role of dopamine reuptake in preventing transmitter pooling and synaptic crosstalk under normal conditions.
High concentrations of dopamine result in rapid activation of D2-IPSCs
The concentration of agonist determines the rate of activation of GPCR-mediated GIRK currents (Sodickson and Bean, 1996; Ingram et al., 1997; Ford et al., 2009). Amperometric measurements have estimated somatic quantal content of noradrenaline vesicles in the LC to be approximately twice that of dopamine vesicles in the VTA (∼37,000 noradrenaline molecules vs ∼14,000 dopamine molecules per vesicle; Jaffe et al., 1998; Huang et al., 2007). Despite the greater number of noradrenaline molecules being released per vesicle, the α2-dependent IPSC was nearly four times slower than D2-IPSC (Fig. 1C,F). This suggests that the α2-IPSC may be mediated by a subsaturating concentration of noradrenaline.
To determine the concentration of catecholamines underlying the two IPSCs, we used rapid-flow application of either dopamine or noradrenaline via a theta tube flow pipette to determine the rising kinetics of the α2- and D2-mediated currents. Application of high, saturating concentrations of dopamine (100 μm, 100 ms) onto VTA macropatches gave rise to an outward current (Fig. 7A) that mimicked the activation kinetics of the D2-IPSC (10% onset: 66.3 ± 4.4 ms, n = 13, p = 0.64 vs D2-IPSCs, Student's t test; Fig. 7B,C). Reducing the concentration of dopamine resulted in slower currents that no longer matched the activation kinetics of D2-IPSCs (10% onset; 10 μm DA: 76.8 ± 5.9 ms, n = 13, p = 0.044 vs D2-IPSCs; 3 μm DA: 92.8 ± 3.7 ms, n = 6, p < 0.001 vs D2-IPSCs, Student's t test; Fig. 7C). This suggests that at least 100 μm dopamine is present at D2-receptors during the initial phase of the D2-IPSCs, confirming previous observations (Ford et al., 2009).
Figure 7.

D2-receptors mediating the IPSC are located near the site of release. A, Average current when 100 μm DA was applied to VTA macropatches for 100 ms (n = 13). Inset, Schematic of the experimental paradigm. B, 100 μm DA mimics the rise kinetics of D2-IPSCs recorded in VTA slices. Right, Expansion of the rising phase (box) with the same time scale as the expansion in F. C, Quantification of the 10% onset of various concentrations of DA applied to VTA patches for 100 ms. The dashed line represents the 10% onset of D2-IPSCs measured in VTA slices, with the gray box indicating the SE (Student's t test). D, Currents resulting from 100 μm NA applied to LC macropatches for 100 ms (n = 6, top) or 1000 ms (n = 9, bottom) have similar onset kinetics as when 100 μm DA was applied to VTA patches. E, Average current when 3 μm NA was applied to LC macropatches for 1000 ms (n = 11). F, 3 μm NA mimics the rise kinetics of α2-IPSCs recorded in LC slices. Right, Expansion of the rising phase (box) with the same time scale as the expansion in B. G, Quantification of the 10% onset of various concentrations of NA applied to LC patches for 1000 ms. The dashed line represents the 10% onset of α2-IPSCs recorded in LC slices, with the gray box indicating the SE (Student's t test). H, Peak concentration versus distance of dopamine (left) and noradrenaline (right) diffusing away from a single vesicle of transmitter release generated by a model of point diffusion in the absence of reuptake. Orange lines indicate the distance at which DA reaches the 100 μm required to mimic D2-IPSC onset kinetics. Blue lines indicate the distance at which NA reaches the 3 μm required to mimic α2-IPSC onset kinetics. I, Scaled diagram demonstrating the spatial range of dopamine and noradrenaline diffusion required to mediate the rising phase of IPSCs. n.s., p > 0.05; *p < 0.05; ***p < 0.001; error bars indicate ± SEM.
Next, noradrenaline was applied onto LC macropatches to determine the concentration necessary to mimic the rise kinetics of the α2-IPSC. Again, the rate of activation of α2-mediated currents was similar to D2-mediated currents from VTA patches when evoked with similar concentrations of agonist (100 μm, 100 ms; 10% onset; NA: 57.9 ± 8.2 ms, n = 6; p = 0.33 vs DA; Fig. 7D, top). In addition, the α2- and D2-currents decayed at a similar rate when exposed to similar applications of agonist (τDecay; NA: 116 ± 16 ms, n = 6; DA: 117 ± 11 ms, n = 13; p = 0.96, Student's t test; Fig. 7D, top). Because α2-IPSCs peak significantly later than D2-IPSCs, noradrenaline was applied for 1000 ms to mimic the longer rise time of the α2-IPSC (Fig. 7D, bottom). When a high concentration of noradrenaline (100 μm, 1000 ms) was applied, α2-mediated currents rose faster than α2-IPSCs (10% onset: 66 ± 14 ms, n = 9, p < 0.001 vs α2-IPSC, Student's t test; Fig. 7G). This suggests that a lower concentration of noradrenaline mediates α2-IPSCs at synapses within LC. The rising phase of the α2-IPSC was best matched by applying 3 μm (1000 ms) noradrenaline (3 μm NA, 10% rise time: 124 ± 14 ms, n = 11, p = 0.10; 10 μm NA, 10% rise time: 82 ± 13 ms, n = 9, p < 0.001 vs α2-IPSC; 1 μm, 10% rise time: 201 ± 30 ms, n = 8, p = 0.04, Student's t test; Fig. 7F,G). Therefore, the concentration of noradrenaline that interacts with α2-receptors during the rising phase of the IPSC is on the order of 30-fold less than the concentration of dopamine that mediates the D2-IPSC.
Discussion
The present study examined two catecholamine-mediated IPSCs and found that they had very different characteristics. Whole-cell recordings revealed that the rise and fall of D2-IPSCs in the VTA was markedly faster than α2-IPSCs in the LC. Both GPCRs activated GIRK conductances that displayed similar kinetics upon rapid-flow application of the respective agonist in macropatch recordings. The difference between the kinetics of the IPSCs was instead dependent on the extent of transmitter diffusion as a result of differences in the level of uptake. Within the VTA, we found that transporters strongly limited dopamine spillover. This allowed for only high, local concentrations to activate proximal D2-receptors. Because the decay time of D2-IPSCs was unaffected by changes in the extent or probability of dopamine release, our results suggest that the time course of dopamine signaling in the VTA does not rely on pooling of transmitter from multiple sites. This suggests that individual release sites contribute independently to D2-receptor activation during the IPSC, which maintains the temporal fidelity of dopamine transmission. In contrast, due to less efficient reuptake in the LC, noradrenaline was able to spill out away from the release site and diffuse a significant distance before activating postsynaptic α2-receptors. Subsequent pooling of extracellular noradrenaline when multiple terminals were active led to a prolonged duration of the α2-IPSC. Because metabotropic transmission is thought to occur through the activation of extrasynaptic GPCRs by transmitter spillover and pooling in the extracellular space (Otis and Mody, 1992; Isaacson et al., 1993; Scanziani, 2000; Kulik et al., 2002; Beenhakker and Huguenard, 2010), these results suggest that the mechanisms underlying metabotropic-receptor-mediated neurotransmission are more diverse than previously believed. Differences in the temporal and spatial tuning of GPCR-mediated transmission may help to explain how dopamine transmission can precisely encode rewards on a subsecond timescale, whereas noradrenaline transmission can slowly modulate broader functions such as attention and vigilance over longer timescales.
The rise time of noradrenaline α2-IPSCs was nearly fourfold slower than that of dopamine D2-IPSCs. This kinetic difference could not be explained by voltage-clamp errors resulting from dendritic filtering, differences in coupling efficiency of GPCRs to GIRK channels, or differences in the kinetics of release between terminals. Instead, the slower rate of activation of the α2-receptor-mediated synaptic current was likely due to lower concentrations of noradrenaline-activating receptors during the IPSC (3 μm NA vs 100 μm DA, Fig. 7C,G). To understand how diffusion contributed to receptor activation, we simulated the spatial profile of catecholamine diffusion from single vesicles in either the VTA or LC (Fig. 7H). The model showed that, in the VTA, the peak concentration of dopamine was maintained at or above 100 μm only within 0.4 μm from the site of release (Fig. 7H, left). D2-receptors located further than 0.4 μm would be exposed to lower concentrations of dopamine. This would be predicted to significantly slow the rate of D2-receptor activation below what was observed for the D2-IPSC (Fig. 7C). When the diffusion of noradrenaline in the LC was simulated, the peak concentration remained >3 μm until 1.7 μm from the release site (Fig. 7H, right). Because α2-receptors located nearer than 1.7 μm would experience a higher peak concentration of noradrenaline, these receptors would have activation kinetics faster than were observed during the α2-IPSC. This simulation suggests that α2-receptors mediating the initial phase of noradrenergic transmission in the LC may be five times further from the site of release than D2-receptors in the VTA (Fig. 7I).
Because the rate of current deactivation was similar for α2- and D2-receptors when agonist was removed (Fig. 7D, top), the longer time course of α2-IPSCs in the slice suggests that a longer-lasting noradrenaline transient led to a prolonged activation of α2-receptors after release. Spillover of neurotransmitter can often prolong transmitter exposure through the activation of extrasynaptic receptors (Carter and Regehr, 2000; DiGregorio et al., 2002; Overstreet and Westbrook, 2003; Cathala et al., 2005; Szabadics et al., 2007; Szapiro and Barbour, 2007; Olah et al., 2009; Beenhakker and Huguenard, 2010; Bennett et al., 2012). To understand how spillover shapes the activation of postsynaptic catecholamine receptors, we lowered the probability of release for both noradrenaline and dopamine. Reducing release probability by decreasing extracellular calcium reduced the decay time of α2-IPSCs, but not D2-IPSCs. Because transmission mediated by pooling is more sensitive to reductions in release than point-to-point mediated transmission (Scanziani, 2000; Arnth-Jensen et al., 2002; DiGregorio et al., 2002), pooling likely plays a significant role in shaping the decay time of only α2-IPSCs. Support for this comes from the fact that slowing extracellular diffusion with dextran did not alter the duration of D2-IPSCs yet slowed the decay of α2-IPSCs. Because dextran did not affect the amplitude of D2-IPSCs, it is likely that D2-receptors are saturated during transmission by the local high concentration of dopamine occurring near the site of release, consistent with our finding from fast-flow experiments with macropatches. However, a potential role for enhanced activation of presynaptic D2-receptors modulating dopamine release under these conditions cannot be excluded.
We found that the only condition that could affect the duration of D2-IPSCs was when transporters were blocked. Reuptake or buffering of dopamine by transporters is therefore the major mechanism that limits spillover or pooling in the VTA. Cocaine had a dramatic effect on delaying the time to peak and prolonging the duration of D2-IPSCs (Beckstead et al., 2004; Ford et al., 2010; Courtney et al., 2012). Ultrastructure studies show that dopamine receptors in the midbrain aggregate at both synaptic and extrasynaptic sites on dopaminergic dendrites (Sesack et al., 1994; Pickel et al., 2002). Because cocaine increases the distance over which dopamine signals (Cragg and Rice, 2004; Rice and Cragg, 2008), blocking reuptake likely allows the concentration profile of dopamine to remain sufficiently high outside the synapse to recruit additional extrasynaptic receptors that normally do not participate in transmission. In the LC, postsynaptic α2-receptors are distributed along the plasma membrane in regions both opposed and unopposed by other membranes (Lee et al., 1998a, 1998b). Because both dextran and cocaine slowed α2-IPSCs, the profile of α2-receptor activation is determined by both diffusion and reuptake, likely because of less efficient uptake in the LC. The reason for the difference in uptake between the LC and VTA is not clear, but may be due to reduced levels of expression of NETs in the LC (Szot et al., 1996; Burchett and Bannon, 1997) and/or a differential distribution of transporters further away from release sites (Beenhakker and Huguenard, 2010). Support for this comes from the observation that DATs are often associated with both the presynaptic and postsynaptic densities of dendrodentritic synapses between midbrain dopamine neurons (Hersch et al., 1997).
Through the course of these experiments, miniature α2-IPSCs were not observed while recording from LC neurons despite the fact that evoked α2-IPSCs were routinely robust in amplitude. This is consistent with volume transmission and the need for pooling of multiple noradrenaline vesicles to drive α2-receptor activation. A recent report, however, described dopamine-mediated spontaneous miniature D2-IPSCs in the VTA (Gantz et al., 2013). This indicates that vesicular dopamine release must occur adjacent to a high density of D2-receptors. Because the time course of miniature D2-IPSCs is similar to evoked IPSCs (Gantz et al., 2013), dopamine synapses likely function independently even during synchronous release from multiple terminals. The density of release sites often regulates the extent of spillover and pooling between synapses (Overstreet and Westbrook, 2003; Balakrishnan et al., 2009). A low number of terminals in the VTA (Wilson et al., 1977; Nirenberg et al., 1996; Ford et al., 2010) may therefore aid in maintaining the synaptic independence between sites to limit dopamine crosstalk.
Autoreceptor inhibition regulates both the overall level of excitability and the patterns of firing across catecholamine neurons (Aghajanian and Bunney, 1977; Egan et al., 1983; Benoit-Marand et al., 2001; Schmitz et al., 2002; Beckstead et al., 2004; Courtney et al., 2012) such that changes in the duration or strength of somatodendritic autoreceptor transmission can dramatically affect catecholamine signaling throughout the brain (Buckholtz et al., 2010; Bello et al., 2011). By regulating the functional extent of spillover, different catecholamines may set the extent that inhibition occurs as result of activity in distant neurons within their local network.
Footnotes
This work was supported by the National Institutes of Health (Grants R00-DA026417 and R01-DA035821 to C.P.F. and Grant T32-NS077888 to N.A.C., the National Alliance for Research on Schizophrenia and Depression (Young Investigator Grant to C.P.F.), and the Mt. Sinai Health Care Foundation (Scholar Award to C.P.F.). We thank Roberto Galan for assistance with modeling simulations and Ben Strowbridge for helpful discussions and a critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Aghajanian GK, Bunney BS. Dopamine “autoreceptors”: pharmacological characterization by microiontophoretic single cell recording studies. Naunyn Schmiedebergs Arch Pharmacol. 1977;297:1–7. doi: 10.1007/BF00508803. [DOI] [PubMed] [Google Scholar]
- Agnati LF, Guidolin D, Guescini M, Genedani S, Fuxe K. Understanding wiring and volume transmission. Brain Res Rev. 2010;64:137–159. doi: 10.1016/j.brainresrev.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD, Trussell LO. Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci. 2009;12:286–294. doi: 10.1038/nn.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Häusser M. Intersynaptic diffusion of neurotransmitter. Trends Neurosci. 1997;20:377–384. doi: 10.1016/S0166-2236(96)20050-5. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Beato M. The time course of transmitter at glycinergic synapses onto motoneurons. J Neurosci. 2008;28:7412–7425. doi: 10.1523/JNEUROSCI.0581-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. 2010;30:15262–15276. doi: 10.1523/JNEUROSCI.3243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011;14:1033–1038. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C, Arroyo S, Berns D, Hestrin S. Mechanisms generating dual-component nicotinic EPSCs in cortical interneurons. J Neurosci. 2012;32:17287–17296. doi: 10.1523/JNEUROSCI.3565-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Farnell L, Gibson WG, Blair D. A quantitative description of the diffusion of noradrenaline in the media of blood vessels following its release from sympathetic varicosities. J Theor Biol. 2004;226:359–372. doi: 10.1016/j.jtbi.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS. Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron. 2001;31:607–616. doi: 10.1016/S0896-6273(01)00377-4. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Treadway MT, Cowan RL, Woodward ND, Li R, Ansari MS, Baldwin RM, Schwartzman AN, Shelby ES, Smith CE, Kessler RM, Zald DH. Dopaminergic network differences in human impulsivity. Science. 2010;329:532. doi: 10.1126/science.1185778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchett SA, Bannon MJ. Serotonin, dopamine and norepinephrine transporter mRNAs: heterogeneity of distribution and response to ‘binge’ cocaine administration. Brain Res Mol Brain Res. 1997;49:95–102. doi: 10.1016/S0169-328X(97)00131-9. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci. 2005;8:1310–1318. doi: 10.1038/nn1534. [DOI] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Courtney NA, Mamaligas AA, Ford CP. Species differences in somatodendritic dopamine transmission determine D2-autoreceptor mediated inhibition of ventral tegmental area neuron firing. J Neurosci. 2012;32:13520–13528. doi: 10.1523/JNEUROSCI.2745-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004;27:270–277. doi: 10.1016/j.tins.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Nicholson C, Kume-Kick J, Tao L, Rice ME. Dopamine-mediated volume transmission in midbrain is regulated by distinct extracellular geometry and uptake. J Neurophysiol. 2001;85:1761–1771. doi: 10.1152/jn.2001.85.4.1761. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 2002;35:521–533. doi: 10.1016/S0896-6273(02)00787-0. [DOI] [PubMed] [Google Scholar]
- Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci. 2010;30:14273–14283. doi: 10.1523/JNEUROSCI.1894-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TM, Henderson G, North RA, Williams JT. Noradrenaline-mediated synaptic inhibition in rat locus coeruleus neurones. J Physiol. 1983;345:477–488. doi: 10.1113/jphysiol.1983.sp014990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Gantz SC, Phillips PE, Williams JT. Control of extracellular dopamine at dendrite and axon terminals. J Neurosci. 2010;30:6975–6983. doi: 10.1523/JNEUROSCI.1020-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Bunzow JR, Williams JT. Spontaneous inhibitory synaptic currents mediated by a G protein-coupled receptor. Neuron. 2013;78:807–812. doi: 10.1016/j.neuron.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris PA, Ciolkowski EL, Pastore P, Wightman RM. Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci. 1994;14:6084–6093. doi: 10.1523/JNEUROSCI.14-10-06084.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geffen LB, Jessell TM, Cuello AC, Iversen LL. Release of dopamine from dendrites in rat substantia nigra. Nature. 1976;260:258–260. doi: 10.1038/260258a0. [DOI] [PubMed] [Google Scholar]
- Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Kuffler SW, Yoshikami D. Post-synaptic potentiation: interaction between quanta of acetylcholine at the skeletal neuromuscular synapse. J Physiol. 1975;251:427–463. doi: 10.1113/jphysiol.1975.sp011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Roth A. Estimating the time course of the excitatory synaptic conductance in neocortical pyramidal cells using a novel voltage jump method. J Neurosci. 1997;17:7606–7625. doi: 10.1523/JNEUROSCI.17-20-07606.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien ML, Phillips PE, Stuber GD, Seipel AT, Wightman RM. Overoxidation of carbon-fiber microelectrodes enhances dopamine adsorption and increases sensitivity. Analyst. 2003;128:1413–1419. doi: 10.1039/b307024g. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Yi H, Heilman CJ, Edwards RH, Levey AI. Subcellular localization and molecular topology of the dopamine transporter in the striatum and substantia nigra. J Comp Neurol. 1997;388:211–227. doi: 10.1002/(SICI)1096-9861(19971117)388:2<211::AID-CNE3>3.0.CO%3B2-4. [DOI] [PubMed] [Google Scholar]
- Huang HP, Wang SR, Yao W, Zhang C, Zhou Y, Chen XW, Zhang B, Xiong W, Wang LY, Zheng LH, Landry M, Hökfelt T, Xu ZQ, Zhou Z. Long latency of evoked quantal transmitter release from somata of locus coeruleus neurons in rat pontine slices. Proc Natl Acad Sci U S A. 2007;104:1401–1406. doi: 10.1073/pnas.0608897104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram S, Wilding TJ, McCleskey EW, Williams JT. Efficacy and kinetics of opioid action on acutely dissociated neurons. Mol Pharmacol. 1997;52:136–143. doi: 10.1124/mol.52.1.136. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-E. [DOI] [PubMed] [Google Scholar]
- Jaffe EH, Marty A, Schulte A, Chow RH. Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J Neurosci. 1998;18:3548–3553. doi: 10.1523/JNEUROSCI.18-10-03548.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe KT, Garris PA, Wiedemann DJ, Wightman RM. Regulation of transient dopamine concentration gradients in the microenvironment surrounding nerve terminals in the rat striatum. Neuroscience. 1992;51:55–64. doi: 10.1016/0306-4522(92)90470-M. [DOI] [PubMed] [Google Scholar]
- Kulik A, Nakadate K, Nyíri G, Notomi T, Malitschek B, Bettler B, Shigemoto R. Distinct localization of GABA(B) receptors relative to synaptic sites in the rat cerebellum and ventrobasal thalamus. Eur J Neurosci. 2002;15:291–307. doi: 10.1046/j.0953-816x.2001.01855.x. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Rosin DL, Van Bockstaele EJ. alpha2A-adrenergic receptors in the rat nucleus locus coeruleus: subcellular localization in catecholaminergic dendrites, astrocytes, and presynaptic axon terminals. Brain Res. 1998a;795:157–169. doi: 10.1016/S0006-8993(98)00266-2. [DOI] [PubMed] [Google Scholar]
- Lee A, Rosin DL, Van Bockstaele EJ. Ultrastructural evidence for prominent postsynaptic localization of alpha2C-adrenergic receptors in catecholaminergic dendrites in the rat nucleus locus coeruleus. J Comp Neurol. 1998b;394:218–229. doi: 10.1002/(SICI)1096-9861(19980504)394:2<218::AID-CNE6>3.0.CO%3B2-5. [DOI] [PubMed] [Google Scholar]
- Markwardt SJ, Wadiche JI, Overstreet-Wadiche LS. Input-specific GABAergic signaling to newborn neurons in adult dentate gyrus. J Neurosci. 2009;29:15063–15072. doi: 10.1523/JNEUROSCI.2727-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/S0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Nielsen TA, DiGregorio DA, Silver RA. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron. 2004;42:757–771. doi: 10.1016/j.neuron.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oláh S, Füle M, Komlósi G, Varga C, Báldi R, Barzó P, Tamás G. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature. 2009;461:1278–1281. doi: 10.1038/nature08503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I. Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J Neurophysiol. 1992;67:227–235. doi: 10.1152/jn.1992.67.1.227. [DOI] [PubMed] [Google Scholar]
- Otis TS, Wu YC, Trussell LO. Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci. 1996;16:1634–1644. doi: 10.1523/JNEUROSCI.16-05-01634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Westbrook GL. Synapse density regulates independence at unitary inhibitory synapses. J Neurosci. 2003;23:2618–2626. doi: 10.1523/JNEUROSCI.23-07-02618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palij P, Stamford JA. Real-time monitoring of endogenous noradrenaline release in rat brain slices using fast cyclic voltammetry: 3. Selective detection of noradrenaline efflux in the locus coeruleus. Brain Res. 1994;634:275–282. doi: 10.1016/0006-8993(94)91930-5. [DOI] [PubMed] [Google Scholar]
- Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-N. [DOI] [PubMed] [Google Scholar]
- Pickel VM, Chan J, Nirenberg MJ. Region-specific targeting of dopamine D2-receptors and somatodendritic vesicular monoamine transporter 2 (VMAT2) within ventral tegmental area subdivisions. Synapse. 2002;45:113–124. doi: 10.1002/syn.10092. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Dopamine spillover after quantal release: Rethinking dopamine transmission in the nigrostriatal pathway. Brain Res Rev. 2008;58:303–313. doi: 10.1016/j.brainresrev.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Richards CD, Nedergaard S, Hounsgaard J, Nicholson C, Greenfield SA. Direct monitoring of dopamine and 5-HT release in substantia nigra and ventral tegmental area in vitro. Exp Brain Res. 1994;100:395–406. doi: 10.1007/BF02738400. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ, Greenfield SA. Characteristics of electrically evoked somatodendritic dopamine release in substantia nigra and ventral tegmental area in vitro. J Neurophysiol. 1997;77:853–862. doi: 10.1152/jn.1997.77.2.853. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Geometric and viscous components of the tortuosity of the extracellular space in the brain. Proc Natl Acad Sci U S A. 1998;95:8975–8980. doi: 10.1073/pnas.95.15.8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/S0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Schmauss C, Sulzer D. Altered dopamine release and uptake kinetics in mice lacking D2 receptors. J Neurosci. 2002;22:8002–8009. doi: 10.1523/JNEUROSCI.22-18-08002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14:88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA, Cull-Candy SG, Takahashi T. Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol. 1996;494:231–250. doi: 10.1113/jphysiol.1996.sp021487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci. 1996;16:6374–6385. doi: 10.1523/JNEUROSCI.16-20-06374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Tamás G, Soltesz I. Different transmitter transients underlie presynaptic cell type specificity of GABAA,slow and GABAA,fast. Proc Natl Acad Sci U S A. 2007;104:14831–14836. doi: 10.1073/pnas.0707204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro G, Barbour B. Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci. 2007;10:735–742. doi: 10.1038/nn1907. [DOI] [PubMed] [Google Scholar]
- Szot P, Reigel CE, White SS, Veith RC. Alterations in mRNA expression of systems that regulate neurotransmitter synaptic content in seizure-naive genetically epilepsy-prone rat (GEPR): transporter proteins and rate-limiting synthesizing enzymes for norepinephrine, dopamine and serotonin. Brain Res Mol Brain Res. 1996;43:233–245. doi: 10.1016/S0169-328X(96)00184-2. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-Z. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM, Fifková E. Monoaminergic synapses, including dendro-dendritic synapses in the rat substantia nigra. Exp Brain Res. 1977;30:161–174. doi: 10.1007/BF00237248. [DOI] [PubMed] [Google Scholar]
- Zoli M, Torri C, Ferrari R, Jansson A, Zini I, Fuxe K, Agnati LF. The emergence of the volume transmission concept. Brain Res Rev. 1998;26:136–147. doi: 10.1016/S0165-0173(97)00048-9. [DOI] [PubMed] [Google Scholar]