

Daily behavioral rhythms in mammals are governed by the central circadian clock located in the suprachiasmatic nucleus (SCN), and calcium is thought to play a role in the oscillation of the SCN. Kon et al. now find that Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity promotes dimerization of CLOCK and BMAL1 and is essential for the cellular oscillation and synchronization among oscillators in the SCN. Kinase-dead CaMKIIα weakened the behavioral rhythmicity and elicited decoupling between the morning and evening activity rhythms.
Keywords: circadian clock, CaMKII, morning and evening oscillators, coupling of oscillators, SCN, CLOCK/BMAL1
Abstract
Daily behavioral rhythms in mammals are governed by the central circadian clock, located in the suprachiasmatic nucleus (SCN). The behavioral rhythms persist even in constant darkness, with a stable activity time due to coupling between two oscillators that determine the morning and evening activities. Accumulating evidence supports a prerequisite role for Ca2+ in the robust oscillation of the SCN, yet the underlying molecular mechanism remains elusive. Here, we show that Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity is essential for not only the cellular oscillation but also synchronization among oscillators in the SCN. A kinase-dead mutation in mouse CaMKIIα weakened the behavioral rhythmicity and elicited decoupling between the morning and evening activity rhythms, sometimes causing arrhythmicity. In the mutant SCN, the right and left nuclei showed uncoupled oscillations. Cellular and biochemical analyses revealed that Ca2+–calmodulin–CaMKII signaling contributes to activation of E-box-dependent gene expression through promoting dimerization of circadian locomotor output cycles kaput (CLOCK) and brain and muscle Arnt-like protein 1 (BMAL1). These results demonstrate a dual role of CaMKII as a component of cell-autonomous clockwork and as a synchronizer integrating circadian behavioral activities.
In mammals, the master circadian pacemaker in the hypothalamic suprachiasmatic nucleus (SCN) generates rest–activity cycles to synchronize with daily changes of environmental conditions (Reppert and Weaver 2002; Crocker and Sehgal 2010; Welsh et al. 2010). Furthermore, in order to adapt to seasonal changes, the activity time, the period between the onset and offset of behavioral activities, is thought to be regulated by two independent clocks in the SCN, termed morning and evening oscillators (Pittendrigh and Daan 1976; de la Iglesia et al. 2000; Jagota et al. 2000; Inagaki et al. 2007; Helfrich-Förster 2009). Decoupling of these oscillators is observed in some species. The golden hamster (Mesocricetus auratus) shows dramatic dissociation of morning and evening activity rhythms under constant light conditions (Pittendrigh and Daan 1976). Similarly, the white-footed mouse (Peromyscus leucopus) or house mouse (Mus musculus) inbred CS strain shows spontaneous dissociation of morning and evening activity rhythms in constant darkness (DD) (Pittendrigh and Daan 1976; Abe et al. 1999). Morning and evening oscillators are normally coupled to maintain the constant length of the activity time, although the mechanism of communication between the oscillators is left undetermined.
Robust behavioral rhythms are integrated by a neuronal network among individual SCN neurons communicating through neurotransmitters and electrical signaling (Yamaguchi et al. 2003; Aton and Herzog 2005; Maywood et al. 2007; Hastings et al. 2008). Behavioral rhythms are severely disrupted in mice carrying a null mutation in vasoactive intestinal peptide (VIP) receptor 2 (VPAC2), a receptor for VIP and pituitary adenylate cyclase-activating peptide (PACAP) (Harmar et al. 2002). Inhibition of the intracellular signaling (e.g., cyclic AMP or G-protein signaling) results in a prolonged free-running period (O’Neill et al. 2008; Doi et al. 2011), but importantly, the loss of function of the cyclic nucleotide/G-protein signaling pathway does not cause dissociation of morning and evening activity rhythms in C57BL/6 mice. Consistently, a modeling study suggests that the mechanism of communication between the oscillators governing morning and evening behaviors is distinct from the coupling mechanism among individual neurons (Indic et al. 2008).
The cell-autonomous circadian oscillation of both the SCN and the peripheral cells is generated by a transcription-based autoregulatory feedback mechanism (Schibler and Sassone-Corsi 2002; Kondratov et al. 2003). Basic helix–loop–helix transcription factors circadian locomotor output cycles kaput (CLOCK) and brain and muscle Arnt-like protein 1 (BMAL1) form a heterodimer that activates transcription of Period (Per) and Cryptochrome (Cry) genes through their E-box enhancer elements. Translated PER and CRY proteins then translocate into the nucleus, where they interfere with CLOCK/BMAL1-dependent transcription and repress their own expression.
In addition to the transcriptional regulation, dynamic fluctuation of ions, especially Ca2+, appears to be essential for circadian clockwork (Nitabach et al. 2005). Individual neurons in a cultured SCN slice exhibit robust circadian fluctuation of intracellular Ca2+ concentration (Ikeda et al. 2003; Enoki et al. 2012), while chelating of intracellular Ca2+ abrogates rhythmic expression of clock genes (Lundkvist et al. 2005). These studies led us to speculate that a Ca2+-dependent feedback system regulates the transcriptional rhythms and raise a fundamental question as to how the Ca2+ signaling is integrated into the transcriptional feedback mechanism. In the present study, we identified Ca2+/calmodulin-dependent protein kinase II (CaMKII) as an essential component of the cellular oscillation machinery. Furthermore, we found that CaMKII plays an important role for synchronization among individual neuronal clocks and for mutual coupling between the morning and evening oscillators that define the activity time of the circadian behaviors in mice.
Results
CaMKII bolsters robust behavioral rhythms with organized coupling of morning and evening oscillators
CaMKII is an important signal-transducing molecule that is activated by an increase of cytosolic Ca2+ (Colomer and Means 2007). CaMKII is encoded by four genes in mice: CamkIIα, CamkIIβ, CamkIIγ, and CamkIIδ, among which CamkIIα and CamkIIβ are expressed predominantly in the brain, while CamkIIγ and CamkIIδ are ubiquitously expressed (Tombes et al. 2003). We confirmed that, when compared with the other isoforms, CaMKIIα is highly expressed in the mouse whole brain (Supplemental Fig. S1) as well as in the SCN neurons governing the behavioral rhythms (Nomura et al. 2003). In order to characterize the role of CaMKII in the behavioral rhythms, we employed recently generated CaMKIIα knock-in mice, the CaMKIIαK42R mutant, in which the kinase activity is completely lost, while the protein level is kept almost normal (Yamagata et al. 2009). Because CaMKII serves as a structural protein in addition to mediating phosphorylation signaling, the kinase-dead knock-in mice enable us to characterize the in vivo function of the kinase activity. This contrasts sharply with studies on CaMKIIα knockout mice with its protein deficiency. The total CaMKII activity in the forebrain of the knock-in mice is reduced to 30%–40% of that of wild-type mice (Yamagata et al. 2009).
The mutant mice showed no significant difference from wild type in total levels of wheel-running activities under 12 h:12 h light–dark cycles (LD) or DD conditions (Supplemental Fig. S2A) and in the dark to light activity ratio (Supplemental Fig. S2B). A χ2 periodogram analysis of the wheel running activity rhythms in DD revealed a prolonged free-running period in the mutant mice (Fig. 1A,B; Supplemental Fig.S2C; Supplemental Table S1). The robustness of the behavioral rhythms was assessed by the periodogram amplitude (Qp), a parameter that reflects the strength or regularity of a rhythm (Sokolov and Bushell 1978). The mutant mice showed a progressive decline of the Qp value from the second through the fourth week (DD2w–DD4w) in DD (Fig. 1C), indicating a reduced stability of the mutant SCN clock when placed in DD. Indeed, three out of 13 mutant mice initially showing rhythmic behaviors eventually became arrhythmic (Fig. 1A, right; Supplemental Table S1). These results demonstrate that CaMKIIα plays an essential role in mediating the high-amplitude and stable behavioral rhythms.
Figure 1.

CaMKII is essential for behavioral rhythms and coupling of morning and evening activities. (A) Representative wheel-running rhythm of CaMKIIαK42R mice. Because CaMKIIαK42R mice showed a temporal change in daily behavioral rhythm, the rhythm parameters were analyzed by using the period indicated as LD, DD1w, DD2w, DD3w, or DD4w. Three out of 13 mutant mice showed arrhythmic behavior, as observed in DD4w in the right panel. Wild-type (WT) littermates were used as control mice. (B) Prolonged period length in behavioral rhythm of CaMKIIαK42R mice. The period length of behavioral rhythm from DD1w to DD3w was analyzed by χ2 periodogram by using ClockLab software (Actimetrics). Plotted are the period lengths of individual animals. Bars indicate mean with SEM. (*) P < 0.01 versus wild type (Student’s t-test). (C) Temporal reduction in periodogram amplitude (Qp) of behavioral rhythm in CaMKIIαK42R mutant mice. The mean value of the Qp in DD1w was set to 100. (*) P < 0.05 versus wild type (Student’s t-test). (D) Free-running period determined by onset or offset of wheel-running activity rhythm in CaMKIIαK42R mutant mice. Plotted are the period lengths of individual animals. Bars indicate mean with SEM. (*) P < 0.05 (Student’s t-test). (E) Prolongation of daily activity time in CaMKIIαK42R mice. The onset and offset times of the activity were defined as the times when the ratio of 1-h activity becomes higher and lower than 2% of the total daily activity, respectively. The daily activity time was defined as the duration from the onset time to the offset time, and the data in the week were averaged and plotted for DD1w–DD4w. (*) P < 0.05; (**) P < 0.005 (Student’s t-test). (F) Temporal analysis of daily activity profile in DD. The ratio (percentage) of 1-h activity per total daily activity was plotted against the circadian time to demonstrate the daily activity profile. Evening (E), night (N), and morning (M) peaks of the wheel-running activity in the K42R mutant are indicated by arrowheads. (G) Temporal delay of morning activity peaks in CaMKIIαK42R mice. Data shown are representative results (A), the mean (F), or mean with SEM (B–E,G) from 11 wild-type or 13 CaMKIIαK42R mice.
Here we found that the free-running period of the mutant mice assessed by their activity offset was significantly longer than that determined by their activity onset (Fig. 1A,D; Supplemental Table S1) and thereby caused a progressive lengthening of the activity time in DD (Fig. 1E). Furthermore, the temporal activity profile of the mutant mice was characterized by a marked increase of the morning activity (Fig. 1F; Supplemental Fig. S2D), whereas the mutant showed a relative decrease of the evening activity and the emergence of a third peak, termed the night peak, between the evening and morning peaks (Fig. 1F; Supplemental Fig. S2D). In the mutant, the evening and morning peaks gradually became temporally decoupled, while the night peak stayed coherent with the evening peak (Fig. 1F,G). These observations collectively demonstrate an essential role for CaMKIIα in tight coupling of the oscillators that govern morning and evening behavioral activities.
Desynchronization of individual cellular rhythms and decoupling of oscillations between left and right nuclei in the SCN of CaMKIIαK42R mice
The unique behavioral phenotype of CaMKIIαK42R mice prompted us to investigate rhythms in pacemaker neurons of the mutant mice. The SCN slices of the mutant mice expressing PER2∷LUC, a fusion protein of PER2 and firefly luciferase (Yoo et al. 2004), were monitored by using a highly sensitive CCD camera for 10 d. We analyzed the bioluminescence signals from the bilateral SCN population in culture and found that the mutant SCN showed a prolonged period of the rhythm compared with wild-type mice (Fig. 2A,B), consistent with the long-period phenotype in the behavioral rhythm (Fig. 1B). Detailed analysis at the single-cell level revealed that the amplitude of individual cellular rhythms damped more rapidly in the mutant SCN than those in wild type (Fig. 2C,D). The Rayleigh test, which evaluates distribution of the peak times (histogram) across the circadian cycle, demonstrated a decrease in the sum of vectorial length of the peak phases (Fig. 2E,F) and an increase in the standard deviation in the mutant mice (Fig. 2E,G). Such desynchronization of individual cellular rhythms in the mutant became prominent with time (Fig. 2C,E–G). These results indicate that the kinase-dead mutation in CaMKIIα attenuated the cellular oscillation of individual SCN cellular clocks and impaired their synchronization.
Figure 2.

Desynchronization of individual cellular rhythms and decoupling of oscillations between left and right nuclei in the SCN of CaMKIIαK42R mice. (A) Bioluminescence rhythm from whole SCN in CaMKIIαK42R mice. (B) Prolonged free-running period of bioluminescence rhythm in the SCN of CaMKIIαK42R mice. (*) P < 0.005 versus wild type (Student’s t-test). Data shown are representative results (A) or the mean with SEM (B) from three independent experiments. (C) Bioluminescence rhythms of single SCN neurons of CaMKIIαK42R mice. (D) Amplitude of the bioluminescence rhythms of single SCN neurons of CaMKIIαK42R mice. The value (the peak value minus the trough one) normalized by the peak value was used as the amplitude in each rhythm. (*) P < 0.1 × 10−25 versus wild type (Student’s t-test). Data shown are the mean with SEM from 60 randomly selected cellular rhythms in the SCN slice. (E) Phase distribution of individual neuronal rhythms. Individual phases are plotted as dots around a circle, and the vector sum of the dots is represented as an arrow inside the circle. (F) Vector sum of individual neuronal phases. (G) Standard deviation of individual neuronal phases. Peak time at ∼102 or 224 h of each rhythm was used as the fourth or ninth peak, respectively. (*) P < 0.05 versus wild type (Student’s t-test). Data shown are representative results (C,E) or the mean with SEM (F,G) from three independent experiments. (H) Bioluminescence rhythms of the left and right nuclei in the SCN of wild-type and CaMKIIαK42R mice. The 10-d rhythm data and the magnified view of the last 4 d of data are shown. (I) Difference in the free-running period between the left and right SCN of CaMKIIαK42R mice. (*) P < 0.05 versus wild type (Student’s t-test). Data shown are representative results (H) or the mean with SEM (I) from three independent experiments.
Regional analysis of the cell populations revealed that coupling between the left and right nuclei was disrupted in the mutant SCN after prolonged culture (days 6–10 vs. days 1–5), whereas the bilateral rhythms of the wild-type SCN were kept synchronized (Fig. 2H, see arrowheads showing peak times of left and right nuclei in the magnified view). The decoupling was caused by dissociation of the free-running periods between the left and right nuclei (Fig. 2I). Together, these results suggest that the desynchronized oscillations between the left and right SCN underlie decoupling between the morning and evening behavioral rhythms in CaMKIIαK42R mice.
Pharmacological inhibition of CaMKII abolishes individual cellular oscillations and intercellular coupling among SCN neurons
The role of CaMKII in the SCN pacemaker neurons was examined by its pharmacological inhibition with a specific inhibitor, KN93. The robust circadian oscillation of the bioluminescence signals from the SCN slice of PER2∷LUC mice was rapidly blunted by administration of 100 µM KN93 (Fig. 3A,B), and washout of the drug restored the gene expression rhythm (Fig. 3A). Single-cell monitoring of the SCN neurons revealed that KN93 treatment progressively attenuated the peak amplitude of the individual cellular rhythms (Fig. 3C,D) and desynchronized them (Fig. 3C). The desynchronization effect was verified by Rayleigh test, which demonstrated a decrease in the vectorial length of the peak phases in KN93-treated neurons and an increase in the standard deviation thereof (Fig. 3E). On the other hand, KN92, an inactive analog of KN93 (Marley and Thomson 1996), showed no effect on the individual rhythm amplitudes and coupling of cellular rhythms (Fig. 3C–E). These results demonstrate essential roles of CaMKII for (1) the maintenance of individual cellular oscillations and (2) the intercellular coupling among oscillators in the SCN.
Figure 3.

CaMKII is essential for cellular oscillation and intercellular coupling among neuronal clocks in the circadian pacemaker. (A) Effect of CaMKII inhibitor on bioluminescence rhythm at the SCN tissue level. The SCN slices were prepared from PER2∷LUC mice, and bioluminescence intensity from the slice culture was recorded. The CaMKII inhibitor was applied from day 4.5 to day 12 during recording of the rhythm. (B) Decrease in PER2 peak expression under the CaMKII inhibitor at the tissue level. First peak expression levels of PER2∷LUC during the drug treatment was set to 100% in each experiment. (*) P < 0.05 versus DMSO-treated slices (Student’s t-test). Data shown are representative results (A) or the mean with SEM (B) from six independent experiments. (C) Effect of the CaMKII inhibitor on bioluminescence rhythm at the single-cell level. SCN slices were prepared from PER2∷LUC knock-in mice, and the CaMKII inhibitor was applied from the fourth to eighth days during recording of individual cellular rhythms. (D) Decrease in PER2 peak expression under the CaMKII inhibitor at the single-cell level. First peak expression level of PER2∷LUC during the drug treatment was set to 100% in each experiment. (*) P < 0.5 × 10−3 (Student’s t-test with Bonferroni correction). Data shown are the mean with SEM from 60 randomly selected cellular rhythms in the SCN slice. (E) Phase distribution of individual neuronal rhythms. Peak time at ∼24 or 130 h of each rhythm was used as a phase marker. (*) P < 0.05 (Student’s t-test). (C; top circles in E) Data shown are representative results of four independent experiments.
CaMKII is essential for normal oscillation of cellular clocks
We focused on the role of CaMKII in the intracellular oscillation mechanism by using cultured Rat-1 fibroblasts, which show prominent circadian rhythms in gene expression with no significant intercellular connection (Rosbash 1998; Kon et al. 2008). Bioluminescence rhythms of Rat-1 cells expressing the Bmal1-luc reporter, which drives rhythmic expression of LUC from the Bmal1 promoter (Kon et al. 2008), were significantly impaired in the presence of 20 µM KN93 (Fig. 4A,B; Supplemental Fig. S3). In contrast, KN92 treatment had no significant effect on the amplitude of the cellular clock (Fig. 4A,B; Supplemental Fig. S3A). We then examined the contribution of CaMKII isoforms by knocking down the specific isoform. We found that simultaneous siRNA-mediated knockdown of the ubiquitously expressed isoforms CamkIIγ (si1 or si2) and CamkIIδ (si3 or si4) markedly attenuated the amplitude of the Bmal1-luc rhythms in NIH3T3 fibroblasts (Fig. 4C,D; Supplemental Fig. S4).
Figure 4.

CaMKII is essential for cellular circadian oscillator and E-box-dependent expression of clock genes. (A) Effects of chronic treatment with CaMKII inhibitor on the cellular circadian rhythm. After treatment with 0.1 µM dexamethasone for 2 h, Rat-1-Bmal1-luc cells were transferred to the recording medium containing 0.1% DMSO (gray lines), 20 µM KN92 (blue lines), or 20 µM KN93 (red lines). (B) Effect of chronic treatment with CaMKII inhibitor on the amplitude of the cellular rhythm. (*) P < 0.3 × 10−8 (Student’s t-test with Bonferroni correction). Relative amplitudes were calculated as described in the Materials and Methods. (C) Effect of siRNA against CaMKII on the cellular rhythm. NIH3T3 cells were transfected with a Bmal1-luc reporter and siRNAs against CaMKIIγ and CaMKIIδ, and then the bioluminescence signals were recorded. (A,C) Normalized cellular rhythms from three independent experiments are shown. (D) Effect of CaMKII knockdown on amplitude of the cellular rhythm. (*) P < 0.03 versus control siRNA (Student’s t-test with Bonferroni correction). (E) Effect of chronic treatment with the CaMKII inhibitor on clock gene expression. Rat-1 cells were cultured as described in A and sampled at the indicated time points after the rhythm induction by dexamethasone. (F) Circadian profile of clock gene expression in NIH3T3 stably expressing the constitutive active form of CaMKII. The signals obtained for each mRNA were normalized by those of Tbp mRNA. (B,D–F) Data are the mean with SEM from three independent experiments.
We explored the role of CaMKII in the transcriptional feedback loops by examining expression profiles of the clock genes in the presence of KN93. Chronic treatment of Rat-1 cells with 10 or 20 µM KN93 dose-dependently weakened the rhythmicities of all of the clock genes examined (Fig. 4E). Among them, expression of E-box-regulated genes, including Per1–3, Dec1, Dec2, Dbp, and Cry1, was markedly down-regulated throughout the day by treatment with 20 µM KN93 (Fig. 4E). Consistently, stable expression of CaMKII(1–290), a constitutively active form of CaMKIIα (Supplemental Fig. S5; Wang and Simonson 1996; Tombes et al. 2003), stimulated expression of the E-box-regulated genes, such as Per1 and Dbp, in NIH3T3 cells (Fig. 4F).
We then examined the role of upstream regulators of CaMKII signaling in the cellular clockwork. Chelating of intracellular Ca2+ by administration of BAPTA-AM reduced the amplitude of the cellular rhythm in Rat-1 cells (Fig. 5A,B). The treatment attenuated E-box-dependent gene expression in Rat-1 cells (Fig. 5C), as was observed for the effect of KN93 (Fig. 4E). Similarly, application of calmidazolium, a specific inhibitor of calmodulin, reduced the amplitude of the cellular rhythm (Fig. 5D,E) and E-box-dependent gene expression (Fig. 5F). These results together indicate that Ca2+–calmodulin–CaMKII signaling provides important input to the cell-autonomous circadian oscillator through activating E-box-mediated transcription.
Figure 5.
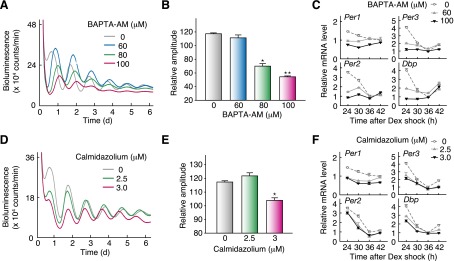
Intracellular Ca2+ and calmodulin are important for cellular oscillation and E-box-dependent expression of clock genes. (A) Effect of BAPTA-AM on the Rat-1 cellular rhythms. (B) Effect of BAPTA-AM on the amplitude of the cellular rhythms. (*) P < 0.5 × 10−5; (**) P < 0.1 × 10−7 (Student’s t-test). Relative amplitudes were calculated as described in the Materials and Methods. (C) Effect of BAPTA-AM on clock gene expression. (D) Effect of calmodulin inhibitor on the Rat-1 cellular rhythms. (E) Effect of calmodulin inhibitor on the amplitude of the cellular rhythms. (*) P < 0.5 × 10−3 (Student’s t-test). Shown are representative data (A,D) or mean with SEM (B, E) from three independent experiments. (F) Effect of the calmodulin inhibitor on clock gene expression. The mRNA signals obtained by RT–PCR analysis were normalized by those of Tbp mRNA. (C,F) Data are mean with SD from two independent experiments.
CaMKII facilitates heterodimerization of CLOCK and BMAL1
The intense effects of Ca2+/CaMKII inhibition on the E-box-dependent gene expression led us to investigate whether CaMKII functionally interacts with E-box activators. Coimmunoprecipitation experiments indicated that CaMKII interacts with CLOCK in the nuclear extracts of mouse liver prepared at Zeitgeber time (ZT) 6 or ZT18 (Fig. 6A). Transient expression of CLOCK and BMAL1 with CaMKII(1–290) increased the relative abundance of the up-shifted band of CLOCK that represents the phosphorylated form (Fig. 6B, left; Supplemental Fig. S6) while having no appreciable effect on BMAL1 phosphorylation (Fig. 6B, right). We focused on the phosphorylation states of endogenous CLOCK and CLOCK-associated BMAL1 in synchronized Rat-1 fibroblasts. Phosphorylation of CLOCK and BMAL1 showed a dynamic circadian variation, as revealed by alteration in relative intensities of the three CLOCK bands (hypershifted, shifted, and nonshifted) (Yoshitane et al. 2009) and the two BMAL1 bands (shifted and nonshifted) (Fig. 6C, lanes 1–4). The rhythmic phosphorylation of CLOCK and BMAL1 was abrogated in the presence of KN93 (Fig. 6C, lanes 5–12). Densitometric analysis verified that the inhibitor treatment down-regulated CLOCK phosphorylation and up-regulated the relative phosphorylation level of BMAL1 across the day (Fig. 6D). Notably, KN93 treatment reduced the protein amounts of BMAL1 that was kept associated with CLOCK (Fig. 6E), while total BMAL1 proteins showed no measurable decrease (Supplemental Fig. S7). Together, these results suggest that CaMKII-mediated phosphorylation of CLOCK facilitates its interaction with BMAL1 and potentiates E-box-dependent gene expression.
Figure 6.

CaMKII promotes phosphorylation of CLOCK and interaction of CLOCK with BMAL1. (A) Association of CLOCK and CaMKII in vivo. The lysates were prepared from the liver sampled at the indicated time points and used for immunoprecipitation by anti-CLOCK (positive control), anti-CaMKIIα/γ/δ or anti-Flag (negative control) antibody, followed by immunoblotting by anti-CLOCK antibody. (B) Effect of CaMKII overexpression on mobility shift of CLOCK and BMAL1. In HEK293 cells, CLOCK and BMAL1 were expressed with the constitutively active form of CaMKII or LacZ. The top panel shows representative raw data, and the bottom panel shows mean with SEM from three (left panel) or six (right panel) independent experiments. (*) P < 0.005 (Student’s t-test). (C) Effect of CaMKII inhibitor on heterodimers of CLOCK and BMAL1. Rat-1 cells were cultured as described in the legend for Figure 4 and sampled at the indicated time points after the rhythm induction by dexamethasone. The cell lysates were used for immunoprecipitation by anti-CLOCK antibody, and the precipitates were immunoblotted by anti-CLOCK (top panel) or anti-BMAL1 (bottom panel) antibody. (D) Ratio of the up-shifted form of CLOCK and BMAL1 under CaMKII inhibition. The ratio means the up-shifted band intensity per total band intensity of CLOCK (left panel) or BMAL1 (right panel). (E) Decreased interaction of CLOCK with BMAL1 under CaMKII inhibition. Representative data (A,C) or the mean with SEM (D,E) from three independent experiments are shown.
CaMKII as a state variable of the circadian oscillator
In the fundamental concept of circadian oscillators, components underlying the rhythm generation should change rhythmically in the amount and/or the activity, which collectively define the state of the oscillator (Aronson et al. 1994; Yamaguchi et al. 2003; Dunlap et al. 2004; O’Neill et al. 2008). CaMKII activity exhibits a circadian variation in a phase close to that of E-box-dependent transcription rhythms (Agostino et al. 2004). We asked whether acute CaMKII inhibition starting and ending at various times of the day affects the phase of the clock in Rat-1 cells stably expressing Bmal1-luc. During a 48-h application of 20 µM KN93, the cellular rhythms were almost completely lost, and removal of the drug (at circadian time [CT] 0, 3, 6, 9, 12, 15, 18, or 21) resumed the oscillation starting at a unique phase irrespective of the time at which the drug was added (Fig. 7A); that is, the cellular clocks were always reset to approximately CT4, a time close to the trough of E-box-dependent gene expression (here we defined CT0 as the peak time of Bmal1-luc-derived bioluminescence signals) (Fig. 7B). These results indicate that inhibition of CaMKII activity resets the phase of the oscillators to the trough level of E-box gene expression.
Figure 7.

The activity level of CaMKII is important for the phase of the circadian oscillator. (A) Effect of transient inhibition of CaMKII on the phase of the cellular clock. Rat-1-Bmal1-luc cells were exposed to 20 µM KN93 (in 0.1% DMSO) or 0.1% DMSO (control) at various circadian time points (CT0 was defined as the peak time of Bmal1-luc expression), and, 48 h after the treatment, the medium was replaced by the conditioned medium that was collected from the culture just prior to the drug treatment. The use of the conditioned medium minimized a phase-shifting effect induced by the medium change (Kon et al. 2008). The eight colored lines represent eight cultures treated with the drug for 48 h starting at different times. The traces were then aligned so that the treatment times overlap. Representative results from two independent experiments are shown. (B) Phase transition curves for the transient inhibition of CaMKII. The horizontal axis indicates the phase at which the drug was administered, and the vertical axis shows the phase to which the clock was reset just after the treatment. The latter phase was calculated from the subsequent rhythm. Data are the mean with variation of two independent experiments. (C) A model for cell-autonomous circadian oscillation. The circadian oscillation in the intracellular Ca2+ levels induces rhythmic activation of calmodulin–CaMKII signaling. CaMKII regulates dimerization of CLOCK and BMAL1 and enhances E-box-dependent gene expression. The transcriptional feedback system may drive rhythmic activities of Ca2+ regulators in order to maintain the cell-autonomous Ca2+ signaling oscillation.
Discussion
CaMKII appears to regulate the circadian clock at multiple levels. Previously, CaMKII was shown to play an important role in the photic input pathway and rhythmic gene expression of clock genes (Yokota et al. 2001; Lundkvist et al. 2005; Harrisingh et al. 2007; Welsh et al. 2010). Photic signal induces a glutamate-mediated cytoplasmic Ca2+ increase in the SCN neurons. Activated CaM/CaMKII then phosphorylates and activates a transcription factor, cAMP response element-binding protein (CREB), which rapidly increases transcription of Per1 to induce phase shifts of the clock (Nomura et al. 2003). The present study identifies a novel pathway in which CaMKII activates CLOCK/BMAL1-mediated E-box-dependent gene expression, which is important for the cell-autonomous circadian oscillation (Figs. 4–7). Intracellular Ca2+ concentration in the rodent SCN shows a circadian change (Ikeda et al. 2003; Enoki et al. 2012), suggesting that cytosolic Ca2+ rhythm is integrated into the cell-autonomous oscillation system. Consistently, intracellular Ca2+ concentration increases in a phase similar to or slightly advanced from that of the activation of CaMKII and E-box-dependent transcription (at CT4–CT8) (Ikeda et al. 2003; Agostino et al. 2004; Welsh et al. 2010). Chelating of intracellular Ca2+ or inhibition of calmodulin blunted cell-autonomous circadian gene expression rhythms even in the nonneuronal cultured cells (Fig. 5). Based on these observations, we propose a novel model in which CaMKII signaling couples Ca2+ dynamics with the E-box-dependent transcriptional feedback mechanism and thereby stabilizes self-sustaining cellular oscillation (Fig. 7C).
In animal behavioral rhythms, dissociation of morning and evening activity rhythms was previously described in several species, and the phenomenon led to an idea that the morning and evening activities are independently regulated by two oscillators (Pittendrigh and Daan 1976). Pioneering studies on the behavioral rhythms of Drosophila clearly demonstrated that the morning and evening activities are independently regulated by different clock neurons communicating with each other in the brain (Grima et al. 2004; Stoleru et al. 2004, 2005). In mammals, multiple oscillating cell groups are observed within the SCN (de la Iglesia et al. 2000; Jagota et al. 2000; Inagaki et al. 2007; Helfrich-Förster 2009). When the morning and evening activity rhythms are dissociated in golden hamsters, the left and right SCN show different phases in clock gene expression rhythm (de la Iglesia et al. 2000). Unilateral lesion of the SCN abolished the splitting and established a single rhythm of behavioral activity (Pickard and Turek 1982). In normal conditions, the left and right SCN are strongly connected with each other even in isolated culture (Michel et al. 2013), and the functional connection is important for coupling of their free-running rhythms, as observed in wild-type SCN slices over 10 d (Fig. 2). In the present study, genetic ablation of CaMKIIα activity in C57BL/6 mice resulted in dissociation of the morning and evening activity rhythms in behavior (Fig. 1) and caused decoupling of oscillations between the left and right SCN (Fig. 2). These studies support the idea that the decoupling of the free-running period between the left and right SCN underlies dissociation of the morning and evening activity rhythms in the behavior. Importantly, dissociation of the behavioral rhythms is not observed in normal C57BL/6, a strain that shows robust behavioral rhythmicity, whereas the other strains, such as CS, exhibit an unstable behavioral rhythm with spontaneous rhythm splitting in DD (Abe et al. 1999). Because loss of CaMKIIα activity induced splitting-like behavior in DD and decreased the amplitude of the rhythm (Fig. 1), CaMKII-mediated tight coupling between the morning and evening oscillators may minimize the dissociation of behavioral activities in C57BL/6 mice and confer robustness of the rhythmicity.
The intercellular coupling of neuronal clocks in the SCN is established by multiple signaling such as neurotransmitters, cAMP signaling, and electrical synapses (Welsh et al. 2010). Inhibition of these molecules results in changes in the free-running period or attenuation of rhythmicities of the behavioral rhythms (Harmar et al. 2002; Long et al. 2005; O’Neill et al. 2008; Doi et al. 2011), the phenotypes similar to what we observed for the CaMKIIαK42R mutant in the present study (Figs. 1, 2). Interestingly, decoupling of the morning and evening activity rhythms is unique to the CaMKII mutant among a wide variety of genetically engineered mice (Fig. 1), raising the possibility that CaMKII signaling plays a major role in functional coupling of the morning and evening oscillators in mice. Because the phase of the cellular clock is tightly regulated by the activity levels of CaMKII (Fig. 7), Ca2+–calmodulin–CaMKII signaling may serve as the core machinery of the cellular clocks that adjust their phases in response to changes in cytosolic Ca2+ level regulated by intercellular communications and environmental signals. This idea is consistent with the previously reported role of CaMKII signaling involved in light entrainment of the circadian clock (Yokota et al. 2001).
Abnormalities in CaMKII are found in patients with several neurological disorders with disrupted clock function, including schizophrenia, bipolar disorders, and Alzheimer’s disease (Colomer and Means 2007; Schibler and Sassone-Corsi 2002). Because several psychotropic drugs such as fluvoxamine and desipramine or lithium modulate CaMKII activity (Celano et al. 2003; Sasaki et al. 2006), recovery of normal clock function through the pharmacological action on CaMKII may prove effective for the treatment of complicated neuronal disorders. Here we demonstrate that CaMKII activity regulates multiple parameters of the circadian clock, such as period length (Figs. 1, 2), oscillation amplitude (Figs. 1–4), phase (Fig. 7), and the interneuronal coupling (Figs. 2, 3). The present study illuminates CaMKII as a druggable target for innovative therapy of disorders that associate with clock dysfunction.
Materials and methods
Real-time monitoring of circadian rhythms of cultured cells
Real-time monitoring of cellular circadian gene expression was performed using Rat-1 or NIH3T3 cells expressing a luciferase reporter under the control of the upstream region of the Bmal1 gene (Kon et al. 2008). Briefly, the cells were plated on 35-mm dishes (1.0 × 106 cells per dish) and cultured at 37°C under 5% CO2 in phenol red-free DMEM (Sigma-Aldrich, catalog no. D2902) supplemented with 10% FBS (Biowest), 3.5 mg/mL glucose, 3.7 mg/mL NaHCO3, 50 U/mL penicillin, and 50 µg/mL streptomycin. Twenty-four hours after the plating, the cells were treated with 0.1 µM dexamethasone for 2 h, and the medium was replaced with a recording medium (DMEM supplemented with 10% FBS, 3.5 mg/mL glucose, 25 U/mL penicillin, 25 µg/mL streptomycin, 0.1 mM luciferin, 10 mM HEPES-NaOH at pH 7.0). The bioluminescence signals from the cells were continually recorded at 37°C under air with a dish-type bioluminescence detector, Kronos (ATTO, AB-2500) or LumiCycle (Actimetrics). For normalization of the bioluminescence levels, raw data were divided by the mean for bioluminescence activities across the 7-d recording. The normalized rhythms were detrended by subtracting 24-h centered moving averages, and the areas under the curves (arbitrary units) were used for calculating the relative amplitudes of the rhythms (DeBruyne et al. 2007).
Real-time monitoring of SCN rhythms
Bioluminescence signals from SCN cultures were recorded as described previously (Inagaki et al. 2007). The coronal SCN slices (100 µm thick) at the middle of the rostro–caudal axis were prepared from PER2∷LUC mice (Yoo et al. 2004) using a microslicer at ZT6–ZT10, and the bioluminescence signals from the slices were recorded by LumiCycle (Actimetrics). Single-cell analysis was performed using Cellgraph (ATTO) equipped with a highly sensitive cryogenic CCD camera (ORCA-II ER or ImagEM, Hamamatsu Photonics). Individual cellular rhythms were analyzed using Aquacosmos software (Hamamatsu Photonics), and distribution of the phases was analyzed by circular statistics software (Oriana, Kovach Computing Services). The bioluminescence signals were recorded after the slice was set on the apparatus (at time 0). In experiments using a drug, the medium was replaced with a medium containing KN92, KN93, or DMSO 96 h after the setting.
RT–PCR analysis and RNAi
Total RNA was prepared from cultured cells or mouse tissues using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. RT–PCR analysis was performed as described previously (Kon et al. 2008). PCR primers used were as follows: for CamkIIγ, 5′-AATCATTAAGATCACAGAACAAC-3′ and 5′-TCTCCTCTGACTGACTGGTG-3′; and for CamkIIδ, 5′-GATGGACTTTCACAGATTCTAC-3′ and 5′-ATTCTGCCACTTCCCATCAC-3′. Using siDirect (http://design.RNAi.jp), siRNAs were designed as follows: CamkIIγ si1, 5′-CCAUGACAGUAUUUCUGAAGA-3′; CamkIIγ si2, 5′-GGCUCAAUGUCCACUAUCACU-3′; CamkIIδ si3, 5′-AAGACAUAGUGGCAAGAGAAU-3′; CamkIIδ si4, 5′-GUAGACUGCUUGAAGAAAUUU-3′; control siA, 5′-CUCGCGACGAGUCGUAUUUC A-3′; and control siB, 5′-GAUCCGUAGCGCGAUUCGAGC-3′. One day after plating NIH3T3 cells on a 35-mm dish (1.0 × 106 cells per dish), siRNA (250 pmol each) was transfected to the cells using Lipofectamine 2000 (Invitrogen).
Immunoprecipitation and immunoblotting analyses
Immunoprecipitation was performed as described previously (Yoshitane et al. 2009). Briefly, anti-CLOCK antibody (CLNT1), anti-CaMKII antibody (Sigma-Aldrich, catalog no. C265), or anti-Flag antibody (Sigma-Aldrich, M2, as a control) was added to the mouse liver nuclear extracts (Fig. 6A) or the cultured cell lysates (Fig. 6B,C). The mixtures were incubated overnight at 4°C, and then the immune complex was precipitated by Protein G-Sepharose (GE Healthcare). In Figure 6B, HEK293T cells were plated on 12-well plates (1.0 × 105 cells per well) and, in the next day, transfected with 400 ng of Myc-CLOCK/pSG5 (a kind gift of Dr. Paolo Sassone-Corsi) and 200 ng of BMAL1/pcDNA3.1 (a kind gift of Dr. Steven M. Reppert), together with 400 ng of CaMKII(1–290)/pcDNA3.1 or 400 ng of LacZ/pcDNA3.1. In Figure 6C, synchronized Rat-1 cells plated on 35-mm dishes were used. The transfected HEK293T and Rat-1 cells were washed with ice-cold PBS and solubilized for 1 h in IPB2 buffer (20 mM HEPES-NaOH, 137 mM NaCl, 2 mM EDTA, 10% [v/v] glycerol, 1% Triton X-100, 50 mM NaF, 1 mM Na3VO4, 1 mM DTT, 1 mM PMSF, 4 µg/mL aprotinin, 4 µg/mL leupeptin at pH 7.8). The cell lysates were centrifuged for 10 min at 17,000 g and subjected to immunoprecipitation and then immunoblotting analysis. The blots were blocked by 1% (w/v) skim milk in Tris-buffered saline (pH 7.4) for 1 h at 37°C and then incubated overnight at 4°C with anti-CLOCK (CLSP3) or anti-BMAL1 (B1BH2) antibody. The primary antibodies were detected with a horseradish peroxidase-conjugated anti-rabbit/mouse IgG antibody (1:5000; Kirkegaard and Perry Laboratories, Inc.), and the signals were visualized with Western lightning plus ECL (PerkinElmer Life Sciences).
Animal experiments
The animal experiments were conducted in accordance with the guidelines of the University of Tokyo. CaMKIIαK42R knock-in mice were generated as described previously (Yamagata et al. 2009). The mice (C57BL/6 background, male, 10 wk old) were housed individually at 23°C in cages (13 × 23 × 15 cm) equipped with a running wheel (diameter 10 cm) with food and water available ad libitum. Wheel-running rhythms were monitored under DD conditions after entrainment in 12 h:12 h LD cycles for at least 2 wk. The number of wheel revolutions was collected every minute into a computer system. Behavioral data were analyzed by using ClockLab software (Actimetrics), and the onset and offset of the daily activity were defined as described in the legend for Figure 1.
Acknowledgments
We are grateful to Professor Masamitsu Iino and Dr. Kazunori Kanemaru for their help and advice with the experiments. We also thank Jun Nakano for critical comments on the manuscript. This work was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) and MEXT, Japan. N.K. and Y.S. were supported by JSPS Research Fellowships for Young Scientists.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.237511.114.
References
- Abe H, Honma S, Honma K, Suzuki T, Ebihara S 1999. Functional diversities of two activity components of circadian rhythm in genetical splitting mice (CS strain). J Comp Physiol 184: 243–251 [DOI] [PubMed] [Google Scholar]
- Agostino PV, Ferreyra GA, Murad AD, Watanabe Y, Golombek DA 2004. Diurnal, circadian and photic regulation of calcium/calmodulin-dependent kinase II and neuronal nitric oxide synthase in the hamster suprachiasmatic nuclei. Neurochem Int 44: 617–625 [DOI] [PubMed] [Google Scholar]
- Aronson BD, Johnson KA, Loros JJ, Dunlap JC 1994. Negative feedback defining a circadian clock: autoregulation of the clock gene frequency. Science 263: 1578–1584 [DOI] [PubMed] [Google Scholar]
- Aton SA, Herzog ED 2005. Come together, right...now: synchronization of rhythms in a mammalian circadian clock. Neuron 48: 531–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celano E, Tiraboschi E, Consogno E, D’Urso G, Mbakop MP, Gennarelli M, de Bartolomeis A, Racagni G, Popoli M 2003. Selective regulation of presynaptic calcium/calmodulin-dependent protein kinase II by psychotropic drugs. Biol Psychiatry 53: 442–449 [DOI] [PubMed] [Google Scholar]
- Colomer J, Means AR 2007. Physiological roles of the Ca2+/CaM-dependent protein kinase cascade in health and disease. Subcell Biochem 45: 169–214 [DOI] [PubMed] [Google Scholar]
- Crocker A, Sehgal A 2010. Genetic analysis of sleep. Genes & Dev 24: 1220–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBruyne JP, Weaver DR, Reppert SM 2007. Peripheral circadian oscillators require CLOCK. Curr Biol 17: R538–R539 [DOI] [PubMed] [Google Scholar]
- de la Iglesia HO, Meyer J, Carpino A Jr, Schwartz WJ 2000. Antiphase oscillation of the left and right suprachiasmatic nuclei. Science 290: 799–801 [DOI] [PubMed] [Google Scholar]
- Doi M, Ishida A, Miyake A, Sato M, Komatsu R, Yamazaki F, Kimura I, Tsuchiya S, Kori H, Seo K, et al. 2011. Circadian regulation of intracellular G-protein signalling mediates intercellular synchrony and rhythmicity in the suprachiasmatic nucleus. Nat Commun 2: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap JC, Loros JJ, Decoursey PJ 2004. Chronobiology: Biological timekeeping. Sinauer Associates, Sunderland, MA [Google Scholar]
- Enoki R, Kuroda S, Ono D, Hasan MT, Ueda T, Honma S, Honma K 2012. Topological specificity and hierarchical network of the circadian calcium rhythm in the suprachiasmatic nucleus. Proc Natl Acad Sci 109: 21498–21503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grima B, Chelot E, Xia R, Rouyer F 2004. Morning and evening peaks of activity rely on different clock neurons of the Drosophila brain. Nature 431: 869–873 [DOI] [PubMed] [Google Scholar]
- Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, Morrison CF, Dorin JR, Piggins HD, Reubi JC, et al. 2002. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109: 497–508 [DOI] [PubMed] [Google Scholar]
- Harrisingh MC, Wu Y, Lnenicka GA, Nitabach MN 2007. Intracellular Ca2+ regulates free-running circadian clock oscillation in vivo. J Neurosci 27: 12489–12499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings MH, Maywood ES, O’Neill JS 2008. Cellular circadian pacemaking and the role of cytosolic rhythms. Curr Biol 18: R805–R815 [DOI] [PubMed] [Google Scholar]
- Helfrich-Förster C 2009. Does the morning and evening oscillator model fit better for flies or mice? J Biol Rhythms 24: 259–270 [DOI] [PubMed] [Google Scholar]
- Ikeda M, Sugiyama T, Wallace CS, Gompf HS, Yoshioka T, Miyawaki A, Allen CN 2003. Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron 38: 253–263 [DOI] [PubMed] [Google Scholar]
- Inagaki N, Honma S, Ono D, Tanahashi Y, Honma K 2007. Separate oscillating cell groups in mouse suprachiasmatic nucleus couple photoperiodically to the onset and end of daily activity. Proc Natl Acad Sci 104: 7664–7669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indic P, Schwartz WJ, Paydarfar D 2008. Design principles for phase-splitting behaviour of coupled cellular oscillators: clues from hamsters with ‘split’ circadian rhythms. J R Soc Interface 5: 873–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagota A, de la Iglesia HO, Schwartz WJ 2000. Morning and evening circadian oscillations in the suprachiasmatic nucleus in vitro. Nat Neurosci 3: 372–376 [DOI] [PubMed] [Google Scholar]
- Kon N, Hirota T, Kawamoto T, Kato Y, Tsubota T, Fukada Y 2008. Activation of TGF-β/activin signalling resets the circadian clock through rapid induction of Dec1 transcripts. Nat Cell Biol 10: 1463–1469 [DOI] [PubMed] [Google Scholar]
- Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP 2003. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes & Dev 17: 1921–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundkvist GB, Kwak Y, Davis EK, Tei H, Block GD 2005. A calcium flux is required for circadian rhythm generation in mammalian pacemaker neurons. J Neurosci 25: 7682–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Jutras MJ, Connors BW, Burwell RD 2005. Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat Neurosci 8: 61–66 [DOI] [PubMed] [Google Scholar]
- Marley PD, Thomson KA 1996. The Ca2+/calmodulin-dependent protein kinase II inhibitors KN62 and KN93, and their inactive analogues KN04 and KN92, inhibit nicotinic activation of tyrosine hydroxylase in bovine chromaffin cells. Biochem Biophys Res Commun 221: 15–18 [DOI] [PubMed] [Google Scholar]
- Maywood ES, O’Neill JS, Chesham JE, Hastings MH 2007. Minireview: the circadian clockwork of the suprachiasmatic nuclei—analysis of a cellular oscillator that drives endocrine rhythms. Endocrinology 148: 5624–5634 [DOI] [PubMed] [Google Scholar]
- Michel S, Marek R, Vanderleest HT, Vansteensel MJ, Schwartz WJ, Colwell CS, Meijer JH 2013. Mechanism of bilateral communication in the suprachiasmatic nucleus. Eur J Neurosci 37: 964–971 [DOI] [PubMed] [Google Scholar]
- Nitabach MN, Holmes TC, Blau J 2005. Membranes, ions, and clocks: testing the Njus-Sulzman-Hastings model of the circadian oscillator. Methods Enzymol 393: 682–693 [DOI] [PubMed] [Google Scholar]
- Nomura K, Takeuchi Y, Yamaguchi S, Okamura H, Fukunaga K 2003. Involvement of calcium/calmodulin-dependent protein kinase II in the induction of mPer1. J Neurosci Res 72: 384–392 [DOI] [PubMed] [Google Scholar]
- O’Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MN 2008. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science 320: 949–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard GE, Turek FW 1982. Splitting of the circadian rhythm of activity is abolished by unilateral lesions of the suprachiasmatic nuclei. Science 215: 1119–1121 [DOI] [PubMed] [Google Scholar]
- Pittendrigh CS, Daan SA 1976. Functional analysis of circadian pacemakers in nocturnal rodents: V. Pacemaker structure: a clock for all seasons. J Comp Physiol 106: 333–355 [Google Scholar]
- Reppert SM, Weaver DR 2002. Coordination of circadian timing in mammals. Nature 418: 935–941 [DOI] [PubMed] [Google Scholar]
- Rosbash M 1998. Why the rat-1 fibroblast should replace the SCN as the in vitro model of choice. Cell 93: 917–919 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Han F, Shioda N, Moriguchi S, Kasahara J, Ishiguro K, Fukunaga K 2006. Lithium-induced activation of Akt and CaM kinase II contributes to its neuroprotective action in a rat microsphere embolism model. Brain Res 1108: 98–106 [DOI] [PubMed] [Google Scholar]
- Schibler U, Sassone-Corsi P 2002. A web of circadian pacemakers. Cell 111: 919–922 [DOI] [PubMed] [Google Scholar]
- Sokolov PG, Bushell WN 1978. The χ2 periodogram: its utility for analysis of circadian rhythms. J Theor Biol 72: 131–160 [DOI] [PubMed] [Google Scholar]
- Stoleru D, Peng Y, Agosto J, Rosbash M 2004. Coupled oscillators control morning and evening locomotor behaviour of Drosophila. Nature 431: 862–868 [DOI] [PubMed] [Google Scholar]
- Stoleru D, Peng Y, Nawathean P, Rosbash M 2005. A resetting signal between Drosophila pacemakers synchronizes morning and evening activity. Nature 438: 238–242 [DOI] [PubMed] [Google Scholar]
- Tombes RM, Faison MO, Turbeville JM 2003. Organization and evolution of multifunctional Ca2+/CaM-dependent protein kinase genes. Gene 322: 17–31 [DOI] [PubMed] [Google Scholar]
- Wang Y, Simonson MS 1996. Voltage-insensitive Ca2+ channels and Ca2+/calmodulin-dependent protein kinases propagate signals from endothelin-1 receptors to the c-fos promoter. Mol Cell Biol 16: 5915–5923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Takahashi JS, Kay SA 2010. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol 72: 551–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, et al. 2009. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIα in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci 29: 7607–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Isejima H, Matsuo T, Okura R, Yagita K, Kobayashi M, Okamura H 2003. Synchronization of cellular clocks in the suprachiasmatic nucleus. Science 302: 1408–1412 [DOI] [PubMed] [Google Scholar]
- Yokota S, Yamamoto M, Moriya T, Akiyama M, Fukunaga K, Miyamoto E, Shibata S 2001. Involvement of calcium–calmodulin protein kinase but not mitogen-activated protein kinase in light-induced phase delays and Per gene expression in the suprachiasmatic nucleus of the hamster. J Neurochem 77: 618–627 [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. 2004. PERIOD2∷LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci 101: 5339–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitane H, Takao T, Satomi Y, Du NH, Okano T, Fukada Y 2009. Roles of CLOCK phosphorylation in suppression of E-box-dependent transcription. Mol Cell Biol 29: 3675–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]