

Abstract
NF-κB is a major transcriptional factor regulating many cellular functions including inflammation; therefore, its appropriate control is of high importance. The detailed mechanism of its activation has been well characterized, but that of negative regulation is poorly understood. In this study, we showed AMAP1, an Arf-GTPase activating protein, as a negative feedback regulator for NF-κB by binding with IKKβ, an essential kinase in NF-κB signaling. Proteomics analysis identified AMAP1 as a binding protein with IKKβ. Overexpression of AMAP1 suppressed NF-κB activity by interfering the binding of IKKβ and NEMO, and deletion of AMAP1 augmented NF-κB activity. The activation of NF-κB induced translocation of AMAP1 to cytoplasm from cell membrane and nucleus, which resulted in augmented interaction of AMAP1 and IKKβ. These results demonstrated a novel role of AMAP1 as a negative feedback regulator of NF-κB, and presented it as a possible target for anti-inflammatory treatments.
Nuclear Factor-κB (NF-κB) consists of a family of transcription factors (p65 or RelA, p50, p52, c-Rel, and RelB) that play critical roles in inflammation, immunity, cell proliferation, differentiation and survival1. It generally exists as a homo- or heterodimer in the cytosol that is bound to the inhibitor of κB (IκB) (see review2). In response to a wide variety of stimuli, including inflammatory cytokines, IκB is phosphorylated and degraded via the ubiquitin pathway, which is followed by NF-κB translocation to the nucleus and activation of transcription. Serine phosphorylation of IκB is mediated by a large multi-unit complex containing two catalytic subunits (IKKα and IKKβ) and the regulatory subunit IKKγ or NEMO3,4. Among these subunits, IKKβ is the essential kinase mediating IκB phosphorylation in most cell types, and germline deletion of IKKβ results in embryonic lethality due to massive liver apoptosis, similar to the phenotype of p65-knockout mice5.
NF-κB signaling proteins including these kinases are essential for maintaining living body as shown by IKKβ or p65 knockout mouse with phenotype of embryonic lethality, but they also have been implicated in the pathogenesis of many human diseases, including cancer6,7,8,9. Therefore, controlling their activities appropriately is of high importance. Although the mechanism for its activation was been well analyzed as described above, that for negative regulation is poorly explored.
We recently reported that IKKβ has a kinase-independent role in regulating widespread cellular responses and cell signaling10, implicating that there are still undiscovered roles of IKKβ. To explore the roles more extensively, we identified A Multiple-domain Arf-GAP Protein 1 (AMAP1) as a binding partner of IKKβ by proteomic analyses. AMAP1, also called ASAP1 or DDEF1, is an Arf-GTPase-activating protein that functions on membrane surfaces to catalyze the hydrolysis of GTP bound to Arf, and plays major roles in the regulation of membrane remodeling and cytoskeletal organization, cellular migration and tumor invasion and metastasis11,12,13,14. More importantly, clinical studies have indicated that AMAP1 is dramatically up-regulated in advanced cancers15,16,17,18,19,20,21,22. The roles of AMAP1 in the inflammatory responses are of high interest considering the close relationship between caner and inflammation2; however, the roles have been poorly understood. Interestingly, Haque et al. reported the inhibitory effect of AMAP1 (ASAP1) on the production of proinflammatory mediators23, but the detailed mechanism remains unknown.
In this report, we carefully examined the involvement of AMAP1 in the regulation of NF-κB, and found that it has an unanticipated role to interfere the IKKβ−NEMO binding, which results in negative-feedback regulation of NF-κB activity. This report describes the detailed role of AMAP1 in the inflammatory responses, and AMAP1 can be a new target for developing anti-inflammatory treatments.
Results
AMAP1 interacts with IKKβ
To discover novel roles of IKKβ, we started with identifying proteins that bind to IKKβ. Three kinds of FLAG-tagged IKKβ were overexpressed in HEK293T cells, and binding proteins were immunoprecipitated using anti-FLAG antibody. The samples were subjected to proteomic analyses using the nano-LC/MALDI-TOF system. Of the proteins identified repeatedly, we focused on AMAP1 since it is clinically important due to its augmented expression in cancer15,16,19,22,24, which is one of the major inflammatory diseases7,9. To confirm the physical association of IKKβ and AMAP1, we performed co-immunoprecipitation (Co-IP) assays using the lysate of HEK293T cells co-overexpressed with HA-tagged AMAP1 and FLAG-tagged IKKβ. IKKβ and AMAP1 were detected in the precipitates with either the anti-HA antibody or the anti-FLAG antibody, respectively (Fig. 1a).
Figure 1. AMAP1 interacts with IKKβ in vivo.

(a) The binding of AMAP1 and IKKβ. FLAG-tagged IKKβ and HA-tagged AMAP1 were expressed in HEK293T cells and immunoprecipitated with anti-FLAG and anti-HA antibodies. AMAP1 and IKKβ were detected in the precipitate by immunoblotting with anti-FLAG and anti-HA antibodies. (b) Schematic representation of IKKβ and its mutants. (c) FLAG-tagged IKKβ/mutants and HA-tagged AMAP1 were expressed in HEK293T cells and immunoprecipitated with anti-FLAG antibody. AMAP1 was detected in the precipitate by immunoblotting with anti-HA antibody. (d) Schematic representation of AMAP1 and its mutants. (e) FLAG-tagged IKKβ and GST-tagged AMAP1/mutants were expressed in HEK293T cells and immunoprecipitated with anti-FLAG antibody. AMAP1 and its mutants were detected in the precipitate by immunoblotting with anti-GST antibody. The data shown are from one representative experiment of the three that were performed. Full-length blots are presented in Supplementary Figure 4.
Next, we determined the binding sites of these proteins. Based on the functional modules of IKKβ, including an amino-terminal kinase domain (KD), a ubiquitin-like domain (ULD), an elongated α-helical scaffold/dimerization domain (SDD) and a carboxyl-terminal NEMO Binding Domain (NBD)25, we divided IKKβ into 4 overlapping FLAG-tagged fragments. The constructs are IKKβ-Δ1 comprising the KD, ULD and SDD, IKKβ-Δ2 comprising the KD and ULD, IKKβ-Δ3 comprising only the KD, and IKKβ-Δ4 comprising the ULD, SDD and NBD (Fig. 1b), and each construct was expressed in HEK293T cells along with HA-tagged AMAP1. We found that AMAP1 was co-precipitated only with IKKβ-Δ4, but not with any other construct of IKKβ (Fig. 1c), indicating that AMAP1 binds to IKKβ at the C-terminal region containing the NBD. Further, we examined which region of AMAP1 is essential for its binding to IKKβ. Each of 6 GST-tagged fragments of AMAP1, which are Bar (Bin-Amphiphysin-Rvs), PH (Pleckstrin Homology), Arf-GAP, ANK (Ankyrin repeat), PRD (Proline Rich Domain) and SH3 (Src Homology3) domains (Fig. 1d), was expressed in HEK293T cells along with FLAG-tagged IKKβ. We found only SH3 domain construct was co-precipitated with IKKβ as shown in Fig. 1e. To confirm it further, we made construct of SH3-domain deleted AMAP1 (AMAP1ΔSH3). Cells were expressed with full-length AMAP1 or AMAP1ΔSH3 along with FLAG-tagged IKKβ, and we found only full-length AMAP1, not AMAP1ΔSH3, was co-precipitated with IKKβ (Supplementary Fig. S1 online). Taken together, these results indicated that the SH3 domain of AMAP1 is responsible for the binding with IKKβ, which implicates that the GTPase activity of AMAP1 is not involved in the interaction with IKKβ.
AMAP1 negatively regulates NF-κB
The C-terminal region of IKKβ identified as the binding site for AMAP1 contains the binding domain for NEMO/IKKγ, which is the regulatory subunit of the IKK complex3. It prompted us to verify the effect of AMAP1 on the association of IKKβ and NEMO because it is required for NF-κB activation26. As shown in Fig. 2a, overexpression of AMAP1 inhibited the binding of IKKβ and NEMO, and it was in a dose-dependent manner (Supplementary Fig. S2 online). In addition, the increased level of NEMO could mitigate the interaction between AMAP1 and IKKβ (Fig. 2b). Taken together, there is competition between AMAP1 and NEMO in IKKβ interaction. As expected from these results, overexpression of AMAP1 suppressed NF-κΒ activity (Fig. 2c), and it was confirmed by less translocation of p65 into nucleus after IL-1β stimulation in AMAP1-overexpressed cells (Fig. 2d). Additionally, suppression of AMAP1 by shRNA enhanced NF-κB activity (Fig. 2e) and augmented p65 translocation into the nucleus after IL-1β stimulation (Fig. 2f). Thus, AMAP1 interferes the binding of IKKβ and NEMO, and negatively regulates NF-κB.
Figure 2. AMAP1 negatively regulates NF-κB.

(a) AMAP1 interfered with the association of IKKβ and NEMO. FLAG-tagged IKKβ and NEMO were expressed in HEK293T cells along with control vector or HA-tagged AMAP1 and immunoprecipitated with anti-FLAG antibody. AMAP1, IKKβ and NEMO were detected in the precipitates by immunoblotting with anti-FLAG, anti-HA and anti-NEMO antibodies. (b) The induction of NEMO reduced the interaction of IKKβ and AMAP1. FLAG-tagged IKKβ and HA-tagged AMAP1 were expressed in HEK293T cells along with control or NEMO vector and immunoprecipitated with anti-FLAG antibody. AMAP1, IKKβ and NEMO were detected in the precipitates by immunoblotting with anti-FLAG, anti-HA and anti-NEMO antibodies. (c) NF-κB activity from nuclear extracts of overexpressed AMAP1 and control cells with or without IL-1β (2.5 ng/mL) treatment for 30 min. (d) Immunoblots of AMAP1 (whole cell) and p65 (nuclear extract) of HEK293T cells transfected with pcDNA3.1-HA-AMAP1 (AMAP1 overexpression) or pcDNA3.1 (control) and the cumulative, quantitative densitometry data. (e) NF-κB activity from nuclear extracts of shAMAP1 and shControl cells with or without IL-1β (2.5 ng/mL) treatment for 30 min. (f) Immunoblots of AMAP1 (whole cell) and p65 (nuclear extract) of HEK293T cells transfected with AMAP1 shRNA (shAMAP1) or Control shRNA (shControl) with or without IL-1β (2.5 ng/mL) and the cumulative, quantitative densitometry data. The data from three independent experiments are shown. Error bar: ±SD. * P < 0.05, ** P < 0.01 in a two-sided, Student's t-test. Full-length blots are presented in Supplementary Figure 4.
NF-κB activation induces AMAP1 translocation to cytoplasm and augments the AMAP1-IKKβ interaction
In the experiments mentioned above, we sometimes observed that more amount of AMAP1 was detected after treatment with IL-1β (Supplementary Fig. S3a online). This observation coincides with previous report showing LPS significantly boosted AMAP1 protein levels within 15 min23. However, considering that 15 min is too short for synthesis or degradation of proteins, it was highly possible that AMAP1 might not be completely dissolved in cell lysis buffer because AMAP1 can be localized in lipid rafts11,12,13,14 and nucleus27, both of which might not be fully dissolved by 1% Triton X-100 of cell lysis buffer used in this study. Indeed, we also observed similar change of AMAP1 by LPS stimulation, but it disappeared when cells were lysed in 1% SDS cell lysis buffer (Supplementary Fig. S3b online). Therefore, we hypothesized that NF-κB activation induces AMAP1 translocation to the cytoplasm from cell membrane or nucleus, which could result in detection of more AMAP1 in cell lysate by 1% Triton X-100. As shown in Fig. 3a, fractionation by sucrose gradient indicated that IL-1β stimulation induced the translocation of AMAP1 from lipid rafts to the cytoplasm. We also performed confocal immunocytochemistry with using human umbilical vein endothelial cells (HUVECs) since HEK293T cells were not appropriate for observation of intracellular localization due to their large nuclei and thin cytoplasm. It was hard to see AMAP1 staining on cellular membrane, but we observed that IL-1β stimulation induced translocation of AMAP1 in nuclei to the cytoplasm (Fig. 3b). Finally, we repeated the sucrose gradient fractionation with HUVECs, and observed the translocation of AMAP1 from the membrane in response to IL-1β stimulation (Fig. 3c), similarly with HEK293T cells.
Figure 3. IL-1β induces AMAP1 translocation and the AMAP1 and IKKβ interaction.

(a) Representative AMAP1 immunoblots of fractions from sucrose gradient centrifugation of HEK293T cells treated with or without IL-1β (2.5 ng/mL) for 30 min and the cumulative, quantitative densitometry data from three independent experiments are shown. (b) HUVECs were treated with or without IL-1β (2.5 ng/mL) for 30 min and then stained with anti-AMAP1 antibody, WGA and DAPI (n = 3). Scale bar = 50 μm. (c) Representative AMAP1 immunoblots of fractions from sucrose gradient centrifugation of HUVECs treated with or without IL-1β (2.5 ng/mL) for 30 min and the cumulative, quantitative densitometry data from three independent experiments are shown. Representative blots from three immunoprecipitation experiments using anti-AMAP1 (d) and anti-IKKβ (e) antibodies in HUVECs treated with or without IL-1β (2.5 ng/mL) for 30 min. The graph represents the cumulative, quantitative densitometry data of the IKKβ blot in anti-AMAP1 antibody precipitates (d), of the AMAP1 blot in anti-IKKβ antibody precipitates (e). Error bar: ±SD. * P < 0.05, *** P < 0.005 in a two-sided, Student's t-test. Full-length blots are presented in Supplementary Figure 4.
Considering that IKKβ is nearly entirely cytoplasmic protein28,29, the translocation of AMAP1 to cytoplasm in response to IL-1β stimulation raised the question as to whether the activation of NF-κB would strengthen the interaction between AMAP1 and IKKβ. As shown in Fig. 3d and e, more IKKβ was co-precipitated with anti-AMAP1 antibody after IL-1β stimulation, and conversely, more AMAP1 was co-precipitated with anti-IKKβ antibody after IL-1β stimulation, revealing that NF-κB activation enhances the interaction between AMAP1 and IKKβ.
Discussion
Most biological processes require positive and negative regulatory mechanisms to maintain equilibrium. The results from this study suggest a novel mechanism for NF-κB termination and contribute new knowledge to the mechanisms of NF-κB regulation. So far, the re-synthesis of IκB proteins induced by the activation of NF-κB is a widely accepted mechanism for terminating the NF-κB response. These proteins enter the nucleus, remove NF-κB from the DNA and re-localize it to the cytosol30,31,32,33. Here, we show that NF-κB activation induces AMAP1 translocation to cytoplasm from nucleus or lipid rafts and accelerates binding of IKKβ and AMAP1, which inhibits the association of IKKβ and NEMO, eventually leading to inactivation of NF-κB. Taken together, AMAP1 works as an inhibitory feedback mechanism for regulating the NF-κB pathway (Fig. 4).
Figure 4. Proposed model for AMAP1 as a binding protein of IKKβ and negative regulator of NF-κB.
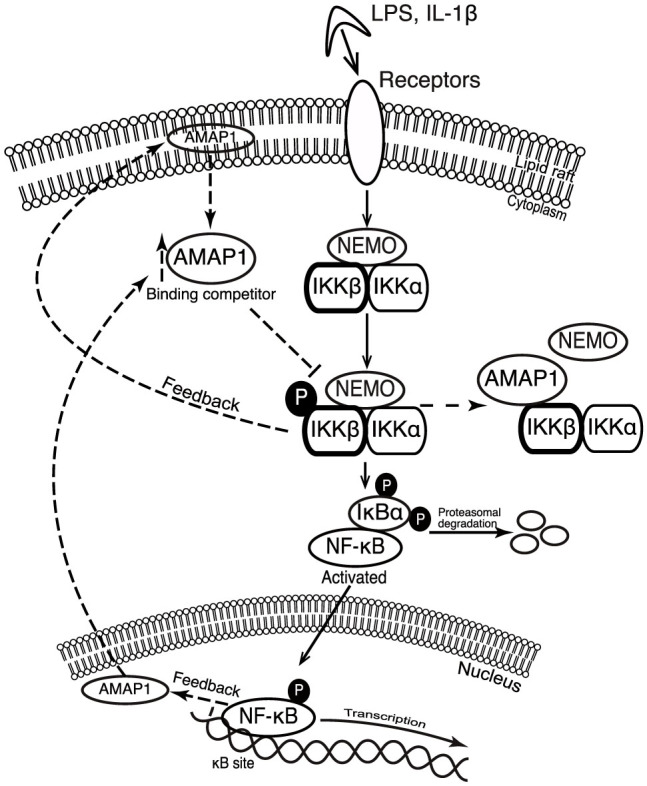
Dashed lines indicate the feedback mechanism.
Recent studies have established strong support for the critical role of NF-κB in cancer. Activation of NF-κB controls multiple cellular processes in cancer, including inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance and radioresistance7,9. Considering the role of AMAP1 explored in this study, augmented expression of AMAP1 observed in cancer might indicate overwhelming NF-κB activation and self-repairing against cancer.
In summary, this study rediscovered AMAP1 as a negative regulator of NF-κB activity in a feedback mechanism, and AMAP1 could be a novel target for treatment or prevention against inflammatory diseases including cancer.
Methods
Reagents
The mammalian expression vectors that were used included pcDNA3.1-HA-AMAP1 and pcDNA3.1 as a control for AMAP1 overexpression, a series of mammalian expression vectors for IKKβ and its mutants (pIRES-AcGFP-hIKK2-WT, pIRES-AcGFP-hIKK2-Δ1, pIRES-AcGFP-hIKK2-Δ2, pIRES-AcGFP-hIKK2-Δ3 and pIRES-AcGFP-hIKK2-Δ4) and a series of mammalian expression vectors for AMAP1 and its mutants (pEBG/AMAP1-FL, pEBG/AMAP1-Bar, pEBG/AMAP1-PH, pEBG/AMAP1-ArfGAP, pEBG/AMAP1-ANK, pEBG/AMAP1-PRD and pEBG/AMAP1-SH3). The pcDNA3-HA-human-NEMO plasmid (Addgene plasmid 13512) to express NEMO was obtained from Professor Kunliang Guan (University of California, San Diego, USA)34. Small hairpin RNA (shRNA) expression constructs for silencing AMAP1 (shAMAP1) and control shRNA (shControl) were purchased from GeneCopoeia, Inc. (Rockville, MD, USA).
The antibodies that were used included anti-IKKβ (Millipore, Billerica, MA, USA); anti-AMAP1 (named anti-DDEF1, Santa Cruz Biotechnology, Dallas, Texas, USA); anti-HA (Sigma-Aldrich, St. Louis, MO, USA); monoclonal ANTI-FLAG® M2 (Sigma-Aldrich); a series of antibodies to anti-IKKβ, anti-phosphor-IκBα, anti-IκBα, anti-p65, anti-β-actin, anti-GAPDH, anti-TATA box-binding protein, anti-vimentin, anti-mouse IgG HRP-linked antibody and anti-rabbit IgG HRP-linked antibody (Cell Signaling Technology, Boston, MA, USA (CST)); anti-mouse IgG HRP and anti-rabbit IgG HRP (eBioscience, Inc. San Diego, CA, USA); Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 594 goat anti-mouse IgG (Invitrogen, Foster City, CA, USA); and Alexa Fluor 488 WGA. The reagents that were used included interleukin-1β (Wako Pure Chemical Industries, Ltd., Osaka, Japan (Wako)), LPS (Sigma-Aldrich) and NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Rockford, IL, USA).
Cell culture
The human embryonic kidney 293T cell line (HEK293T) was maintained in DMEM (Wako) containing 10% FBS (Gibco, Foster City, CA, USA), 1% penicillin G/streptomycin (Sigma-Aldrich) at 37°C under 5% CO2. Human umbilical vein endothelial cells (HUVECs) were maintained in Endothelial cell Basal Medium-2 (EBM-2) (Lonza, Basel, Switzerland) at 37°C under 5% CO2.
Proteomics analysis
A Nano-LC/MALDI–TOF system was used with three types of IKKβ (wild-type IKKβ, a dominant-positive mutant of IKKβ and a dominant-negative mutant of IKKβ) that were overexpressed in HEK293T cells and immunoprecipitated by a specific antibody. Each sample was reduced with 45 mM of DTT (Wako), alkylated with 100 mM of iodoacetamide (Sigma-Aldrich) and digested with 2000 ng of trypsin (Promega, Madison, WI, USA). One-dimensional peptide fractionation was performed with a DiNa Direct Nano-Flow LC/MALDI system (KYA Tech., Tokyo, Japan) using a reverse-phase (RP) trap column (HiQ Sil C18-3, 0.8 mm i.d. × 3 mm) and an RP analytical column (HiQ Sil C18-3 Gradient, 0.15 mm i.d. × 50 mm). The peptides was subjected to the trap column and then to the analytical column using a gradient of 0–50% solvent B in solvent A over 65 minutes [solvent A: 0.1% trifluoroacetic Acid (TFA), 2% acetonitrile; solvent B: 0.1%TFA, 70% acetonitrile] followed by 50–100% solvent B for 15 minutes at a flow rate 200 nL/minute. The RP column eluent was spotted onto a MALDI sample plate using a DiNa Direct Nano-flow LC/MALDI system (KYA Tech.) and analyzed using a 4800 mass spectrometer (Applied Biosystems Inc., Foster City, CA, USA). The peptides were fragmented under collision-induced dissociation conditions to give fragment ions that produced sequence information for the peptide. The software packages used for data acquisition and analysis included GPS explorer (Applied Biosystems Inc.) and Mascot (Matrix Science, Boston, MA, USA), respectively. The parameters of tolerance for the searches were set to 100 ppm for the MS and 0.2 Da for the MS/MS analyses.
Co-immunoprecipitation (Co-IP)
HEK293T cells (8 × 105) were cotransfected with 0.5 μg of AMAP1 plasmids and 0.5 μg of IKKβ plasmids using Effectene reagent (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. After 48 h, the cells were washed with PBS and lysed with Cell Lysis Buffer (CST). One milligram of the lysate was incubated with 1 μg of control IgG, anti-HA-tag or anti-FLAG antibody overnight at 4°C with agitation. Twenty-five microliters of protein G Sepharose (GE Healthcare, Uppsala, Sweden) was added, and the samples were rotated at 4°C for 1 h. The beads were washed three times with Cell Lysis Buffer and one time with Tris buffer pH 7.5. Proteins that were bound to the beads were eluted into SDS sample buffer, and the eluted material was analyzed by immunoblotting using anti-HA, anti-FLAG and anti-GST antibodies. Co-precipitation of endogenous AMAP1 with IKKβ was performed using anti-AMAP1 and anti-IKKβ antibodies.
Measurement of p65 activity
HEK293T cells were lysed with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) for nuclear and cytoplasmic extractions. Transcription factor kits were used to detect the active NF-κΒ in the nuclear extract for NF-κΒ p65 according to the manufacturer's protocol. Briefly, the biotinylated NF-κΒ consensus sequence was bound to streptavidin-coated ELISA wells. Because only the active form of NF-κΒ (p65) binds to the DNA sequence, nonspecific binding was minimized. The p65 that was bound to the consensus sequence was incubated with anti-p65 antibody and then with a secondary, HRP-conjugated antibody. A chemiluminescent substrate was added to the wells, and the resulting signal was detected using a luminometer.
Sucrose gradient
Cells were treated with 5 ng/mL IL-1β or PBS for 30 min and lysed in cell lysis buffer (CST) containing 1 mM PMSF. An identical amount of protein from each sample was mixed with 90% sucrose in MES buffer (6 mL final volume, sucrose concentration 51.7–58.7%) and transferred to a Beckman ultracentrifuge tube. Four milliliters of 35% sucrose followed by 3 mL of 5% sucrose were overlaid, the samples were spun in a Beckman Coulter ultracentrifuge (39,000 rpm; approximately 180,000 × g in a SW40Ti rotor, 20 h), and 26 fractions were collected from the top of the gradient. For the detection of the lipid raft fractions, all fractions were dot-blotted with HRP-labeled cholera toxin B (Sigma-Aldrich) to detect Ganglioside GM1.
Immunofluorescence
HUVECs were grown on a 4-well chamber slide (Thermo Scientific) and treated with or without IL-1β 2.5 ng/mL for 30 min. The cells were washed with PBS, fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. The cells were then incubated with blocking solution (5% goat normal serum in PBS) and labeled with anti-AMAP1 and anti-vimentin antibodies that were coupled to Alexa Fluor 488- and 584-conjugated secondary antibodies. The stained cells were mounted using Mounting Medium with DAPI (Vector Laboratories Inc., Burlingame, CA, USA). Images were captured and exported using a confocal microscope (Olympus FV1000; 60× oil-immersion lens, FLUOVIEW 3.0 software).
Statistical analysis
The data are presented as the mean of ±SD. Groups were compared using a two-tailed, Student's t-test. P < 0.05 was considered significant.
Author Contributions
D.N.-T. and N.A. conceived and designed the research. D.N.-T., M.K., A.Y. and N.A. performed the experiments. A.Y. and A.H. made the constructs of AMAP1. H.S., E.N., K.K., H.A., T.K., T.K. and M.Y. supervised the project.
Supplementary Material
Supplementary Figures S1-S4
Acknowledgments
This work was performed under the Sponsored Research Program contracted between Kyoto University and Otsuka Pharmaceutical Co., Ltd., and was also supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We also thank Prof. Kunliang Guan for the pcDNA3-HA-human-NEMO plasmid.
References
- Oeckinghaus A. & Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspect. Biol. 1, a000034, 10.1101/cshperspect.a000034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 441, 431–436, 10.1038/nature04870 (2006). [DOI] [PubMed] [Google Scholar]
- Rothwarf D. M., Zandi E., Natoli G. & Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395, 297–300, 10.1038/26261 (1998). [DOI] [PubMed] [Google Scholar]
- Mercurio F. et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278, 860–866 (1997). [DOI] [PubMed] [Google Scholar]
- Li Z. W. et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 189, 1839–1845 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A. et al. The AKT/I kappa B kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-kappa B and beta-catenin. Oncogene 24, 1021–1031, 10.1038/sj.onc.1208296 (2005). [DOI] [PubMed] [Google Scholar]
- Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harbor Perspect. Biol. 1, a000141, 10.1101/cshperspect.a000141 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R. et al. High expression levels of IKKalpha and IKKbeta are necessary for the malignant properties of liver cancer. Int. J. Cancer 126, 1263–1274, 10.1002/ijc.24854 (2010). [DOI] [PubMed] [Google Scholar]
- Chaturvedi M. M., Sung B., Yadav V. R., Kannappan R. & Aggarwal B. B. NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene 30, 1615–1630, 10.1038/onc.2010.566 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida N. et al. IKKbeta regulates essential functions of the vascular endothelium through kinase-dependent and -independent pathways. Nat. Commun. 2, 318, 10.1038/ncomms1317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman C., Short S. M., Subramanian R. R., Zetter B. R. & Roberts T. M. DEF-1/ASAP1 is a GTPase-activating protein (GAP) for ARF1 that enhances cell motility through a GAP-dependent mechanism. J. Biol. Chem. 277, 7962–7969, 10.1074/jbc.M109149200 (2002). [DOI] [PubMed] [Google Scholar]
- Inoue H., Ha V. L., Prekeris R. & Randazzo P. A. Arf GTPase-activating protein ASAP1 interacts with Rab11 effector FIP3 and regulates pericentrosomal localization of transferrin receptor-positive recycling endosome. Mol. Biol. Cell. 19, 4224–4237, 10.1091/mbc.E08-03-0290 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Yerushalmi G. M., Grigera P. R. & Parsons J. T. Mislocalization or reduced expression of Arf GTPase-activating protein ASAP1 inhibits cell spreading and migration by influencing Arf1 GTPase cycling. J. Biol. Chem. 280, 8884–8892, 10.1074/jbc.M412200200 (2005). [DOI] [PubMed] [Google Scholar]
- Randazzo P. A. et al. The Arf GTPase-activating protein ASAP1 regulates the actin cytoskeleton. Proc. Natl. Acad. Sci. U. S. A. 97, 4011–4016, 10.1073/pnas.070552297 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J. M. et al. CIN85, a Cbl-interacting protein, is a component of AMAP1-mediated breast cancer invasion machinery. Embo J. 26, 647–656, 10.1038/sj.emboj.7601534 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T. et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene 29, 2393–2403, 10.1038/onc.2010.6 (2010). [DOI] [PubMed] [Google Scholar]
- Onodera Y. et al. Rab5c promotes AMAP1-PRKD2 complex formation to enhance beta1 integrin recycling in EGF-induced cancer invasion. J. Cell. Biol. 197, 983–996, 10.1083/jcb.201201065 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S. et al. Requirement for Arf6 in breast cancer invasive activities. Proc. Natl. Acad. Sci. U. S. A. 101, 6647–6652, 10.1073/pnas.0401753101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y. et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. Embo J. 24, 963–973, 10.1038/sj.emboj.7600588 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabe H., Onodera Y., Mazaki Y. & Hashimoto S. ArfGAP family proteins in cell adhesion, migration and tumor invasion. Curr. Opin. Cell. Biol. 18, 558–564, 10.1016/j.ceb.2006.08.002 (2006). [DOI] [PubMed] [Google Scholar]
- Morishige M. et al. GEP100 links epidermal growth factor receptor signalling to Arf6 activation to induce breast cancer invasion. Nat. Cell. Biol. 10, 85–92, 10.1038/ncb1672 (2008). [DOI] [PubMed] [Google Scholar]
- Boulay P. L., Cotton M., Melancon P. & Claing A. ADP-ribosylation factor 1 controls the activation of the phosphatidylinositol 3-kinase pathway to regulate epidermal growth factor-dependent growth and migration of breast cancer cells. J. Biol. Chem. 283, 36425–36434, 10.1074/jbc.M803603200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A. et al. An ADP ribosylation factor-GTPase activating protein negatively regulates the production of proinflammatory mediators in response to lipopolysaccharide. Cancer Immunol. Immunother. 60, 1439–1446, 10.1007/s00262-011-1048-9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabe H. et al. The EGFR-GEP100-Arf6-AMAP1 signaling pathway specific to breast cancer invasion and metastasis. Traffic 10, 982–993, 10.1111/j.1600-0854.2009.00917.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G. et al. Crystal structure of inhibitor of kappaB kinase beta. Nature 472, 325–330, 10.1038/nature09853 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M. J. et al. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science (New York, N.Y.) 289, 1550–1554 (2000). [DOI] [PubMed] [Google Scholar]
- Lin D. et al. ASAP1, a gene at 8q24, is associated with prostate cancer metastasis. Cancer Res. 68, 4352–4359, 10.1158/0008-5472.CAN-07-5237 (2008). [DOI] [PubMed] [Google Scholar]
- Birbach A. et al. Signaling molecules of the NF-kappa B pathway shuttle constitutively between cytoplasm and nucleus. J. Biol. Chem. 277, 10842–10851, 10.1074/jbc.M112475200 (2002). [DOI] [PubMed] [Google Scholar]
- Verma U. N., Yamamoto Y., Prajapati S. & Gaynor R. B. Nuclear role of I kappa B Kinase-gamma/NF-kappa B essential modulator (IKK gamma/NEMO) in NF-kappa B-dependent gene expression. J. Biol. Chem. 279, 3509–3515, 10.1074/jbc.M309300200 (2004). [DOI] [PubMed] [Google Scholar]
- de Martin R. et al. Cytokine-inducible expression in endothelial cells of an I kappa B alpha-like gene is regulated by NF kappa B. Embo J. 12, 2773–2779 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskill S. et al. Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell 65, 1281–1289 (1991). [DOI] [PubMed] [Google Scholar]
- Sun S. C., Ganchi P. A., Ballard D. W. & Greene W. C. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science (New York, N.Y.) 259, 1912–1915 (1993). [DOI] [PubMed] [Google Scholar]
- Pahl H. L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866, 10.1038/sj.onc.1203239 (1999). [DOI] [PubMed] [Google Scholar]
- Tang E. D. Negative Regulation of the Forkhead Transcription Factor FKHR by Akt. J. Biol. Chem. 274, 16741–16746, 10.1074/jbc.274.24.16741 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1-S4