

Abstract
The cross-regulation of G protein-coupled receptors (GPCRs) plays an important role in the immune response. Studies from several laboratories have suggested that a hierarchy of sensitivities to cross-desensitization exists for the chemoattractant GPCRs. We carried out experiments to study the capacity of the formyl peptide receptor-1 (FPR1) to desensitize chemokine receptors CCR1 and CCR2. Our results show that activation of FPR1 resulted in the desensitization and partial internalization of CCR1, but not CCR2, in both primary human monocytes and HEK293 cells co-expressing CCR1, CCR2, and FPR1 (HR1R2F cells). The desensitization of CCR1 by FPR1 stimulation was not due to the simple depletion of the Ca2+ stores, but was dependent on activation of PKC. Furthermore, we found that the cross-desensitization of CCR1 by FPR1 was associated with CCR1 phosphorylation, and moderate reduction of CCR1 cell surface expression. In contrast, CCR2 was not phosphorylated or internalized following FPR1 activation. Additional studies showed that optimal cross-talk between FPR1 and CCR1 were dependent on the functional activity of PKCβ. These results provide a mechanistic basis for the capacity of certain GPCR ligands to exert rapid and selective cross-inactivation of other chemoattractant receptors, and suggest that FPR1 is able to exert “traffic control” in the migration of inflammatory cells by rapidly inhibiting the cell responses to potentially “low priority” chemoattractants such as CCR1 agonists without inhibiting the response to “higher priority” CCR2 chemoattractants.
Introduction
Several of the chemokine receptors are expressed by leukocytes, and these must collectively coordinate their migration to sites of inflammation and microbial infection, in response to various locally produced chemotactic ligands. The classical chemoattractant receptor (formyl-peptide receptor (FPR1)) and the receptors for chemokines are key participants in the innate and acquired defense systems, and guide leukocytes to sites of inflammation. CCR1 is a chemokine receptor which may play a role in early immune responses and is expressed by T and B cells (1), monocytes and dendritic cells (2), eosinophils (3) and bone marrow progenitor cells (4). CCR1 can be activated by several chemokine ligands including CCL3 and CCL5 (5). While CCR1 is well established to contribute to the accumulation of T cells and monocytes in chronic inflammatory disease states, the role of CCR1 in acute inflammation, or in early acquired immune responses, is not entirely clear. A second chemokine receptor, CCR2, is expressed by monocytes, T cells, NK cells, basophils, mast cells, dendritic cells and B cells (6–8), and is activated primarily by the ligand CCL2. In host defense against bacterial infections, inflammatory monocytes respond rapidly to microbial stimulation by expression of CCR2, and traffic in response to elevated CCL2 secretion. In murine models of infection with bacterial, protozoal, and fungal pathogens, CCR2-mediated recruitment of monocytes is required to suppress pathogen growth. In addition, the high-affinity receptor for bacterial and mitochondrial N-formylpeptides (FPR1), is highly expressed by monocytes and neutrophils (9–11), and the locally produced formyl peptides are potent stimuli to attract monocyte/macrophages and neutrophils to the site of pathogen infection and tissue damage (10, 11).
The proper guidance of leukocytes to the site of inflammation requires that inflammatory cells recognize appropriate chemoattractant signals since agonists for chemoattractant receptors can be produced by multiple sources, including bacteria and host cells within and surrounding the inflammatory stimulus site, and proper guidance of inflammatory cells is required. Our laboratory and others have shown that G protein-coupled receptors exert mutual functional regulation through the process of heterologous desensitization. In this study, we evaluated the capacity of FPR1 to cross-talk with the chemokine receptors CCR1 and CCR2, which chemoattract monocytes. We show that CCR1, CCR2 and FPR1 are co-expressed in primary human monocytes and FPR1 activation rapidly desensitizes CCR1, but not CCR2, in a PKCβ-dependent signaling pathway.
Materials and Methods
Isolation of peripheral blood mononuclear cells
Human peripheral blood mononuclear cells (PBMC) were isolated from blood by using Ficoll-Paque plus® (Amersham Biosciences) density gradient centrifugation. The CD14+ monocytes were isolated using the Midi-MACS® magnetic separation system and CD14+ isolation kit (Miltenyi Biotec, Auburn, CA) from PBMCs according to the manufacturer’s directions. Briefly, cells were incubated with 80 μl of MACS buffer (PBS containing 2mM EDTA and 0.5% BSA) and 20 μl of anti-CD14 beads per 107 cells. After incubation, the cells were washed with buffer, resuspended and loaded onto the LS magnetic column. The columns were then washed 3 times with MACS buffer, and the cells were eluted from the column using RPMI-1640 containing 10% fetal bovine serum, 25 mM Hepes, 2 mM L-glutamine or Hanks Balanced Salt Solution.
Flow Cytometry
Agonist or inhibitor-treated cells were incubated with undiluted goat serum for 30 mins at 4°C, then washed in FACS buffer (1% BSA in PBS). Cells were resuspended with FACS buffer containing anti-CCR1-PE, anti-CD3-APC-Cy7, or anti-CD14-Pacific Blue (BD Biosciences Pharmingen, Palo Alto, CA), or anti-CCR2-PE antibody (R&D systems, Minneapolis, MN), and incubated at 4°C for 30 mins. After washing twice with FACS buffer, the cells were resuspended in FACS buffer, and analyzed using FACS Calibur, FACS Aria, or LSRII flow cytometers (BD Biosciences). A minimun of 10,000 gated cells were analyzed per sample for the relevant cell surface receptor expression. For quantification analysis of CCR1 and CCR2, the mean fluorescent intensities of CCR1-PE or CCR2-PE labeled cells were converted to the number of antibody bound receptors with Quantibrite PE calibration beads (Becton Dickinson, San Jose, CA) according to the manufacturer’s instructions.
Cell Lines and Cell Culture
HR1R2F, human embryonic kidney cells stably expressing pcDNA3.0-FPR1, CCR1, and CCR2, were cultured in D10 medium (DMEM medium containing 10% fetal bovine serum, 25mM Hepes, 2mM L-glutamine) supplemented with 500 μg/ml neomycin. Constructs of human CCR1 and CCR2 were obtained from Missouri S&T cDNA Resource Center (Rolla, MO). Human fMLF receptor FPR1 construct was a gift from Dr. J.M. Wang (NCI-Frederick, MD). HEK293 cells were transfected with the constructs of CCR1, CCR2 and FPR1 using lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were cultured in D10 medium for 2 weeks with 500 μg/ml neomycin. Then the CCR1, CCR2 and FPR1 positive cells were sorted using flow cytometry. Cells with high or low levels of receptor expression were fractioned as 75%–100% and 25–50% of the mean fluorescence intensity of CCR1-PE. For certain experiments, HR1RF cells were further transfected with PKCβ-GFP (OriGene Technologies, Inc., MD) using lipofectamine 2000 (Invitrogen, CA). Cells were cultured in D10 medium supplemented with 1mg/ml neomycin for 1 week, then cells were sorted using Influx cell sorter (Becton Dickinson).
Intracellular calcium flux
Cells were loaded with 5μM fura-2 AM and 2.5mM probenecid (Molecular Probes, Invitrogen, Carlsbad, CA) in either RPM1640 medium or Hanks Balanced Salt Solution with calcium/Mg2+ for 30 mins at room temperature in the dark. After washing the samples, the cells were transferred into constantly stirred cuvettes containing Hanks’ Balanced Salt Solution with Ca2+/Mg2+ and maintained at 37°C in a fluorescence spectrophotometer (Aminco Bowman AB2 spectrofluorometer, SLM Aminco, Rochester, NY or FluoroMax-3, Jobin Yvon Inc. Edison NJ). Stimulants at different concentrations were added to each cuvette at the indicated time points, and intracellular calcium was monitored by measuring Fura-2 fluorescence at 510 nm, using 340/380-nm dual-wavelength excitation. Calibration was performed at the end of analysis by the addition of 0.05% Triton X-100 for maximal Fura-2 ratio (Rmax) and then 5 mM EGTA for minimal Fura-2 ratio (Rmin). The intracellular calcium concentration was then calculated from the 340/380nm fluorescence ratio according to the method of Grynkiewicz et al. (12).
Chemotaxis
Chemotaxis analysis was carried out in response to recombinant CCL3, CCL2 or fMLF (PeproTech, Rocky Hill, NJ) diluted in chemotaxis medium (RPMI-1640 containing 1% bovine serum albumin and 25 mmol/L HEPES pH 7.0). Chemoattractants were placed in the bottom chambers, and cells were placed in the upper chambers, of 48-well chemotaxis chambers (NeuroProbe Inc., Gaithersburg, MD), and the chambers were separated by a polycarbonate membrane (10 μm pore size) precoated with 50 μg/mL of mouse collagen I. After two hours of incubation at 37° chamber, the membranes were removed from the C in a humidified CO2 chemotaxis chambers and non-adherent cells were gently mechanically removed from the upper side of the membrane. The cells on the lower side of the membrane were stained using the HEMA3 staining system (Fisher Diagnostics, Middletown, VA). The number of migrated cells in 3 high-powered fields (×40) was counted by light microscopy. All experiments were repeated at least 3 times. Results are expressed as the chemotaxis index (CI), which is calculated as the ratio of cells per high power (40×) field in the presence of chemoattractant divided by the number of cells per field in the absence of chemoattractant.
Analysis of receptor phosphorylation
HR1R2F cells were serum starved in DMEM medium without phosphate and pyruvate, containing 1% bovine serum albumin for 24 hrs. Following treatment with 1μM fMLF, the cells were lysed in Lysis Buffer (50mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, complete mini protease inhibitors, phosphatase inhibitor cocktail II (Calbiochem, La Jolla, CA) and 1% Triton X-100). After centrifugation at 13,000 rpm for 15 mins, rabbit polyclonal anti-CCR1, or CCR2 antibodies (Santa Cruz Biotechnology Inc.) and washed Protein G-Sepharose (GE, Amersham, Carlsbad, CA) were added to the supernatants for immunoprecipitation. The immunoprecipitated complexes were subjected to SDS-PAGE and western blot analysis with the PhosphoDetect™ Phosphoserine Detection Kit (Millipore). The level of phosphorylation was analyzed by measuring absorbance and using ImageJ 1.43u image analysis software. Following subtraction of background absorbance, the data were normalized with GAPDH as a loading control. The normalized absorbance of the control preparation was arbitrarily set to 100.
Fluorescence Microscopy
HR1R2F cells cultured in 96-well plates were starved in serum-free DMEM for 16 hrs, followed by treatment with 0.5 μM fMLF for 20 min. Cells were fixed and the immunofluorescence was assessed following staining with Becton Dickinson Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit following the manufacturer’s protocol. Phospho-PKD/PKμ (Ser 744/748), phospho-PKCα/β II (Thr638/641), phospho-PKCδ (Thr505); phospho-PKC θ (Thr538), phospho-PKC ζ/λ (Thr410/403) (Cell signaling technology Inc.) were prepared in the BD washing buffer and added to the cells at 4°C, then stained with alexa-488 labeled secondary antibodies for 2 hrs at 4°C, and imaged with a Nikon inverted fluorescence microscope using a 20× objective lens.
Statistical analysis
Statistical analyses were performed using unpaired Student’s t-test or ANOVA.
Results
Activation of FPR1 cross-desensitizes CCR1, but not CCR2, in human PBMCs and monocytes
We have previously demonstrated that activation of receptors for bacterial formyl peptide fMLF in human monocytes led to the functional cross-desensitization of chemokine receptor CCR5 (9). To determine whether FPR1, CCR1 and CCR2 are cross-regulated, we first analyzed the pattern of receptor expression in CD14-positive monocytes. We found that 99.5% of CCR1-positive cells, and 99.7% of CCR2-positive cells, co-express FPR1. Analysis of receptor-induced calcium mobilization responses showed that following pretreatment with fMLF, CCL3 failed to induce a normal calcium flux response, while CCL2 retained a normal response (Fig. 1A), suggesting that the activation of FPR1 cross-desensitizes CCR1 but not CCR2. In reversing the order of agonist administration, neither CCL3 (Fig. 1B) nor CCL2 (Fig. 1C) were able to desensitize the cellular response to fMLF, showing that the cross-talk between FPR1 and CCR1 is not bi-directional.
Figure 1. Pretreatment with fMLF suppresses CCL3 but not CCL2 induced calcium flux in PBMCs.

(A) The calcium response of Fura-2 loaded PBMCs was measured following treatment with 12.5μM fMLF, followed by 15.6 nM CCL3 and then 13.9 nM CCL2. The calcium response of PBMCs was also determined following initial treatment with either 15.6 nM CCL3 (B) or 13.9 nM CCL2 (C) followed by administration of 12.5 μM fMLF.
More extensive analysis (Fig. 2a) showed that CCL3 induced a potent CCR1 calcium mobilization response in monocytes (EC50 of 1.75 nM). However, following FPR1 activation the CCR1 response was substantially reduced (EC50 increased to over 100 nM), which is consistent with FPR1-induced cross desensitization of CCR1. In contrast, the functional activity of CCR2 remained essentially normal following FPR1 activation (Fig. 2b). Similar results were obtained from non-fractionated PBMCs. Additional analysis (Fig. 2C) shows that the fMLF-induced inhibition of the response to CCL3 is fMLF dose-dependent, with greater inhibition achieved with increasing fMLF concentrations.
Figure 2. Suppression of calcium flux and induction of receptor internalization of CCR1 by fMLF pretreatment in monocytes.

Fura-2 AM loaded monocytes were treated with 1 μM fMLF for five minutes and subsequently treated with the designated concentrations of CCL3 (A) or CCL2 (B). Alternatively, cells were pretreated with the designated concentrations of fMLF (C), and subsequently treated with 12.5 nM CCL3. Change in intracellular calcium of untreated cells (solid circle) and fMLF-pretreated cells (triangle) is computed as peak [Ca2+]i – baseline [Ca2+]i and normalized to the change observed at the highest concentration of chemokine without any fMLF pretreatment. Data are displayed as mean ± SD of three to four independent donor responses. *p<0.01, untreated cells vs. fMLF-pretreated cells at each concentration. CCR1 and CCR2 internalization after fMLF treatment in monocytes was measured using flow cytometry analysis (D, E). Monocytes were untreated (circle), treated with 1 μM fMLF (square), or 12.5 nM of CCL3 (panel D, triangle), or 11.1 nM of CCL2 (panel E, triangle). Aliquots of treated monocytes were collected into cold FACS buffer at the indicated times and CCR1 (D) or CCR2 (E) cell surface expression was assessed by flow cytometry. Data is displayed as mean ± SD of three independent donors. *p<0.05, for comparison with baseline of CCR1 or CCR2 cell surface expression.
Because cross-desensitization of GPCRs may be associated with internalization of the target receptor (9, 13, 15, 16), we determined whether the desensitized CCR1 undergoes internalization. The results (Fig. 2D) showed that treatment with 1μM fMLF induced internalization of approximately 35% of CCR1 at 5 min, and 40 to 50% of the receptor was internalized by 30 min (Fig. 2D). In contrast, the cell surface expression level of CCR2 was reduced by approximately 20% at 30 min (Fig. 2E). These data show that the FPR1-induced cross-desensitization of CCR1 was associated with partial internalization of the receptor, while less internalization (or desensitization) of CCR2 was detected.
Both CCR1 and CCR2 utilize phospholipase C and the ER calcium store in monocytes
The major signaling pathway driving calcium mobilization during Gαi-linked GPCR activation is the Gβγ-mediated activation of PLCβ isoforms, with subsequent generation of DAG and IP3. IP3 then releases the ER calcium stores through the activation of ER membrane resident IP3 receptors (17, 18). However, there are multiple phospholipase C isoforms which have been shown to serve as signaling mediators for chemokine receptor-induced calcium mobilization (19, 20). We determined whether the lack of CCR2 desensitization after fMLF pretreatment might be due to the use of distinct calcium signaling mechanisms for CCR2 and CCR1. We pretreated fresh monocytes with U73122, a highly selective phospholipase C antagonist (21) and found (Fig. 3A & B) that the treatment completely inhibited both CCL2 and CCL3-induced calcium mobilization, indicating that phospholipase C is critical for the calcium flux responses mediated by CCR1 and CCR2.
Figure 3. CCR1 and CCR2 calcium signaling in primary human monocytes.

(A, B) Role of phospholipase C in the initiation of CCR1 and CCR2 calcium signaling in human monocytes. Fura-2 AM loaded monocytes were left untreated or pretreated with 10 μM U73122 for 5 min. Calcium responses were elicited by 12.5 nM CCL3 (A, C, E) or 11.1 nM CCL2 (B, D, F). (C, D) Role of extracellular and intracellular calcium stores was determined by addition of 5mM EGTA to the cells 30s before agonist addition. Responses in the presence and absence of 5 mM EGTA were measured. (E, F) Role of the ER calcium stores was determined by 5 min. pretreatment with 50 μM of DBHQ prior to agonist addition. Cells with and without DBHQ were then stimulated by the chemokines. Representative data from one of three independent donors are presented.
The magnitude of the calcium mobilization response can be potentially dependent on both intra- and extracellular stores of calcium. We assessed the response of monocytes in the presence of EGTA, a calcium chelating agent, in order to eliminate the contribution of extracellular calcium stores to the chemokine response. The results show (Fig. 3C & D) that the initial absolute magnitude of the overall CCL3 and CCL2 responses were essentially unchanged in the presence of EGTA. Thus, dissimilar usage of extra and intracellular calcium cannot explain the difference in susceptibility to FPR1-mediated cross-desensitization of CCR1 and CCR2.
We also characterized the role of ER calcium stores in the response through CCR1 and CCR2. We pretreated human monocytes with di-tert-butylhydroquinone (DBHQ), a compound that inhibits the sarco/endoplasmic reticulum calcium (SERCa) -ATPase pump responsible for calcium reuptake into the ER from the cytosol (22, 23). The results (Fig. 3E & F) show that depletion of this store led to nearly complete abrogation of the calcium mobilization response to CCL3 and CCL2. Therefore, CCR1 and CCR2 both utilize the phospholipase C/inositol triphosphate (IP3) pathway to induce ER calcium store mobilization in human monocytes, and the difference in the susceptibility of CCR1 and CCR2 to heterologous desensitization by FPR1 does not appear to be due to the existence of divergent signaling mechanisms for these two receptors.
We then examined the possibility that the rapid CCR1 desensitization by FPR1 might be due to cytosolic calcium store depletion. We pretreated monocytes with fMLF for 5–10 min in the absence of EGTA and assessed the release of ER calcium stores with DBHQ. The results (data not shown) show that there was no significant difference in the release of stored calcium. In fact, these experiments showed a consistent trend toward a more rapid, and more robust, calcium release, when compared to the release in the untreated (baseline) cells. This suggests that the calcium efflux pathways from the ER calcium stores were modestly potentiated by fMLF pretreatment. These results suggest that calcium store depletion is not responsible for the observed heterologous desensitization of CCR1 by FPR1.
Susceptibility of CCR1 and CCR2 to cross-desensitization is not related to the level of cell surface expression
We examined the relationship between receptor expression levels and the calcium mobilization response following activation of CCR1 and CCR2. We found that when utilizing the same agonist concentration, CCL2 induced an approximately 2-fold higher calcium flux response than CCL3 (Fig. 4A), and the expression level of CCR2 was about 2-fold greater than CCR1 (Fig. 4B). To investigate whether the receptor expression level correlates directly with receptor desensitization susceptibility, we generated a cell line designated HR1R2F, which co-expresses CCR1, CCR2 and FPR1. This cell line eliminates the contribution of other potential receptors which might be activated by either fMLF, CCL3 or CCL2. The results show (Fig. 4C & D) that in HR1R2 cells, fMLF desensitized the calcium response of CCR1 but not CCR2. Quantitative flow cytometry showed that the expression levels of CCR1 and CCR2 are similar (data not shown). These results suggest that the relative receptor expression levels of CCR1 and CCR2 are not a major factor in determining the sensitivity to cross-talk with FPR1.
Figure 4. Expression of CCR1 and CCR2 in monocytes, and desensitization of CCR1 but not CCR2 by fMLF in HR1R2F cells.

(A) Fura-2 AM loaded monocytes stimulated with the indicated concentrations of CCL3 (solid triangle) or CCL2 (solid circle). The change in intracellular calcium was calculated and normalized as described above. Data is displayed as mean ± SD of two independent donor responses. *p<0.05 for CCL2 response vs. CCL3 response at each chemokine concentration. (B) The expression levels of CCR1 and CCR2 in monocytes were measured by flow cytometry. Monocytes were labeled with PE-conjugated anti-CCR1 or anti-CCR2 monoclonal antibodies and receptor density was determined based on fluorescence intensity calibration with Quantibrite PE Beads (Becton Dickinson). Data are presented as the expression of receptor as a percentage of CCR2, and are the mean ± SD of three independent donors, * = p<0.001. (C) The calcium flux response with or without 5 μM fMLF pretreatment for 5 min was measured with 5 nM CCL3 or buffer (control). (D) The calcium flux response was measured with or without pretreatment for 5 min with or without 5 μM fMLF, and induced with 5 nM CCL2 or PBS (control).
We further sorted the HR1R2F cells into two populations with high (HR1R2F-H) or low CCR1 (HR1R2F-L) expression (Fig. 5A &B). Flow cytometry shows that these cell populations express essentially identical levels of FPR1 (average mean fluorescence intensity for HR1R2F-H: 381; for HR1R2F-L: 405). The calcium mobilization response to CCL3 is significantly reduced in both HR1R2F-L (Fig. 5C) and HR1R2F-H (Fig. 5D) cell populations following fMLF pre-treatment. We also found that pretreatment with fMLF resulted in reduced efficacy of the CCL3 chemotactic response using either of the HR1R2F cell populations to CCL3 (Fig. 5E). These results suggest that FPR1 is capable of cross-desensitizing CCR1 under conditions where the expression of the receptor is comparatively high. Thus, the sensitivity of CCR1 to FPR1 desensitization is not dependent on the level of its cell surface expression. In contrast, the fMLF pretreatment resulted in a minimal reduction in the chemotaxis response to CCL2 (chemotaxis response for HR1R2F-H control: 6.4 +/− 1.0, with fMLF: 6.2 +/− 0.5; HR1R2F-L control: 6.2 +/− 0.4; with fMLF: 6.1 +/− 0.5).
Figure 5. Cross desensitization of CCR1 by fMLF in HR1R2F-L and HR1R2F-H cell lines.

(A,B) The HR1R2F parental cell line was sorted on the basis of CCR1 expression into high- and low-receptor bearing cell lines. HR1R2F cell lines were immunostained with PE-anti-CCR1 or PE-anti-FPR1, and the relative expression levels were determined by flow cytometry. (C,D) The calcium flux response induced by 6.25 nM CCL3 in 1 μM fMLF-pretreated or non-pretreated HR1R2F-L (C) or HR1R2F-H (D) cell lines was measured. (E) Chemotaxis induced by CCL3 in untreated (open bar) or fMLF-pretreated (grey bar) HR1R2F cells with high or low expression level of CCR1. Chemotaxis is reported as the chemotaxis index. Data are displayed as mean ± SD of three independent experiments, * = p<0.001.
Phosphorylation of CCR1 following FPR1 activation
To better understand the molecular mechanism of the CCR1 desensitization, we examined the receptor phosphorylation of CCR1 and CCR2 after the cells were treated with fMLF, after labeling HR1R2F cells with 32P-orthophosphate. CCR1 and CCR2 were immunoprecipitated from the cell lysates and subjected to SDS-PAGE. The results (Fig. 6) show that CCR1, but not CCR2, was phosphorylated following the fMLF treatment. These results suggest that a signaling process is initiated by FPR1 activation that results in the phosphorylation of CCR1, while CCR2 remains unaffected.
Figure 6. Phosphorylation of CCR1 but not CCR2 after FPR1 activation.
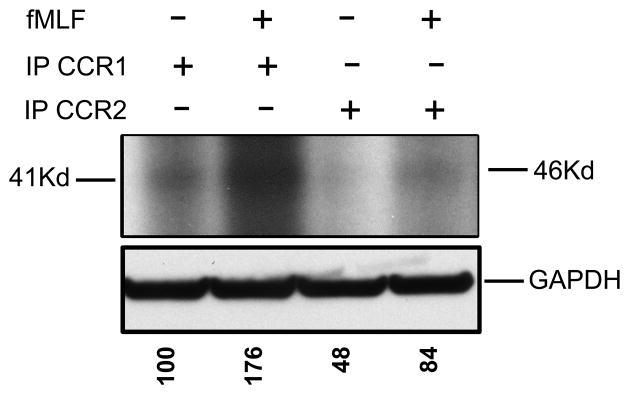
The HR1R2F cells were radiolabeled with 32P-orthophosphate and left untreated or pretreated with 1 μM fMLF. The cell lysates were immunoprecipiated with anti-CCR1 or CCR2 antibodies. The protein samples were dissolved in SDS-loading buffer, and subjected to SDS-PAGE. Phosphorylation of the CCR1 and CCR2 was assessed by autoradiography. The levels of GAPDH were determined by western blot analysis. The normalized absorbance of each of the phosphorylated bands is presented below the GAPDH panel (results are representative of three independent experiments).
Biochemical basis for cross-desensitization of CCR1
We examined the capacity of fMLF pre-treatment to inhibit the capacity of cells to manifest a chemotactic response to CCL3. The results show (Fig. 7A) that HR1R2F cells pre-treated with fMLF exhibited a significant reduction in the chemotactic response over a broad dose range of CCL3 (Fig. 7A). Moreover, we examined the capacity of the PKC inhibitor staurosporine to block the cross-desensitization of CCR1, and the results show (Fig 7B) that staurosporine significantly blocked the fMLF-induced inhibition of the chemotactic response to CCL3 at doses as low as 10nM. These results confirm that staurosporine-sensitive kinases play an important role in the signaling processes which are responsible for FPR1-induced cross-desensitization of CCR1.
Figure 7. Staurosporine inhibits the fMLF-induced cross-desensitization of CCR1-driven chemotactic activity of HR1R2F cells.

(A) The chemotaxis response of HR1R2F cells to CCL3 at the indicated agonist concentrations was determined. Cells were pretreated with (triangles) or without (circles) 1 μM fMLF for 5 min prior to analysis of the chemotactic response to CCL3. (B) HR1R2F cells were pretreated with staurosporine at the designated concentrations for 5 min prior to treatment with 1 μM fMLF for 5 min, and the chemotactic response of these cells to 5nM CCL3 was determined. The results are expressed as the chemotactic response where the cells which were not staurosporine or fMLF treated were set arbitrarily to 100%. *p<0.05 compared to the chemotaxis response in the absence of staurosporine.
In an effort to further characterize the requirement for PKC activity in the cross-desensitization of CCR1, we evaluated the response of the major PKC isoforms following fMLF-activation. HR1R2F cells were treated with 0.5μM fMLF for 20 min, and expression of phosphorylated PKD, PKC α/βII, PKC δ, PKC θ, or PKC ζ/λ was assessed. The results show (Fig. 8) that the fMLF-treatment resulted in the activation of both PKCα/βII and PKCθ. Under the conditions of this experiment we detected little activation of either PKCδ or PKC ζ/λ. Based on these results, we examined the kinetics of PKCβI, βII, and θ activation following fMLF treatment using western blot analysis. Our results show (Fig. 9A) that PKCβI phosphorylation is apparent by 10 min, and the induction of PKCβII and PKCθ phosphorylation is detectable with 0.5 and 1 μM fMLF, respectively (Fig. 9B, C). The results from the western blot analysis also show that these cells constitutively exhibit a detectable level of both PKCβI and PKCβII.
Figure 8. FPR1 mediated activation of PKCβ and PKCθ.

HR1R2F cells were treated with 0 (left panels) or 0.5 μM (right panels) fMLF for 20 min, followed by staining with the designated anti-phospho-PKC antibodies as described in the Materials and Methods section. Nuclei were stained with DAPI. A higher magnification of a selected region of panel G and H is presented (panels U–X). Scale bars are shown in panels Q–T (100 μm) and U–X (25 μm).
Figure 9. FPR1 mediated PKC activation, and role of PKCβ in the cross-phosphorylation of CCR1.

HR1R2F cells were treated with 0.5 μM fMLF for the designated times (A) or doses of fMLF for 20 min (B & C), followed by western blot analysis of phosphor-PKC using antibodies for phosphorylated PKCβ1 (Thr 642) (A), anti-phospho-PKCβII (Thr638/641) (B), and anti-phospho-PKC θ (Thr538) (C) antibodies. GAPDH was also probed as loading controls. (D) Cells were also pretreated with 1 μM PKCβ pseudosubstrate inhibitor (PPβi) 20 min, followed by treatment with vehicle or 0.5 μM fMLF or 10 nM CCL3 for 20 min. The assessment of phosphorylation of CCR1 was then carried out as described in the Materials and Methods. CCR1 and GAPDH were also analyzed to control for loading. The normalized absorbance of each of the phosphorylated bands is presented below the GAPDH panel (panels A & B). The densitometry results for 3 replicate experiments are shown for panel D).
In an effort to assess the role of PKCβ in the cross-talk between FPR1 and CCR1, we examined the role of PKCβ in the FPR1-induced cross-phosphorylation of CCR1. HR1R2F cells were first pre-treated with a PKCβ pseudosubstrate inhibitor (PPβi), and these cells were then induced with either fMLF or CCL3. The results of western blot analysis show (Fig. 9D) that PPβi treatment substantially blocked the fMLF-induced phosphorylation of CCR1. Next, we wished to determine the role of PKCβ in the FPR1-induced cross-desensitization of CCR1 activity. Cells were pre-treated with either staurosporine or PPβi, then the cells were either treated with fMLF or buffer, and the CCL3-induced calcium flux response was determined. The results show (Fig. 10A) that fMLF-treated cells exhibit a weak calcium response to CCL3, and both staurosporine and PPβi partially returned the response to a control level. Additional experiments were carried out with cells transfected to over-express PKCβ-GFP, and the results show (Fig. 10B) that these cells exhibit a reduced calcium mobilization response to CCL3. Moreover, pre-treatment of cells over-expressing PKCβ with fMLF resulted in a response to CCL3 which was somewhat less than the response of normal cells pre-treated with fMLF (Fig. 10B).
Figure 10. PKCβ but not PKCθ mediates fMLF-induced CCR1 desensitization in HR1R2F cells.

(A) Cells were labeled with Fura-2, and then treated with vehicle, staurosporine or PKCβ pseudosubstrate inhibitor (PPβi) for 2 min. A calcium mobilization response was then induced with 5 nM CCL3. (B) Cells expressing GFP or PKCβII-GFP were labeled with Fura-2, followed by treatment of 1 μM fMLF for 2 min, followed by induction with 5 nM CCL3. (C) HR1R2F cells were pretreated with inhibitor vehicle control (ctrl), PKCβ pseudosubstrate inhibitor (PPBi), or PKCθ pseudosubstrate inhibitor (PPθi) for 2 min, followed by treatment with 1μM fMLF (+fMLF) or without fMLF (Ctrl). The calcium response to 5 nM CCL3 was then determined.
Finally, we wished to more directly examine the role of PKCβ in the cross-talk between FPR1 and CCR1. Cells were first treated with the PKCβ-selective inhibitor PPβi, followed by fMLF pre-treatment, and the functional activity of CCR1 was measured by a calcium mobilization response. The results show (Fig. 10C) that the PPβi treatment partially reduced the inhibitory effect of the fMLF treatment, suggesting that PKCβ is required for optimal cross-talk between FPR1 and CCR1. On the other hand, the data also demonstrate that the fMLF-induced cross-desensitization of CCR1 remained normal in cells initially treated with the PKCθ-selective inhibitor PPθi, suggesting that PKCθ does not play an apparent role in this cross-talk interaction.
Discussion
A protective inflammatory response is dependent on the appropriate migration of leukocytes from one anatomic site to another. The traffic of these cells is dependent on their ability to decipher a complex mixture of chemotactic signals that accumulate at a given anatomical location, and “prioritize” among the chemoattractants so that the cells migrate to the most appropriate target site. A variety of processes have been proposed to participate in this “prioritization” including the regulation of adhesion molecules on leukocyte and/or endothelial cell surface (24, 25), the production of cytokines such as TNFα which can implement a “stop” signal (26–28), and homologous desensitization of chemoattractant receptors once the concentration of a given chemoattractant in a tissue microenvironment reaches a sufficient concentration (29). We believe an important additional part of this prioritization process is mediated by the mechanism of heterologous desensitization and selective cross-talk between chemoattractant receptors (Reviewed in 16). Gi protein-coupled chemoattractant receptors exhibit a hierarchy in initiating cross-desensitization, and this is inversely correlated with their susceptibility to heterologous desensitization (30). For example, there is a hierarchy in the cross-desensitization between μ-opioid and chemokine receptors since μ-opioid receptors can desensitize CCR5, but not FPR1 or CXCR4 (31, 32). It appears that FPR1 is a strong desensitizer and is relatively resistant as a target for cross-desensitization (9, 14, 30, 33). Moreover, FPR1 induces a desensitizing signal cascade which is effective for the inactivation of several unrelated chemoattractant receptors, including C5aR, CXCR1, PAFR, BLT1 & 2, CCR5, and CXCR4 (9, 33–36). In contrast, FPR1 is only weakly desensitized following activation of the C5a and CXCL8 receptors, and is not significantly altered by PAF or LTB4 receptor signaling. The present studies show that the activation of FPR1 induces cross-desensitization of CCR1, but not CCR2, and this result agrees with a previous report that fMLF treatment suppresses CCL3 induced calcium flux in human monocytes (9). Our data also suggest that CCR2 is relatively resistant to regulation by cross-desensitization.
The heterologous desensitization phenomenon has now been studied for a number of GPCRs, and there is a great deal of diversity in the biochemical consequences for the target receptor. While many cross-desensitized GPCRs fail to undergo internalization, some GPCRs are at least partially internalized as a consequence of the heterologous desensitization process. For example, the κ-opioid receptor is partially internalized following cross-desensitization induced by CXCR4 (15), and cross-desensitization of CCR5 following activation of FPR1 in monocytes and dendritic cells is associated with virtually complete staurosporine-sensitive internalization (9, 14). The biochemical basis for the internalization of CCR5 in these studies is uncertain since desensitization of Gi-linked GPCRs is typically dependent on PKCs, and not on G protein-coupled receptor kinases (GRKs), as would normally be the case for homologous desensitization (37). Moreover, PKC-mediated phosphorylation is not typically linked to β-arrestin association with the receptor complex (38).
Previous reports have shown that cross-desensitization between Gi-linked GPCRs is dependent on the activation of one or more members of the PKC family of protein kinases (reviewed in 16). For example, the μ-opioid receptor is a strong inducer of PKCζ activation, and this GPCR induces cross-desensitization of both CCR1 and CCR5, and previous reports show that opioid-induced cross-talk with CCR1 and CCR5 are dependent on the activation of this atypical PKC (39, 40). In contrast, the activation of CXCR1 induces activation and rapid outer membrane translocation of PKCε, and this protein kinase appears to mediate cross-desensitization of CCR5 (41). In the present studies we find that fMLF induces strong activation of both PKCβ and PKCε, a result which is consistent with previous reports showing fMLF-induced activation of PKC activity in human neutrophils (42, 43). The data presented here suggests that FPR1-mediated heterologous desensitization of CCR1 is dependent on PKCβ activity. This is based on data showing that inhibition of PKCβ activity substantially reduces cross-talk with CCR1 based on reduced cross-phosphorylation of CCR1, and attenuated inhibition of the CCR1 calcium mobilization response. It should be noted that the selective pseudosubstrate inhibitor PPβi gave substantial inhibition of the cross-phosphorylation of CCR1 (Fig. 9D), but only partially restored the CCL3-induced calcium mobilization response (Fig. 10C). It is possible that optimal cross-talk between FPR1 and CCR1 may require the participation of additional PKC isoforms. Nevertheless, we have previously suggested that the variable use of individual PKC isoforms may allow the GPCRs to selectively cross-talk with targeted GPCRs, and in this way successful cross-talk interactions are dependent on the precise PKC isoform which is activated, and the susceptibility of the target receptor to that kinase (40).
In this regard, since FPR1 cross-desensitizes both CCR5 and CCR1, these receptors may both possess PKCβ-target sequences, while CCR2b does not. CCR2b is most likely the splice variant of CCR2 which is expressed on the outer membrane of these cells, since CCR2a is poorly expressed in part because of the presence of a retention signal motif in the C-terminal region of this CCR2a (44). Previous work has shown that the PKC target site in the C-terminus of CCR5 is at S337 (45), and this corresponds to S341 in CCR1 and S345 in CCR2b. We have not determined the PKC phorphorylation site in CCR1, but it would be expected to be S341 since the adjacent sequence of S341 is homologous to S337 in CCR5, and these sequences are consistent with common PKCβ phosphorylation sites (including arginine at position −3). In contrast, the sequence of CCR2b adjacent to S345 has little homology to either CCR1 or CCR5, and does not contain common sequence elements for a PKC phosphorylation site (46).
In the present study, we considered the possibility that CCR1 and CCR2 might utilize distinct calcium stores, and that this might provide an explanation for the difference in susceptibility to cross-desensitization. We found that both CCR1 and CCR2 use a PLC signaling pathway to activate the same ER calcium store, and fMLF-treated monocytes recover their ER calcium stores quickly. These data demonstrate that calcium store depletion is not responsible for the observed heterologous desensitization of CCR1 by formyl peptide receptors.
The relative resistance of both FPR1 and CCR2 to cross-desensitization may reflect the strength of these receptors in performing leukocyte recruitment during inflammation. We suggest that the production of ligands for these receptors, in the context of many other (potentially competing) chemoattractants, will direct the successful migration of receptor-bearing cells. In the case of FPR1, there are a number of microbial and endogenous agonists that are likely to be present at the sites of inflammation, including mitochondrial components, cathepsin G, and annexin 1, and collectively the localized concentration of FPR1 agonists may approach or surpass micromolar concentrations (reviewed in 47). The cross-talk contributes to the traffic control of leukocytes which must emigrate from a blood vessel, navigate through numerous chemoattractants and regulatory factors, and finally arrive at the correct anatomical location. The capacity of cells to traffic in response to CCR2 or CCR1 ligands is likely to be influenced by the collection of other chemoattractants, and the combination of these diverse ligands is likely to differ depending on the nature of the inflammatory response, the presence of microbial agents, and the tissue type. Indeed, in certain situations, CCR1 may be more important than CCR2 for the mobilization of inflammatory cells. For example, the migration of monocytes to the synovium appears to be directed most strongly by CCR1, and not CCR2 (48). In addition, the resistance to cross-desensitization of more than a single receptor allows the migration to be controlled by more than a single chemoattractant, and this may be an important property of the leukocyte migration since the “collective strength” of more than one chemoattractant may be necessary to successfully guide leukocyte chemotaxis. Studies by Foxman et al. (49) suggest that neutrophils can migrate in sequence to one chemoattractant and then another, allowing for combinations of the attractants to guide cells “in series” toward the eventual target. While it must be pointed out that the process of heterologous desensitization is only one of several mechanisms that may regulate leukocyte chemotaxis in inflammation, we report here that the cross-desensitization process is rapid, and occurs even at low concentrations of the inducing ligand. Both speed and receptor sensitivity are likely to be essential for effective regulation of migration, since the complex mix of chemoattractants may change with time following the initiation of the inflammatory response, especially after leukocytes are recruited to the site and each of these recent immigrant cell populations produces a new collection of their own chemoattractants.
Acknowledgments
The authors thank Dr. Xiaoxuan Fan for expert flow cytometry assistance, and the support of the Temple University Flow Cytometry Facility. We also thank Dr. Todd Quinton for his assistance with the calcium mobilization measurements, and Dr. Satya Kunapuli for access to the SLM Aminco Bowman spectrofluorometer. Finally, we thank Dr. Martin Bootman for advice during these studies.
Abbreviations used in this paper
- DBHQ
2,5-di-tert-butylhydroquinone
- GPCR
G protein coupled receptor
- IP3
inositol 1,4,5-trisphosphate
- NAADP
nicotinic acid dinucleotide phosphate
- PLC
phospholipase C
- PKA
protein kinase A
- PKC
protein kinase C
- U73122
1-[6-[((17b)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione
Footnotes
This work was supported in part by grants from the National Institutes of Health, National Institute on Drug Abuse DA-14230, DA-25532, P30DA-13429, P01DA-23860, and DA-06650.
References
- 1.Murphy PM. Chemokine receptors: structure, function and role in microbial pathogenesis. Cytokine Growth Factor Rev. 1996;7:47–64. doi: 10.1016/1359-6101(96)00009-3. [DOI] [PubMed] [Google Scholar]
- 2.Sozzani S, Luini W, Borsatti A, Polentarutti N, Zhou D, Piemonti L, D’Amico G, Power CA, Wells TN, Gobbi M, Allavena P, Mantovani A. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J Immunol. 1997;159:1993–2000. [PubMed] [Google Scholar]
- 3.Tiffany HL, Alkhatib G, Combadiere C, Berger EA, Murphy PM. CC chemokine receptors 1 and 3 are differentially regulated by IL-5 during maturation of eosinophilic HL-60 cells. J Immunol. 1998;160:1385–1392. [PubMed] [Google Scholar]
- 4.Su S, Mukaida N, Wang J, Zhang Y, Takami A, Nakao S, Matsushima K. Inhibition of immature erythroid progenitor cell proliferation by macrophage inflammatory protein-1alpha by interacting mainly with a C-C chemokine receptor, CCR1. Blood. 1997;90:605–611. [PubMed] [Google Scholar]
- 5.Richardson RM, Pridgen BC, Haribabu B, Snyderman R. Regulation of the human chemokine receptor CCR1. Cross-regulation by CXCR1 and CXCR2. J Biol Chem. 2000;275:9201–9208. doi: 10.1074/jbc.275.13.9201. [DOI] [PubMed] [Google Scholar]
- 6.Frade JM, Mellado M, del Real G, Gutierrez-Ramos JC, Lind P, Martinez AC. Characterization of the CCR2 chemokine receptor: functional CCR2 receptor expression in B cells. J Immunol. 1997;159:5576–5584. [PubMed] [Google Scholar]
- 7.Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J Immunol. 1992;148:2423–2428. [PubMed] [Google Scholar]
- 8.Locati M, Zhou D, Luini W, Evangelista V, Mantovani A, Sozzani S. Rapid induction of arachidonic acid release by monocyte chemotactic protein-1 and related chemokines. Role of Ca2+ influx, synergism with platelet-activating factor and significance for chemotaxis. J Biol Chem. 1994;269:4746–4753. [PubMed] [Google Scholar]
- 9.Shen W, Li B, Wetzel MA, Rogers TJ, Henderson EE, Su SB, Gong W, Le Y, Sargeant R, Dimitrov DS, Oppenheim JJ, Wang JM. Down-regulation of the chemokine receptor CCR5 by activation of chemotactic formyl peptide receptor in human monocytes. Blood. 2000;96:2887–2894. [PubMed] [Google Scholar]
- 10.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 11.Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999;189:657–662. doi: 10.1084/jem.189.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 13.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 14.Le Y, Wetzel MA, Shen W, Gong W, Rogers TJ, Henderson EE, Wang JM. Desensitization of chemokine receptor CCR5 in dendritic cells at the early stage of differentiation by activation of formyl peptide receptors. Clin Immunol. 2001;99:365–372. doi: 10.1006/clim.2001.5021. [DOI] [PubMed] [Google Scholar]
- 15.Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, Adler MW, Rogers TJ. Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the κ-opioid receptor. J Neuroimmunol. 2008;197:114–121. doi: 10.1016/j.jneuroim.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steele AD, Szabo I, Bednar F, Rogers TJ. Interactions between opioid and chemokine receptors: heterologous desensitization. Cytokine Growth Factor Rev. 2002;215:1–14. doi: 10.1016/s1359-6101(02)00007-2. [DOI] [PubMed] [Google Scholar]
- 17.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signaling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 18.Berridge MJ, Bootman MD, Roderick HL. Calcium signaling: dynamics, homeostasis and remodeling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 19.Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D. Roles of phospholipase C β2 in chemoattractant-elicited responses. Proc Natl Acad Sci USA. 1997;94:7971–7975. doi: 10.1073/pnas.94.15.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-β2 and –β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 21.Smith RJ, Sam LM, Justen JM, Bundy GL, Bala GA, Bleasdale JE. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J Pharmacol Exp Ther. 1990;253:688–697. [PubMed] [Google Scholar]
- 22.Moore GA, McConkey DJ, Kass GE, O’Brien PJ, Orrenius S. 2,5-Di-(tert-butyl)-1,4-benzohydroquinone rapidly elevates cytosolic Ca2+ concentration by mobilizing the inositol 1,4,5-trisphosphate-sensitive Ca2+ pool. FEBS Lett. 1987;224:331–336. [Google Scholar]
- 23.Kass GE, Duddy SK, Moore GA, Orrenius S. 2,5-Di(tert-butyl)-1,4-benzohydroquinone - a novel inhibitor of liver microsomal Ca2+ sequestration. J Biol Chem. 1989;264:15192–15198. [Google Scholar]
- 24.Hecht I, Cahalon L, Hershkoviz R, Lahat A, Franitza S, Lider O. Heterologous desensitization of T cell functions by CCR5 and CXCR4 ligands: inhibition of cellular signaling, adhesion and chemotaxis. Int Immunol. 2003;15:29–38. doi: 10.1093/intimm/dxg002. [DOI] [PubMed] [Google Scholar]
- 25.Heit B, Colarusso P, Kubes P. Fundementally different roles for LFA-1, Mac-1 and α4-integrin in neutrophil chemotaxis. J Cell Sci. 2005;118:5205–5220. doi: 10.1242/jcs.02632. [DOI] [PubMed] [Google Scholar]
- 26.Franitza S, Hershkoviz R, Kam N, Lichtenstein N, Vaday GG, Alon R, Lider O. TNF-α associated with extracellular matrix fibinectin provides a stop signal for chemotactically migrating T cells. J Immunol. 2000;165:2738–2747. doi: 10.4049/jimmunol.165.5.2738. [DOI] [PubMed] [Google Scholar]
- 27.Vaday GG, Franitza S, Schor H, Hecht I, Brill A, Cahalon L, Hershkoviz R, Lider O. Combinatorial signals by inflammatory cytokines and chemokines mediate leukocyte interactions with extracellular matrix. J Leukoc Biol. 2001;69:885–892. [PubMed] [Google Scholar]
- 28.Lokuta MA, Huttenlocher A. TNF-α promotes a stop signal that inhibits neutrophil polarization and migration via a p38 MAPK pathway. J Leukoc Biol. 2005;78:210–219. doi: 10.1189/jlb.0205067. [DOI] [PubMed] [Google Scholar]
- 29.Lin F, Butcher EC. Modeling the role of homologous receptor desensitization in cell gradient sensing. J Immunol. 2008;181:8335–8343. doi: 10.4049/jimmunol.181.12.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali H, Richardson RM, Haribabu B, Snyderman R. Chemoattractant receptor cross-desensitization. J Biol Chem. 1999;274:6027–6030. doi: 10.1074/jbc.274.10.6027. [DOI] [PubMed] [Google Scholar]
- 31.Grimm MC, Ben Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, Ali H, Richardson R, Snyderman R, Oppenheim JJ. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med. 1998;188:317–325. doi: 10.1084/jem.188.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OMZ, Oppenheim JJ, Rogers TJ. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol. 2003;74:1074–1082. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- 33.Li BQ, Wetzel MA, Mikovits JA, Henderson EE, Rogers TJ, Gong W, Le Y, Ruscetti FW, Wang JM. The synthetic peptide WKYMVm attenuates the function of the chemokine receptors CCR5 and CXCR4 through activation of formyl peptide receptor-like 1. Blood. 2001;97:2941–2947. doi: 10.1182/blood.v97.10.2941. [DOI] [PubMed] [Google Scholar]
- 34.Tomhave ED, Richardson RM, Didsbury JR, Menard L, Snyderman R, Ali H. Cross-desensitization of receptors for peptide chemoattractants. Characterization of a new form of leukocyte regulation. J Immunol. 1994;153:3267–3275. [PubMed] [Google Scholar]
- 35.Blackwood RA, Hartiala KT, Kwoh EE, Transue AT, Brower RC. Unidirectional heterologous receptor desensitization between both the fMLP and C5a receptor and the IL-8 receptor. J Leukoc Biol. 60:88–93. doi: 10.1002/jlb.60.1.88. [DOI] [PubMed] [Google Scholar]
- 36.Campbell JJ, Foxman EF, Butcher EC. Chemoattractant receptor cross-talk as a regulatory mechanism in leukocyte adhesion and migration. Eur J Immunol. 1997;27:2571–2578. doi: 10.1002/eji.1830271016. [DOI] [PubMed] [Google Scholar]
- 37.Bohm SK, Grady EF, Bunnett NW. Regulatory mechanisms that modulate signaling by G protein-coupled receptors. Biochem J. 1997;322:1–18. doi: 10.1042/bj3220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 39.Zhang N, Hodge D, Rogers TJ, Oppenheim JJ. Ca2+-independent protein kinase Cs mediate heterologous desensitization of leukocyte chemokine receptors by opioid receptors. J Biol Chem. 2003;278:12729–12736. doi: 10.1074/jbc.M300430200. [DOI] [PubMed] [Google Scholar]
- 40.Song C, Rahim RT, Davey PC, Bednar F, Bardi G, Zhang L, Zhang N, Oppenheim JJ, Rogers TJ. Protein kinase Czeta mediates micro-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. J Biol Chem. 2011;286:20354–20365. doi: 10.1074/jbc.M110.177303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nasser MW, Marjoram RJ, Brown SL, Richardson RM, Nasser MW, Marjoram RJ, Brown SL, Richardson RM. Cross-desensitization among CXCR1, CXCR2, and CCR5: role of protein kinase C-epsilon. J Immunol. 2005;174:6927–6933. doi: 10.4049/jimmunol.174.11.6927. [DOI] [PubMed] [Google Scholar]
- 42.Spisani S, Falzarano S, Traniello S, Nalli M, Selvatici R. A ‘pure’ chemoattractant formylpeptide analogue triggers a specific signalling pathway in human neutrophil chemotaxis. FEBS J. 2005;272:883–891. doi: 10.1111/j.1742-4658.2004.04497.x. [DOI] [PubMed] [Google Scholar]
- 43.Selvatici R, Falzarano S, Mollica A, Spisani S. Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur J Pharmacol. 2006;534:1–11. doi: 10.1016/j.ejphar.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 44.Wong LM, Myers SJ, Tsou CL, Gosling J, Arai H, Charo IF. Organization and differentiatial expression of the human monocyte chemoattractant protein 1 receptor gene. J Biol Chem. 1997;272:1038–1045. doi: 10.1074/jbc.272.2.1038. [DOI] [PubMed] [Google Scholar]
- 45.Pollok-Kopp B, Schwarze K, Baradari VK, Oppermann M. Analysis of ligand-stimulated CC chemokine receptor 5 (CCR5) phosphorylation in intact cells using phosphosite-specific antibodies. J Biol Chem. 2003;278:2190–2198. doi: 10.1074/jbc.M209844200. [DOI] [PubMed] [Google Scholar]
- 46.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 47.Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Grow Fact Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 48.Lebre MC, Vergunst CE, Choi IYK, Aarrass S, Oliveira ASF, Wyant T, Horuk R, Reedquist KA, Tak PP. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rhematoid arthritis. Plos One. 2011;6:e21772. doi: 10.1371/journal.pone.0021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]