

Abstract
Objective:
To examine whether β-amyloid (Aβ) and APOE ε4 status independently contribute or interact to influence longitudinal cognitive decline in clinically normal older individuals (CN).
Methods:
Data from 490 CNs were aggregated across 3 observational cohort studies (Harvard Aging Brain Study, Alzheimer's Disease Neuroimaging Initiative, and Australian Imaging Biomarkers and Lifestyle Study of Ageing; median age = 75.0 years, 255 female), and the contributions of APOE ε4 and Aβ on longitudinal change over a median of 1.49 years were examined. Cognitive decline was assessed with the Mini-Mental State Examination (MMSE) and Logical Memory (immediate and delayed recall scores).
Results:
High Aβ participants were more likely to be APOE ε4+ than low Aβ participants. CNs who were both high Aβ and APOE ε4+ showed greater decline in Logical Memory immediate recall (p < 0.087), Logical Memory delayed recall (p < 0.024), and MMSE (p < 0.034) compared to all other groups (low Aβ/APOE ε4−, low Aβ/APOE ε4+, and high Aβ/APOE ε4−). No other pairwise contrast was significant for any cognitive measure.
Conclusions:
Clinically normal individuals who are APOE ε4+ and have high Aβ showed the highest cognitive decline. These results suggest that Aβ and APOE ε4 are not redundant contributors of decline in aging but rather interact to promote decline during the short follow-up period examined in this study. Longer follow-up periods will be essential to fully elucidate the influence of Alzheimer disease risk factors on cognitive decline in aging.
Approximately one-third of clinically normal older individuals (CN) have evidence of β-amyloid (Aβ) accumulation,1 a pathology linked to Alzheimer disease (AD). Aβ accumulation in CNs is associated with subtle reductions in cross-sectional cognition2 and heightened risk of subsequent clinical impairment.3–5 A major risk factor for AD is the presence of the APOE ε4 allele, which is associated with a decade or more decrease in AD symptom onset.6 Although APOE ε4 influences AD risk through increased Aβ accumulation,7,8 it is also possible that APOE ε4 additionally has an independent contribution, or interacts with Aβ, to influence decline. However, the few recent studies that have simultaneously investigated Aβ and APOE ε4 in CNs with respect to cognitive or functional outcomes have not converged to reveal a consistent pattern.9–12
As secondary prevention trials are being planned in asymptomatic at-risk populations, it is critical to understand the relative contributions of both Aβ and APOE ε4 status on short-term cognitive decline, as these factors may influence eligibility criteria and need for stratification or covariate adjustment in investigating treatment effects. Large cohorts of CNs with longitudinal data are required to evaluate potential interactions in these risk factors. We explore the influence of Aβ and APOE ε4 on decline in a large cohort of CNs by combining data from 3 independent AD studies that included PET amyloid imaging: Harvard Aging Brain Study (HABS), Alzheimer's Disease Neuroimaging Initiative (ADNI), and Australian Imaging Biomarkers and Lifestyle Study of Ageinga (AIBL).
METHODS
Inclusion criteria.
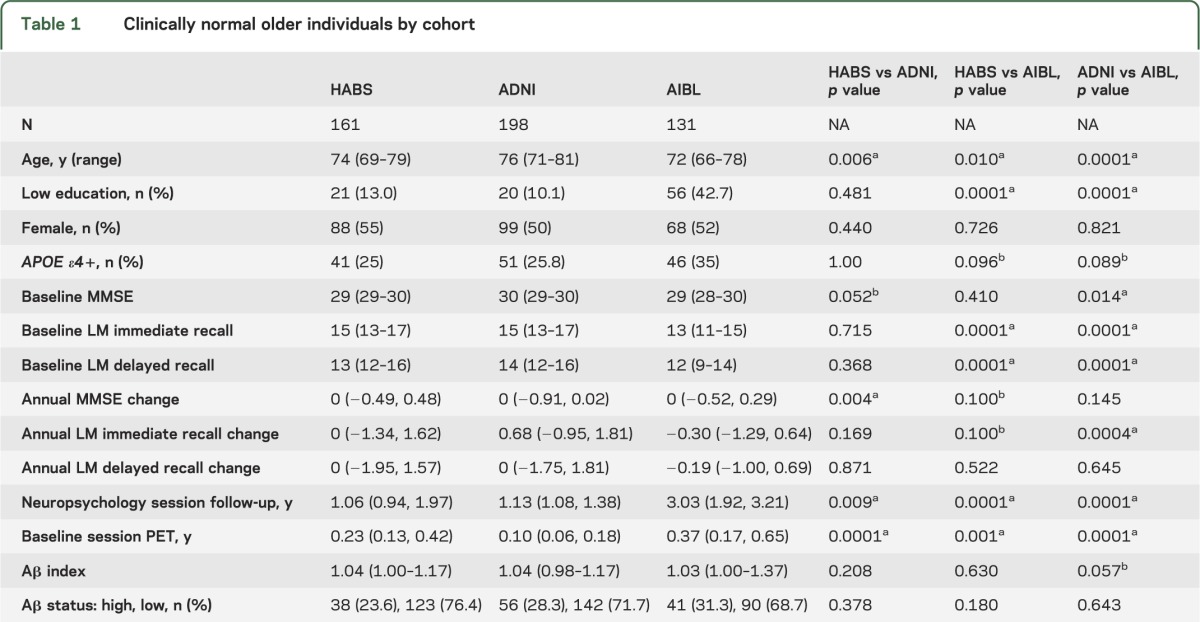
Cohort-specific inclusion criteria can be found in previous publications.13–15 Enrollment for CNs used in these analyses began in 2010 for HABS, 2010 for ADNI (when florbetapir imaging was included; some ADNI CNs were also previously enrolled in an earlier phase of the study), and 2006 for AIBL. All participants included in this analysis were categorized as clinically normal, had a Clinical Dementia Rating (CDR) 0, and Mini-Mental State Examination (MMSE) ≥26 at the baseline testing session used in these analyses. Participants were included regardless of subjective complaint status, as long as their CDR global score was 0. Participants were included if they completed a PET amyloid imaging scan within 1 year of a testing session (referred to here as baseline), had at least 1 follow-up cognitive session after amyloid imaging, and had APOE genotyping. APOE 2/4 CNs were excluded (<2%), given that the effect of this genotype on AD risk is unclear. Testing sessions greater than 1 year prior to amyloid imaging data were discarded (thus, baseline sessions in this analysis are not necessarily the cohort-defined baseline). Overall, 490 CNs were included in these analyses (table 1).
Table 1.
Clinically normal older individuals by cohort
Standard protocol approvals, registrations, and consents.
Institutional review boards approved study procedures across participating institutions. Written informed consent was obtained from all participants.
Cognitive outcomes.
Change in the MMSE and Logical Memory I and IIa (i.e., immediate and delayed recall) were examined (the only cognitive scores available across all cohorts). HABS CNs completed these tests approximately every year, whereas AIBL CNs underwent testing every 1.5 years. ADNI CNs completed these tests approximately every year, with an additional MMSE assessment 6 months after the ADNI-defined baseline visit. All available testing sessions following the analysis-defined baseline session were used (HABS: n = 86 completed 2 visits, n = 67 3 visits, and n = 8 4 visits. ADNI: n = 152 2 visits, n = 31 3 visits, and n = 15 4 visits. AIBL: n = 41 2 visits and n = 90 3 visits). Baseline scores for MMSE and Logical Memory were taken from the same analysis-defined baseline session for each CN.
Structural MRI.
Details regarding MRI acquisition for ADNI and AIBL have been described elsewhere.16,17 For HABS, MRI scanning was completed at the Massachusetts General Hospital Martinos Center on a Siemens TIM Trio 3T System with a 12-channel head coil. Structural T1-weighted volumetric magnetization-prepared, rapid acquisition gradient echo scans were collected (repetition time/echo time/inversion time = 6,400/2.8/900 msec, flip angle = 8°, 1 × 1 × 1.2 mm resolution).
Structural scans were used to define regions of interest (ROIs) to derive global Aβ indices. To define ROIs in each participant's native space, structural scans from HABS and AIBL were processed in our laboratory using FreeSurfer v5.1.18,19 Since extracted amyloid PET data from ADNI are available online using FreeSurfer-derived ROIs, our laboratory did not reprocess ADNI MRI data.
Aβ imaging.
Aβ status was derived using Pittsburgh compound B (PiB) for HABS/AIBL and florbetapir for ADNI. Details regarding Aβ imaging acquisition and processing are available elsewhere.15,17,20 To increase consistency across cohorts, all data were analyzed as standard uptake value ratios, using a whole cerebellum reference region.
ADNI.
Global florbetapir index values were downloaded (http://adni.loni.ucla.edu/data-samples/access-data/). These values were derived using a previously published pipeline, using summed images with additional postprocessing to account for differences that may exist in data collected at different ADNI sites.20,21 In brief, summed images corresponding to 50–70 minutes postinjection were coregistered to each participant's MRI using SPM5, enabling alignment of FreeSurfer ROIs to the summed PET image.22,23 PET values were extracted across 4 large bilateral regions: frontal (orbitofrontal cortex/inferior frontal gyrus/middle frontal gyrus/superior frontal gyrus/frontal pole), cingulate (anterior cingulate/posterior cingulate/isthmus cingulate), parietal (precuneus/inferior parietal cortex/superior parietal cortex/supramarginal gyrus), and lateral temporal (middle temporal/superior temporal gyri). These values were averaged and normalized by the whole cerebellum to yield a global Aβ index for each participant.
HABS.
PiB-PET data were collected 0–60 minutes postinjection. These images were realigned and frames corresponding to 40–60 minutes postinjection were summed. The first 8 minutes of data were summed and used to guide coregistration between PET and MRI using FreeSurfer's bbregister, a surface-based coregistration algorithm. ROI extraction, averaging, and normalization were identical to the process implemented in ADNI.
AIBL.
PiB summed images corresponding to 50–70 minutes postinjection were downloaded (https://ida.loni.ucla.edu/login.jsp?project=AIBL) and coregistered to each participant's structural MRI scan using FreeSurfer's bbregister. ROI extraction, averaging, and normalization were identical to the process implemented in ADNI.
Gaussian mixture modeling.
We employed a Gaussian mixture model approach to classify CNs as high or low Aβ (e-analysis on the Neurology® Web site at Neurology.org). In brief, CNs with greater than 50% probability of belonging to their cohort's high Aβ distribution were labeled high Aβ, whereas CNs with greater than 50% probability of belonging to their cohort's low Aβ distribution were classified as low Aβ.
Statistical models.
Analyses were performed using R v3.0. Group differences were assessed with Wilcoxon rank sum tests for continuous variables and χ2 tests for dichotomous variables.
To investigate contributions of Aβ and APOE ε4 to longitudinal change in MMSE and Logical Memory scores, we implemented 2 linear mixed regression models for each cognitive outcome (e-methods): inclusion of interactions of Aβ with time and APOE ε4 status with time in the same model and inclusion of interactions of Aβ with time and APOE ε4 status with time along with their joint interaction with time. All models included main effects of baseline age, education, sex, and cohort and their interactions with time, as well as a random intercept for each participant. To explore interactions between Aβ and APOE ε4 status, decline across all pairwise group contrasts was performed (low Aβ/APOE ε4−, low Aβ/APOE ε4+, high Aβ/APOE ε4−, and high Aβ/APOE ε4+). All p values were 2-sided, and no multiple comparisons correction was performed.
RESULTS
Cohort characteristics.
We examined 490 CNs with a median neuropsychological session follow-up period of 1.49 years (interquartile range 1.07–2.24 years; table 1). Compared to ADNI and HABS, AIBL CNs were younger and had lower education. ADNI CNs were older than HABS. There were more APOE ε4 carriers in AIBL compared to the other cohorts (AIBL enriches by APOE ε4 status17). Baseline MMSE was higher in ADNI compared to AIBL and HABS. Logical Memory scores were lower in AIBL than ADNI and HABS. HABS had the shortest follow-up duration, whereas AIBL had the longest follow-up duration.
Aβ distributions.
Gaussian mixture models were fit to each cohort's distribution of Aβ index values, and in each cohort a 2-distribution model was optimal (figure e-1). Classification using this method revealed a similar proportion of high and low Aβ CNs across cohorts; however, classification certainty was lowest in ADNI (florbetapir) compared to ADNI/AIBL (PiB) (e-analysis and figure e-2).
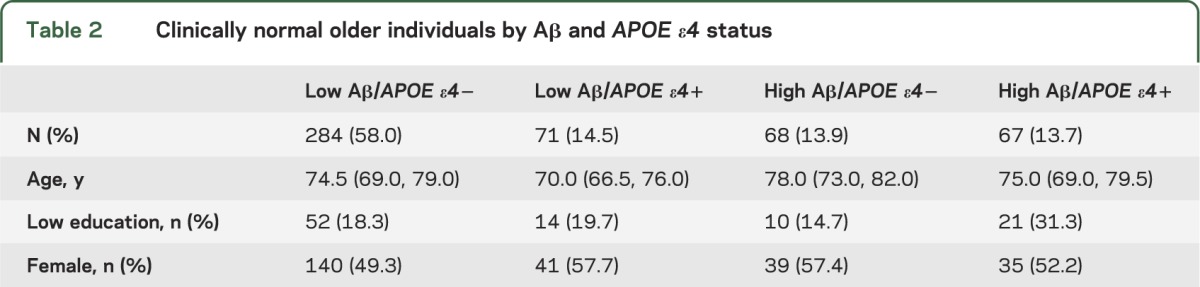
Based on this Aβ classification, CNs were divided into groups based on joint Aβ and APOE ε4 status (table 2). As expected, high Aβ CNs were more likely to be APOE ε4+ (p < 0.0001). Low Aβ/APOE ε4+ CNs were younger than all other groups (p values < 0.002) and high Aβ/APOE ε4− CNs were older than all other groups (p values < 0.02). Low Aβ/APOE ε4− participants were also younger than high Aβ/APOE ε4− (p = 0.0002). High Aβ/APOE ε4+ CNs had lower education than low Aβ/APOE ε4− (p = 0.028) and high Aβ/APOE ε4− CNs (p = 0.036). There were no group differences in sex.
Table 2.
Clinically normal older individuals by Aβ and APOE ε4 status
Longitudinal change models.
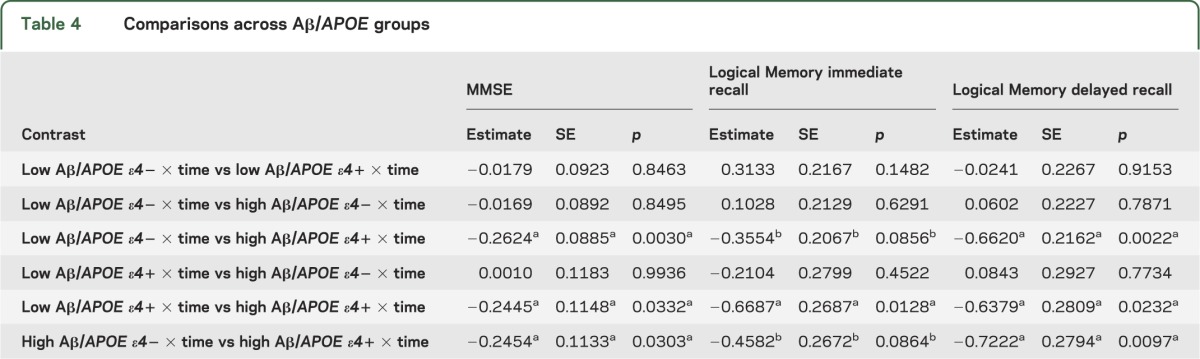
Terms reflecting associations with longitudinal cognitive change are summarized in table 3. In models containing interactions between APOE ε4 and time as well as Aβ and time as simultaneous predictors, neither term was significant. However, when the 3-way interaction between APOE, Aβ, and time was included, this term was significant for both Logical Memory immediate and delayed recall. To understand these interactions, we directly contrasted groups based on Aβ and APOE ε4 status (low Aβ/APOE ε4−, low Aβ/APOE ε4+, high Aβ/APOE ε4−, and high Aβ/APOE ε4+ groups; figure 1 and table 4). High Aβ/APOE ε4+ CNs showed significantly greater decline than all other groups for Logical Memory delayed recall, whereas all pairwise contrasts with the high Aβ/APOE ε4+ group were significant or marginally significant for Logical Memory immediate recall. Although the interaction term between Aβ and APOE ε4 status was not significant (p = 0.11) for MMSE, all pairwise contrasts with high Aβ/APOE ε4+ were significant. No other pairwise difference was significant.
Table 3.
Summary of linear mixed models
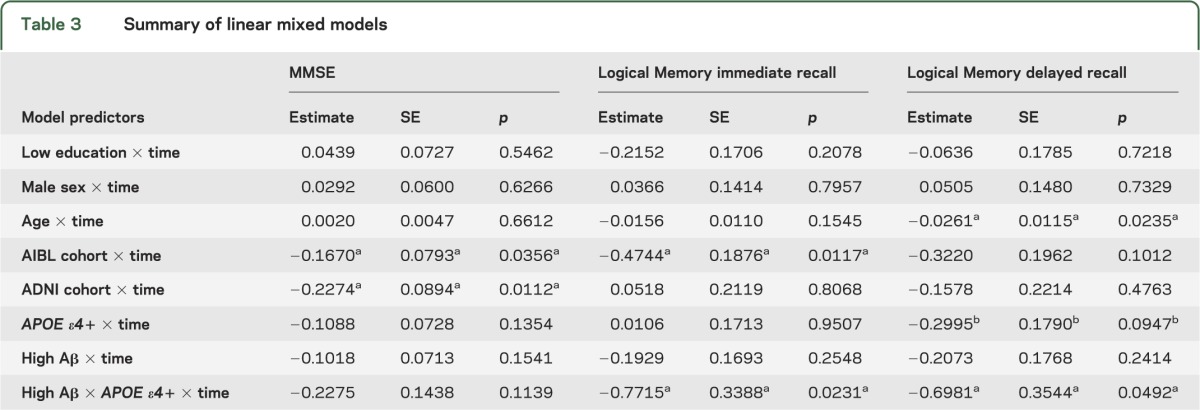
Figure 1. Decline in Logical Memory by joint APOE ε4 and Aβ status.
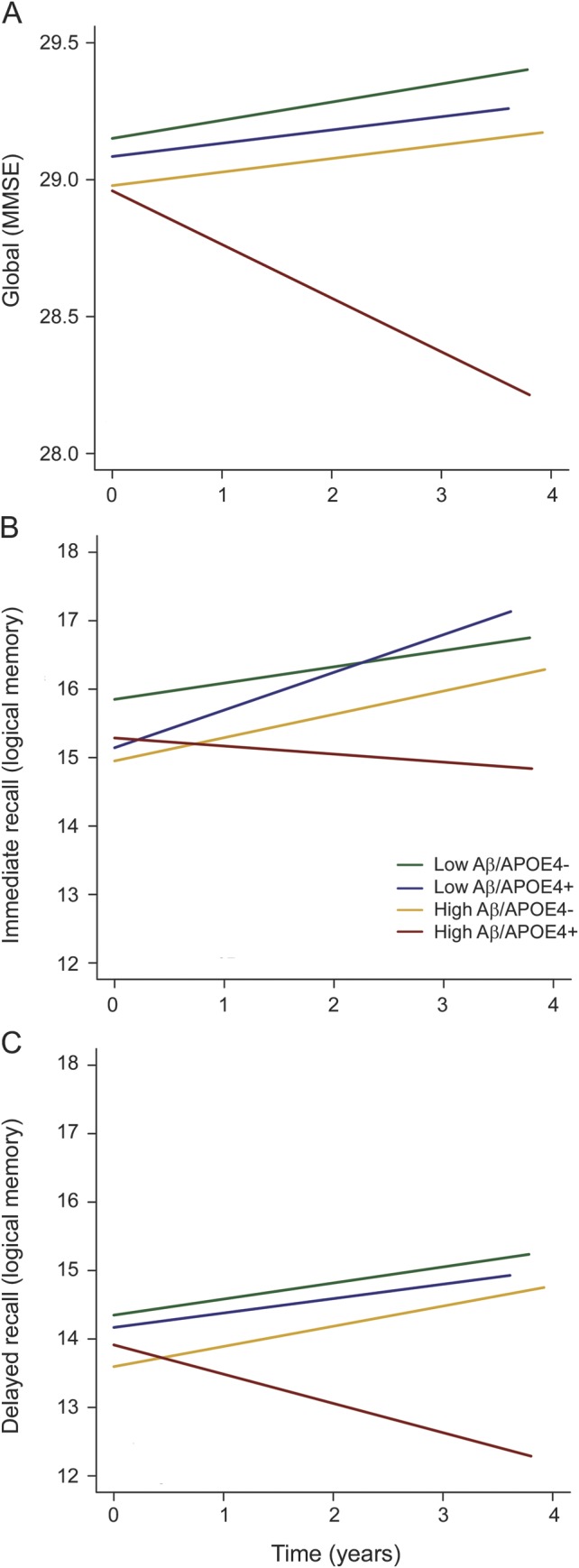
Estimates from linear mixed models predicting change in Logical Memory scores for groups based on joint APOE ε4/Aβ status. Decline in the high Aβ/APOE ε4+ group is greater than other groups for (A) Mini-Mental State Examination (MMSE), (B) Logical Memory immediate recall, and (C) delayed recall. Each plotted line extends to the longest follow-up period within that group. Aβ = β-amyloid.
Table 4.
Comparisons across Aβ/APOE groups
DISCUSSION
In a large dataset of clinically normal individuals, we found that both Aβ and APOE ε4 are contributors to cognitive decline over a short follow-up period. Specifically, there were significant interactions between Aβ and APOE ε4 status in predicting change on both immediate and delayed Logical Memory scores and a marginally significant interaction for change in MMSE. Across all 3 measures, this interaction revealed greater decline in high Aβ/APOE ε4+ participants, whereas minimal decline was present in the other groups.
Although our ability to identify independent contributions of Aβ and APOE ε4 may be limited by the high association between these risk factors, the presence of an interaction between Aβ and APOE ε4 status in predicting longitudinal decline suggests that these variables do not merely reflect redundant sources of information. There are several possible mechanisms that may promote cognitive decline specifically in high Aβ/APOE ε4+ CNs. First, it is possible that APOE ε4 may have Aβ-independent effects on neuronal integrity, and that these effects may make individuals more vulnerable to toxic effects of Aβ. For instance, these Aβ-independent effects of APOE ε4 may impact synaptogenesis, synaptic plasticity, tau phosphorylation, mitochondrial activity, neuroinflammation, or neurodevelopment.24 The presence of Aβ-independent effects of APOE ε4 is further supported by functional imaging differences in young human APOE ε4+ (before the age at which Aβ accumulation occurs)25–27 as well as in older APOE ε4+ participants lacking evidence of fibrillar Aβ accumulation.28,29 In isolation, effects of APOE ε4 may not be consequential to cognition, but become consequential when co-occurring with elevated Aβ. Second, it is possible that Aβ and APOE ε4 in conjunction impart greater levels of neuronal toxicity. Given that the apoE4 protein is less effective than apoE3/2 in responding to neuronal injury,30 neural injury related to Aβ may be enhanced within APOE ε4 carriers. Third, it is possible that high Aβ/APOE ε4+ CNs have had underlying Aβ for longer than high Aβ/APOE ε4− CNs and are therefore further along the AD trajectory than their APOE ε4− counterparts. Studies that examine incident Aβ positivity will be essential to understand the contribution of Aβ positivity duration on longitudinal cognitive decline. Finally, high Aβ/APOE ε4+ CNs may have higher quantities of underlying pathology than high Aβ/APOE ε4− CNs, in terms of neurofibrillary tangles,31 cerebrovascular disease,32 vascular Aβ,33 or cerebral Aβ.34 Future studies that incorporate multiple markers of pathology in addition to Aβ and APOE ε4 status will be crucial to understanding mechanisms underlying cognitive decline within at-risk CNs.
Our finding of greater decline in high Aβ/APOE ε4+ CNs is seemingly at odds with 2 recent studies examining longitudinal change within CNs. Specifically, one study showed independent effects of Aβ and APOE ε4 in memory decline11 while another showed that Aβ, but not APOE ε4, was independently associated with functional decline.12 However, given the smaller sample sizes in these studies compared to the current analysis (Lim et al.11: n = 141; Roe et al.12: n = 201), it is possible that these analyses were underpowered to detect an interaction between Aβ and APOE ε4. Although an interaction between Aβ and APOE ε4 status was not identified in either aforementioned longitudinal study, studies examining cross-sectional relationships between Aβ, APOE ε4, and cognition within CNs have suggested the presence of an interaction.9,10 Although recent longitudinal datasets may be limited by smaller sample sizes than cross-sectional studies, a longitudinal design may be advantageous since it accounts for individual differences that are not due to pathologic processes. By combining data across multiple observational studies, we were able to aggregate a longitudinal dataset large enough to enable investigation of the interaction between Aβ and APOE ε4 status within CNs.
Although combining data across HABS, ADNI, and AIBL provided a large number of CNs with known APOE ε4 and Aβ status, it is important to consider study design differences that may complicate the interpretation of our results. For instance, AIBL specifically recruited CNs with subjective cognitive complaints, and previous work suggests that associations between AD markers may be strongest in this group.35 Investigating the contribution of subjective cognitive complaints was beyond the scope of the current article but is currently under investigation by our group. Another potential confound is different frequencies of neuropsychological testing across the examined cohorts. In particular, practice effects may vary depending on this frequency and we did not have enough observations per participant to model nonlinear slopes that may account for these practice effects (most participants only have 2 measurements). Additional factors that may be influential to our results are exclusion criteria and recruitment based on factors such as APOE ε4 and socioeconomic status.
To address differences in amyloid imaging acquisitions across cohorts, we employed a data-driven Gaussian mixture modeling approach to the Aβ values from each cohort separately. There is currently no universally accepted method for defining Aβ cutoff values, and little consistency exists across laboratories (with methods ranging from hierarchical clustering between CNs and patients,17 iterative outlier removal within CNs,36 defined in CNs < age 40,37,38 postmortem verification39). Aβ cutoff values are especially problematic in elderly CNs, given that the proportion of slightly elevated participants (who are the most difficult to classify) will be higher in CN populations compared to AD. Nevertheless, our resulting classification has similarities to cutoffs derived using different approaches. For instance, the cutoff we derived for ADNI of 1.126 is similar to the cutoff of 1.11 defined in young participants (age < 56) and verified with postmortem examination of older CNs and patients.20,37,39 It is also noteworthy that the classification certainty within ADNI (florbetapir) was lower than the classification certainty within HABS and AIBL (PiB). This may be due to the more limited range of florbetapir values, making CNs with slightly elevated values more difficult to classify. For the current analysis, we classified CNs using a 50% probability cutoff, which does not take into account the increased uncertainty present with florbetapir and may result in misclassification in CNs with slightly elevated florbetapir values. However, given the current paucity of studies investigating the relevance of slightly elevated Aβ values in CNs, additional studies will be necessary to determine the ability of different amyloid imaging tracers to differentiate biologically relevant signal from noise in CNs with slightly elevated values.
Our analyses have several additional limitations. The median follow-up period was short (1.49 years) and may be insufficient to adequately capture independent effects of Aβ and APOE ε4. Given the limited overlap in measures of cognition across studies, we were only able to examine change in MMSE and Logical Memory. More sensitive measures across different cognitive domains may be more insightful in detecting subtle early decline within CNs. Biases in participant recruitment also exist, given that the majority of CNs used in these analyses are highly educated, which may limit the generalizability of these findings to more representative samples. Ongoing follow-up of these and other large cohorts will provide further insights into the contributions of both Aβ and APOE ε4 to decline in CNs.
Contributions of AD risk factors to decline in aging are increasingly relevant given proposals for secondary prevention trials targeting CNs. The Anti-Amyloid Treatment in Asymptomatic Alzheimer's disease (A4) trial will assess the efficacy of an antiamyloid therapy in high Aβ CNs using cognitive endpoints and biomarker data. Our findings suggest that it may be important to account for APOE ε4 even among high Aβ participants and to potentially stratify enrollment on the basis of APOE ε4 across treatment arms. Furthermore, the Alzheimer's Prevention Initiative has been awarded funding to execute an antiamyloid trial targeting APOE ε4 homozygotes.40 Our findings also suggest that it may be important to account for Aβ status among APOE ε4 carriers. Finally, the heterogeneity observed in this study highlights the need for large samples of CNs to determine the impact of AD risk factors on longitudinal decline and to observe a treatment effect.
Supplementary Material
ACKNOWLEDGMENT
(1) Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found on the Neurology® Web site at Neurology.org. (2) Data used in the preparation of this article were obtained from the Australian Imaging Biomarkers and Lifestyle Flagship Study of Ageing (AIBL) funded by the Commonwealth Scientific and Industrial Research Organisation (CSIRO), which was made available at the ADNI database (www.loni.ucla.edu/ADNI). The AIBL researchers contributed data but did not participate in analysis or writing of this report. AIBL researchers can be found on the Neurology® Web site at Neurology.org. (3) Data used in the preparation of this article were obtained from the Harvard Aging Brain Study (HABS) funded by the National Institute on Aging (NIA). The HABS researchers contributed data but did not participate in analysis or writing of this report. A complete listing of HABS investigators can be found on the Neurology® Web site at Neurology.org.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- AIBL
Australian Imaging Biomarkers and Lifestyle Study of Ageing
- CDR
Clinical Dementia Rating
- CN
clinically normal older individuals
- HABS
Harvard Aging Brain Study
- MMSE
Mini-Mental State Examination
- PiB
Pittsburgh compound B
- ROI
region of interest
Footnotes
AUTHOR CONTRIBUTIONS
E. Mormino: conceptualization of the study, analysis and interpretation of data, drafting and revising manuscript. R. Betensky: analysis and interpretation of data, revising manuscript. T. Hedden: analysis and interpretation of data, revising manuscript. A. Schultz: analysis and interpretation of data. A. Ward: analysis and interpretation of data. W. Huijbers: analysis and interpretation of data. D. Rentz: analysis and interpretation of data, revising manuscript. K. Johnson: interpretation of data, revising manuscript. R. Sperling: conceptualization of the study, interpretation of data, revising manuscript.
STUDY FUNDING
HABS: This work was funded by NIA grants P01AG036694, P50AG005134, R01AG037497, R01AG034556, K24AG035007, K01AG040197, and F32AG044054; NCI grant R01 CA075971; The Alzheimer's Association Grant ZEN-10-174210; NIBIB Grant P41EB015896; and the Harvard NeuroDiscovery Center. This research was carried out in part at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Biotechnology Resource Grant supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH. ADNI: The data collection and sharing for this project was funded by ADNI (NIH grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, NV; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, Rev October 16, 2012, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514. AIBL: This study was supported by the Science and Industry Endowment Fund (Australia), the Commonwealth Scientific and Industrial Research Organisation (Australia), The National Health and Medical Research Council of Australia Program and Project Grants, the Austin Hospital Medical Research Foundation, operational infrastructure support from the Victorian State Government, the Alzheimer's Drug Discovery Foundation, and the Alzheimer's Association.
DISCLOSURE
E. Mormino received funding from NIH grants F32AG044054 and P01 AG036694. R. Betensky received funding from NIH grants R01 CA075971, R03 CA165070, UL 1RR025758, P50 NS051343, P50 NS051343, P30 CA006516, P50 AG005134, P01 AG036694, R01 NS070834, R01 NS070834, and R01 AG026484. T. Hedden received funding from NIH grants K01 AG040197, P01 AG036694, and R01 AG034556. A. Schultz, A. Ward, and W. Huijbers report no disclosures relevant to the manuscript. D. Rentz receives research support from the NIH grants P01 AG036694, R01 MH090291, U01 AG024904, R01 AG027435, R01 AG037497, and P50 AG005134, and Alzheimer Association grant IIRG-08-90934. K. Johnson has served as paid consultant for Bayer, Bristol-Myers Squibb, GE Healthcare, Janssen Alzheimer's Immunotherapy, Siemens Medical Solutions, and Genzyme. He is a site coinvestigator for Lilly/Avid, Bristol-Myers Squibb, Pfizer, Janssen Immunotherapy, and Navidea. He has spoken at symposia sponsored by Janssen Alzheimer's Immunotherapy and Pfizer. T. Benzinger has served on an advisory board for Eli Lilly and has received research funding from Avid Radiopharmaceuticals. These relationships are not related to the content in the manuscript. K. Johnson receives funding from NIH grants R01EB014894, R21 AG038994, R01 AG026484, R01 AG034556, P50 AG00513421, U19 AG10483, P01 AG036694, R13 AG042201174210, R01 AG027435, and R01 AG037497 and the Alzheimer's Association grant ZEN-10-174210. R. Sperling has served as a paid consultant for Bristol-Myers Squibb, Eisai, Janssen Alzheimer Immunotherapy, Pfizer, Merck, and Roche, and as an unpaid consultant to Avid, Eli Lilly. She is a site coinvestigator for Avid, Bristol-Myers Squibb, Pfizer, and Janssen Alzheimer Immunotherapy clinical trials. She has spoken at symposia sponsored by Eli Lilly, Pfizer, and Janssen Alzheimer Immunotherapy. These relationships are not related to the content in the manuscript. R. Sperling receives research support for NIH grants U01 AG032438, U01 AG024904, R01 AG037497, R01 AG034556, K24 AG035007, P50 AG005134, U19 AG010483, R01 AG027435, and P01 AG036694 and the Alzheimer's Association grant ZEN-10-174210. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging and the Alzheimer's Association workgroup. Alzheimers Dement 2011;7:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedden T, Oh H, Younger AP, Patel TA. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 2013;80:1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology 2012;78:1576–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 2009;66:1469–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol 2011;69:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923 [DOI] [PubMed] [Google Scholar]
- 7.Kok E, Haikonen S, Luoto T, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 2009;65:650–657 [DOI] [PubMed] [Google Scholar]
- 8.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci USA 2009;106:6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kantarci K, Lowe V, Przybelski SA, et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology 2012;78:232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim YY, Ellis KA, Ames D, et al. Abeta amyloid, cognition, and APOE genotype in healthy older adults. Alzheimers Dement 2013;9:538–545 [DOI] [PubMed] [Google Scholar]
- 11.Lim YY, Ellis KA, Pietrzak RH, et al. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology 2012;79:1645–1652 [DOI] [PubMed] [Google Scholar]
- 12.Roe CM, Fagan AM, Grant EA, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology 2013;80:1784–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aisen PS, Petersen RC, Donohue MC, et al. Clinical Core of the Alzheimer's Disease Neuroimaging Initiative: progress and plans. Alzheimers Dement 2010;6:239–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr 2009;21:672–687 [DOI] [PubMed] [Google Scholar]
- 15.Hedden T, Mormino EC, Amariglio RE, et al. Cognitive profile of amyloid burden and white matter hyperintensities in cognitively normal older adults. J Neurosci 2012;32:16233–16242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jack CR, Jr, Bernstein MA, Fox NC, et al. The Alzheimer's disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283 [DOI] [PubMed] [Google Scholar]
- 18.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis, I: segmentation and surface reconstruction. Neuroimage 1999;9:179–194 [DOI] [PubMed] [Google Scholar]
- 19.Fischl B, Liu A, Dale AM. Automated manifold surgery: constructing geometrically accurate and topologically correct models of the human cerebral cortex. IEEE Trans Med Imaging 2001;20:70–80 [DOI] [PubMed] [Google Scholar]
- 20.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 2009;132:1310–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006;31:968–980 [DOI] [PubMed] [Google Scholar]
- 23.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355 [DOI] [PubMed] [Google Scholar]
- 24.Wolf AB, Valla J, Bu G, et al. Apolipoprotein E as a beta-amyloid-independent factor in Alzheimer's disease. Alzheimers Res Ther 2013;5:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dennis NA, Browndyke JN, Stokes J, et al. Temporal lobe functional activity and connectivity in young adult APOE varepsilon4 carriers. Alzheimers Dement 2010;6:303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filippini N, MacIntosh BJ, Hough MG, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci USA 2009;106:7209–7214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiman EM, Chen K, Alexander GE, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci USA 2004;101:284–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jagust WJ, Landau SM. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci 2012;32:18227–18233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheline YI, Morris JC, Snyder AZ, et al. APOE allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. J Neurosci 2010;30:17035–17040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahley RW, Huang Y. Apolipoprotein ε sets the stage: response to injury triggers neuropathology. Neuron 2012;76:871–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghebremedhin E, Schultz C, Thal DR, et al. Gender and age modify the association between APOE and AD-related neuropathology. Neurology 2001;56:1696–1701 [DOI] [PubMed] [Google Scholar]
- 32.McCarron MO, Delong D, Alberts MJ. APOE genotype as a risk factor for ischemic cerebrovascular disease: a meta-analysis. Neurology 1999;53:1308–1311 [DOI] [PubMed] [Google Scholar]
- 33.Greenberg SM, Briggs ME, Hyman BT, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke 1996;27:1333–1337 [DOI] [PubMed] [Google Scholar]
- 34.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993;90:9649–9653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chetelat G, Villemagne VL, Bourgeat P, et al. Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol 2010;67:317–324 [DOI] [PubMed] [Google Scholar]
- 36.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008;65:1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer’s disease and cognitively normal subjects. J Nucl Med 2012;53:378–384 [DOI] [PubMed] [Google Scholar]
- 38.Mormino EC, Brandel MG, Madison CM, et al. Not quite PiB-positive, not quite PiB-negative: slight PIB elevations in elderly normal control subjects are biologically relevant. Neuroimage 2012;59:1152–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011;305:275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer's Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis 2011;26(suppl 3):321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.