

Significance
Epidemics of microbial infections are a considerable threat to human and animal health. Analysis of 3,615 genome sequences, coupled with virulence studies in animals, permitted us to delineate the nature and timing of molecular events that contributed to an ongoing global human epidemic of infections caused by group A Streptococcus, the “flesh-eating” pathogen. We clarified decades-long uncertainty about the timing and sequence of genomic alterations that underpinned the global epidemic. Analyses of this type are crucial for developing better strategies to predict and monitor strain emergence and epidemics, formulate effective protective public health maneuvers, and develop or modify vaccines.
Keywords: pathogenesis, phylogeography, mobile genetic element, flesh-eating disease, molecular clock
Abstract
We sequenced the genomes of 3,615 strains of serotype Emm protein 1 (M1) group A Streptococcus to unravel the nature and timing of molecular events contributing to the emergence, dissemination, and genetic diversification of an unusually virulent clone that now causes epidemic human infections worldwide. We discovered that the contemporary epidemic clone emerged in stepwise fashion from a precursor cell that first contained the phage encoding an extracellular DNase virulence factor (streptococcal DNase D2, SdaD2) and subsequently acquired the phage encoding the SpeA1 variant of the streptococcal pyrogenic exotoxin A superantigen. The SpeA2 toxin variant evolved from SpeA1 by a single-nucleotide change in the M1 progenitor strain before acquisition by horizontal gene transfer of a large chromosomal region encoding secreted toxins NAD+-glycohydrolase and streptolysin O. Acquisition of this 36-kb region in the early 1980s into just one cell containing the phage-encoded sdaD2 and speA2 genes was the final major molecular event preceding the emergence and rapid intercontinental spread of the contemporary epidemic clone. Thus, we resolve a decades-old controversy about the type and sequence of genomic alterations that produced this explosive epidemic. Analysis of comprehensive, population-based contemporary invasive strains from seven countries identified strong patterns of temporal population structure. Compared with a preepidemic reference strain, the contemporary clone is significantly more virulent in nonhuman primate models of pharyngitis and necrotizing fasciitis. A key finding is that the molecular evolutionary events transpiring in just one bacterial cell ultimately have produced millions of human infections worldwide.
Understanding of the nature and timing of many critical molecular events underlying emergence and dissemination of microbial pathogens that cause epidemic disease remains elusive. Comprehensive delineation of the key molecular contributors to these processes is essential for developing better strategies to recognize and predict virulent strain emergence and epidemics, formulate protective public health policies and maneuvers, and develop or modify vaccines. In recent years, fast and inexpensive massively parallel DNA sequencing has facilitated genetically based enhanced understanding of these and other infectious disease problems.
Group A Streptococcus (GAS, also known as Streptococcus pyogenes), a Gram-positive bacterial pathogen, causes human infections worldwide (1–20). For example, GAS is responsible for more than 600 million infections globally each year, including a conservative estimate of 10,000–15,000 severe invasive infections annually in the United States (4). This organism has a long-recognized proclivity to cause epidemic waves, the reasons for which are largely unknown. After several decades of declining incidence, a striking resurgence of severe invasive infections caused by serotype Emm protein 1 (M1) GAS was reported in many countries in the late 1980s and early 1990s (1–3, 5–20). The resurgence received widespread public interest in part because of the untimely death of Jim Henson, the Muppeteer, of invasive GAS, and because of severe infections occurring in other notable public figures in Canada and elsewhere (21). This resurgence of severe life-threatening GAS infections was a potent reminder that we know relatively little about the evolutionary genetic events and epidemiological forces that underpin temporal variation in bacterial disease frequency and severity. Lack of precise understanding of these topics significantly hobbles our ability to understand and predict bacterial strain emergence and epidemics.
GAS serves as a model pathogen for studying the evolutionary genomics of epidemic disease in part because it causes abundant human infections; comprehensive, population-based strain collections are available from diverse countries; the organism has a relatively small genome size (∼1.8 Mb); and high-quality full-genome sequences are available for strains of diverse M protein serotypes (22, 23). In addition, humans are the pathogen’s only natural host, which means that genetic and epidemiologic events are not obscured by processes occurring in nonhuman hosts or the environment. The primary goal of this study was to test the hypothesis that large-scale genome sequencing and animal infection models would shed new light on heretofore vague molecular events contributing to the evolutionary genomics of epidemic disease in general, and specifically in GAS. Our findings are based on the largest bacterial whole-genome sequencing project reported thus far.
Results and Discussion
Genome-Wide Polymorphism Levels and Genes with Evidence of Diversifying Selection.
We studied 3,615 serotype M1 organisms from diverse locations on two continents (Table 1, Dataset S1, and Fig. S1). Most of the strains were collected as part of comprehensive, population-based studies conducted in the areas sampled. To determine the genome-wide polymorphisms present in these 3,615 strains, we first resequenced the genome of reference serotype M1 strains SF370 (23) and MGAS5005 (24), using Illumina-based technology. Although each of these genomes had been sequenced initially to high quality (Q40 consensus), using technology available at the time (Sanger dideoxy terminator sequencing), resequencing to ∼125-fold coverage permitted us to correct a total of 235 errors in the genome assemblies (180 for SF370 and 55 for MGAS5005). The previously deposited genome sequences have been updated.
Table 1.
Summary of 3,615 strains studied
| Geographic region | Time span | Number of strains |
| Ontario, Canada | 1996–2009 | 346 |
| Denmark | 1973–2013 | 436 |
| East Germany | 1969–1991 | 155 |
| Finland | 1988–2011 | 1106 |
| Iceland | 1988–2010 | 50 |
| Norway | 1997–2009 | 215 |
| Sweden | 1973–2012 | 482 |
| Georgia, USA | 1995–2010 | 340 |
| Minnesota, USA | 1997–2010 | 474 |
| Other | ∼1920s–1980s | 11 |
Information for the complete list of 3,615 strains is found in Dataset S1. Fig. S1 contains specific years included in the study. East Germany refers to the former East Germany. Canada strains were from Ontario. US strains were from Georgia (Atlanta area) and Minnesota. “Other” includes strains of predominantly historic interest.
Comparison of the two corrected core genomes using a combination of bioinformatics analyses [variant ascertainment algorithm (VAAL), MUMmer, and ClustalW; see SI Materials and Methods], and targeted Sanger sequencing revealed that these two strains differed by 933 polymorphisms, including 896 SNPs and 37 indels. Approximately two-thirds of these polymorphisms (n = 576/896 SNPs) are located in a contiguous 36-kb region of the genome we previously described (24); the remaining one-third of polymorphisms (n = 320/896 SNPs) are distributed throughout the core genome [i.e., the ∼1.7-Mb portion of the chromosome common among GAS serotypes that remains after exclusion of mobile genetic elements (MGEs)]. One-half of the polymorphisms (n = 286/576) in this 36-kb region are concentrated in a highly divergent 2.6-kb region between the slo and metB genes (Fig. 1).
Fig. 1.

Comparison of SNP distribution in strain SF370 and strain MGAS5005. Polymorphisms between the SF370 and MGAS5005 genomes were identified using a combination of sequence alignment and polymorphism discovery tools: VAAL, MUMmer, and ClustalX. (A) Distribution of SNPs across the MGAS5005 genome. Plotted is the SNP density (y axis) in a 5-kb window iterated every 100 bp across the MGAS5005 genome (x-axis). The distribution of SNPs is nonrandom, with 576 (64.3%) of 896 SNPs occurring in the 36-kb region between the purA and nadC genes. (B) Distribution of SNPs across the MGAS5005 genome 36-kb region of recombination. Plotted is the SNP density in a 100-bp window iterated every 50 bp across the 36-kb region of recombination. The distribution of SNPs is nonrandom, with 286 (49.7%) of 586 SNPs occurring in the 2.6-kb sequence between the slo and metB genes.
Analysis of the 3,615 genome sequences, using VAAL, identified 13,221 SNP loci (3,727 synonymous, 7,609 nonsynonymous, and 1,982 intergenic SNPs) and 1,598 indels (1,027 in coding sequences and 571 in intergenic regions) in the core genome. Inference of genetic relationships based on the core genome SNPs divided the strains into two subpopulations that mirrored the region of recombination group designation, including 172 strains that are SF370-like and 3,443 strains that are MGAS5005-like (Fig. 2A). Excluding the horizontally acquired polymorphisms in the 36-kb region of recombination, all 3,615 organisms are closely related genetically (Fig. 2B), differing strain-to-strain on average by only 48 SNPs and 8 indels distributed throughout the core genome. The average genetic distance between strains of the postresurgence MGAS5005-like population and strains of the preresurgence SF370-like population is 113 core-genome SNPs. The postresurgence strains differ, on average, by only 41 SNPs and 7 indels in the core genome, whereas the preresurgence strains differ, on average, by 104 SNPs and 15 indels in the core genome. Five genes that encode known virulence factors and are likely under positive selection had levels of allelic variation significantly greater than average: M protein (emm), regulator of protease B (ropB), streptococcal inhibitor of complement (sic), and the two-component-system control of virulence regulator (covR) and sensor (covS).
Fig. 2.

Genetic relationships among the 3,615 serotype M1 strains. Genetic relationships among the strains were inferred by the method of neighbor joining, using SplitsTree. Strains with an SF370-like 36-kb purA-to-nadC region are shown in blue, and strains that are MGAS5005-like (i.e., strains that have a mosaic chromosome with a recombined M12-like purA-to-nadC region) are shown in red. (A) Genetic relationships inferred on the basis of all 13,221 core chromosomal SNPs identified among the 3,615 strains. Including SNPs within the 36-kb purA-to-nadC region, the SF370-like strains are genetically distinct from the MGAS5005-like strains. (B) Genetic relationships inferred based on the 12,355 core chromosomal SNPs that remain after exclusion of the horizontally acquired SNPs in the 36-kb purA-to-nadC region of recombination. Exclusion of the SNPs in the 36-kb region of recombination collapses the vast majority of the genetic distance between the older SF370-like progenitor strains and the contemporary MGAS5005-like descendant strains. Excluding SNPs in the 36-kb purA-to-nadC region and rooting the tree using GAS serotype M3 strain MGAS315 as an outgroup places SF370 and older SF370-like strains on one side of the tree (upper left), and most SF370-like strains from the ∼mid-1970s onward and all MGAS5005-like strains on the opposite side (lower right). Both trees are shown at the same scale.
Variation in MGE Content.
Prophages and integrative conjugative elements (ICEs) are critical actors in pathogen–host interactions because in many bacteria (including GAS), they encode one or more virulence factor or factors mediating resistance to antimicrobial agents. Most genomes of GAS are polylysogenic and generally contain 3 or more distinct prophages and one or more ICEs (25). Common GAS prophage-encoded virulence factors include pyrogenic toxin superantigens, extracellular DNases, and a secreted phospholipase A2 (22–33). GAS ICE-encoded antimicrobial agents typically include those mediating resistance to tetracycline or macrolides (34).
To assess variation in MGE content in the strain samples studied here, we used a two-step process. First, to detect loss of known M1 gene content, sequence reads for each strain were mapped to the MGAS5005 and/or SF370 reference genomes. Second, to detect gain of known GAS gene content not represented in the SF370 or MGAS5005 genomes, the sequence reads were mapped to a GAS pseudopangenome constructed using 21 complete genomes representing 13 M-protein serotypes (see Materials and Methods for detail and Fig. S2 for an example). Nearly all strain-to-strain differences in gene content were associated with differences in MGE content (Fig. 3). The great majority of strains causing disease since the mid-1980s had the same MGE content as MGAS5005; that is, they contained prophage 5005.1 encoding superantigen SpeA and prophages 5005.2 and 5005.3 encoding DNases Spd3 and streptococcal DNase D2, (SdaD2), respectively. However, three other abundantly represented patterns of variation in prophage content and several less-common variant patterns also were found (Fig. 3). A few rare-variant MGE patterns indicated ICE acquisitions consistent with sporadic reports of serotype M1 strains resistant to tetracycline or macrolides (35).
Fig. 3.

MGE content of MGAS5005-like M1 strains. Shown is the phylogeny inferred by neighbor-joining for 3,443 MGAS5005-like contemporary M1 strains based on 12,355 concatenated core SNPs. SNPs located in the 36-kb region of recombination and the MGEs were excluded to constrain the inferred phylogeny to vertically inherited SNPs. Labeled is the branch leading to the tree root and SF370-like strains, which are not shown because of space constraints. Strains are colored by MGE content, as indicated in the bottom figure inset. There were 13 MGE content patterns found in 5 or more strains of the cohort. MGE patterns found in 4 or fewer strains are shown collectively as open circles/triangles (n = 69). The vast majority of the strains (84%) have the same MGE content as strain MGAS5005 (i.e., no difference in gene content was detected relative to MGAS5005). Invasive isolates are shown as circles, and pharyngitis isolates as triangles. Evident in the tree are numerous independent occurrences of MGE acquisition and loss, as well as commonality in MGE content among strains as a consequence of vertical inheritance. MGE content of the 172 SF370-like strains is shown in Fig. 5.
Molecular Evolutionary Genetics of Contemporary M1 Clone Emergence.
In a model we put forth to explain the evolutionary genetic events that resulted in an abundant postresurgent contemporary clone of serotype M1 GAS (24), we hypothesized that an ancestral serotype M1 strain initially acquired phages encoding speA and sdaD2. This organism subsequently experienced a horizontal gene transfer event involving the acquisition of a 36-kb chromosomal region encoding streptolysin O and NAD+-glycohydrolase virulence factors. Near identity of this DNA sequence with the corresponding region in the genomes of two serotype M12 strains led us to hypothesize that a serotype M12 strain served as the donor of this DNA segment (24). However, because of inadequate strain sample size and a lack of sufficient full-genome sequence data, we were unable to unambiguously define the precise order of these critical molecular events. Others have proposed alternative evolutionary scenarios based on analysis of very few strains, many with uncertain provenance (36).
Our large genome sequence dataset permitted us to generate a comprehensive M1 population genetic structure (Fig. 3), against which the timing of key events of the different models could be tested. The great majority (n = 165; 96%) of the SF370-like strains were isolated before or in 1988, whereas all MGAS5005-like organisms were isolated in or after 1988 (Fig. 4). The emergence of MGAS5005-like strains corresponds with reports of increased M1 GAS infection frequency and severity on different continents (1–3, 5–20). We found that the ancestral SF370-like organisms had either the speA1 or speA2 allele, whereas the descendant MGAS5005-like strains have only the speA2 allele (Fig. 5). These two alleles differ by a single nonsynonymous nucleotide substitution that results in a Gly110Ser amino acid replacement (37). This amino acid residue is located immediately adjacent to a region of SpeA-containing cysteine residues that participate in the formation of a disulfide loop required for T-cell mitogenicity (38). Relative to SpeA1, the SpeA2 variant has significantly higher affinity for class II major histocompatibility complex-DQ molecules (39).
Fig. 4.
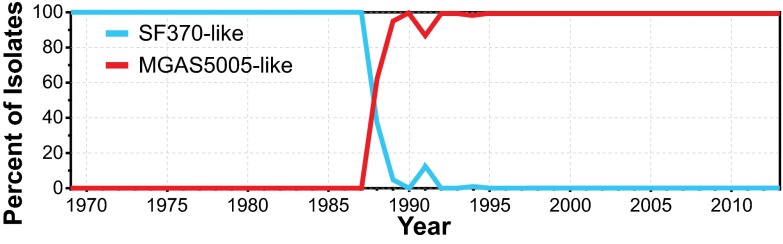
Temporal distribution of the SF370-like and MGAS5005-like M1 strains. Over the short period of only a few years (1987–1989), contemporary MGAS5005-like strains emerged rapidly and essentially displaced antecedent SF370-like M1 strains across a broad geographic region, if not globally.
Fig. 5.

Timing of molecular genetic events leading to the emergence of the contemporary MGAS5005-like M1 strains. Illustrated is the phylogeny of 198 strains based on 1,594 concatenated core SNPs inferred by neighbor-joining. SNPs located in the 36-kb region of recombination and the MGEs were excluded to constrain the inference to primarily vertically inherited SNPs. Shown as circles are all 172 strains with an SF370-like 36-kb region. Nearly all of these strains (n = 165) were isolated before 1989. Shown as squares are reference strain MGAS5005 isolated in 1996 and all strains with an MGAS5005-like 36-kb region isolated in 1988 (n = 25), the first year such strains are present in the study sample. (A) Prophage content is illustrated for each strain by color, as shown in the inset figure on the right. The majority of the strains (n = 134/198, 68%), including the majority (n = 50/81, 62%) of the SF370-like strains from the 1970s, have the same prophage content as strain MGAS5005. (B) Year of isolation for each strain is illustrated by color, as shown in the inset on the right. All isolates before 1988 have an SF370-like 36-kb region. Strains with an MGAS5005-like 36-kb region branch from a progenitor cluster of SF370-like strains that already contain the same prophage content as MGAS5005 (bottom right on the trees). Inspection of the trees shows that an SF370-spd3 or 5005.2-spd3-encoding prophage is present in all isolates going back to the 1920s; a 5005.3-sdaD2-encoding prophage is present in strains as early as 1969. Strains having the complete MGAS5005 complement of prophage, including the 5005.1-speA2 allele, are present as early as 1973. In contrast, the first strains with an MGAS5005-like 36 kb-region are not present in the comprehensive M1 population studied herein until 1988.
Analysis of the genome sequence data using assembly, comparison, and phylogenetic inference tools (EDENA, BLAST, MOSAIK, and TABLET) revealed three critical points. First, importantly, the entire 40-kb prophage encoding the speA2 allele found in contemporary MGAS5005-like strains is virtually identical in nucleotide sequence to the speA1-encoding prophage present in the SF370-like strains from the 1920s. Second, the two prophages are integrated at the same chromosomal location in SF370- or MGAS5005-like strains. Third, all speA-encoding strains from the mid-1970s onward have the speA2 allele, a finding confirmed by PCR amplification and targeted DNA sequencing. This discovery, along with the overall phylogeny of the serotype M1 population, indicates that the speA2 allele evolved from the speA1 allele in a single genetic event that occurred well before the emergence of severe invasive infections and subsequently became abundantly represented in contemporary M1 strains by clonal expansion (Fig. 5). Inasmuch as the speA2 allele has never been found in strains other than contemporary M1 organisms (37, 40, 41), we conclude that identity-by-descent (vertical) inheritance from a common precursor cell is the only reasonable explanation for the abundant presence of speA2 among contemporary M1 organisms.
Similar to the speA prophage findings, the spd3-containing prophage present in old or contemporary strains is integrated at the same chromosomal location and has a low level of polymorphism throughout its ∼33-kb sequence among the vast majority of all M1 strains present in the population from the early 1970s onward (Fig. 5). As with the speA-positive strains, the phylogenetic structure of the 3,615 genomes indicates that acquisition of the spd3-encoding prophage occurred well before the resurgence in M1 invasive infection.
A key unresolved issue in the molecular evolutionary events resulting in contemporary M1 strains is the timing of acquisition of prophage 5005.3 encoding the SdaD2 DNase virulence factor. Our analysis discovered that prophage 5005.3 was abundantly present among the M1 strains isolated temporally (∼late 1960s) before the speA2 allele evolved from speA1 (∼early 1970s; Fig. 5). Thus, the phylogeny indicates that acquisition of prophage 5005.3 encoding sdaD2 occurred before acquisition of prophage 5005.1 encoding speA. Importantly, analogous to prophages 5005.1 and 5005.2, regardless of the year of isolation, the 5005.3 prophage had very little genetic diversity and was integrated at the same chromosomal location. These findings also strongly argue for a clonal expansion and identity-by-descent evolutionary pathway.
Finally, the core genome phylogeny, coupled with information about the year of isolation of strains we studied (Fig. 5), unambiguously shows that acquisition of the new 36-kb region of recombination occurred after the phage-acquisition events described earlier. Hence, we must conclude that in the evolutionary path leading to the most recent common ancestor of contemporary epidemic M1 organisms, this horizontal gene transfer event was the final major molecular event preceding their emergence and explosive geographic dissemination.
The comprehensive M1 genomic population structure coupled with the year of isolation data (i.e., the historic record) places the 36-kb purA–nadC recombination event as occurring after the early 1970s, preceding isolation of the first MGAS5005-like strains in the late 1980s. To more precisely determine the time of occurrence of this 36-kb horizontal recombination event, a best-fit rooted time-tree (a chronogram) was generated for all 3,443 MGAS5005-like strains, using Path-O-Gen. This analysis estimated that the year of origin of the most recent common ancestor of MGAS5005-like strains was 1983 (Fig. 6).
Fig. 6.

Estimation of 1983 as the year of origin of the contemporary MGAS5005-like M1 strains. Phylogeny among all 3,443 MGAS5005-like strains was inferred on the basis of 11,333 concatenated core SNPs by neighbor-joining. SNPs in the 36-kb region of recombination and in MGEs were excluded to constrain the inference to primarily vertically inherited SNPs. A chronogram (time-tree) with best-fit root was generated from the neighbor-joining tree and the year of isolation. Illustrated is a straight-line best fit of the root-to-tip divergence (i.e., the number of core SNPs between the individual strains and the best-fit root) for each of the strains. The good correlation coefficient (i.e., a value >0.8) indicates that the data fit very well, with the assumption of a uniform molecular clock. The slope of the line is the estimated evolutionary rate in SNPs per core genome (∼1.7 million nucleotide sites) per year. The X-intercept (1983) is the estimated time of origin of the most recent common ancestor.
To summarize, the relative lack of genetic diversification among contemporary serotype M1 strains indicates that the precursor clone arose recently and that its progeny spread intercontinentally to many localities in an epidemic wave of pharyngitis and serious invasive infections occurring over the last 25 y.
Temporal Dissemination of Subclones.
Few genome-scale data sets are available for comprehensive, population-based samples of bacterial pathogens that bear on temporal dissemination patterns and rates. Our study was designed to help alleviate this knowledge deficit. Understanding the temporal and geographic dynamics of pathogen transmission in the human population is relevant to many aspects of basic, clinical, and translational research, including molecular pathogenesis, vaccine development and deployment, and public health. We studied geographically and temporally diverse strain samples, most of which are population-based and comprehensive. These strain sample features facilitate our ability to test the hypothesis that temporal structure exists in clone distribution. Consistent with our hypothesis, strong patterns of temporal structure were evident in the phylogeny (Fig. 7A). This was readily apparent for individual geographic regions. For example, all invasive strains in Finland recovered between 1988 and 2000 are present only in the upper of the two major trunks of the contemporary M1 phylogenetic tree (Fig. 7B). In contrast, the vast majority of strains from the 2003–2011 disease wave are located in the lower trunk of the tree (Fig. 7B).
Fig. 7.

Temporal structure of the M1 population. Illustrated is the phylogeny for all 3,443 MGAS5005-like strains inferred by neighbor-joining, based on 12,355 vertically inherited core SNPs. The branch point leading to the tree root and SF370-like strains is labeled. (A) Strains from all 9 geographic regions are color coded by year of isolation, as indicated in the lower inset. Evident on the branches is a correlation between increasing root-to-tip genetic distance and increasing year of isolation. (B) To further illustrate this finding, invasive isolates from Finland, collected as part of a prospective population-based surveillance program, are color coded according to the peaks of infection, as indicated in the epidemic curve shown in the lower inset.
Population Genomics of Pharyngitis and Sterile-Site Strains.
We know relatively little about the molecular population genomic relationship between bacterial strains of the same species causing distinct types of infections. Our sample included 597 M1 pharyngitis strains from Finland, most of which were recovered in a population-based surveillance study conducted between 1994 and 1997 (13), years for which we also had a comprehensive country-wide sample of invasive strains (42). For most M protein serotypes of GAS, including M1 strains, there is proportionally a far larger number of pharyngitis cases compared with invasive infections occurring in the same geographic area. On the basis of core SNPs, we discovered that as a population, the pharyngitis strains clustered phylogenetically, with invasive strains causing disease during the same period in Finland (Fig. 8A). In addition, the pharyngitis and invasive M1 strains contained the same complement of toxin-encoding prophages (Fig. 3). Therefore, the invasive and pharyngitis isolates are not distinct populations based on either the core or dispensable genetic content. Rather, they derive from a common genetic pool. We conclude that the explanation most parsimonious with the phylogeny is that invasive isolates evolve repeatedly from the population of strains causing pharyngitis in the same period and geographic region. This should not be interpreted to mean the pharyngitis and invasive strains as populations are genetically identical, because they are not. For example, we found differences in allele frequencies between these two populations in genes such as hasA and hasB that are involved in hyaluronic acid capsule biosynthesis (Fig. 8B and Fig. S3). We also identified a high frequency of occurrence of frameshift (inactivating) mutations in the hasA gene in pharyngitis strains (Fig. 8B). These mutations are known to down-regulate hyaluronic acid capsule expression and result in increased virulence. A divergent signal of diversifying selection between pharyngitis and invasive isolates was also discovered in population genomic investigation of a limited number of GAS serotype M3 strains (43).
Fig. 8.
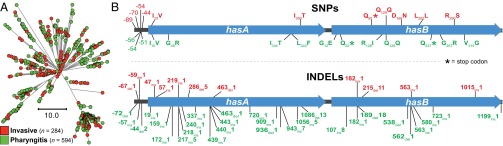
SNPs and indels present in hasA and hasB in Finland invasive infection isolates and pharyngitis isolates. (A) Illustrated is the inferred phylogeny for all 878 contemporary MGAS5005-like Finland isolates collected between 1988 and 1998 based on 1,477 core SNPs by neighbor-joining. Invasive isolates are shown in red and pharyngitis in green. Invasive infection and pharyngitis isolates comingle in all branches of the phylogeny; they do not constitute separate, genetically distinct populations. (B) Diagrammed are the hasA and hasB GAS hyaluronic acid capsule synthesis genes and the adjacent upstream intergenic region. Polymorphisms found among all of the MGAS5005-like Finland invasive isolates (n = 504) are shown in red above the genes and among all of the pharyngitis isolates (n = 594) in green below the genes. The SNP distribution is illustrated in the upper panel, and the indel distribution in the lower panel. The predicted consequences of coding-region SNPs are indicated. After normalizing for the number of strains in each of the sets, polymorphisms in the pharyngitis isolates were found at 1.7 times the frequency they occurred in the invasive infection isolates.
Comparative Strain Virulence in Infected Nonhuman Primates.
The extensive reported clinical and epidemiologic data are consistent with the idea that preresurgence serotype M1 strains recovered before ∼1988 are less virulent than postresurgence strains causing human infections after ∼1988 (1–3, 5–20). However, this idea has not been rigorously tested. To directly test this hypothesis, we used two nonhuman primate models of infection to compare the virulence of preresurgence reference strain SF370 with postresurgence strain MGAS2221. The cynomolgus macaque has been used extensively for experimental studies because it closely approximates human–GAS molecular interactions (27, 44–46). Two groups of monkeys were infected in the upper respiratory tract with either strain SF370 or strain MGAS2221 and monitored for 21 d. Strain SF370 was used because it was the first GAS genome to be sequenced, has been used extensively in studies over the last 13 y, and contains the old variant of the 36-kb region of recombination. Similarly, the genome of strain MGAS2221 has been sequenced, and the strain has been used in a variety of experimental studies. Neither strain has function-altering mutations in major regulatory genes such as covR, covS, ropB, or mga. We discovered that strain MGAS2221 was significantly more virulent in this pharyngitis infection model, as assessed by bacterial burden recovered over the course of the experiment (Fig. 9A).
Fig. 9.

Postresurgence strain MGAS2221 is significantly more virulent than SF370 in nonhuman primate models of pharyngitis and necrotizing fasciitis. (A) Cynomolgus macaques were inoculated in the upper respiratory tract, and mean cfus recovered from the oropharynx are shown with P value for Friedman test. (B–D) Cynomolgus macaques were inoculated intramuscularly in the anterior thigh. Cfus per gram of infected tissue, necrotizing fasciitis lesion volume, and histopathology score obtained at necropsy are shown. P values for the Mann–Whitney test are shown.
We next tested the hypothesis that strain MGAS2221 was significantly more virulent during invasive infection, as assessed using a well-described nonhuman primate model of necrotizing fasciitis (27, 44, 45). Significantly more bacteria were recovered from animals infected with MGAS2221 compared with those infected with strain SF370 (Fig. 9B), a finding that strongly supports the enhanced-virulence hypothesis. In addition, strain MGAS2221 caused significantly larger lesions and more severe histopathology than strain SF370 (Fig. 9 C and D).
Concluding Comment
In recent years, large-scale genome sequencing has been used to address previously unapproachable problems in diverse areas such as cancer genetics and human molecular evolution (47–50). Although progress has been made with some pathogens (51–55), relatively little analogous work has been performed to understand molecular factors underpinning strain emergence and the comprehensive genomic landscape of bacterial epidemics. Our study, the largest bacterial whole-genome sequencing project reported thus far, was designed to provide knowledge about these areas of infectious disease research. Specifically, we tested the hypothesis that large-scale genome sequencing of population-based strain samples, coupled with animal infection models, would provide clarifying data bearing on heretofore vague molecular events contributing to the evolutionary genetic events underpinning epidemic disease.
Much theorizing and speculation and some data have been presented in efforts to explain the precipitous increase in frequency and severity of invasive infections caused by serotype M1 GAS during the mid-1980s (14, 24, 36, 56–60). Analyses using techniques that index variation within a relatively small area of the GAS chromosome or very small numbers of strains supported the idea that many contemporary M1 isolates were clonally related (8, 24, 36, 40, 41, 56, 58, 59). These studies led to several distinct hypotheses (24, 36, 60) to explain the molecular genetic events underpinning the emergence of the contemporary M1 clone. A key difference between models was the order of the molecular events leading to now-extant contemporary serotype M1 strains. Delineation of the nature and order of precise genetic changes underlying strain emergence and altered virulence is of particular importance, as it helps link cause and effect and generate testable hypotheses. Moreover, previous studies did not permit a detailed understanding of the genome-wide genetic relationships among currently circulating M1 strains. In one model (24), acquisition of the 36-kb region of recombination from an M12 donor organism was the final event, whereas Maamary et al. (36) concluded that acquisition of the SpeA-encoding prophage was the triggering event. The present study permitted us to clearly delineate the crucial evolutionary genetic pathway. The only reasonable interpretation of the aggregate population genomic data obtained from analysis of these 3,615 genomes from widespread areas is that acquisition of the 36-kb region of recombination was the final major genetic event preceding virulent clone emergence and intercontinental spread. Thus, the claim (36) that horizontal transfer of the M12-like 36-kb region was a relatively early event preceding acquisition of the SpeA-encoding phage in the evolutionary path of contemporary M1 strains is not supported by the comprehensive population genomic structure and is wrong.
Our work permitted us to delineate the critical chain of molecular events that gave rise to the organisms causing the contemporary epidemic of serotype M1 GAS human infections. The molecular events included phage-mediated virulence gene acquisition, single nucleotide variant accumulation, and horizontal transfer of the 36-kb region encoding the virulence factors Nga and Slo (61–73), likely mediated by generalized transduction (24). It is now clear from our analysis that a complex multistep molecular process occurring sequentially over decades culminated in a single organism with increased virulence that fulminantly disseminated globally and displaced other GAS M1 lineages. The genome analysis permitted us to estimate the acquisition of the 36-kb region encoding Nga and Slo to a horizontal gene transfer and recombination event that occurred in 1983. In this regard, it is important to note that infections caused by M1 strains clearly increased significantly in frequency and severity in the United States between 1980–1988 (16), and in the United Kingdom between 1983 and 1985 (9). Moreover, it has been well documented (2, 3, 5, 7, 8, 11, 12, 14, 19) that epidemic M1 infections occurred in Norway, Sweden, Finland, Denmark, Australia, and Canada in the ensuing years. The result of the genetic events we describe here was an explosive intercontinental epidemic of serotype M1 human infections including pharyngitis and severe invasive diseases such as necrotizing fasciitis, the “flesh-eating” infection. GAS causes >600 million cases of human infections yearly, and extensive epidemiologic studies have shown that serotype M1 organisms are frequently the most abundant M protein serotype (3, 4, 7, 9, 14, 74–76). In conclusion, the molecular evolutionary events transpiring in just one bacterial cell ultimately produced many millions of human infections worldwide.
Our retrospective study analyzed strains stored in large population-based repositories, in some cases over decades. The availability of strain samples of this type was essential to the success of our investigation. Given that it is possible to sequence the genomes of thousands of bacterial strains, regardless of species, in short order at an economically feasible cost, it is now reasonable with appropriate sampling and epidemiologic strategies to identify strain emergence and patterns of dissemination in near-real-time. These types of studies, conducted in synchrony with detailed analyses of relevant emergent phenotypic properties (e.g., virulence, antimicrobial agent resistance, dissemination capacity), will undoubtedly provide new information useful for significantly enhanced basic, clinical, and translational research on human, veterinary, and plant pathogens. This approach will permit identification of pathogenic clones during, rather than after, emergence and widespread dissemination. In addition, extensive full-genome analysis of many emerging or emerged pathogens may give rise to generalizable mechanisms underlying the evolutionary genomics of epidemics.
Materials and Methods
Bacterial Strain Samples.
This investigation is based on 3,018 invasive and 597 pharyngitis serotype M1 strains that were recovered primarily from nine distinct geographic regions on two continents (Table 1, Dataset S1, and Fig. S1). Most strains were isolated between January 1994 and December 2012 as part of comprehensive population-based surveillance studies in the time frames shown in Fig. S1. The samples from Iceland and Finland were recovered from 1988 through 2011. Strains from Georgia (metropolitan Atlanta area) and Minnesota (statewide) were collected as part of the Active Bacterial Core Surveillance Program administered by the Centers for Disease Control and Prevention (www.cdc.gov/abcs/index.html). These two states were chosen for study because they are geographically distant and because their census population sizes approximate those of several of the Nordic countries in our study. The sample also included a small number of strains of historic interest from several other countries and times that have been described elsewhere (24, 40, 41). Reference strain SF370 (23) was purchased from ATCC.
Whole-Genome Sequencing and Polymorphism Analysis.
DNA extracted from each of the 3,615 strains was prepared for multiplexed sequencing, using Illumina Nextera XT kits (27). Genome sequence data were obtained predominantly with an Illumina HiSeq2000 instrument according to the manufacturer’s instructions. Most runs included 96 bar-coded sequencing libraries in each of the eight lanes of a flowcell (i.e., 768 strains sequenced concurrently). A few genomes were sequenced with an Illumina MiSeq instrument. Polymorphisms (SNPs and indels) were identified relative to corrected reference serotype M1 strains MGAS5005 (GenBank accession number CP000017) and/or SF370 (GenBank accession number AE004092) (23, 24), using VAAL software and methods described in SI Materials and Methods. The nature of the SNPs (coding/noncoding, synonymous/nonsynonymous, etc.) was determined using SNPfx.pl (a PERL script developed in-house). Sequence data for the 3,615 genomes have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (accession number SRA036051).
Gene Content and Mobile Genetic Element Analysis.
The known GAS pangenome core and accessory gene content were determined on the basis of 21 complete GAS genomes of 13 emm types, using PanOCT and reciprocal BLAST best-hit. A GAS pseudopangenome sequence (2.84 Mb, 3,167 genes) was generated by concatenating all additional accessory genes onto the MGAS5005 genome. Concatenated accessory genes included 50 bp of flanking sequence to facilitate subsequent read mapping. Sequence reads for all 3,615 serotype M1 strains were mapped to the GAS pseudopangenome using Mosaik (https://code.google.com/p/mosaik-aligner/), and gene fragments per kilobase per million mapped reads (FPKM) values were determined using Cufflinks. FPKM values relative to MGAS5005 were used to make gene presence or absence calls and then to determine MGE content for all 3,615 strains. Reads not aligning to the GAS pseudopangenome were assembled de novo using EDENA (SI Materials and Methods), and resultant contigs greater than 100 nucleotides were used to query the NCBI nonredundant database, using BLAST. Additional bioinformatic analyses are described in SI Materials and Methods.
Experimental Animal Infections.
The virulence of strains SF370 and MGAS2221 was assessed in pharyngitis and necrotizing fasciitis models, using cynomolgus macaques (Macaca fasicularis) (Charles River BRF), as previously described (44–46). Strains SF370 (n = 3 animals) or MGAS2221 (n = 4 animals) were used. For the pharyngitis studies, animals were anesthetized, inoculated in the upper respiratory tract with 1 × 108 cfu, and sampled sequentially for 21 d. For the necrotizing fasciitis experiments, animals were anesthetized and inoculated intramuscularly in the anterior thigh to a uniform depth with 1 × 108 cfu/kg of GAS. The animals were observed continuously for ∼24 h, euthanized, and necropsied. The study protocols were approved by the Houston Methodist Research Institute Animal Care and Use Committee.
Supplementary Material
Acknowledgments
We thank Connie Cantu, Chandni Valson, Joseph Ferretti, Alicia Alonso, Anne Ramstad Alme, Gunnhild Roedal, Kirsten Burmeister, Anna Syk, Ingrid Andersson, Christina Johansson, Gunnel Mollerberg, Johanna Makinen, Tuula Siljander, Anna Muotiala, Helena Seppala, the FiRe–Finnish Study Group for Antimicrobial Resistance, and Bernard Beall and the Active Bacterial Core surveillance of the Centers for Disease Control and Prevention’s Emerging Infections Programs network. We thank Kathryn Stockbauer for critical comment and editorial assistance and David Morens and Frank DeLeo for suggestions to improve the manuscript. This project was supported in part with funds from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, Houston Methodist Hospital, and the Fondren Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the Sequence Read Archive (accession no. SRA036051).
2Deceased September 18, 2013.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1403138111/-/DCSupplemental.
References
- 1.Beall B, Facklam R, Hoenes T, Schwartz B. Survey of emm gene sequences and T-antigen types from systemic Streptococcus pyogenes infection isolates collected in San Francisco, California; Atlanta, Georgia; and Connecticut in 1994 and 1995. J Clin Microbiol. 1997;35(5):1231–1235. doi: 10.1128/jcm.35.5.1231-1235.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bucher A, et al. Spectrum of disease in bacteraemic patients during a Streptococcus pyogenes serotype M-1 epidemic in Norway in 1988. Eur J Clin Microbiol Infect Dis. 1992;11(5):416–426. doi: 10.1007/BF01961856. [DOI] [PubMed] [Google Scholar]
- 3.Carapetis J, Robins-Browne R, Martin D, Shelby-James T, Hogg G. Increasing severity of invasive group A streptococcal disease in Australia: Clinical and molecular epidemiological features and identification of a new virulent M-nontypeable clone. Clin Infect Dis. 1995;21(5):1220–1227. doi: 10.1093/clinids/21.5.1220. [DOI] [PubMed] [Google Scholar]
- 4.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5(11):685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 5.Chelsom J, Halstensen A, Haga T, Høiby EA. Necrotising fasciitis due to group A streptococci in western Norway: Incidence and clinical features. Lancet. 1994;344(8930):1111–1115. doi: 10.1016/s0140-6736(94)90629-7. [DOI] [PubMed] [Google Scholar]
- 6.Colman G, Tanna A, Efstratiou A, Gaworzewska ET. The serotypes of Streptococcus pyogenes present in Britain during 1980-1990 and their association with disease. J Med Microbiol. 1993;39(3):165–178. doi: 10.1099/00222615-39-3-165. [DOI] [PubMed] [Google Scholar]
- 7.Davies HD, et al. Ontario Group A Streptococcal Study Group Invasive group A streptococcal infections in Ontario, Canada. N Engl J Med. 1996;335(8):547–554. doi: 10.1056/NEJM199608223350803. [DOI] [PubMed] [Google Scholar]
- 8.Demers B, et al. Severe invasive group A streptococcal infections in Ontario, Canada: 1987-1991. Clin Infect Dis. 1993;16(6):792–800. doi: 10.1093/clind/16.6.792. discussion 801-792. [DOI] [PubMed] [Google Scholar]
- 9.Gaworzewska E, Colman G. Changes in the pattern of infection caused by Streptococcus pyogenes. Epidemiol Infect. 1988;100(2):257–269. doi: 10.1017/s095026880006739x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikebe T, et al. Working Group for Group A Streptococci in Japan Changing prevalent T serotypes and emm genotypes of Streptococcus pyogenes isolates from streptococcal toxic shock-like syndrome (TSLS) patients in Japan. Epidemiol Infect. 2003;130(3):569–572. [PMC free article] [PubMed] [Google Scholar]
- 11.Kaul R, McGeer A, Low DE, Green K, Schwartz B. Population-based surveillance for group A streptococcal necrotizing fasciitis: Clinical features, prognostic indicators, and microbiologic analysis of seventy-seven cases. Ontario Group A Streptococcal Study. Am J Med. 1997;103(1):18–24. doi: 10.1016/s0002-9343(97)00160-5. [DOI] [PubMed] [Google Scholar]
- 12.Martin PR, Høiby EA. Streptococcal serogroup A epidemic in Norway 1987-1988. Scand J Infect Dis. 1990;22(4):421–429. doi: 10.3109/00365549009027073. [DOI] [PubMed] [Google Scholar]
- 13.Muotiala A, Seppälä H, Huovinen P, Vuopio-Varkila J. Molecular comparison of group A streptococci of T1M1 serotype from invasive and noninvasive infections in Finland. J Infect Dis. 1997;175(2):392–399. doi: 10.1093/infdis/175.2.392. [DOI] [PubMed] [Google Scholar]
- 14.Musser J, Krause R. The revival of group A streptococcal diseases, with a commentary on staphylococcal toxic shock syndrome. In: Krause R, editor. Emerging Infections. New York: Academic Press; 1998. pp. 185–218. [Google Scholar]
- 15.O’Brien KL, et al. Epidemiology of invasive group a streptococcus disease in the United States, 1995-1999. Clin Infect Dis. 2002;35(3):268–276. doi: 10.1086/341409. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz B, Facklam RR, Breiman RF. Changing epidemiology of group A streptococcal infection in the USA. Lancet. 1990;336(8724):1167–1171. doi: 10.1016/0140-6736(90)92777-f. [DOI] [PubMed] [Google Scholar]
- 17.Sharkawy A, et al. Ontario Group A Streptococcal Study Group Severe group a streptococcal soft-tissue infections in Ontario: 1992-1996. Clin Infect Dis. 2002;34(4):454–460. doi: 10.1086/338466. [DOI] [PubMed] [Google Scholar]
- 18.Stevens DL, et al. Severe group A streptococcal infections associated with a toxic shock-like syndrome and scarlet fever toxin A. N Engl J Med. 1989;321(1):1–7. doi: 10.1056/NEJM198907063210101. [DOI] [PubMed] [Google Scholar]
- 19.Strömberg A, Romanus V, Burman LG. Outbreak of group A streptococcal bacteremia in Sweden: An epidemiologic and clinical study. J Infect Dis. 1991;164(3):595–598. doi: 10.1093/infdis/164.3.595. [DOI] [PubMed] [Google Scholar]
- 20.Tyrrell GJ, et al. M types of group a streptococcal isolates submitted to the National Centre for Streptococcus (Canada) from 1993 to 1999. J Clin Microbiol. 2002;40(12):4466–4471. doi: 10.1128/JCM.40.12.4466-4471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. http://en.wikipedia.org/wiki/Necrotizing_fasciitis. Accessed February 11, 2014.
- 22.Beres SB, et al. Genome sequence of a serotype M3 strain of group A Streptococcus: Phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. 2002;99(15):10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferretti JJ, et al. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc Natl Acad Sci USA. 2001;98(8):4658–4663. doi: 10.1073/pnas.071559398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumby P, et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J Infect Dis. 2005;192(5):771–782. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 25.Beres SB, Musser JM. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS ONE. 2007;2(8):e800. doi: 10.1371/journal.pone.0000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banks DJ, et al. Progress toward characterization of the group A Streptococcus metagenome: Complete genome sequence of a macrolide-resistant serotype M6 strain. J Infect Dis. 2004;190(4):727–738. doi: 10.1086/422697. [DOI] [PubMed] [Google Scholar]
- 27.Fittipaldi N, et al. Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol. 2012;180(4):1522–1534. doi: 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 28.Green NM, et al. Genome sequence of a serotype M28 strain of group a streptococcus: Potential new insights into puerperal sepsis and bacterial disease specificity. J Infect Dis. 2005;192(5):760–770. doi: 10.1086/430618. [DOI] [PubMed] [Google Scholar]
- 29.Holden MT, et al. Complete genome of acute rheumatic fever-associated serotype M5 Streptococcus pyogenes strain manfredo. J Bacteriol. 2007;189(4):1473–1477. doi: 10.1128/JB.01227-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McShan WM, et al. Genome sequence of a nephritogenic and highly transformable M49 strain of Streptococcus pyogenes. J Bacteriol. 2008;190(23):7773–7785. doi: 10.1128/JB.00672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyoshi-Akiyama T, Watanabe S, Kirikae T. Complete genome sequence of Streptococcus pyogenes M1 476, isolated from a patient with streptococcal toxic shock syndrome. J Bacteriol. 2012;194(19):5466. doi: 10.1128/JB.01265-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smoot JC, et al. Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc Natl Acad Sci USA. 2002;99(7):4668–4673. doi: 10.1073/pnas.062526099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng PX, et al. Complete Genome Sequence of emm1 Streptococcus pyogenes A20, a Strain with an Intact Two-Component System, CovRS, Isolated from a Patient with Necrotizing Fasciitis. Genome Announc. 2013;1(1) doi: 10.1128/genomeA.00149-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varaldo PE, Montanari MP, Giovanetti E. Genetic elements responsible for erythromycin resistance in streptococci. Antimicrob Agents Chemother. 2009;53(2):343–353. doi: 10.1128/AAC.00781-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter SS, et al. Macrolide-resistant Streptococcus pyogenes in the United States, 2002-2003. Clin Infect Dis. 2005;41(5):599–608. doi: 10.1086/432473. [DOI] [PubMed] [Google Scholar]
- 36.Maamary PG, et al. Tracing the evolutionary history of the pandemic group A streptococcal M1T1 clone. FASEB J. 2012;26(11):4675–4684. doi: 10.1096/fj.12-212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson K, Schlievert PM, Selander RK, Musser JM. Characterization and clonal distribution of four alleles of the speA gene encoding pyrogenic exotoxin A (scarlet fever toxin) in Streptococcus pyogenes. J Exp Med. 1991;174(5):1271–1274. doi: 10.1084/jem.174.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grossman D, Cook RG, Sparrow JT, Mollick JA, Rich RR. Dissociation of the stimulatory activities of staphylococcal enterotoxins for T cells and monocytes. J Exp Med. 1990;172(6):1831–1841. doi: 10.1084/jem.172.6.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kline JB, Collins CM. Analysis of the superantigenic activity of mutant and allelic forms of streptococcal pyrogenic exotoxin A. Infect Immun. 1996;64(3):861–869. doi: 10.1128/iai.64.3.861-869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musser JM, et al. Streptococcus pyogenes causing toxic-shock-like syndrome and other invasive diseases: Clonal diversity and pyrogenic exotoxin expression. Proc Natl Acad Sci USA. 1991;88(7):2668–2672. doi: 10.1073/pnas.88.7.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musser JM, et al. Genetic diversity and relationships among Streptococcus pyogenes strains expressing serotype M1 protein: Recent intercontinental spread of a subclone causing episodes of invasive disease. Infect Immun. 1995;63(3):994–1003. doi: 10.1128/iai.63.3.994-1003.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoe NP, et al. Rapid selection of complement-inhibiting protein variants in group A Streptococcus epidemic waves. Nat Med. 1999;5(8):924–929. doi: 10.1038/11369. [DOI] [PubMed] [Google Scholar]
- 43.Shea PR, et al. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci USA. 2011;108(12):5039–5044. doi: 10.1073/pnas.1016282108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olsen RJ, Musser JM. Molecular pathogenesis of necrotizing fasciitis. Annu Rev Pathol. 2010;5:1–31. doi: 10.1146/annurev-pathol-121808-102135. [DOI] [PubMed] [Google Scholar]
- 45.Olsen RJ, et al. Decreased necrotizing fasciitis capacity caused by a single nucleotide mutation that alters a multiple gene virulence axis. Proc Natl Acad Sci USA. 2010;107(2):888–893. doi: 10.1073/pnas.0911811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Virtaneva K, et al. Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc Natl Acad Sci USA. 2005;102(25):9014–9019. doi: 10.1073/pnas.0503671102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 48.Abecasis GR, et al. 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poznik GD, et al. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science. 2013;341(6145):562–565. doi: 10.1126/science.1237619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casali N, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014;46(3):279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chewapreecha C, et al. Dense genomic sampling identifies highways of pneumococcal recombination. Nat Genet. 2014;46(3):305–309. doi: 10.1038/ng.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holt KE, et al. Tracking the establishment of local endemic populations of an emergent enteric pathogen. Proc Natl Acad Sci USA. 2013;110(43):17522–17527. doi: 10.1073/pnas.1308632110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mather AE, et al. Distinguishable epidemics of multidrug-resistant Salmonella Typhimurium DT104 in different hosts. Science. 2013;341(6153):1514–1517. doi: 10.1126/science.1240578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Z, et al. Neutral genomic microevolution of a recently emerged pathogen, Salmonella enterica serovar Agona. PLoS Genet. 2013;9(4):e1003471. doi: 10.1371/journal.pgen.1003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aziz RK, et al. Mosaic prophages with horizontally acquired genes account for the emergence and diversification of the globally disseminated M1T1 clone of Streptococcus pyogenes. J Bacteriol. 2005;187(10):3311–3318. doi: 10.1128/JB.187.10.3311-3318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aziz RK, Kotb M. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis. 2008;14(10):1511–1517. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cleary PP, et al. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet. 1992;339(8792):518–521. doi: 10.1016/0140-6736(92)90339-5. [DOI] [PubMed] [Google Scholar]
- 59.Cleary PP, LaPenta D, Vessela R, Lam H, Cue D. A globally disseminated M1 subclone of group A streptococci differs from other subclones by 70 kilobases of prophage DNA and capacity for high-frequency intracellular invasion. Infect Immun. 1998;66(11):5592–5597. doi: 10.1128/iai.66.11.5592-5597.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vlaminckx BJ, et al. Dynamics in prophage content of invasive and noninvasive M1 and M28 Streptococcus pyogenes isolates in The Netherlands from 1959 to 1996. Infect Immun. 2007;75(7):3673–3679. doi: 10.1128/IAI.01695-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bricker AL, Carey VJ, Wessels MR. Role of NADase in virulence in experimental invasive group A streptococcal infection. Infect Immun. 2005;73(10):6562–6566. doi: 10.1128/IAI.73.10.6562-6566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bricker AL, Cywes C, Ashbaugh CD, Wessels MR. NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol Microbiol. 2002;44(1):257–269. doi: 10.1046/j.1365-2958.2002.02876.x. [DOI] [PubMed] [Google Scholar]
- 63.Chiarot E, et al. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio. 2013;4(1):e00387–00312. doi: 10.1128/mBio.00387-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Limbago B, Penumalli V, Weinrick B, Scott JR. Role of streptolysin O in a mouse model of invasive group A streptococcal disease. Infect Immun. 2000;68(11):6384–6390. doi: 10.1128/iai.68.11.6384-6390.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madden JC, Ruiz N, Caparon M. Cytolysin-mediated translocation (CMT): A functional equivalent of type III secretion in gram-positive bacteria. Cell. 2001;104(1):143–152. doi: 10.1016/s0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 66.Michos A, et al. Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J Biol Chem. 2006;281(12):8216–8223. doi: 10.1074/jbc.M511674200. [DOI] [PubMed] [Google Scholar]
- 67.O’Seaghdha M, Wessels MR. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog. 2013;9(6):e1003394. doi: 10.1371/journal.ppat.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raskova H, Vanecek J. Effects of intraventricular injections of streptolysin O in unanesthetized cats. Science. 1957;126(3276):700–701. doi: 10.1126/science.126.3276.700. [DOI] [PubMed] [Google Scholar]
- 69.Riddle DJ, Bessen DE, Caparon MG. Variation in Streptococcus pyogenes NAD+ glycohydrolase is associated with tissue tropism. J Bacteriol. 2010;192(14):3735–3746. doi: 10.1128/JB.00234-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sierig G, Cywes C, Wessels MR, Ashbaugh CD. Cytotoxic effects of streptolysin o and streptolysin s enhance the virulence of poorly encapsulated group a streptococci. Infect Immun. 2003;71(1):446–455. doi: 10.1128/IAI.71.1.446-455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stevens DL, Salmi DB, McIndoo ER, Bryant AE. Molecular epidemiology of nga and NAD glycohydrolase/ADP-ribosyltransferase activity among Streptococcus pyogenes causing streptococcal toxic shock syndrome. J Infect Dis. 2000;182(4):1117–1128. doi: 10.1086/315850. [DOI] [PubMed] [Google Scholar]
- 72.Tatsuno I, Isaka M, Minami M, Hasegawa T. NADase as a target molecule of in vivo suppression of the toxicity in the invasive M-1 group A Streptococcal isolates. BMC Microbiol. 2010;10:144. doi: 10.1186/1471-2180-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Timmer AM, et al. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J Biol Chem. 2009;284(2):862–871. doi: 10.1074/jbc.M804632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luca-Harari B, et al. Strep-EURO Study Group Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2009;47(4):1155–1165. doi: 10.1128/JCM.02155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shulman ST, et al. North American Streptococcal Pharyngitis Surveillance Group Seven-year surveillance of north american pediatric group a streptococcal pharyngitis isolates. Clin Infect Dis. 2009;49(1):78–84. doi: 10.1086/599344. [DOI] [PubMed] [Google Scholar]
- 76.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: Systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9(10):611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.