

Significance
We have developed a new class of host-targeted biologics to prevent influenza by engineering multivalent carbohydrate-binding modules that bind with high affinity to sialic acid, the critical component of the influenza virus cell surface receptor. Mouse studies reveal a remarkable efficacy: a single 1-μg dose of the lead biologic given 7 d before a lethal challenge with 2009 pandemic H1N1 influenza virus provides complete protection. This new approach has the potential to be a front-line defense against any current and future influenza virus, overcoming viral escape to vaccines and antivirals. In addition, the biologics may have broader application against other respiratory pathogens.
Keywords: drug discovery, recombinant protein therapy
Abstract
There is a need for new approaches for the control of influenza given the burden caused by annual seasonal outbreaks, the emergence of viruses with pandemic potential, and the development of resistance to current antiviral drugs. We show that multivalent biologics, engineered using carbohydrate-binding modules specific for sialic acid, mask the cell-surface receptor recognized by the influenza virus and protect mice from a lethal challenge with 2009 pandemic H1N1 influenza virus. The most promising biologic protects mice when given as a single 1-μg intranasal dose 7 d in advance of viral challenge. There also is sufficient virus replication to establish an immune response, potentially protecting the animal from future exposure to the virus. Furthermore, the biologics appear to stimulate inflammatory mediators, and this stimulation may contribute to their protective ability. Our results suggest that this host-targeted approach could provide a front-line prophylactic that has the potential to protect against any current and future influenza virus and possibly against other respiratory pathogens that use sialic acid as a receptor.
Influenza viruses continue to be a threat to human health and a burden on health services (1). The emergence of highly pathogenic H5N1 viruses and recent introductions of H7N9 viruses from avian sources (2), and their potential to acquire human transmissibility, increase the threat (3–5). Although vaccines remain a cornerstone of prevention, significant time is required to develop an effective vaccine against a new virus strain. Anti-influenza drugs approved by the Food and Drug Administration, such as the viral neuraminidase inhibitors oseltamivir (Tamiflu) and zanamivir (Relenza) and the M2 ion-channel blocker adamantanes (amantadine and rimantadine), are available, but their effectiveness can be compromised by the virus’s ability to mutate and become drug resistant (6, 7).
The influenza virus binds to sialic acid receptors present on the respiratory tract epithelium via its surface HA glycoprotein, an event that triggers viral endocytosis (8). Other respiratory pathogens, such as parainfluenza viruses (9), some coronaviruses (10), and Streptococcus pneumoniae (11), also use sialic acid as a receptor. Human influenza viruses such as the 2009 pandemic H1N1 virus recognize α-2,6–linked sialic acid receptors present in the upper respiratory tract, whereas avian influenza viruses such as H5N1 predominantly recognize α-2,3–linked sialic acid receptors, which are present in the human lower respiratory tract as well (12, 13). The recently emerged human H7N9 influenza virus is unusual in recognizing both types of receptors and therefore has the possibility of sustained human-to-human transmission and pandemic potential (14, 15).
We hypothesized that masking such receptors in the respiratory tract with proteins specific for sialic acid could provide a novel host-targeted therapeutic route to prevent infection. Numerous sialic acid-binding proteins are known, but most have low affinity for sialic acid (e.g., the HA monomer that has ∼2.5 mM affinity for its receptor but gains affinity by being present in high copy number on the virus surface) (16). We have shown previously that engineered multivalent polypeptides containing up to four tandemly linked copies of the sialic acid-recognizing carbohydrate-binding module (CBM) from Vibrio cholerae nanH sialidase display low (nanomolar) binding affinity compared with the 30-μM affinity of the single domain (17).
Here we report the engineering and characterization of further sialic acid-recognizing multivalent CBMs (mCBMs) together with in vitro and in vivo evidence of their potential in preventing influenza infection. Significantly, our lead mCBM demonstrates protective in vivo efficacy when given to mice as a single 1-μg dose 7 d in advance of a lethal virus challenge with pandemic 2009 H1N1 influenza virus, indicating that these mCBMs show great promise as biologics for the prophylaxis of influenza and potentially other respiratory pathogens that recognize sialic acid receptors.
Results
Design, Engineering, and Characterization of Biologics.
Multivalent sialic acid-binding proteins were engineered using either the sialic acid-binding domain from V. cholerae NanH sialidase (18) (VcCBM) (Fig. 1A) or the homologous domain from S. pneumoniae NanA sialidase (SpCBM) (Fig. 1B). Multivalency was achieved either through tandem repeats (17) or by fusing one or two tandemly linked CBM domains to an oligomerization domain (TD) derived from Pseudomonas aeruginosa sialidase (Fig. 1C) (19), a domain that self-associates to form a trimer. In all constructs, domains were linked using a five-amino acid peptide sequence (Table S1). To assess the binding affinity of the multivalent forms of VcCBM and SpCBM to sialic acid, surface plasmon resonance (SPR) was performed using an immobilized biotinylated α-2,3-sialyllactose-polyacrylamide ligand. All mCBMs bind sialic acid with high affinity, with dissociation constants <2 nM at 25 °C, showing that they gain affinity through an avidity effect (Fig. 1D and Table S2). Glycan array screening of VcCBM (17) and SpCBM (Fig. S1) showed that they recognize glycans containing terminal α-2,3– or α-2,6–linked sialic acids, and crystallographic analysis with bound sialyllactose reveals that only the terminal sialic acid interacts with each domain (18).
Fig. 1.

Building blocks of the multivalent CBM forms and their affinities for sialic acid. (A) VcCBM, residues 25–216 of the V. cholerae sialidase [Protein Data Bank (PDB) ID: 1W0P] with α-2,3-sialyllactose drawn as spheres. (B) SpCBM, residues 121–305 of S. pneumoniae NanA sialidase with α-2,3-sialyllactose (PDB ID: 4C1W). (C) The TD, residues 333–438, of the P. aeruginosa sialidase (PDB ID: 2W38) in rainbow colors; the other two monomers are in gold and silver colors. (D) Multivalent forms: their molecular weights, valences and binding affinities for α-2,3-sialyllactose as determined by SPR at 25 °C [KD values for VcCBM, Vc2CBM, and Vc3CBM have been reported previously (17)]. Tandem repeat CBMs and oligomeric CBMs fused to the TD are linked by a five-amino acid linker (details are given in SI Materials and Methods).
To confirm that mCBMs target cell-surface sialic acids, Madin–Darby canine kidney (MDCK) and human lung carcinoma A549 cells were either left untreated or treated with a broad-specificity sialidase using the catalytic domain of S. pneumoniae NanA sialidase (20) before incubation with the hexameric forms, Vc2CBMTD and Sp2CBMTD, followed by immunostaining with Alexa Fluor 488-labeled anti-rabbit IgG antibody. Live imaging studies showed that both hexameric CBMs bind specifically at the cell surface of both cell types and that binding was largely abrogated after cells were pretreated with the promiscuous sialidase to remove sialic acids (Fig. S2A). This result was verified further by quantification of the GFP-fluorescence intensity of sialidase-treated and untreated cells (Fig. S2B).
In Vitro Testing of Biologics Against Influenza Viruses.
The ability of the mCBMs to block virus infection was tested first in vitro (Table S3). MDCK cells were preincubated with different concentrations of mCBMs before infection with 100–200 pfu of three different influenza A viruses [A/WSN/1933 (H1N1), A/PR/8/1934 (H1N1), and A/Udorn/1972 (H3N2)] and an influenza B virus (B/Hong Kong/1973). Cell protection was quantified by reduction of virus plaques in cells treated with mCBMs as compared with untreated, infected cells. Cell protection was observed with all mCBMs tested, with EC50 values ranging from 0.39–4.1 µM for mCBMs with valences between 3 and 6, compared with ∼300 μM when the monovalent CBM was used (Table S3). A viral replication inhibition assay also was performed using MDCK cells with different concentrations of mCBMs before cells were infected with different influenza A viruses [multiplicity of infection (MOI) = 0.01 pfu per cell]. Reduction in viral replication was assayed by immunostaining treated cells with goat anti-influenza A virus antibodies. The mCBMs inhibited viral replication in all strains tested. EC50 values were similar for A/WSN/1933 (H1N1) (0.5–4.25 µM) and A/Udorn/1972 (H3N2) (0.45–5 µM) and were up to 10-fold higher when tested against A/PR/8/1934 (H1N1) (1.34–44.5 µM). The higher EC50 values observed for A/PR/8/1934 (H1N1) possibly may reflect the long passage history of the virus in different host systems, so that A/PR/8/1934 (H1N1) may not be the optimal strain for assessing the ability of biologics to block virus infection in vitro. Furthermore, imaging studies using an anti-influenza A nucleoprotein (NP) antibody demonstrated blocking of influenza virus in MDCK cells that were pretreated with hexameric mCBMs as compared with untreated cells infected with influenza A/WSN/1933 (H1N1) virus (Fig. S2C). Detection of viral NP was substantially reduced in CBM-treated MDCK cells, confirming the efficacy of mCBMs in reducing or preventing viral attachment in vitro.
We also tested the effect of mCBMs on MDCK cells over a period of 24 h to determine cell viability and found that these proteins were well tolerated at maximum feasible concentrations (5 mg/mL) when measured against a sodium azide control group (Table S3). Moreover, the therapeutic index calculated for each mCBM demonstrated values from 9 to 119, indicating that these biologics could be tolerated in vivo.
Efficacy of Biologics in Vivo.
Next, we explored the ability of Vc4CBM to block a lethal infection of A/WSN/1933 (H1N1) virus in BALB/c mice. Vc4CBM (500 µg) was administered intranasally as a single dose immediately before viral challenge. The Vc4CBM-treated mice survived and regained weight after a small initial weight loss with a two-logarithm reduction in lung virus titers 7 d post inoculation (p.i.) (Fig. S3 A and B). This initial promising result led us to explore the protective effects of the trimeric (VcCBMTD, SpCBMTD, 400 µg) and hexameric (Vc2CBMTD, Sp2CBMTD, 100 µg) forms given intranasally 1 d before a lethal challenge with A/WSN/1933 (H1N1) virus (Fig. S3 C and D). Of the four mCBMs, the hexameric forms showed almost no detectable virus in lungs at 7 d p.i., with Sp2CBMTD-treated mice demonstrating negligible weight loss (Fig. S3C). In contrast, VcCBMTD led to a higher viral titer and a greater weight loss than seen in the control; this result may have been caused by an impure preparation. At this point we decided to focus on the hexameric forms because of their greater efficacy.
We subsequently evaluated the biologics in BALB/c mice in challenge experiments against lethal doses [10 times the 50% mouse lethal dose (MLD50)] of mouse-adapted A/California/04/2009 (H1N1) pandemic influenza virus, exploring single intranasal doses (10, 50, 250, or 500 μg) of Vc4CBM, Vc2CBMTD, or Sp2CBMTD given on day −1, 0, or +1 (day 0 being the day of viral challenge). Survival studies continued to day 21 p.i. (Fig. 2). None of the mCBMs protected mice when given 24 h after a lethal viral challenge (Fig. S4). When administered on day −1, Vc4CBM (500 μg) gave only 40% survival. When administered on day 0, Vc4CBM (500 μg) gave 100% survival, in line with the A/WSN/1933 (H1N1) virus challenge, but gave only 40% survival at lower doses. In contrast, when administered as single doses on day −1 or day 0, the hexameric forms gave significant protection compared with untreated, infected mice. Vc2CBMTD gave 80% survival at the lower doses of 10 or 50 μg. Sp2CBMTD provided the best protection, with all mice surviving at all doses administered on day −1 and all mice surviving after administration of the 50- or 250-μg dose on day 0 and 60% surviving at the 10-μg dose. All animals that survived virus challenge lost weight during infection, reaching a maximum loss around day 8 p.i., but soon started to regain weight after this period; the most beneficial results were observed when Sp2CBMTD was administrated at a 250-µg dose on day 0 (Fig. 2B).
Fig. 2.

Effect of prophylactic administration of mCBMs in BALB/c mice given a lethal challenge with mouse-adapted A/California/04/2009 (H1N1) influenza virus. (A) (Upper) Survival and (Lower) weight changes of mice (n = 5) after a single intranasal dose of Vc4CBM (50, 250, or 500 µg), Vc2CBMTD (10, 50, or 250 µg), or Sp2CBMTD (10, 50, or 250 µg) given on day −1 before viral challenge. (B) (Upper) Survival curves for mice given the same single intranasal doses of one of the three biologics on day 0, immediately before viral challenge. (Lower) Corresponding weight-loss curves. Each value represents mean body weight ± SD for five mice.
We next explored the effect of repeat dosing with Sp2CBMTD administered further in advance of infection. Single 50-μg doses of Sp2CBMTD were administered intranasally to BALB/c mice once, twice, or three times up to 1 wk before a lethal challenge (10 MLD50) with mouse-adapted A/California/04/2009 (H1N1) virus on day 0. There was 100% survival with all dosing regimens, and, strikingly, there was little or no weight loss when two or three doses were administered (Fig. 3A). Then the lowest effective dose of Sp2CBMTD was explored. Single 10-, 1-, or 0.1-μg doses of Sp2CBMTD were administered on day −7, −3, or −1 in advance of a lethal challenge with mouse-adapted A/California/04/2009 (H1N1) virus on day 0. All mice survived the 10- and 1-μg dosing regimens. Significantly, even a single 1-μg dose given on day −7 resulted in a maximum weight loss of only 8% on day 8 p.i., which soon was restored (Fig. 3B), with mice continuing to thrive with no adverse clinical signs to day 21 p.i.. In contrast, at 0.1-μg dosing, 80%, 20%, or 0% of mice survived when Sp2CBMTD was administered on day −1, −3, or −7, respectively (Fig. 3B), suggesting that the timing and dosing of mCBM administration are significant in determining the level of protection in mice given lethal doses of influenza virus. Administration of these low doses did not result in the initial weight losses observed when higher doses were given in the earlier studies (Fig. S3), suggesting greater toleration at low dose. Viral lung titers measured on days 3, 6, and 9 p.i. showed that virus had cleared from the lungs by day 9 in mice given single 50- and 10-μg doses, whereas virus titers in mice given 1- and 0.1-μg doses were similar to those in control (infected, untreated) mice (Fig. 3C). Significantly, high titers of serum anti-HA antibodies were present in surviving mice after treatment at all dosing regimens, with reciprocal HA inhibition titers ranging from 160–320 with repeat dosing and from 320–640 with single dosing, suggesting that viral replication is sufficient to elicit an immune response.
Fig. 3.

Survival and weight changes of BALB/c mice administered Sp2CBMTD before a lethal challenge with mouse-adapted A/California/04/2009 (H1N1) influenza virus. (A) BALB/c mice (n = 5) were given single, double, or triple intranasal doses of Sp2CBMTD (50 µg) on days −7, −3, or −1 before viral challenge on day 0. Mice also were given a triple intranasal dose of Sp2CBMTD alone to determine the toxicity of the biologic (shown in blue in the right panel). (B) Survival and weight changes of mice (n = 5) after single intranasal doses of Sp2CBMTD (10, 1, or 0.1 µg) given on days −7, −3, or −1 before viral challenge. (Upper) Survival curves for each administration day. (Lower) Corresponding weight-loss curves. In all cases, control animals were infected and mock-treated with PBS only. Each value represents mean body weight ± SD for five mice. (C) Virus lung titers on days 3, 6, and 9 p.i. following intranasal administration of single dose of Sp2CBMTD on day −7, −3, or −1 before lethal viral challenge. Values represent the mean viral titers (expressed as log10 50% tissue culture infectious dose/mL) ± SD from three mice. *P < 0.001 compared with control (infected, untreated) mice.
Detection of Biologics in Lung Tissue.
To determine whether mCBMs target cell surfaces in the lung, tissue from mice intranasally administered a single dose of Sp2CBMTD (400 μg) and culled 1 d later was stained using anti-SpCBM and anti-IgG Alexa Fluor 488 antibodies. Sp2CBMTD was detected readily on the surface of lung alveoli epithelial cells (Fig. S5A), demonstrating that mCBMs target cell surfaces along the respiratory tract. The in vitro studies suggest that this targeting is most likely to occur through binding sialic acid receptors. We then investigated whether Sp2CBMTD (50 μg), given as a pretreatment, can be detected in virally infected mouse lungs. Lung tissue from mice infected with a lethal dose of influenza A/WSN/1933 virus (5 × 103 pfu) and culled at day 2 or day 7 p.i. was stained for Sp2CBMTD. Immunofluorescence images of infected lung tissue demonstrated clear evidence of the presence of Sp2CBMTD in the lower respiratory tract with strong binding to alveolar epithelial cell surfaces of lung tissues on day 2 p.i. (Fig. S5B). Lung tissue harvested at day 7 p.i. also showed the presence of Sp2CBMTD (Fig. S5B), albeit at a much lower level.
Immunogenicity of the Biologics.
One concern with using pathogen-derived biologics is the potential for immunogenicity that may reduce the effectiveness of repeat administration. SpCBM and VcCBM were chosen specifically from human pathogens that may have evolved immunotolerance to the host. To determine if preexisting immunity to either of the single CBM domains is present in the human population, we carried out ELISAs using immobilized VcCBM and SpCBM antigens, human sera from a mixed-age population of males and females, and an anti-human IgG-HRP conjugate. Analysis of all blood samples against positive adenovirus controls demonstrated that there were no significant levels of anti-VcCBM or anti-SpCBM antibodies in the pool of human sera tested (Fig. S6). The generation of anti-SpCBM antibodies was explored in mouse sera (at day 21 p.i.) from the challenge study that used an intranasally administered single 10-μg or 1-μg dose of Sp2CBMTD and mouse-adapted A/California/04/2009 (H1N1) pandemic influenza virus as described above. Prophylactic administration of Sp2CBMTD was shown to elicit serum IgG, IgM, and IgA antibodies in a dose-dependent manner, with the 1-μg Sp2CBMTD dose eliciting negligible levels of serum IgA and IgM (Fig. S7). Immunogenicity of the mCBMs may be an issue with repeat use, but the proteins could be modified to be less immunogenic if necessary (21).
Stimulation of Inflammatory Mediators by the Biologics.
The original hypothesis in the biologic design was the masking of sialic acid receptors; however, the remarkable protective ability of a single low dose (1 μg) of Sp2CBMTD given 7 d in advance of infection raises the possibility that the biologic also may prime the immune system into an antiviral state. Accordingly, we explored the induction of a limited set of inflammatory cytokines/chemokines by intranasal administration of Vc2CBMTD or Sp2CBMTD in mice and found that there is a significant difference between the two biologics, with Sp2CBMTD stimulating higher levels of IL-1β, MIP-2 (mouse homolog of IL-8), IFN-γ, and TNF-α than seen with either Vc2CBMTD or PBS (Fig. 4). The S. pneumoniae CBM forms part of a larger region of the NanA sialidase that previously has been reported to stimulate certain cytokines in human and mouse brain cells (22), although other segments of NanA may be responsible for this activity. It is possible that our biologics modulate cellular processes, including cytokine induction, that contribute to the protective ability of Sp2CBMTD, but the identification of these processes will require further extensive exploration.
Fig. 4.
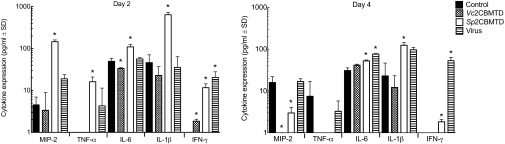
Effect of mCBMs on pulmonary expression of chemokines and cytokines. Inflammatory mediators MIP-2, TNF-α, IL-6, IL-1β, IFN-γ, GM-CSF, and IL-2 were assayed by ELISA using lung homogenates obtained from BALB/c mice on days 2 and 4 after nasal administration of Vc2CBMTD (100 μg), Sp2CBMTD (100 μg), A/WSN/1933 (H1N1) influenza virus (5 × 103 pfu), or PBS (control). All except GM-CSF and IL-2 were detected in lung homogenates. Bars indicate the mean concentration (pg/mL) ± SD from five mice. *P < 0.05 compared with the results for the control (uninfected, untreated) group.
Discussion
There is an urgent need for new therapeutic approaches to control influenza. Significant effort is being devoted to developing new vaccine approaches to influenza (23, 24), new inhibitors of viral targets (25), and novel strategies targeted at host factors (26, 27). One host-targeted strategy is to suppress the availability of sialic acid receptors on the respiratory tract epithelium that are recognized by the influenza virus HA glycoprotein. DAS181 (Fludase) is a biologic formed by linking the catalytic domain of the promiscuous sialidase from Actinomyces viscosus to the epithelium-anchoring domain of human amphiregulin (28). DAS181 removes sialic acids from the epithelial surface and, when administered intranasally, protects against infection with avian H5N1 and pandemic 2009 H1N1 viruses in mice (29, 30). DAS181 is currently in Phase II development (27). Clarithromycin, a macrolide antibiotic used to treat tonsillitis and pharyngitis, has potential as an influenza therapeutic; it reduces the expression of sialic acid receptors on the surface of airway epithelial cells and decreases the number of acidic endosomes in the cell, reducing viral endocytosis (31).
In this study, we have demonstrated that masking sialic acid receptors using engineered high-affinity mCBMs based on the accessory sialic acid-binding domains from V. cholerae and S. pneumoniae sialidases can protect cells in vitro against infection from different strains of influenza A and B viruses. The mCBMs also conferred protection in vivo in BALB/c mice against lethal doses of the influenza A/WSN/1933 virus and of the 2009 pandemic H1N1 influenza A virus. This protection is most evident in studies performed using Sp2CBMTD with mouse-adapted A/California/04/2009 (H1N1) virus, with the biologic administered prophylactically as a single intranasal dose as low as 1 μg up to 7 d before viral challenge. Even prophylactic administration of a 0.1-μg dose of Sp2CBMTD afforded 80% protection when given the day before viral challenge. Although some weight loss occurred in the mice given these single doses, the repeat-dosing experiments using either two or three 50-μg doses suggest that regular dosing regimens at much lower levels of mCBMs may be the optimal regimen to minimize weight loss. Moreover, the generation of serum anti-HA antibodies observed in mice given mCBMs in advance or on the day of viral challenge suggests that, in addition to affording protection, the biologics allow “vaccination” to occur upon exposure to virus, potentially providing protection against future exposure. Interestingly, the mCBMs did not protect mice from influenza infection when administered as a single dose 24 h after lethal viral challenge. One explanation may be that viral HA levels were sufficiently high to compete for sialic acid-binding sites on the surface of respiratory epithelial cells, and that a single mCBM dose may not have been adequate to prevent further attachment and replication from newly formed viral particles. It is possible that an alternative dosing strategy could be considered as therapeutic treatment for influenza infection if mCBM dosing were administered more regularly in the early stages of infection.
In the current study, immunofluorescence of lung tissue demonstrated that only very small amounts of Sp2CBMTD, given as a single 50-μg dose, could be observed up to 8 d after administration. This finding suggests that a single lower dose, such as 1 μg given 7 d in advance, is unlikely to be effective in blocking a lethal viral challenge and suggests that other factors may contribute to Sp2CBMTD’s efficacy. Interestingly, the enhanced stimulation of certain inflammatory cytokines and chemokines observed in mice given Sp2CBMTD in the absence of pathogen warrants further exploration to determine the extent to which this activity contributes to Sp2CBMTD’s mode of action. Sialic acids are widely expressed on the surface of all cells in all vertebrates and are involved in regulating multiple cellular functions, including the development of immunity (32). Intrinsic sialic acid-recognizing proteins, often themselves multivalent, are known to mediate and modulate cellular interactions (33), so it is highly likely that Sp2CBMTD binds some as yet unknown receptor to elicit the modulation of the immune response.
We have demonstrated that biologics targeted to the mammalian host have certain advantages that merit further exploration. Our biologics have the capacity to bind to and mask different sialic acid receptors found in the upper and lower respiratory tract and therefore may provide protection throughout the human airway if a suitable delivery system is used. There are many challenges ahead in bringing these biologics to the clinic, but we believe that this class of therapeutics has the potential to be a powerful option for the control of influenza. The biologics have a possible broader application in blocking other respiratory pathogens that use sialic acid in pathogenesis, including S. pneumoniae, a leading cause of secondary bacterial infection often associated with influenza and responsible for increased morbidity and mortality (34).
Materials and Methods
mCBMs.
mCBMs were engineered using PCR-based cloning techniques. Constructs were propagated in Escherichia coli DH5a cells and were transformed into an E. coli BL21 Gold (DE3) expression host. Affinity and size-exclusion chromatography were used for purification of the mCBMs. Full details are given in SI Materials and Methods.
Detection of Sialic Acid Binding.
SPR, glycan array screening, crystallography, and fluorescence imaging were used to establish the affinity, specificity, and cell-surface binding of mCBMs to sialic acids. Full details are given in SI Materials and Methods.
In Vitro Assays.
Cell protection and virus replication inhibition assays were performed using MDCK cell monolayers incubated with mCBMs and different strains of influenza virus at 37 °C. EC50 values were determined from dose–response curves. The viability of MDCK cells after addition of mCBMs also was assayed over 24 h. Fluorescence imaging of MDCK and A549 cells pretreated with mCBMs and infected with influenza virus was performed also. Full details are given in SI Materials and Methods.
Mouse Infection Studies.
Mouse-adapted A/WSN/1933 (H1N1) and A/California/04/2009 (H1N1) influenza viruses were grown in MDCK cells and embryonated chicken eggs, respectively. BALB/c mice were lightly anesthetized with isoflurane before intranasal administration of mCBMs (given as a single dose on day −1, 0, or +1 or as repeated doses given once, twice or three times on days −7, −3, or −1) before a lethal virus challenge on day 0. Control (infected, untreated) and toxicity control (uninfected, treated) mice were tested also. Mice were weighed on the indicated days and assessed for visual clinical signs of disease. When necessary, lungs were harvested and were inflated for preparation of frozen tissue sections for immunofluorescence or homogenized in PBS and centrifuged. Clarified lung homogenates were tested for infectious virus as determined by plaque assays on MDCK cells and levels of cytokine/chemokine using a commercial ELISA kit. Immune sera samples from mice surviving to day 21 p.i. were tested for the presence of anti-HA antibodies and anti-mCBM antibodies using a hemagglutination inhibition assay or ELISA, respectively. Full details are given in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Bernie Precious, David Jackson, and Rick Randall for help with tissue culture and supply of viruses; Kristi Prevost for conducting animal experiments; Gill Broadbent and Keith Charlton for useful discussions; and Janice Bramham and Martin Wear for SPR analysis at the University of Edinburgh. This work was supported by a Biotechnology and Biological Sciences Research Council (UK) Follow-on-Fund award, a Medical Research Council (UK) Developmental Pathway Funding Scheme award, and by an award from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, under contract number HHSN266200700005C.
Footnotes
Conflict of interest statement: H.C. and G.L.T. are inventors on a patent held by the University of St Andrews that describes the CBM technology.
Data deposition: The atomic coordinates of SpCBM have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4C1W).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404205111/-/DCSupplemental.
References
- 1.Salomon R, Webster RG. The influenza virus enigma. Cell. 2009;136(3):402–410. doi: 10.1016/j.cell.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao R, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368(20):1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 3.Imai M, et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature. 2012;486(7403):420–428. doi: 10.1038/nature10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herfst S, et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science. 2012;336(6088):1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russell CA, et al. The potential for respiratory droplet-transmissible A/H5N1 influenza virus to evolve in a mammalian host. Science. 2012;336(6088):1541–1547. doi: 10.1126/science.1222526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Y, et al. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet. 2013;381(9885):2273–2279. doi: 10.1016/S0140-6736(13)61125-3. [DOI] [PubMed] [Google Scholar]
- 7.Baz M, et al. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N Engl J Med. 2009;361(23):2296–2297. doi: 10.1056/NEJMc0910060. [DOI] [PubMed] [Google Scholar]
- 8.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T, et al. Receptor specificities of human respiroviruses. J Virol. 2001;75(10):4604–4613. doi: 10.1128/JVI.75.10.4604-4613.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwegmann-Wessels C, Herrler G. Sialic acids as receptor determinants for coronaviruses. Glycoconj J. 2006;23(1-2):51–58. doi: 10.1007/s10719-006-5437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trappetti C, et al. Sialic acid: A preventable signal for pneumococcal biofilm formation, colonization, and invasion of the host. J Infect Dis. 2009;199(10):1497–1505. doi: 10.1086/598483. [DOI] [PubMed] [Google Scholar]
- 12.Shinya K, et al. Avian flu: Influenza virus receptors in the human airway. Nature. 2006;440(7083):435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 13.Walther T, et al. Glycomic analysis of human respiratory tract tissues and correlation with influenza virus infection. PLoS Pathog. 2013;9(3):e1003223. doi: 10.1371/journal.ppat.1003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong X, et al. Receptor binding by an H7N9 influenza virus from humans. Nature. 2013;499(7459):496–499. doi: 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, et al. Biological features of novel avian influenza A (H7N9) virus. Nature. 2013;499(7459):500–503. doi: 10.1038/nature12379. [DOI] [PubMed] [Google Scholar]
- 16.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. 1998;37(20):2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Connaris H, Crocker PR, Taylor GL. Enhancing the receptor affinity of the sialic acid-binding domain of Vibrio cholerae sialidase through multivalency. J Biol Chem. 2009;284(11):7339–7351. doi: 10.1074/jbc.M807398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moustafa I, et al. Sialic acid recognition by Vibrio cholerae neuraminidase. J Biol Chem. 2004;279(39):40819–40826. doi: 10.1074/jbc.M404965200. [DOI] [PubMed] [Google Scholar]
- 19.Xu G, Ryan C, Kiefel MJ, Wilson JC, Taylor GL. Structural studies on the Pseudomonas aeruginosa sialidase-like enzyme PA2794 suggest substrate and mechanistic variations. J Mol Biol. 2009;386(3):828–840. doi: 10.1016/j.jmb.2008.12.084. [DOI] [PubMed] [Google Scholar]
- 20.Xu G, Li X, Andrew PW, Taylor GL. Structure of the catalytic domain of Streptococcus pneumoniae sialidase NanA. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64(Pt 9):772–775. doi: 10.1107/S1744309108024044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cantor JR, et al. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108(4):1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee A, et al. Activation of brain endothelium by pneumococcal neuraminidase NanA promotes bacterial internalization. Cell Microbiol. 2010;12(11):1576–1588. doi: 10.1111/j.1462-5822.2010.01490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ekiert DC, Wilson IA. Broadly neutralizing antibodies against influenza virus and prospects for universal therapies. Curr Opin Virol. 2012;2(2):134–141. doi: 10.1016/j.coviro.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osterhaus A, Fouchier R, Rimmelzwaan G. Towards universal influenza vaccines? Philos Trans R Soc Lond B Biol Sci. 2011;366(1579):2766–2773. doi: 10.1098/rstb.2011.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boltz DA, Aldridge JR, Jr, Webster RG, Govorkova EA. Drugs in development for influenza. Drugs. 2010;70(11):1349–1362. doi: 10.2165/11537960-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludwig S. Disruption of virus-host cell interactions and cell signaling pathways as an anti-viral approach against influenza virus infections. Biol Chem. 2011;392(10):837–847. doi: 10.1515/BC.2011.121. [DOI] [PubMed] [Google Scholar]
- 27.Moss RB, et al. A phase II study of DAS181, a novel host directed antiviral for the treatment of influenza infection. J Infect Dis. 2012;206(12):1844–1851. doi: 10.1093/infdis/jis622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malakhov MP, et al. Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection. Antimicrob Agents Chemother. 2006;50(4):1470–1479. doi: 10.1128/AAC.50.4.1470-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belser JA, et al. DAS181, a novel sialidase fusion protein, protects mice from lethal avian influenza H5N1 virus infection. J Infect Dis. 2007;196(10):1493–1499. doi: 10.1086/522609. [DOI] [PubMed] [Google Scholar]
- 30.Triana-Baltzer GB, et al. Novel pandemic influenza A(H1N1) viruses are potently inhibited by DAS181, a sialidase fusion protein. PLoS ONE. 2009;4(11):e7788. doi: 10.1371/journal.pone.0007788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamaya M, et al. Clarithromycin inhibits type a seasonal influenza virus infection in human airway epithelial cells. J Pharmacol Exp Ther. 2010;333(1):81–90. doi: 10.1124/jpet.109.162149. [DOI] [PubMed] [Google Scholar]
- 32.Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446(7139):1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 34.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J Infect Dis. 2008;198(7):962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.