

Significance
We identified a significant regional flux of methane over a large area of shale gas wells in southwestern Pennsylvania in the Marcellus formation and further identified several pads with high methane emissions. These shale gas pads were identified as in the drilling process, a preproduction stage not previously associated with high methane emissions. This work emphasizes the need for top-down identification and component level and event driven measurements of methane leaks to properly inventory the combined methane emissions of natural gas extraction and combustion to better define the impacts of our nation’s increasing reliance on natural gas to meet our energy needs.
Keywords: unconventional gas, greenhouse gas, hydraulic fracturing
Abstract
The identification and quantification of methane emissions from natural gas production has become increasingly important owing to the increase in the natural gas component of the energy sector. An instrumented aircraft platform was used to identify large sources of methane and quantify emission rates in southwestern PA in June 2012. A large regional flux, 2.0–14 g CH4 s−1 km−2, was quantified for a ∼2,800-km2 area, which did not differ statistically from a bottom-up inventory, 2.3–4.6 g CH4 s−1 km−2. Large emissions averaging 34 g CH4/s per well were observed from seven well pads determined to be in the drilling phase, 2 to 3 orders of magnitude greater than US Environmental Protection Agency estimates for this operational phase. The emissions from these well pads, representing ∼1% of the total number of wells, account for 4–30% of the observed regional flux. More work is needed to determine all of the sources of methane emissions from natural gas production, to ascertain why these emissions occur and to evaluate their climate and atmospheric chemistry impacts.
Methane is a very important component of the Earth’s atmosphere: it represents a significant component of the natural and anthropogenically forced greenhouse effect, with a global warming potential 28–34 times greater than CO2 using a 100-y horizon and even greater on shorter time scales (1, 2). It also is an important sink for the hydroxyl radical, the dominant agent that defines the atmosphere’s cleansing capacity (3), has a significant impact on tropospheric ozone, and is one of the important sources of water vapor in the stratosphere, which in turn impacts stratospheric ozone and climate (4). The recent observation that global methane concentrations have begun increasing (5), after a decade of static or decreasing emissions in the late 1990s to ∼2007, has renewed interest in pinpointing the causes of global methane trends. Recently natural gas has been explored as a potential bridge to renewable energy, owing in part to the reduction in carbon emissions produced from electricity generation by natural gas compared with coal (6–9). Advances in drilling and well stimulation techniques have allowed access to previously locked reservoirs of natural gas, such as the Marcellus shale formation in Pennsylvania, which has led to a boom in natural gas production in the last decade (10). This has led to estimations of the carbon footprint of natural gas to examine the impact of increasing our reliance on natural gas for various energy needs (11–16). An important unresolved issue is the contribution of well-to-burner tip CH4 emission to the greenhouse gas footprint of natural gas use. Given that CH4 is a much more potent greenhouse gas than CO2, quantifying CH4 emissions has become critical in estimating the long- and short-term environmental and economic impacts of increased natural gas use. According to a recent study, if total CH4 emissions are greater than approximately 3.2% of production, the immediate net radiative forcing for natural gas use is worse than for coal when used to generate electricity (8).
The first estimates for CH4 emissions from shale gas development were reported in late 2010 and are based on uncertain emission factors for various steps in obtaining the gas and getting it to market (17, 18). In the short time since these first estimates, many others have published CH4 emission estimates for unconventional gas (including tight-sand formations in addition to shales), giving a range of 0.6–7.7% of the lifetime production of a well emitted “upstream” at the well site and “midstream” during processing and 0.07–10% emitted during “downstream” transmission, storage, and distribution to consumers (reviewed in refs. 18 and 19). The highest published estimates for combined upstream and midstream methane emissions (2.3–11.7%) are based on actual top-down field-scale measurements at specific regions (20, 21). Whereas a recent shale gas study (22) based on field sites across the United States to which the authors were given access scaled actual measurements up to the national level and found lower emissions than US Environmental Protection Agency (EPA) estimates, an equally recent study (23) used atmospheric measurements of greenhouse gases across the United States to inform a model and found CH4 emissions, cumulatively and specifically from fossil fuel production activities, to be underestimated by the EPA.
The current range of observed CH4 emissions from US natural gas systems (2.3–11.7%), if it were representative of the national scale, applied to the reported 2011 unassociated gas production number yields a range of CH4 emissions between 5.6 and 28.4 Tg CH4, whereas the EPA reports 6.7 Tg CH4 from natural gas systems in 2011 and only 28 Tg CH4 total anthropogenic emissions (24). Natural gas systems are currently estimated to be the top source of anthropogenic CH4 emission in the United States, followed closely by enteric fermentation, but the top-down observations suggest that natural gas may play a more substantial role than previously thought (24). Inadequate accounting of greenhouse gas emissions hampers efforts to identify and pursue effective greenhouse gas reduction policies.
Although it is clear that analysis of the effect of natural gas use would benefit from better measurements of emissions from unconventional gas wells, the inaccessible and transient nature of these leaks makes them difficult to identify and quantify, particularly at a scale at which they are useful for bottom-up inventories or mitigation strategies (i.e., leak rates of individual components or activities). Previous techniques have used either bottom-up inventories of the smallest scale of contributions or top-down apportionment of observed large-scale regional enhancements over a complex area to identify the source of the enhancements (11, 17, 20–23, 25). Although the latter suggest that the leak rate may be higher than what bottom-up inventories have allocated, they give little to no information about where in the upstream production process these leaks occur, thus hampering the interpretation of these data for bottom-up inventories or mitigation purposes.
Here we use an aircraft-based approach that enables sampling of methane emissions between the regional and component level scales and can identify plumes from single well pads, groups of well pads, and larger regional scales, giving more information as to the specific CH4 emission sources. We implemented three types of flights over 2 d in June 2012: investigative (I), mass-balance flux (MB), and regional flux (RF). Details of each flight are presented in Table 1. Our results indicate a large regional CH4 flux in southwestern PA. We show that the methane emission flux from the drilling phase of operation can be 2 to 3 orders of magnitude greater than inventory estimates, providing an example and improved understanding of the differences between observed data and bottom-up inventories.
Table 1.
Meteorological conditions and time duration of each aircraft flight experiment
| Flight type | Flight no. | Date | Start time (EDT) | Duration, min | Wind speed, m/s | Wind direction |
| RF | 1 | 6/20/2012 | 10:00 | 96 | 3.0 | 276 |
| RF | 2 | 6/21/2012 | 8:55 | 89 | 3.7 | 270 |
| MB | 1 | 6/20/2012 | 11:55 | 30 | 3.1 | 236 |
| MB | 2 | 6/20/2012 | 15:15 | 56 | 3.3 | 239 |
| MB | 3 | 6/21/2012 | 16:00 | 60 | 5.5 | 252 |
| MB | 4 | 6/21/2012 | 14:05 | 73 | 4.7 | 226 |
| I | 1 | 6/20/2012 | 12:25 | 5 | 3.0 | 258 |
| I | 2 | 6/21/2012 | 15:22 | 6 | 4.7 | 227 |
| I | 3 | 6/21/2012 | 9:14 | 15 | 4.2 | 257 |
Flights are classified into three flight types: RF, MB, and I (defined in text). Investigative flights were short and occurred between and during the longer RF and MB flights. Flights are identified by their flight type and flight number (e.g., RF-1, MB-3, etc.). Note that flights MB-1 through MB-3 are near pad Delta and flight MB-4 is near pad Tau.
Results and Discussion
We conducted measurements in southwestern PA in the Marcellus shale formation region in June 2012. For two morning flights we calculated a regional flux of 2.0–13.0 g CH4 s−1 km−2 for RF-1 over a box that approximates the size of our flight path (dashed box in Fig. 1) that we define as the original sampling area (OSA) and 2.0–14.9 g CH4 s−1 km−2 for RF-2. These ranges represent our analysis of the combined effect of all sources of uncertainty, which is dominated by the range of accumulation time scales over which the enhancement may have occurred (i.e., a maximum of 18 h commencing with the time of collapse of the boundary layer the day before, to a minimum of 5–6 h for air to flush through the sampling area). These estimates are not statistically different from the range of estimates obtained by summing up bottom-up emissions estimates for oil and gas development, coal mining, and other sources for the OSA depicted by the dashed orange box in Fig. 1 (corresponding to a ∼6-h time scale) and for the 18-h upwind accumulation area (UAA) shown in Fig. S1: 2.3–4.6 g CH4 s−1 km−2. Methane emissions from natural gas contribute 22–62% of the estimated bottom-up flux in this region. Using our top-down flux measurements, the assumed range of methane from natural gas contribution (22–62%), and industry reported production rates, we estimate a possible range for the fugitive methane emission rate of 2.8–17.3% of production in this region, which applies only to these two specific study dates.
Fig. 1.

Regional enhancement of methane at 250 m AGL on the morning of June 20th. The dashed orange box represents the OSA, 2,844 km2, and the gray dots show well locations.
It is important to note that we could find no evidence from state records or from our analysis of photographs taken during flights of wells in flowback after hydraulic fracturing in the area during the sampling time (discussed in SI Text). Flowback is the period after fracturing when a portion of the fracturing fluid used returns up the wellbore, flushing out with it substantial amounts of natural gas. We used data submitted voluntarily by oil and gas operators to FracFocus.org to identify one potential flowback event (for a pad not sampled in this study) and included the emissions in our bottom-up inventory. We would expect the regional emission rate to be greater if more wells were in flowback (11, 17, 18).
Although our top-down and bottom-up flux estimates are not statistically different, the top-down flux estimate encompasses a range of larger magnitude fluxes compared with the bottom-up method, and the upper limit for the fraction of production emitted is large enough to provide ample motivation to pursue investigation of possible significant methane emission processes not included in the bottom-up inventory. To quantify emission rates from significant sources of CH4 emissions in this shale gas drilling region we conducted mass-balance flights (MB-1–MB-4) for well pads with observed enhancements large enough to use the aircraft-based mass-balance technique, as described in ref. 26. In the region between Washington, PA and south to the border of WV we observed multiple high concentration methane plumes and investigated two areas where initial observations revealed well pads with potentially high methane emission rates. The high density of pads in this region and the prevailing wind direction (SW) during the time of measurement combined to make plume attribution to single pads difficult. In cases in which fluxes from individual pads could not be isolated, we averaged the calculated flux from a wider region over the number of pads that could have possibly contributed. Fig. 2 shows the downwind methane concentrations in a vertical plane perpendicular to the mean wind direction from an isolated pad designated “Delta” (shown in Fig. 1 near the northern hotspot). Attribution of the flux to that (or any specific) source involved maneuvering in a circular pattern around the prospective source, with observed enhanced methane concentrations only on the downwind side, as shown in Fig. S2. Fig. 3 shows the downwind methane concentrations that include signal enhancement from a pad “Tau” (shown in Fig. 1 near the southern hotspot), as well as from other upwind pads, coal-bed methane wells, and a significant plume from an adjacent coal mine. The high density of potential upwind sources around Tau made attribution to specific sources impossible, although it is probable that some of this flux comes from at least one pad in the drilling stage (Tau). Combining results of MB-1 thru MB-3 yielded an average of 236 g CH4 s−1 per pad for seven high emitting pads, corresponding to 34 g CH4 s−1 per well. Individual MB flight results are presented in Table 2. Note that these seven pads, with ∼40 wells, representing approximately 1% of the wells in the 2,844-km2 OSA region, contributed a combined emission flux of 1.7 kg CH4 s−1, equal to 4.3–30% of our top-down measured flux.
Fig. 2.

Interpolated methane concentration ∼1 km downwind of pad Delta, showing isolated methane plume near the center of the transect.
Fig. 3.

Interpolated methane concentration from several pads near pad Tau. A distinct methane plume from a nearby coal mine occurs around 3 km.
Table 2.
Results from four MB experiments and the number of pads and wells contributing to the flux
| Flight | Flight MB-1 | Flight MB-2* | Flight MB-3 | Flight MB-4 | Average ± σ |
| Total flux (g CH4/s) | 380 | 248 | 1,880 | 1,490 | — |
| Total pads contributing | 2 | 1 | 7 | — | — |
| Flux (g CH4/s) per pad | 190 | 248 | 269 | — | 236 ± 41 |
| Total permitted wells | 15 | 8 | 41 | — | — |
| Flux (g CH4/s) per well | 25 | 31 | 46 | — | 34 ± 11 |
Flights 1–3 were conducted near pad Delta and flight 4 near pad Tau. Flux per pad and per well is obtained by dividing the total flux by either the total number of pads or total number of wells.
Isolated pad Delta.
The methane emissions from the gas wells reported in Table 2 are surprisingly high considering that all of these wells were still being drilled, had not yet been hydraulically fractured, and were not yet in production. The Pennsylvania Department of Environmental Protection (27) confirmed that total vertical depth had not yet been reached in these wells at the time of the sampling, and our photographic evidence recorded equipment typical during the drilling phase, as shown in Fig. S3. Because of the large number of wells in our study region we were not able to review all well files to determine the total number of wells being drilled during the time of study. EPA greenhouse gas inventories report a total of 51.3 kg CH4 per well from the entire drilling period that typically lasts 2 wk (24). Using, as limits, a 2-wk and a 2-d (the duration of our observations) drilling phase time scale, this leads to an estimated flux of 0.04–0.30 g CH4 s−1 per well, 2 to 3 orders of magnitude lower than our observed average flux per well (for the high emitters we studied) of 34 g CH4 s−1. Although we only quantitatively sampled pads where we saw significant enhancement above the background, it is important to note that we could detect little to no emission from many other pads, particularly in the region north of the OSA, from Washington north to Pittsburgh. Thus, we do not intend for our regional flux estimate to be taken as necessarily representative of the Marcellus as a whole but only for the region defined as the OSA for these days. We also note that some sources were too intermittent to determine a flux via the aircraft mass-balance method. At a compressor station north of Washington we observed methane concentrations up to 45 ppm, but there was no consistent plume between consecutive passes downwind of the station.
Bottom-up emission factor studies usually assume no emission from gas wells during this prehydraulic fracturing period (11–16). Release from gas kicks—gas entry into the wellbore during vertical drilling despite efforts to keep the wellbore at a higher pressure than surrounding rock, a technique known as overbalanced drilling—is one possible explanation. However, it is generally assumed that gas kicks are not significant emission sources and are transient (28), although we observed comparable emissions on consecutive days. Alternatively, underbalanced drilling methods may have been used on these wells, where lower pressure in the wellbore allows fluids and gas from the various geological formations (i.e., coal deposits) being drilled through to seep into the wellbore and up to the surface, resulting in emission of hydrocarbons, including methane, during the drilling phase if the emissions are not contained or flared (28). Note that although these well pads were not permitted as coal bed methane wells the entire southwest region of Pennsylvania contains underlying coal deposits. The underbalanced drilling hypothesis is supported by aerial pictures that show a lack of a shale shaker or mud pits at these sites that are typically used in overbalanced drilling. Whatever the source of high emissions from the pads we identified as in the drilling stage, these emissions, equaling 0.6g CH4 s−1 km−2, are not included in our bottom-up estimate (or any other bottom-up estimate). The addition of this emission to our bottom-up inventory would shift the estimates slightly higher, but because our original results were not statistically different owing to the large range of estimates from our top-down approach, our conclusions are no different.
During the morning RF-2 flight we acquired whole-air samples using the National Oceanic and Atmospheric Administration (NOAA) programmable flask package, which were analyzed for hydrocarbons and CH4. We found that relative to other studies of shale-well natural gas, the air samples in this region exhibited much lower mole ratios of propane and n-butane to methane, at 0.007 ± 0.001 and 0.0018 ± 0.0003, respectively. Previous reports indicate molar ratios of ∼0.05 for propane (28, 29) and ∼0.01 for n-butane (30). However, the observed n-butane to propane ratio, 0.27 ± 0.01, is very similar to values reported in previous work, which average 0.24 (31). These findings suggest that the shale natural gas signal is being diluted by an essentially pure CH4 source. Although this is not the only possibility, these results support the hypothesis that the methane plumes derive from underbalanced drilling methods as wells are drilled through formations such as shallow coal pockets producing coal-bed methane during the drilling phase. Coal-bed methane is typically composed of very high percentages of CH4 (∼98%), with trace heavier hydrocarbons (32).
Conclusions
This work shows that it is possible to interrogate and quantify emissions from individual pads and pad clusters at scales relevant to bottom-up inventories and mitigation strategies and to estimate the emission rate for a region encompassing a large number of well pads using the aircraft measurement approach. The range of regional leak rates found here for the OSA (3–17%) is similar to leak rates found by recent studies across the United States in the CO Denver-Julesburg Basin (20) and the UT Uintah Basin (21). Additionally, although a leakage rate was not calculated, a study over large areas of TX, OK, and KS (25) found surprisingly high methane emissions, indicating that high fugitive emission rates are likely to be a national-scale issue, although the mechanisms of these fugitive leaks may be different at each site. Although a recent study (22) found production sites, to which they were given access, to be emitting less CH4 than EPA inventories suggest, these regional scale findings and a recent national study (23) indicate that overall sites leak rates can be higher than current inventory estimates. Additionally, a recent comprehensive study of measured natural gas emission rates versus “official” inventory estimates found that the inventories consistently underestimated measured emissions and hypothesized that one explanation for this discrepancy could be a small number of high-emitting wells or components (33).
These high leak rates illustrate the urgent need to identify and mitigate these leaks as shale gas production continues to increase nationally (10). The identification presented here of emissions during the drilling stage 2 to 3 orders of magnitude larger than inventory estimates indicates the need to examine all aspects of natural gas production activity to improve inventory estimates and identify potential opportunities for mitigation strategies and that top-down measurements provide an important complement to bottom-up inventory determinations. Shale gas production is expected to increase globally as many shale gas plays are starting to be explored (34). If a midrange value of the reported fraction of production that is emitted, 7%, is applied to the projected global peak shale gas production rate, 23 trillion ft3 per year (34), it would correspond to 24 Tg CH4 emitted per year, or ∼4% of the current global total (natural and anthropogenic) CH4 emission rate (35). Further studies are needed to enable better understanding of the operational details that lead to the largest emissions, how they might be better controlled, and to provide a more detailed picture of the expected life cycle-integrated emissions from unconventional gas wells.
Methods
Measurements were conducted between June 18, 2012 and June 21, 2012 over southwestern PA using Purdue’s Airborne Laboratory for Atmospheric Research, a modified Beechcraft Duchess aircraft. This aircraft is equipped with a 50-Hz Best Air Turbulence probe, described by ref. 36, that measures wind vectors and pressure, a 50-Hz microbead thermistor that measures temperature, a 50-Hz global positioning system/inertial navigation system, and a 0.5-Hz high precision Picarro CO2/CH4/H2O cavity ring down spectrometer (CRDS). The CRDS has ∼0.05% (1 ppb) precision for methane determined during in-flight calibration, and comparable accuracy, using three NOAA Earth System Research Laboratory tanks with CH4 concentrations of 1.8030, 2.2222, and 2.5995 ppm. A programmable flask package (PFP) provided by NOAA for whole-air sampling was also installed on the aircraft. The PFP consists of 12 flasks that hold air pressurized to 2.7 atm in 0.7-L bottles. Flasks are analyzed for 55 species, including CH4, and hydrocarbons, by NOAA.
We calculated a regional flux on two mornings by integrating the enhancement in CH4 above the background in the OSA (enhancement area of 2,844 km2). The height of the box was defined as the boundary layer height, which was determined from the earliest [∼10:00 AM Eastern Daylight Time (EDT)] vertical profiles of potential temperature, H2O, CH4, and CO2. Boundary layer heights were observed to be 370 m above ground level (AGL) for flight RF-1 and 500 m AGL for flight RF-2 and assumed to be constant during the data collection period for each flight. The raw CH4 data from flight at a constant altitude (∼250 m AGL) around the area of interest was interpolated using the EasyKrig3.0 program (37). For RF-1 the observed concentrations are presented in Fig. 1, and the flight data for RF-2 are shown in Fig. S4. The 2D interpolation output was turned into a 3D matrix of CH4 values by assuming the CH4 concentration decreased linearly with height up to the boundary layer top, with background concentrations of 1.89 ppm CH4. This assumption was based on the observed vertical profiles that depict an approximately linear decrease of the CH4 mole ratio with altitude. We compared integration of CH4 under the actual vertical profile and a linear regression of the vertical profile, shown in Fig. S5, and found less than a 7% difference, which supports use of the linear approximation for the whole study region. Fig. S5 shows a vertical profile obtained during flight RF-1 at ∼10:00 AM EDT. The profile extends into the residual layer above the stable boundary layer. The residual layer represents well mixed (i.e., clean, air from the previous day as the boundary layer collapsed and is used to estimate the CH4 background concentration, 1.89 ppm on both days). The CH4 enhancement was then calculated by removing the background value and converting to mol·m−3. Multiplying the enhancement by the pixel volume, 29,386.5 m3 (171.6 m longitudinal∙171.25 m latitudinal∙1 m vertical), and integrating over all pixels in the sample area produces the total enhancement in moles, which can be converted to units of g or kg. To obtain a flux, the enhancement was then divided by a chosen time scale, discussed below, and divided by the total area of the OSA, 2,844 km2, to obtain the flux in g s−1km−2.
Uncertainty was assessed by examining the range of reasonable assumptions to calculate the CH4 enhancement and the time scale of the accumulation. A simpler CH4 enhancement estimate was done by assuming a spatially uniform CH4 enhancement in the box taken from the observed CH4 vertical profile after it had been smoothed. The CH4 enhancement differed by approximately ±30% using this technique. In addition, the effect of background CH4 estimate was quantified by using reasonable upper limits in background concentration from background air observed in the southwest and west of the OSA during both flights, which was generally higher than the concentrations observed in the residual layer. We estimate the upper limit to the background concentration to be 2.00 ppm. In this scenario a 20% difference in the calculated CH4 enhancement is observed. The time scale was changed to reflect different possibilities for accumulation. The lower limit to the accumulation time scale used (6 h for RF-1, 5 h for RF-2) was the time for the observed winds to flush the box. The flush time of the box represents the physical minimum time for enhanced air to be replaced with assumed cleaner upwind air, at the observed wind speeds. This assumption is supported by the observation that both RF-1 and RF-2 show cleaner air in the upwind area at the time of flight (W corner of the OSA box; Fig. 1 and Fig. S4), consistent with much smaller density of wells, as can be seen in Fig. 1 and Fig. S4. The longest time scale used (18 h) represents the time from the collapse of the boundary layer the day before (∼6:00 PM) to the time observations were made. These component uncertainties are then propagated to produce the total range of the flux estimate.
A complicating factor affecting our ability to directly compare the top-down flux estimate with the bottom-up inventory is the influence of advective transport. At night, surface winds are typically low and unsustained, leading to very slow transport of air masses, and winds on the morning of our flights were low (2–3 m/s). However, for an 18-h accumulation, it is likely that these observations include mixing with air containing emissions (and/or cleaner air) from a region upwind (SW) of the measurement region. To investigate the potential impact of the upwind area we used the NOAA Hybrid Single Particle Lagrangian Integrated Trajectory Model (HYSPLIT) to predict the maximum size of the upwind area (ready.arl.noaa.gov/HYSPLIT.php). Starting at the time of observations (10:00 AM EDT) we ran an 18-h matrix back trajectory encompassing the area of observations. The 18-h time scale was chosen because it presents the largest estimate of potential upwind influence. We chose the isobaric mode with an effective altitude that is constant at 50 m to represent transport within the stable surface layer. The resulting area of influence, which we call the UAA, covers 14,597 km2 and is shown in Fig. S1. This area is five times larger than the original sample area. An appropriate comparison with a bottom-up inventory will, therefore, have to include an estimate for an area encompassing the entire 18-h back trajectory region (UAA) and an estimate for the OSA. The average emissions over the UAA corresponds to a lower limit for the bottom-up flux, because the top-down measurements likely did not sample completely mixed air, and in this case the upwind area contains cleaner air, which dilutes the emissions. Likewise, the OSA represents an upper limit for comparison with the bottom-up flux because the top-down measurements similarly did not sample air exclusively influenced by the OSA (which has a higher density of emission sources), and accumulation may have effectively occurred over a time scale greater than the estimated 5–6 h.
Bottom-up inventories including energy sector, agriculture, landfill, and other miscellaneous emissions were produced for both the OSA and the UAA and are described in SI Text. Energy sector emissions were computed using the following national and state databases: Pennsylvania Department of Environment databases of oil, gas, and coal production and locations; West Virginia Department of Environment databases of oil, gas, and coal production and locations; Ohio Department of Natural Resources databases of oil, gas, and coal production; Energy Information Administration databases of state to state pipeline transmission and location; Department of Labor database of Employment and Production; and the Pipeline and Hazardous Material Safety Administration database of pipeline transmission. Default gas compositions were used (38), and all conversions between volume and mass assume standard gas conditions: 15 °C and 1 atm. Emission factors from ref. 11 are used to calculate routine fugitive emissions from natural gas production and processing and for life cycle fugitive emissions from coal and oil energy sectors. Emissions from natural gas transmission and distribution and well flowback events are calculated from emission factors provided in refs. 16 and 17, respectively. For comparison, a bottom-up inventory of natural gas sector emissions using only ref. 16 emission factors was also completed for the OSA. Methane emissions from the agriculture sector were calculated from total animal counts in the counties of interest (39) multiplied by methane emission factors from refs. 40 and 41. Other methane emissions were included from EPA-reported greenhouse gas emissions from landfills and other miscellaneous sources (42). Table 3 shows the total emissions from the bottom-up inventory for the OSA and UAA, as well as the comparison inventory for the OSA. More detailed emissions are presented for the OSA in Table S1 and the UAA in Table S2.
Table 3.
Total expected emissions from all sources and percent contribution to the total emission for the OSA and the UAA using Howarth et al. (11) emission factors and for the OSA using NETL (16) emission factors
| Area | Source | Expected emissions, g CH4 s−1 km−2 | Contribution, % |
| OSA (Howarth EFs) | Natural gas | 0.85 (low)–2.23 (high) | 21.9–42.0 |
| Oil | 0 | 0 | |
| Coal | 2.96 | 55.7–76.3 | |
| Flowback | 0.05–0.10 | 1.3–1.9 | |
| AFO | 0.015 | 0.3–0.4 | |
| Other | 0 | 0 | |
| Total (average) | 3.88–5.31 (4.60) | ||
| UAA (Howarth et al. EFs) | Natural gas | 0.76 (low)–1.70 (high) | 42.0–61.6 |
| Oil | 0 | 0 | |
| Coal | 1.01 | 36.6–55.8 | |
| Flowback | 0.01–0.02 | 0.6–0.7 | |
| AFO | 0.015 | 0.5–0.8 | |
| Other | 0.019 | 0.7–1.0 | |
| Total (average) | 1.81–2.76 (2.29) | ||
| OSA (NETL EFs) | Natural gas | 1.41 | 31.4–31.8 |
| Oil | 0 | 0 | |
| Coal | 2.96 | 65.9–66.7 | |
| Flowback | 0.05–0.10 | 1.1–2.3 | |
| AFO | 0.015 | 0.3 | |
| Other | 0 | 0 | |
| Total (average) | 4.42–4.49 (4.46) |
AFO, animal feeding operation; EFs, emission factors; NETL, National Energy Technology Laboratory.
The comparison of uncaptured natural gas emissions as a percentage of total natural gas produced has been used as a standard of comparison between studies. We used the bottom-up inventories to compute the proportion of our observed top-down flux that would be expected to come from the natural gas sector. As shown in Table 3, the total contribution of methane emissions from the natural gas sector is assumed to be between 22% and 62% in this region. This range was used to calculate the contributing portion of natural gas emissions from the extrema in the top-down flux to be divided by the local unassociated production rates of 50.1 g CH4 s−1 km−2 for the OSA and 15.9 g CH4 s−1 km−2 for the UAA, as shown in Table 4. We report emission rates in Table 4 and estimate a fugitive emission rate between 2.8% and 17.3% of natural gas production for this region on these particular days. This estimate should be compared with other estimates with caution because these estimates generally use more comprehensive temporal data (16, 17, 19, 20). Nevertheless, the upper range of this emission rate is surprisingly high, particularly because there were no major or widespread activities such as flowback events or well workovers of which we are aware that are typically associated with higher methane emission rates.
Table 4.
Natural gas portion of the top-down flux as a percentage of the unassociated natural gas production rate
| Parameter | 18-h Estimate | 5 to 6-h Estimate | ||
| Low | High | Low | High | |
| Top-down flux, g CH4 s-1 km-2 | 2.0 | 4.2 | 6.6 | 14.0 |
| CH4 from natural gas, % | 22 | 62 | 22 | 62 |
| Natural gas production rate, g CH4 s-1 km-2 | 15.9 | 50.1 | ||
| Natural gas flux/ production rate, % | 2.8 | 16.4 | 2.9 | 17.3 |
The mass-balance technique used here is described in ref. 26. Briefly, CH4 concentration data are collected at varying altitudes downwind of a source approximately perpendicular to the prevailing wind direction. Downwind transects were flown to the top of the boundary layer, determined from vertical profiles of potential temperature, H2O, CH4, and CO2, or more commonly, until the signal reached background levels. The observation of plumes that did not extend all of the way to the boundary layer top is attributed to the fact that downwind transects were typically flown 2–5 km downwind of a source, corresponding to too short a transport time scale for complete vertical mixing but necessary to isolate sources in a landscape with a dense distribution of potential sources. Fig. S6 shows raw CH4 transect data 1.1 km downwind of pad Delta during flight MB-2. Interpolation of the raw transect data to create a 2D matrix of CH4 values was done using EasyKrig3.0 (37). Fig. 2 shows the output from the interpolation of the raw data in Fig. S6. After the interpolated CH4 and horizontal wind matrices are obtained, the flux is calculated according to Eq. 1.
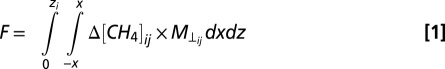 |
Here the limit zi is the top of the boundary layer, or the height at which the plume stops, and the limits x and −x are the horizontal limits determined from an arbitrary reference point in the middle of the transects. ΔCH4 is obtained by converting CH4 concentrations from ppm to mol·m−3 using measured temperature and pressure, then subtracting an average background CH4 value, calculated by averaging the edges of the interpolated matrix, from each point in the interpolated CH4 matrix, denoted by the subscripts ij. M⊥ is the component of the mean wind that is perpendicular to the plane downwind of the source, which has also been interpolated from observations. Integrating across x and z and multiplying by dx, 110 m, and dz, 20 m, gives the flux in mol/s per cell, which can then be converted to units of g/s or kg/s. In cases in which there were multiple well pads contributing, the number of potential upwind pads was determined from visually inspecting the CH4 data and spatial distribution of pads in the upwind direction.
Hydrocarbon concentration values were obtained from flask samples taken during flight RF-2. Of the 12 flasks, 2 were sampled in the free troposphere and excluded from hydrocarbon ratio calculations. The lowest hydrocarbon mole ratios in a single flask within the boundary layer were used as an estimate of background values and subtracted from the remaining nine flasks to obtain delta hydrocarbon values. The least-squares regression, forced through zero, between delta values of hydrocarbons, was used to find the hydrocarbon ratio. The uncertainty in the ratio reported is the uncertainty in the least-squares regression slope.
Supplementary Material
Acknowledgments
We thank Karen Edelstein and Bongghi Hong for their help in compiling the bottom-up inventory and Carolina Siso for flask VOC analysis. The data presented here can be made available by the authors, by contacting pshepson@purdue.edu. Funding was provided by the David R. Atkinson Center for a Sustainable Future at Cornell University.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316546111/-/DCSupplemental.
References
- 1. Stocker T, et al. (2013) in Climate Change 2013: The Physical Science Basis: Technical Summary, eds Joussaume S, Penner J, Fredolin T (Cambridge University Press, New York), Table 8.7.
- 2.Shindell DT, et al. Improved attribution of climate forcing to emissions. Science. 2009;326(5953):716–718. doi: 10.1126/science.1174760. [DOI] [PubMed] [Google Scholar]
- 3.Thompson AM. The oxidizing capacity of the Earth's atmosphere: Probable past and future changes. Science. 1992;356(5060):1157–1165. doi: 10.1126/science.256.5060.1157. [DOI] [PubMed] [Google Scholar]
- 4.Solomon S, et al. Contributions of stratospheric water vapor to decadal changes in the rate of global warming. Science. 2010;327(5970):1219–1223. doi: 10.1126/science.1182488. [DOI] [PubMed] [Google Scholar]
- 5.Frankenberg C, et al. Global column-averaged methane mixing ratios from 2003 to 2009 as derived from SCIAMACHY: Trends and variability. J Geophys Res. 2011;116(D4):D04302. [Google Scholar]
- 6.Pacala S, Socolow R. Stabilization wedges: Solving the climate problem for the next 50 years with current technologies. Science. 2004;305(5686):968–972. doi: 10.1126/science.1100103. [DOI] [PubMed] [Google Scholar]
- 7.Myhrvold NP, Caldeira K. Greenhouse gases, climate change and the transition from coal to low-carbon electricity. Environ Res Lett. 2012 doi: 10.1088/1748-9326/7/1/014019. [DOI] [Google Scholar]
- 8.Alvarez RA, Pacala SW, Winebrake JJ, Chameides WL, Hamburg SP. Greater focus needed on methane leakage from natural gas infrastructure. Proc Natl Acad Sci USA. 2012;109(17):6435–6440. doi: 10.1073/pnas.1202407109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkatesh A, Jaramillo P, Griffin WM, Matthews HS. Uncertainty in life cycle greenhouse gas emissions from United States natural gas end-uses and its effects on policy. Environ Sci Technol. 2011;45(19):8182–8189. doi: 10.1021/es200930h. [DOI] [PubMed] [Google Scholar]
- 10.US Energy Information Administration EIA annual energy outlook 2013. 2013. Available at www.eia.gov/forecasts/aeo/pdf.0383(2013).pdf. Accessed August 7, 2013.
- 11.Howarth RW, Santoro RL, Ingraffea A. Methane and the greenhouse-gas footprint of natural gas from shale formations. Clim Change. 2011;106(4):679–690. [Google Scholar]
- 12.Hultman N, Rebois D, Scholten M, Ramig C. The greenhouse impact of unconventional gas for electricity generation. Environ Res Lett. 2011 doi: 10.1088/1748-9326/6/4/044008. [DOI] [Google Scholar]
- 13.Jiang M, et al. Life cycle greenhouse gas emissions of Marcellus shale gas. Environ Res Lett. 2011 doi: 10.1088/1748-9326/6/3/034014. [DOI] [Google Scholar]
- 14.Stephenson T, Valle JE, Riera-Palou X. Modeling the relative GHG emissions of conventional and shale gas production. Environ Sci Technol. 2011;45(24):10757–10764. doi: 10.1021/es2024115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burnham A, et al. Life-cycle greenhouse gas emissions of shale gas, natural gas, coal, and petroleum. Environ Sci Technol. 2012;46(2):619–627. doi: 10.1021/es201942m. [DOI] [PubMed] [Google Scholar]
- 16.National Energy Technology Laboratory 2011. Life Cycle Greenhouse Gas Inventory of Natural Gas Extraction, Delivery and Electricity Production (DOE/NETL Publication 2011/1522). Available at www.netl.doe.gov/File%20Library/Research/Energy%20Analysis/Life%20Cycle%20Analysis/NG-GHG-LCI.pdf. Accessed December 11, 2012.
- 17.US Environmental Protection Agency Greenhouse gas emissions reporting from the petroleum and natural gas industry: Background technical support document. 2010. Available at www.epa.gov/ghgreporting/documents/pdf/2010/Subpart-W_TSD.pdf. Accessed December 15, 2011.
- 18.Howarth RW, Shindell D, Santoro RL, Ingraffea A, Phillips N, Townsend-Small A. 2011. Methane Emissions from Natural Gas Systems. National Climate Assessment, Feb. Report No. 2011-003 (Office of Science and Technology Policy, Washington, DC)
- 19.Weber CL, Clavin C. Life cycle carbon footprint of shale gas: Review of evidence and implications. Environ Sci Technol. 2012;46(11):5688–5695. doi: 10.1021/es300375n. [DOI] [PubMed] [Google Scholar]
- 20.Pétron G, et al. Hydrocarbon emissions characterization in the Colorado Front Range: A pilot study. J Geophys Res. 2012 doi: 10.1029/2011JD016360. [DOI] [Google Scholar]
- 21.Karion A, et al. Methane emissions estimate from airborne measurements over a western United States natural gas field. Geophys Res Lett. 2013 doi: 10.1002/grl.50811. [DOI] [Google Scholar]
- 22.Allen DT, et al. Measurements of methane emissions at natural gas production sites in the United States. Proc Natl Acad Sci USA. 2013;110(44):17768–17773. doi: 10.1073/pnas.1304880110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller SM, et al. Anthropogenic emissions of methane in the United States. Proc Natl Acad Sci USA. 2013;110(50):20018–20022. doi: 10.1073/pnas.1314392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.US Environmental Protection Agency 2013. Inventory of US Greenhouse Gas Emissions and Sinks: 1990-2011. EPA Publication 430-R-13-001 (US Environmental Protection Agency, Washington, DC) [PubMed]
- 25.Katzenstein AS, Doezema LA, Simpson IJ, Blake DR, Rowland FS. Extensive regional atmospheric hydrocarbon pollution in the southwestern United States. Proc Natl Acad Sci USA. 2003;100(21):11975–11979. doi: 10.1073/pnas.1635258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mays KL, et al. Aircraft-based measurements of the carbon footprint of Indianapolis. Environ Sci Technol. 2009;43(20):7816–7823. doi: 10.1021/es901326b. [DOI] [PubMed] [Google Scholar]
- 27.Pennsylvania Department of Environmental Protection 2012. Internal File Review: Permitted Wells: 12524400-02, 12524425, 125124516, 12524517, 12524529-41, 12524552, 12524553, 1252455-58, 12524563, 12524564, 12524589, 12524600, 12524617, 12524618, 12524627, 12524628, 12524696, 12524713, 12524714, 12524762 (Pennsylvania Department of Environmental Protection, Southwest Regional Office, Pittsburgh)
- 28.American Society of Mechanical Engineers Shale Shaker Committee . Drilling Fluids Processing Handbook. Amsterdam, NY: American Society of Mechanical Engineers Shale Shaker Committee; 2005. [Google Scholar]
- 29.US Environmental Protection Agency ANNEX 3: Methodological descriptions for additional source or sink categories. 2012. Available at www.epa.gov/climatechange/ghgemissions/usinventoryreport.html. Accessed February 19, 2013.
- 30.Braziel ER. Infrastructure projects connect Marcellus shale to ethane, NGL markets. 2011. Available at www.aogr.com/index.php/magazine/cover-story/infrastructure-projects-connect-marcellus-shale-to-ethane-ngl-markets. Accessed November 26, 2012.
- 31.Osborn SG, McIntosh JC. Chemical and isotopic tracers of the contribution of microbial gas in Devonian organic-rich shales and reservoir sandstones, northern Appalachian Basin. Appl Geochem. 2010;25(3):456–471. [Google Scholar]
- 32.Kotarba MJ. Composition and origin of coalbed gases in the Upper Silesian and Lublin basins, Poland. Org Geochem. 2001;32(1):163–180. [Google Scholar]
- 33.Brandt AR, et al. Energy and environment. Methane leaks from North American natural gas systems. Science. 2014;343(6172):733–735. doi: 10.1126/science.1247045. [DOI] [PubMed] [Google Scholar]
- 34.Mohr SH, Evans GM. Long term forecasting of natural gas production. Energy Policy. 2011;39(9):5550–5560. [Google Scholar]
- 35.Dlugokencky EJ, Nisbet EG, Fisher R, Lowery D. Global atmospheric methane: budget, changes and dangers. Phil Trans R Soc A. 2011;369(1943):2058–2072. doi: 10.1098/rsta.2010.0341. [DOI] [PubMed] [Google Scholar]
- 36.Garman KE, et al. An airborne and wind tunnel evaluation of a wind turbulence measurement system for aircraft-based flux measurements. J Atmos Ocean Technol. 2006;23(12):1696–1708. [Google Scholar]
- 37.Chu D. The GLOBEC kriging software package–EasyKrig3.0. 2004. Available at http://globec.whoi.edu/software/kriging/easy_krig/easy_krig.html. Accessed January 7, 2011.
- 38.American Petroleum Institute Compendium of greenhouse gas emissions methodologies for the oil and natural gas industry. 2009. Available at www.api.org/ehs/climate/new/upload/2009_ghg_compendium.pdf. Accessed December 14, 2012.
- 39.Hong B, Swaney DP, Howarth RW. A toolbox for calculating net anthropogenic nitrogen inputs (NANI) Environ Model Softw. 2011;26(5):623–633. [Google Scholar]
- 40.Jorgensen H. Methane emission by growing pigs and adult sows as influenced by fermentation. Livest Sci. 2007;109(1-3):216–219. [Google Scholar]
- 41.Zhou JB, Jiang MM, Chen GQ. Estimation of methane and nitrous oxide emission from livestock and poultry in China during 1949-2003. Energy Policy. 2007;35(7):3759–3767. [Google Scholar]
- 42.US Environmental Protection Agency EPA greenhouse gas reporting program 2010 summary. 2010. Available at www.epa.gov/ghgreporting/ghgdata/2010data.html. Accessed May 8, 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.