

Abstract
With the completion of the human genome sequence, attention turned to identifying and annotating its functional DNA elements. As a complement to genetic and comparative genomics approaches, the Encyclopedia of DNA Elements Project was launched to contribute maps of RNA transcripts, transcriptional regulator binding sites, and chromatin states in many cell types. The resulting genome-wide data reveal sites of biochemical activity with high positional resolution and cell type specificity that facilitate studies of gene regulation and interpretation of noncoding variants associated with human disease. However, the biochemically active regions cover a much larger fraction of the genome than do evolutionarily conserved regions, raising the question of whether nonconserved but biochemically active regions are truly functional. Here, we review the strengths and limitations of biochemical, evolutionary, and genetic approaches for defining functional DNA segments, potential sources for the observed differences in estimated genomic coverage, and the biological implications of these discrepancies. We also analyze the relationship between signal intensity, genomic coverage, and evolutionary conservation. Our results reinforce the principle that each approach provides complementary information and that we need to use combinations of all three to elucidate genome function in human biology and disease.
Quest to Identify Functional Elements in the Human Genome
Completing the human genome reference sequence was a milestone in modern biology. The considerable challenge that remained was to identify and delineate the structures of all genes and other functional elements. It was quickly recognized that nearly 99% of the ∼3.3 billion nucleotides that constitute the human genome do not code for proteins (1). Comparative genomics studies revealed that the majority of mammalian-conserved and recently adapted regions consist of noncoding elements (2–10). More recently, genome-wide association studies have indicated that a majority of trait-associated loci, including ones that contribute to human diseases and susceptibility, also lie outside protein-coding regions (11–16). These findings suggest that the noncoding regions of the human genome harbor a rich array of functionally significant elements with diverse gene regulatory and other functions.
Despite the pressing need to identify and characterize all functional elements in the human genome, it is important to recognize that there is no universal definition of what constitutes function, nor is there agreement on what sets the boundaries of an element. Both scientists and nonscientists have an intuitive definition of function, but each scientific discipline relies primarily on different lines of evidence indicative of function. Geneticists, evolutionary biologists, and molecular biologists apply distinct approaches, evaluating different and complementary lines of evidence. The genetic approach evaluates the phenotypic consequences of perturbations, the evolutionary approach quantifies selective constraint, and the biochemical approach measures evidence of molecular activity. All three approaches can be highly informative of the biological relevance of a genomic segment and groups of elements identified by each approach are often quantitatively enriched for each other. However, the methods vary considerably with respect to the specific elements they predict and the extent of the human genome annotated by each (Fig. 1).
Fig. 1.

The complementary nature of evolutionary, biochemical, and genetic evidence. The outer circle represents the human genome. Blue discs represent DNA sequences acted upon biochemically and partitioned by their levels of signal [combined 10th percentiles of different ENCODE data types for high, combined 50th percentiles for medium, and all significant signals for low (see Reconciling Genetic, Evolutionary, and Biochemical Estimates and Fig. 2)]. The red circle represents, at the same scale, DNA with signatures of evolutionary constraint (GERP++ elements derived from 34mammal alignments). Overlaps among the sequences having biochemical and evolutionarily evidence were computed in this work (Fig. 3 and SI Methods). The small purple circle represents protein-coding nucleotides (Gencode). The green shaded domain conceptually represents DNA that produces a phenotype upon alteration, although we lack well-developed summary estimates for the amount of genetic evidence and its relationship with the other types. This summary of our understanding in early 2014 will likely evolve substantially with more data and more refined experimental and analytical methods.
Some of these differences stem from the fact that function in biochemical and genetic contexts is highly particular to cell type and condition, whereas for evolutionary measures, function is ascertained independently of cellular state but is dependent on environment and evolutionary niche. The methods also differ widely in their false-positive and false-negative rates, the resolution with which elements are defined, and the throughput with which they can be surveyed. Moreover, each approach remains incomplete, requiring continued method development (both experimental and analytical) and increasingly large datasets (additional species, assays, cell types, variants, and phenotypes). It is thus not surprising that the methods vary considerably with respect to the specific elements they identify. However, the extent of the difference is much larger than simply technical limitations would suggest, challenging current views and definitions of genome function.
Many examples of elements that appear to have conflicting lines of functional evidence were described before the Encyclopedia of DNA Elements (ENCODE) Project, including elements with conserved phenotypes but lacking sequence-level conservation (17–20), conserved elements with no phenotype on deletion (21, 22), and elements able to drive tissue-specific expression but lacking evolutionary conservation (23, 24). However, the scale of the ENCODE Project survey of biochemical activity (across many more cell types and assays) led to a significant increase in genome coverage and thus accentuated the discrepancy between biochemical and evolutionary estimates. This discrepancy led to much debate both in the scientific literature (25–31) and in online forums, resulting in a renewed need to clarify the challenges of defining function in the human genome and to understand the sources of the discrepancy.
To address this need and provide a perspective by ENCODE scientists, we review genetic, evolutionary, and biochemical lines of evidence, discuss their strengths and limitations, and examine apparent discrepancies between the conclusions emanating from the different approaches.
Genetic Approach.
Genetic approaches, which rely on sequence alterations to establish the biological relevance of a DNA segment, are often considered a gold standard for defining function. Mutations can be naturally occurring and identified by screening for phenotypes generated by sequence variants (13, 32) or produced experimentally by targeted genetic methods (33) or nongenetic interference (34). Transfection studies that use reporter assays in cell lines (35, 36) or embryos (37) can also be used to identify regulatory elements and measure their activities. Genetic approaches tend to be limited by modest throughput, although speed and efficiency is now increasing for some methods (36, 38–40). The approach may also miss elements whose phenotypes occur only in rare cells or specific environmental contexts, or whose effects are too subtle to detect with current assays. Loss-of-function tests can also be buffered by functional redundancy, such that double or triple disruptions are required for a phenotypic consequence. Consistent with redundant, contextual, or subtle functions, the deletion of large and highly conserved genomic segments sometimes has no discernible organismal phenotype (21, 22), and seemingly debilitating mutations in genes thought to be indispensible have been found in the human population (41).
Evolutionary Approach.
Comparative genomics provides a powerful approach for detecting noncoding functional elements that show preferential conservation across evolutionary time. A high level of sequence conservation between related species is indicative of purifying selection, whereby disruptive mutations are rejected, with the corresponding sequence deemed to be likely functional. Evidence of function can also come from accelerated evolution across species or within a particular lineage, revealing elements under positive selection for recently acquired changes that increase fitness; such an approach gains power by incorporating multiple closely related genomes because each species provides information about sequence constraint. Multispecies comparisons have been used in studies of diverse clades, ranging from yeast to mammals. Methods that detect sequences likely under selection have had success in recognizing protein-coding regions, structural RNAs, gene regulatory regions, regulatory motifs, and specific regulatory elements (3, 42–48). The comparative genomics approach can also incorporate information about mutational patterns that may be characteristic of different types of elements.
Although powerful, the evolutionary approach also has limitations. Identification of conserved regions depends on accurate multispecies sequence alignments, which remain a substantial challenge. Alignments are generally less effective for distal-acting regulatory regions, where they may be impeded by regulatory motif turnover, varying spacing constraints, and sequence composition biases (17, 49). Analyzing aligned regions for conservation can be similarly challenging. First, most transcription factor-binding sequences are short and highly degenerate, making them difficult to identify. Second, because detection of neutrally evolving elements requires sufficient phylogenetic distance, the approach is well suited for detecting mammalian-conserved elements, but it is less effective for primate-specific elements and essentially blind to human-specific elements. Third, certain types of functional elements such as immunity genes may be prone to rapid evolutionary turnover even among closely related species. More generally, alignment methods are not well suited to capture substitutions that preserve function, such as compensatory changes preserving RNA structure, affinity-preserving substitutions within regulatory motifs, or mutations whose effect is buffered by redundancy or epistatic effects. Thus, absence of conservation cannot be interpreted as evidence for the lack of function.
Finally, although the evolutionary approach has the advantage that it does not require a priori knowledge of what a DNA element does or when it is used, it is unlikely to reveal the molecular mechanisms under selection or the relevant cell types or physiological processes. Thus, comparative genomics requires complementary studies.
Biochemical Approach.
The biochemical approach for identifying candidate functional genomic elements complements the other approaches, as it is specific for cell type, condition, and molecular process. Decades of detailed studies of gene regulation and RNA metabolism have defined major classes of functional noncoding elements, including promoters, enhancers, silencers, insulators, and noncoding RNA genes such as microRNAs, piRNAs, structural RNAs, and regulatory RNAs (50–53). These noncoding functional elements are associated with distinctive chromatin structures that display signature patterns of histone modifications, DNA methylation, DNase accessibility, and transcription factor occupancy (37, 54–66). For example, active enhancers are marked by specific histone modifications and DNase-accessible chromatin and are occupied by sequence-specific transcription factors, coactivators such as EP300, and, often, RNA polymerase II. Although the extent to which individual features contribute to function remains to be determined, they provide a useful surrogate for annotating candidate enhancers and other types of functional elements.
The ENCODE Project was established with the goal of systematically mapping functional elements in the human genome at high resolution and providing this information as an open resource for the research community (67, 68). Most data acquisition in the project thus far has taken the biochemical approach, using evidence of cellular or enzymatic processes acting on a DNA segment to help predict different classes of functional elements. The recently completed phase of ENCODE applied a wide range of biochemical assays at a genome-wide scale to study multiple human cell types (69). These assays identified genomic sequences (i) from which short and long RNAs, both nuclear and cytoplasmic, are transcribed; (ii) occupied by sequence-specific transcription factors, cofactors, or chromatin regulatory proteins; (iii) organized in accessible chromatin; (iv) marked by DNA methylation or specific histone modifications; and (v) physically brought together by long-range chromosomal interactions.
An advantage of such functional genomics evidence is that it reveals the biochemical processes involved at each site in a given cell type and activity state. However, biochemical signatures are often a consequence of function, rather than causal. They are also not always deterministic evidence of function, but can occur stochastically. For example, GATA1, whose binding at some erythroid-specific enhancers is critical for function, occupies many other genomic sites that lack detectable enhancer activity or other evidence of biological function (70). Likewise, although enhancers are strongly associated with characteristic histone modifications, the functional significance of such modifications remains unclear, and the mere presence of an enhancer-like signature does not necessarily indicate that a sequence serves a specific function (71, 72). In short, although biochemical signatures are valuable for identifying candidate regulatory elements in the biological context of the cell type examined, they cannot be interpreted as definitive proof of function on their own.
What Fraction of the Human Genome Is Functional?
Limitations of the genetic, evolutionary, and biochemical approaches conspire to make this seemingly simple question difficult to answer. In general, each approach can be used to lend support to candidate elements identified by other methods, although focusing exclusively on the simple intersection set would be much too restrictive to capture all functional elements. However, by probing quantitative relationships in data from the different approaches, we can begin to gain a more sophisticated picture of the nature, identity, and extent of functional elements in the human genome.
Case for Abundant Junk DNA.
The possibility that much of a complex genome could be nonfunctional was raised decades ago. The C-value paradox (27, 73, 74) refers to the observation that genome size does not correlate with perceived organismal complexity and that even closely related species can have vastly different genome sizes. The estimated mutation rate in protein-coding genes suggested that only up to ∼20% of the nucleotides in the human genome can be selectively maintained, as the mutational burden would be otherwise too large (75). The term “junk DNA” was coined to refer to the majority of the rest of the genome, which represent segments of neutrally evolving DNA (76, 77). More recent work in population genetics has further developed this idea by emphasizing how the low effective population size of large-bodied eukaryotes leads to less efficient natural selection, permitting proliferation of transposable elements and other neutrally evolving DNA (78). If repetitive DNA elements could be equated with nonfunctional DNA, then one would surmise that the human genome contains vast nonfunctional regions because nearly 50% of nucleotides in the human genome are readily recognizable as repeat elements, often of high degeneracy. Moreover, comparative genomics studies have found that only 5% of mammalian genomes are under strong evolutionary constraint across multiple species (e.g., human, mouse, and dog) (2, 3).
Case for Abundant Functional Genomic Elements.
Genome-wide biochemical studies, including recent reports from ENCODE, have revealed pervasive activity over an unexpectedly large fraction of the genome, including noncoding and nonconserved regions and repeat elements (58–60). Such results greatly increase upper bound estimates of candidate functional sequences (Fig. 2 and Fig. S2). Many human genomic regions previously assumed to be nonfunctional have recently been found to be teeming with biochemical activity, including portions of repeat elements, which can be bound by transcription factors and transcribed (79, 80), and are thought to sometimes be exapted into novel regulatory regions (81–84). Outside the 1.5% of the genome covered by protein-coding sequence, 11% of the genome is associated with motifs in transcription factor-bound regions or high-resolution DNase footprints in one or more cell types (Fig. 2), indicative of direct contact by regulatory proteins. Transcription factor occupancy and nucleosome-resolution DNase hypersensitivity maps overlap greatly and each cover approximately 15% of the genome. In aggregate, histone modifications associated with promoters or enhancers mark ∼20% of the genome, whereas a third of the genome is marked by modifications associated with transcriptional elongation. Over half of the genome has at least one repressive histone mark. In agreement with prior findings of pervasive transcription (85, 86), ENCODE maps of polyadenylated and total RNA cover in total more than 75% of the genome. These already large fractions may be underestimates, as only a subset of cell states have been assayed. However, for multiple reasons discussed below, it remains unclear what proportion of these biochemically annotated regions serve specific functions.
Fig. 2.

Summary of the coverage of the human genome by ENCODE data. The fraction of the human genome covered by ENCODE-detected elements in at least one cell line or tissue for each assay is shown as a bar graph. All percentages are calculated against the whole genome, including the portion that is not uniquely mappable with short reads and thus is invisible to the analysis presented here (see Fig. S1). A more detailed summary can be found in Fig. S2. For transcripts, coverage was calculated from RNA-seq–derived contigs (104) using the count of read fragments per kilobase of exon per million reads (FPKM) and separated into abundance classes by FPKM values. Note that FPKMs are not directly comparable among different subcellular fractions, as they reflect relative abundances within a fraction rather than average absolute transcript copy numbers per cell. Depending on the total amount of RNA in a cell, one transcript copy per cell corresponds to between 0.5 and 5 FPKM in PolyA+ whole-cell samples according to current estimates (with the upper end of that range corresponding to small cells with little RNA and vice versa). “All RNA” refers to all RNA-seq experiments, including all subcellular fractions (Fig. S2). DNAse hypersensitivity and transcription-factor (TFBS) and histone-mark ChIP-seq coverage was calculated similarly but divided according to signal strength. “Motifs+footprints” refers to the union of occupied sequence recognition motifs for transcription factors as determined by ChIP-seq and as measured by digital genomic footprinting, with the fuscia portion of the bar representing the genomic space covered by bound motifs in ChIP-seq. Signal strength for ChIP-seq data for histone marks was determined based on the P value of each enriched region (the –log10 of the P value is shown), using peak-calling procedures tailored to the broadness of occupancy of each modification (SI Methods).
The lower bound estimate that 5% of the human genome has been under evolutionary constraint was based on the excess conservation observed in mammalian alignments (2, 3, 87) relative to a neutral reference (typically ancestral repeats, small introns, or fourfold degenerate codon positions). However, estimates that incorporate alternate references, shape-based constraint (88), evolutionary turnover (89), or lineage-specific constraint (90) each suggests roughly two to three times more constraint than previously (12–15%), and their union might be even larger as they each correct different aspects of alignment-based excess constraint. Moreover, the mutation rate estimates of the human genome are still uncertain and surprisingly low (91) and not inconsistent with a larger fraction of the genome under relatively weaker constraint (92). Although still weakly powered, human population studies suggest that an additional 4–11% of the genome may be under lineage-specific constraint after specifically excluding protein-coding regions (90, 92, 93), and these numbers may also increase as our ability to detect human constraint increases with additional human genomes. Thus, revised models, lineage-specific constraint, and additional datasets may further increase evolution-based estimates.
Results of genome-wide association studies might also be interpreted as support for more pervasive genome function. At present, significantly associated loci explain only a small fraction of the estimated trait heritability, suggesting that a vast number of additional loci with smaller effects remain to be discovered. Furthermore, quantitative trait locus (QTL) studies have revealed thousands of genetic variants that influence gene expression and regulatory activity (94–98). These observations raise the possibility that functional sequences encompass a larger proportion of the human genome than previously thought.
Reconciling Genetic, Evolutionary, and Biochemical Estimates
The proportion of the human genome assigned to candidate functions varies markedly among the different approaches, with estimates from biochemical approaches being considerably larger than those of genetic and evolutionary approaches (Fig. 1). These differences have stimulated scientific debate regarding the interpretation and relative merits of the various approaches (26–29). We highlight below caveats of each approach and emphasize the importance of integration and new high-throughput technologies for refining estimates and better understanding the functional segments in the human genome.
Although ENCODE has expended considerable effort to ensure the reproducibility of detecting biochemical activity (99), it is not at all simple to establish what fraction of the biochemically annotated genome should be regarded as functional. The dynamic range of biochemical signals differs by one or more orders of magnitude for many assays, and the significance of the differing levels is not yet clear, particularly for lower levels. For example, RNA transcripts of some kind can be detected from ∼75% of the genome, but a significant portion of these are of low abundance (Fig. 2 and Fig. S2). For polyadenylated RNA, where it is possible to estimate abundance levels, 70% of the documented coverage is below approximately one transcript per cell (100–103). The abundance of complex nonpolyadenylated RNAs and RNAs from subcellular fractions, which account for half of the total RNA coverage of the genome, is likely to be even lower, although their absolute quantification is not yet achieved. Some RNAs, such as lncRNAs, might be active at very low levels. Others might be expressed stochastically at higher levels in a small fraction of the cell population (104), have hitherto unappreciated architectural or regulatory functions, or simply be biological noise of various kinds. At present, we cannot distinguish which low-abundance transcripts are functional, especially for RNAs that lack the defining characteristics of known protein coding, structural, or regulatory RNAs. A priori, we should not expect the transcriptome to consist exclusively of functional RNAs. Zero tolerance for errant transcripts would come at high cost in the proofreading machinery needed to perfectly gate RNA polymerase and splicing activities, or to instantly eliminate spurious transcripts. In general, sequences encoding RNAs transcribed by noisy transcriptional machinery are expected to be less constrained, which is consistent with data shown here for very low abundance RNA (Fig. 3). Similarly, a majority of the genome shows reproducible evidence of one or more chromatin marks, but some marks are in much lower abundance, are preferentially associated with nonconserved heterochromatin regions (e.g., H3K9me3; Fig. 3B), or are known to act at a distance by spreading (105). Indeed, for any given biochemical assay, the proportion of the genome covered is highly dependent on the signal threshold set for the analysis (Fig. 2 and Fig. S2). Regions with higher signals generally exhibit higher levels of evolutionarily conservation (Fig. 3 and Fig. S3). Thus, one should have high confidence that the subset of the genome with large signals for RNA or chromatin signatures coupled with strong conservation is functional and will be supported by appropriate genetic tests. In contrast, the larger proportion of genome with reproducible but low biochemical signal strength and less evolutionary conservation is challenging to parse between specific functions and biological noise.
Fig. 3.

Relationship between ENCODE signals and conservation. Signal strength of ENCODE functional annotations were defined as follows: log10 of signal intensity for DNase and TFBS, log10 of RPKM for RNA, and log10 of −log10 P value for histone modifications. Annotated regions were binned by 0.1 units of signal strength. (A) The number of nucleotides in each signal bin was plotted. (B) The fraction of the genome in each signal bin covered by conserved elements (by genomic evolutionary rate profiling) (115) was plotted.
Another major variable underlying the differences in genome coverage is assay resolution. Biochemical methods, such as ChIP or DNase hypersensitivity assays, capture extended regions of several hundred bases, whereas the underlying transcription factor-binding elements are typically only 6–15 bp in length. Regulatory motifs and DNase footprints within bound regions show much stronger evidence of constraint than surrounding nucleotides that nevertheless fall within the region. Functional elements predicted from chromatin-state annotations tend to span even larger regions (e.g., the median length of enhancer states is ∼600 bp), although the driver nucleotides can be similarly few. Biochemical activity may also spread from neighboring regions, in genomic coordinates or 3D genome organization, making it even more difficult to establish the potential nucleotide drivers. Nonetheless, immediately consigning a biochemically marked region to the nonfunctional bin for lack of a driver motif would be premature. Genetic tests by deletion or sequence substitution are needed to resolve the question of their functional significance.
Thus, unanswered questions related to biological noise, along with differences in the resolution, sensitivity, and activity level of the corresponding assays, help to explain divergent estimates of the portion of the human genome encoding functional elements. Nevertheless, they do not account for the entire gulf between constrained regions and biochemical activity. Our analysis revealed a vast portion of the genome that appears to be evolving neutrally according to our metrics, even though it shows reproducible biochemical activity, which we previously referred to as “biochemically active but selectively neutral” (68). It could be argued that some of these regions are unlikely to serve critical functions, especially those with lower-level biochemical signal. However, we also acknowledge substantial limitations in our current detection of constraint, given that some human-specific functions are essential but not conserved and that disease-relevant regions need not be selectively constrained to be functional. Despite these limitations, all three approaches are needed to complete the unfinished process of inferring functional DNA elements, specifying their boundaries, and defining what functions they serve at molecular, cellular, and organismal levels.
Functional Genomic Elements and Human Disease
Presently, ∼4,000 genes have been associated with human disease, a likely underestimate given that the majority of disease-associated mutations have yet to be mapped. There is overwhelming evidence that variants in the regulatory sequences associated with such genes can lead to disease-relevant phenotypes. Biochemical approaches provide a rich resource for understanding disease-relevant functional elements, but they are most powerful as part of a multifaceted body of evidence for establishing function. Three specific examples from the β-globin locus illustrate how biochemical data can be integrated with evolutionary constraint and genetic assays of function (Fig. 4). The expression of globin genes at progressive stages of development is controlled by transcription factors binding at multiple cis-regulatory modules (CRMs) (106), but these CRMs differ dramatically in epigenetic signals and evolutionary history. For example, the independently acting enhancer LCR hypersensitive site 2 (HS2) (107) shows strong constraint on the motifs bound by transcription factors and strong DNase footprints. A second CRM, HBG1 3′ enhancer (108), is also bound in vivo by GATA1 (and other proteins) and is active as an enhancer, but shows almost no constraint over mammalian evolution. Last, a third location, HBG1-D (109, 110), shows DNase hypersensitivity but lacks biological activity in enhancer assays. Rather, binding of this and other CRMs in the locus by BCL11A leads to a reorganization of the chromatin interactions and repression of genes encoding the fetally expressed γ-globins in adult erythroid cells. This CRM is virtually devoid of evidence of mammalian constraint, at least in part because the adult-stage silencing of γ-globin genes is specific to primates. These vignettes illustrate the complementary nature of genetic, evolutionary, and biochemical approaches for understanding disease-relevant genomic elements and also the importance of data integration, as no single assay identifies all functional elements.
Fig. 4.
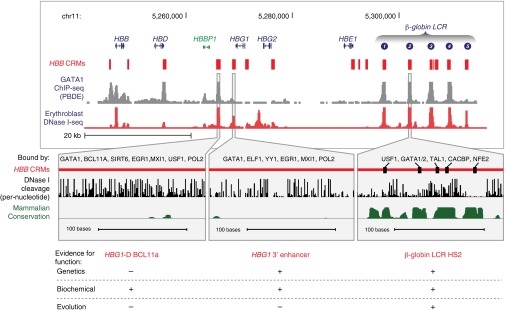
Epigenetic and evolutionary signals in cis-regulatory modules (CRMs) of the HBB complex. (Upper) Many CRMs (red rectangles) (106) have been mapped within the cluster of genes encoding β-like globins expressed in embryonic (HBE1), fetal (HBG1 and HBG2), and adult (HBB and HBD) erythroid cells. All are marked by DNase hypersensitive sites and footprints (Gene Expression Omnibus accession nos. GSE55579, GSM1339559, and GSM1339560), and many are bound by GATA1 in peripheral blood derived erythroblasts (PBDEs). (Lower, Left) A DNA segment located between the HBG1 and HBD genes is one of the DNA segments bound by BCL11A (109, 110) and several other proteins (ENCODE uniformly processed data) to negatively regulate HBG1 and HBG2. It is sensitive to DNase I but is not conserved across mammals. (Center) An enhancer located 3′ of the HBG1 gene (red line) (108) is bound by several proteins in PBDEs and K562 cells (from the ENCODE uniformly processed data) and is sensitive to DNase I, but shows almost no signal for mammalian constraint. (Right) The enhancer at hypersensitive site (HS)2 of the locus control region (LCR) (red line) (107) is bound by the designated proteins at the motifs indicated by black rectangles. High-resolution DNase footprinting data (116) show cleavage concentrated between the bound motifs, which are strongly constrained during mammalian evolution, as shown on the mammalian phastCons track (48).
Conclusion
In contrast to evolutionary and genetic evidence, biochemical data offer clues about both the molecular function served by underlying DNA elements and the cell types in which they act, thus providing a launching point to study differentiation and development, cellular circuitry, and human disease (14, 35, 69, 111, 112). The major contribution of ENCODE to date has been high-resolution, highly-reproducible maps of DNA segments with biochemical signatures associated with diverse molecular functions. We believe that this public resource is far more important than any interim estimate of the fraction of the human genome that is functional.
By identifying candidate genomic elements and placing them into classes with shared molecular characteristics, the biochemical maps provide a starting point for testing how these signatures relate to molecular, cellular, and organismal function. The data identify very large numbers of sequence elements of differing sizes and signal strengths. Emerging genome-editing methods (113, 114) should considerably increase the throughput and resolution with which these candidate elements can be evaluated by genetic criteria. Given the limitations of our current understanding of genome function, future work should seek to better define genome elements by integrating all three methods to gain insight into the roles they play in human biology and disease.
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: In addition to data already released via the ENCODE Data Coordinating Center, the erythroblast DNase-seq data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession nos. GSE55579, GSM1339559, and GSM1339560).
Authored by members of the ENCODE Consortium.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318948111/-/DCSupplemental.
References
- 1.Lander ES, et al. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Waterston RH, et al. Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 3.Lindblad-Toh K, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478(7370):476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ponting CP, Hardison RC. What fraction of the human genome is functional? Genome Res. 2011;21(11):1769–1776. doi: 10.1101/gr.116814.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones FC, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484(7392):55–61. doi: 10.1038/nature10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grossman SR, et al. 1000 Genomes Project Identifying recent adaptations in large-scale genomic data. Cell. 2013;152(4):703–713. doi: 10.1016/j.cell.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraser HB. Gene expression drives local adaptation in humans. Genome Res. 2013;23(7):1089–1096. doi: 10.1101/gr.152710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeong S, et al. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell. 2008;132(5):783–793. doi: 10.1016/j.cell.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Carroll SB. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell. 2008;134(1):25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 10.Chan YF, et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010;327(5963):302–305. doi: 10.1126/science.1182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleinjan DA, van Heyningen V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76(1):8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinjan DA, Lettice LA. Long-range gene control and genetic disease. Adv Genet. 2008;61:339–388. doi: 10.1016/S0065-2660(07)00013-2. [DOI] [PubMed] [Google Scholar]
- 13.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22(9):1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward LD, Kellis M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dermitzakis ET, Clark AG. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: Conservation and turnover. Mol Biol Evol. 2002;19(7):1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- 18.Costas J, Casares F, Vieira J. Turnover of binding sites for transcription factors involved in early Drosophila development. Gene. 2003;310:215–220. doi: 10.1016/s0378-1119(03)00556-0. [DOI] [PubMed] [Google Scholar]
- 19.Moses AM, et al. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLOS Comput Biol. 2006;2(10):e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludwig MZ, Patel NH, Kreitman M. Functional analysis of eve stripe 2 enhancer evolution in Drosophila: Rules governing conservation and change. Development. 1998;125(5):949–958. doi: 10.1242/dev.125.5.949. [DOI] [PubMed] [Google Scholar]
- 21.Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302(5644):413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- 22.Ahituv N, et al. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007;5(9):e234. doi: 10.1371/journal.pbio.0050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGaughey DM, et al. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res. 2008;18(2):252–260. doi: 10.1101/gr.6929408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vakhrusheva OA, Bazykin GA, Kondrashov AS. Genome-Level Analysis of Selective Constraint without Apparent Sequence Conservation. Genome Biol Evol. 2013;5(3):532–541. doi: 10.1093/gbe/evt023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doolittle WF. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci USA. 2013;110(14):5294–5300. doi: 10.1073/pnas.1221376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graur D, et al. On the immortality of television sets: “Function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol Evol. 2013;5(3):578–590. doi: 10.1093/gbe/evt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eddy SR. The C-value paradox, junk DNA and ENCODE. Curr Biol. 2012;22(21):R898–R899. doi: 10.1016/j.cub.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Eddy SR. The ENCODE project: Missteps overshadowing a success. Curr Biol. 2013;23(7):R259–R261. doi: 10.1016/j.cub.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Mattick JS, et al. The extent of functionality in the human genome. HUGO J. 2013;7(1):2. [Google Scholar]
- 30.Niu DK, Jiang L. Can ENCODE tell us how much junk DNA we carry in our genome? Biochem Biophys Res Commun. 2012;430(4):1340–1343. doi: 10.1016/j.bbrc.2012.12.074. [DOI] [PubMed] [Google Scholar]
- 31.Germain PL, Ratti E, Boem F. Junk or functional DNA?: ENCODE and the function controversy. Biology & Philosophy. 2014 doi: 10.1007/s10539-014-9441-3. [DOI] [Google Scholar]
- 32.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amsterdam A, et al. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13(20):2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berns K, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428(6981):431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 35.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kheradpour P, et al. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23(5):800–811. doi: 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457(7231):854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30(3):265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melnikov A, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30(3):271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfeiffer BD, et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proc Natl Acad Sci USA. 2008;105(28):9715–9720. doi: 10.1073/pnas.0803697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacArthur DG, et al. 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stark A, et al. Harvard FlyBase curators; Berkeley Drosophila Genome Project Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450(7167):219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423(6937):241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 44.Xie X, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434(7031):338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas JW, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424(6950):788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- 46.Cliften P, et al. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301(5629):71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- 47.Boffelli D, et al. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299(5611):1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- 48.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elnitski L, et al. Distinguishing regulatory DNA from neutral sites. Genome Res. 2003;13(1):64–72. doi: 10.1101/gr.817703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aravin AA, Hannon GJ, Brennecke J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318(5851):761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- 53.Olovnikov I, Aravin AA, Fejes Toth K. Small RNA in the nucleus: The RNA-chromatin ping-pong. Curr Opin Genet Dev. 2012;22(2):164–171. doi: 10.1016/j.gde.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51(6):975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 55.Agarwal S, Rao A. Long-range transcriptional regulation of cytokine gene expression. Curr Opin Immunol. 1998;10(3):345–352. doi: 10.1016/s0952-7915(98)80174-x. [DOI] [PubMed] [Google Scholar]
- 56.Lakshmanan G, Lieuw KH, Grosveld F, Engel JD. Partial rescue of GATA-3 by yeast artificial chromosome transgenes. Dev Biol. 1998;204(2):451–463. doi: 10.1006/dbio.1998.8991. [DOI] [PubMed] [Google Scholar]
- 57.Noonan JP, McCallion AS. Genomics of long-range regulatory elements. Annu Rev Genomics Hum Genet. 2010;11:1–23. doi: 10.1146/annurev-genom-082509-141651. [DOI] [PubMed] [Google Scholar]
- 58.Nardone J, Lee DU, Ansel KM, Rao A. Bioinformatics for the ‘bench biologist’: How to find regulatory regions in genomic DNA. Nat Immunol. 2004;5(8):768–774. doi: 10.1038/ni0804-768. [DOI] [PubMed] [Google Scholar]
- 59.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 60.Li CC, Ramirez-Carrozzi VR, Smale ST. Pursuing gene regulation ‘logic’ via RNA interference and chromatin immunoprecipitation. Nat Immunol. 2006;7(7):692–697. doi: 10.1038/ni0706-692. [DOI] [PubMed] [Google Scholar]
- 61.Weinmann AS, Farnham PJ. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods. 2002;26(1):37–47. doi: 10.1016/S1046-2023(02)00006-3. [DOI] [PubMed] [Google Scholar]
- 62.Johnson KD, Bresnick EH. Dissecting long-range transcriptional mechanisms by chromatin immunoprecipitation. Methods. 2002;26(1):27–36. doi: 10.1016/S1046-2023(02)00005-1. [DOI] [PubMed] [Google Scholar]
- 63.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ozsolak F, et al. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22(22):3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Horak CE, Snyder M. Global analysis of gene expression in yeast. Funct Integr Genomics. 2002;2(4-5):171–180. doi: 10.1007/s10142-002-0065-3. [DOI] [PubMed] [Google Scholar]
- 67.ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 68.Birney E, et al. ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng Y, et al. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 2009;19(12):2172–2184. doi: 10.1101/gr.098921.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Henikoff S, Shilatifard A. Histone modification: Cause or cog? Trends Genet. 2011;27(10):389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 72.Weiner A, et al. Systematic dissection of roles for chromatin regulators in a yeast stress response. PLoS Biol. 2012;10(7):e1001369. doi: 10.1371/journal.pbio.1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomas CA., Jr The genetic organization of chromosomes. Annu Rev Genet. 1971;5:237–256. doi: 10.1146/annurev.ge.05.120171.001321. [DOI] [PubMed] [Google Scholar]
- 74.Gregory TR. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev Camb Philos Soc. 2001;76(1):65–101. doi: 10.1017/s1464793100005595. [DOI] [PubMed] [Google Scholar]
- 75.Keightley PD. Rates and fitness consequences of new mutations in humans. Genetics. 2012;190(2):295–304. doi: 10.1534/genetics.111.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ehret CF, De Haller G. Origin, development and maturation of organelles and organelle systems of the cell surface in Paramecium. J Ultrastruct Res. 1963;23(Suppl 6):1–42. doi: 10.1016/s0022-5320(63)80088-x. [DOI] [PubMed] [Google Scholar]
- 77.Ohno S. So much “junk” DNA in our genome. Brookhaven Symp Biol. 1972;23:366–370. [PubMed] [Google Scholar]
- 78.Lynch M. The Origins of Genome Architecture. Sunderland, MA: Sinauer Associates; 2007. [Google Scholar]
- 79.Kamal M, Xie X, Lander ES. A large family of ancient repeat elements in the human genome is under strong selection. Proc Natl Acad Sci USA. 2006;103(8):2740–2745. doi: 10.1073/pnas.0511238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lowe CB, Bejerano G, Haussler D. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc Natl Acad Sci USA. 2007;104(19):8005–8010. doi: 10.1073/pnas.0611223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lowe CB, et al. Three periods of regulatory innovation during vertebrate evolution. Science. 2011;333(6045):1019–1024. doi: 10.1126/science.1202702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McClintock B. Controlling elements and the gene. Cold Spring Harb Symp Quant Biol. 1956;21:197–216. doi: 10.1101/sqb.1956.021.01.017. [DOI] [PubMed] [Google Scholar]
- 83.de Souza FS, Franchini LF, Rubinstein M. Exaptation of transposable elements into novel cis-regulatory elements: Is the evidence always strong? Mol Biol Evol. 2013;30(6):1239–1251. doi: 10.1093/molbev/mst045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishihara H, Smit AF, Okada N. Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Res. 2006;16(7):864–874. doi: 10.1101/gr.5255506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Clark MB, et al. The reality of pervasive transcription. PLoS Biol. 2011;9(7):e1000625. doi: 10.1371/journal.pbio.1000625. discussion e1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jacquier A. The complex eukaryotic transcriptome: Unexpected pervasive transcription and novel small RNAs. Nat Rev Genet. 2009;10(12):833–844. doi: 10.1038/nrg2683. [DOI] [PubMed] [Google Scholar]
- 87.Lindblad-Toh K, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438(7069):803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 88.Parker SC, Hansen L, Abaan HO, Tullius TD, Margulies EH. Local DNA topography correlates with functional noncoding regions of the human genome. Science. 2009;324(5925):389–392. doi: 10.1126/science.1169050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meader S, Ponting CP, Lunter G. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res. 2010;20(10):1335–1343. doi: 10.1101/gr.108795.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ward LD, Kellis M. Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science. 2012;337(6102):1675–1678. doi: 10.1126/science.1225057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scally A, Durbin R. Revising the human mutation rate: Implications for understanding human evolution. Nat Rev Genet. 2012;13(10):745–753. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- 92.Lohmueller KE, et al. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS Genet. 2011;7(10):e1002326. doi: 10.1371/journal.pgen.1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ward LD, Kellis M. Response to comment on “Evidence of abundant purifying selection in humans for recently acquired regulatory functions”. Science. 2013;340(6133):682. doi: 10.1126/science.1233366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dimas AS, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325(5945):1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Montgomery SB, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464(7289):773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Battle A, et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24(1):14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Degner JF, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482(7385):390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pickrell JK, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 2011;5(3):27. [Google Scholar]
- 100.Lovén J, et al. Revisiting global gene expression analysis. Cell. 2012;151(3):476–482. doi: 10.1016/j.cell.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Islam S, et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21(7):1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marinov GK, et al. From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing. Genome Res. 2014 doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 104.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Talbert PB, Henikoff S. Spreading of silent chromatin: Inaction at a distance. Nat Rev Genet. 2006;7(10):793–803. doi: 10.1038/nrg1920. [DOI] [PubMed] [Google Scholar]
- 106.King DC, et al. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;15(8):1051–1060. doi: 10.1101/gr.3642605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tuan DY, Solomon WB, London IM, Lee DP. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human “beta-like globin” genes. Proc Natl Acad Sci USA. 1989;86(8):2554–2558. doi: 10.1073/pnas.86.8.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bodine DM, Ley TJ. An enhancer element lies 3′ to the human A gamma globin gene. EMBO J. 1987;6(10):2997–3004. doi: 10.1002/j.1460-2075.1987.tb02605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu J, et al. Transcriptional silencing of gamma-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24(8):783–798. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sankaran VG, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365(9):807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gerstein MB, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trynka G, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. 2013;45(2):124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Carr PA, Church GM. Genome engineering. Nat Biotechnol. 2009;27(12):1151–1162. doi: 10.1038/nbt.1590. [DOI] [PubMed] [Google Scholar]
- 115.Davydov EV, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLOS Comput Biol. 2010;6(12):e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hesselberth JR, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6(4):283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.