

Significance
The classical active penetration model for Toxoplasma invasion was established in studies of infection of nonphagocytic host cells by virulent strains of the parasite. Here, we show that avirulent Toxoplasma parasites use a noncanonical invasion pathway when infecting macrophages. Instead of active penetration at cell surface, avirulent Toxoplasma parasites are initially phagocytosed by macrophages and, subsequently, form a parasite vacuole from a phagosomal compartment. This phagosome to vacuole invasion (PTVI) pathway is associated with more efficient infection of macrophages. We hypothesize that PTVI represents a Trojan horse strategy for phagocyte infection and may lead to enhanced systemic dissemination and immune stimulation, acute infection control, and chronic infection establishment.
Keywords: virulence, Trojan horse, apicomplexa, phagocytes
Abstract
Unlike most intracellular pathogens that gain access into host cells through endocytic pathways, Toxoplasma gondii initiates infection at the cell surface by active penetration through a moving junction and subsequent formation of a parasitophorous vacuole. Here, we describe a noncanonical pathway for T. gondii infection of macrophages, in which parasites are initially internalized through phagocytosis, and then actively invade from within a phagosomal compartment to form a parasitophorous vacuole. This phagosome to vacuole invasion (PTVI) pathway may represent an intermediary link between the endocytic and the penetrative routes for host cell entry by intracellular pathogens. The PTVI pathway is preferentially used by avirulent strains of T. gondii and confers an infectious advantage over virulent strains for macrophage tropism.
Phagocytosis is one of the most ancient defense mechanisms for the host to destroy invasive pathogens. However, most intracellular pathogens exploit this very pathway for internalization and survival in phagocytes (1). A notable exception to this paradigm is the infection pathway used by apicomplexan parasites—exemplified by Toxoplasma gondii. The current consensus model of T. gondii infection suggests that the parasite actively invades host cells by forming a moving junction (MJ) at the host cell surface (2). Penetration of T. gondii through this junction is largely driven by its own actin motor complex (3). The parasitophorous vacuoles formed by this pathway are nonfusogenic with the host endocytic system, thus evading lysosome-mediated destruction (4–6). However, recent reports including the findings that host F-actin participates in entry by T. gondii (7), and that the parasite is still able to infect cells, albeit much less efficiently, even without some key components of the invasion machinery (8), suggest the existence of alternative infection pathways for Toxoplasma. The active penetration model was defined largely by using nonphagocytic host cells and hypervirulent strains of the parasite. Unlike their virulent counterparts, the interaction of avirulent Toxoplasma strains with macrophage and dendritic cell results in heightened innate cytokine and chemokine production (9) and the development of a “hypermotile” host cellular phenotype (10), which promotes the control of acute infection and mediates dissemination into sites of parasite latency (11, 12). Here, we investigated whether avirulent parasites interact with phagocytic host cells in a fundamentally different way from the outset. We found that the avirulent Toxoplasma strains infect macrophages initially via phagocytosis and subsequent active penetration from within the phagosome to form a parasitophorous vacuole. This hybrid invasion pathway may represent an intermediary link between the endocytic and the penetrative routes for host cell entry by intracellular pathogens.
Results
To investigate whether avirulent Toxoplasma (PTG, type II strain) uses the active penetration pathway to invade macrophages similar to the virulent Toxoplasma (RH, type I strain), the moving junction—a hallmark of active penetration—was examined during synchronized infection into RAW264.7 macrophages. As expected, virulent RH parasites were observed penetrating host plasma membrane through a moving junction marked by rhoptry neck protein 4 (RON4) staining (Fig. 1A, Left). In contrast, avirulent PTG barely formed moving junctions during initial contact. The parasites instead elicited extensive membrane ruffles and actin polymerization underneath the contacting sites (Fig. 1A, Right). More than 70% of the adherent PTG induced membrane protrusion and actin nucleation, whereas only 5.9% of adherent RH did (Fig. 1B). The same phenotype was also seen in infections by using other sources of macrophages including naïve mouse peritoneal macrophages and human adherent PBMCs (Fig. S1). Electron microscope imaging showed that surface-attached PTG resided in a zipper-like phagocytic cup, suggesting that the parasites are being phagocytosed by the macrophage (Fig. 1C). Indeed, internalized live PTG were recruited into host Rab5a, FcγR, or lysosomal associated membrane protein 1 (LAMP-1) positive endocytic compartments similar to the phagosomes formed by dead parasites (Fig. 1D). Following initial attachment, both live and dead PTG parasites were mostly associated with phagocytic cups and were internalized at the same rate (Fig. 1E, Left). As expected, the dead parasites progressively accumulated in phagolysosomes and were unable to form PVMs (Fig. 1E, Center and Right). To know whether phagocytosed live PTG followed the same fate, we traced the relative distribution of live tachyzoites in a LAMP-1+ phagosomal compartment and in a dense granule protein 7 (GRA7+) parasitophorous vacuole membrane (PVM), respectively. In contrast to the kinetics of dead parasites, the frequency of the live parasites accumulated in LAMP-1+ compartments did not exceed 15% over an extended time, whereas GRA7+ PVMs were accumulated in infected resting macrophages (Fig. 1E, Center and Right), suggesting that phagocytosed live PTG tachyzoite transiently passes through a phagosomal compartment before the formation of the PVM. Alternatively, only a minority of internalized PTG acquired access to this compartment, whereas a majority of live parasites successfully established parasitophorous vacuoles, bypassing a LAMP-1+ intermediate compartment.
Fig. 1.

Type II T. gondii (PTG strain) is internalized mainly through phagocytosis instead of active penetration during macrophage infection. (A) RAW264.7 cells were infected with GFP-RH and GFP-PTG for 10 min at 37 °C after temperature switch from 4 °C. Epifluorescence images of infected cells show F-actin (visualized by phalloidin-AF555 staining), PVM (stained with α-ROP1 Ab), and moving junction (stained with α-RON4 Ab), respectively. The arrowhead points to a moving junction formed by RH (the magnified MJ shown in Inset), and arrows pinpoint adherent PTG inducing host actin polymerization. (B) Frequency of actin nucleation by extracellularly attached PTG and RH 10 min after infection. Data shown are representative of 20 experiments performed in triplicates with mean ± SD. (C) Electron microscopy image showing PTG induced phagocytic cup formation (black arrows) in vaccine strain-activated mouse peritoneal macrophages 15 min after pulse infection. (D) Localization of phagosomal markers Rab5a, FcγR, and LAMP-1 around the internalized dead and live PTG in resting RAW264.7 cell. Arrows point to the internalized parasites associated with the indicated phagocytic markers. Arrowhead points to a PV formed by live PTG (stained by α-GRA7 Ab). (E) Kinetics of phagocytic cup formation (F-actin+), residence in phagosome (LAMP-1+) and PVM formation (GRA7+) by live and dead PTG. At each time point, parasites in the three cellular compartments were scored simultaneously and distribution within each cellular compartment was calculated relative to the total number of parasites scored. (Scale bars: 5 µm.)
To distinguish between these two possibilities, we investigated whether the internalized live PTG tachyzoites are able to form a moving junction and establish a parasitophorous vacuole from a LAMP-1+ compartment. Indeed, as shown by RON4 staining, PTG established a moving junction intracellularly as evidenced by inaccessibility to anti-surface antigen 1 (SAG1) antibody staining performed before cell permeabilization (Fig. 2A). Moreover, we observed constricted parasites with their apical portion residing in a GRA7+ nascent parasitophorous vacuole with the other half of the parasite still contained within a LAMP-1+ phagosome (Fig. 2B). This result indicated that phagocytosed PTG is able to form moving junction and establish parasitophorous vacuole by a process resembling active penetration, but was initiated from the phagosomal membrane rather than at the host cell surface. Previous studies have described a similar process of entry into bone marrow-derived macrophages by using live video microscopy, although this phenomenon was seen only with a small portion of virulent type I strain RH parasites (13). Many intracellular pathogens (such as Listeria and Francisella) were reported able to escape from phagosome into the cytosol for multiplication, a process that depends on phagosome acidification and can be blocked by inhibition of vacuolar-ATPase (14, 15). To determine whether PVM formation by phagocytosed PTG depends on phagosome maturation, RAW264.7 cells were pretreated with Bafilomycin A1 (Baf A1) followed by infection with parasites in the absence of the drug. Transient inhibition of phagosome acidification decreased PVM formation of PTG strain by 56.4% (Fig. 2C). As expected, PVM formation by virulent RH strain was insensitive to Baf A1 preinhibition because the active penetration pathway avoided direct interaction with host phagocytic system (Fig. 2D, Left). However, upon opsonization of RH with an antibody against surface SAG1 antigen, which artificially reroutes RH invasion from active penetration to FcγR-mediated phagocytosis, PVM formation by opsonized parasites became Baf A1 sensitive (Fig. 2D, Right), suggesting that once phagocytosed by resting macrophages, both PTG and RH parasites remain competent for PVM formation by MJ-mediated invasion. This conclusion was further supported by the observed stripping of opsonized antibody from the RH surface following passage through the moving junction (Fig. 2E). We propose to name this noncanonical infection pathway as phagosome to vacuole invasion (PTVI) (Fig. 2F). During PTVI, live parasites are uptaken by macrophages through phagocytosis. Once internalized, the parasite starts active invasion by forming moving junction at the phagosome membrane to establish a parasitophorous vacuole.
Fig. 2.

Phagocytosed PTG undergoes phagosome to vacuole invasion (PTVI) to establish parasitophorous vacuoles in resting macrophages. (A) Internalized PTG form moving junctions intracellularly. Arrow points to an extracellular PTG positively stained by surface antigen SAG1 antibody. Arrowhead points to an intracellular PTG (SAG1−) forming a moving junction as visualized by α-RON4 staining. (B) Intracellular PTG form moving junctions from within phagosome. Arrowhead points to a constriction indicating the localization of the moving junction. Nascent PVM was stained by α-GRA7 Ab, and phagosome was stained by α-LAMP-1 Ab. (C) Sensitivity of PVM formation by PTG strain to Baf A1 inhibition of phagosomal maturation. RAW264.7 cells were pretreated with 25 nM Baf A1 for 1 h followed by PTG infection for 40 min in absence of the drug. Frequency of GRA7+ vacuoles within internalized PTG is shown, and data are presented as mean ± SD in triplicates. ***P < 0.001, Student t test. (D) PVM formation by RH is insensitive to Baf A1 pretreatment of host cells but becomes sensitive after opsonization with the mouse α-SAG1 IgG2a. The data were analyzed as in C. (E) Stripping of opsonized antibody from phagocytosed RH surface as parasites transit from phagosome to vacuole. The number 1-, 2-, and 3-coded RH parasites show opsonized Ab at the three different stages of stripping during invasion, with opsonized Ab intact on surface of parasite 1 still residing in a phagosome (LAMP-1+), completely stripped from the surface of parasite 3, which has established a vacuole (ROP1+) and undergoing stripping from the surface of parasite 2 during passage through the MJ. Opsonized α-SAG1 Ab was visualized by donkey anti-mouse AF568 (blue), PVM was stained with α-ROP1 (white) and phagosome with α-LAMP-1(red). The yellow dotted line indicates the putative localization of the moving junction. Please note that magnification of the cropped image of parasite 2 is higher than that of parasite 1 and 3. (F) Schematic depiction of PTVI pathway, the two-step infection process, is initiated with phagocytosis of T. gondii followed by parasite active invasion from within phagosome. (Scale bars: 5 µm.)
The above data clearly demonstrated that PTG tachyzoites mainly use the PTVI pathway to infect resting macrophages. A question of great interest is whether PTG parasites are able to undertake a PTVI pathway in IFNγ-activated macrophages, because they impose formidable barriers to avirulent Toxoplasma strains by exerting toxoplasmacidal activity mediated by immunity-related GTPases and toxoplasmastatic activity mediated by iNOS (16). To address this question, we examined PTG and RH infections in peritoneal macrophages obtained from mice primed with a vaccine strain by applying the same approaches used in naïve macrophages studies. Similar to what we observed in naïve macrophages, virulent RH parasites were still resistant to phagocytosis by these highly activated macrophages. Of cell-attached RH tachyzoites, only 8% of them induced phagocytic cups (Fig. 3 A and B) and the rest of the parasites underwent active penetration through established moving junctions at host plasma membrane (Fig. 3C, Left). As expected, more than 80% of adherent PTG parasites induced phagocytic cups (Fig. 3 A and B) and formed moving junctions intracellularly in activated primary macrophages (Fig. 3C, Right and Fig. S2). Next, we examined whether the PVM formation by PTG parasites depends on phagosomal maturation in IFNγ-activated RAW264.7 cells. Transient inhibition of phagosomal acidification with Baf A1 reduced PVM formation of internalized PTG tachyzoites by 42.9%. Concomitantly internalized parasites stuck in LAMP-1+ phagosomes increased by 41.4% compared with DMSO treatment control (Fig. 3, Left and Center). These results indicated that blockade of phagosomal maturation interrupts phagosome-to-vacuole transition of internalized parasites in activated macrophages similar to the effect seen in resting cells. The accumulation of Irgb6, an IFNγ-inducible GTPase, was not affected by Baf A1 pretreatment on once established PVMs (Fig. 3D, Right), indicating that PVMs formation and IRGs trafficking onto PVMs are sequential steps. Collectively, the data presented strongly demonstrated that the avirulent PTG strain infects IFNγ-activated macrophages through the PTVI pathway, although the PVMs formed are subject to IRG-mediated disruption later (17, 18).
Fig. 3.

Phagocytosed PTG tachyzoites are able to establish parasitophorous vacuoles through PTVI pathway in highly activated mouse macrophages. (A) Representative epifluorescent images illustrating that attached PTG parasites (Right) were phagocytosed in highly activated mouse macrophages, whereas RH tachyzoites (Left) were not. Primed mouse peritoneal macrophages were used for ex vivo infections. The host F-actin was stained with Phalloidin-Alexa Fluor 555 and PVMs with α-GRA7 antisera. (B) Bar diagram showing the frequency of phagocytic cup induction by attached parasites 10 min after infection. (C) Representative images showing that phagocytosed PTG parasite forms a moving junction within highly activated mouse peritoneal macrophages (Right), whereas RH forms a MJ at host plasma membrane (Left). Arrowhead pinpoints the moving junction of each parasite. A magnified MJ is shown on right of each image. Host cells were stained with phalloidin-AF350. Moving junctions were visualized with α-RON4 staining, and the apical end of parasite was labeled with α-MIC2 staining. (D) Effect of Baf A1-pretreatment on PVM formation, phagosomal residence of internalized GFP-PTG tachyzoites, and Irgb6 accumulation on PVMs in IFNγ-activated RAW264.7 cells. Parasite PVM, host phagosome, and Irgb6 were triple stained with rabbit α-ROP1, rat α-LAMP-1, and goat α-Irgb6 Ab, respectively. Frequency of internalized PTG parasites distributed within ROP1+ vacuoles (Left), LAMP-1+ phagosomes (Center) and Irgb6-coated ROP1+ PVMs (Right) was determined by scoring 10 fields (60× objective) per coverslip (four coverslips for each treatment) 50 min after infection. Data are presented as mean ± SEM: ***P < 0.001, **P < 0.01, Student t test. (Scale bars: 5 µm.)
The avirulent PTG strain of T. gondii undergoes PTVI pathway, whereas the virulent RH strain resists phagocytosis in both resting and activated macrophages in vitro, suggesting that the PTVI infection pathway may be mainly used by avirulent strains of T. gondii. There are three major clonal lineages of T. gondii (type I, II, and III) that dominate infections of human and other species in Europe and North America (19). Type I strains are the most virulent because a single tachyzoite can kill a mouse. The types II and III strains are much less virulent with LD50 more than 3 orders of magnitude higher than type I strains (20). The potential of the three lineages for PTVI infection was compared by monitoring Toxoplasma-induced actin nucleation and Baf A1 sensitivity of PVM formation in resting RAW264.7 cells. The results showed that type II strain ME49 has the highest PTVI potential, type I strain GT-1 has the lowest potential, and type III strain CTG has an intermediate potential (Fig. 4 A and B), suggesting that avirulent Toxoplasma strains prefer to use the PTVI pathway for macrophage infection. We next wanted to know whether the preferential use of the PTVI pathway enhances infectivity of avirulent T. gondii strains in macrophages. We therefore directly compared the infection rate of PTG and RH strains, which preferentially use the PTVI and the classical active penetration pathway respectively, in a competitive infection assay where GFP-PTG and mCherry-RH were mixed with RAW264.7 or MEF simultaneously (moi = 0.5 for each strain). No significant difference was found between PTG and RH strains during fibroblast infections in terms of the number of PVMs formed within a 40-min time span (Fig. 5B, Right). However, for infection of macrophages, PTG parasites showed a 450% higher infection rate and 360% more PVM formation than RH strain 40 min after infection regardless of whether the strains were mixed or separated (Fig. 5 A and B, Left). An even more dramatic increase of ex vivo infection efficiency (747%) by PTG parasites was seen in primary macrophages, but not in B cells and T cells harvested from the peritoneum of naïve mice (Fig. 5 C and D and Fig. S3). The same macrophage infection preference and high infection efficiency by avirulent Toxoplasma strain was also observed during in vivo infections where naïve mice were i.p infected by equal numbers of GFP-PTG and mCherry-RH tachyzoites separately or in combination. Although infection rates of B cells and T cells by the two strains were equally low (approximately 1%), the infection rates of macrophages by PTG strain reached more than 30% on average and were 680% and 225% higher than RH strain in the single and the dual infections, respectively (Fig. 5 E and F and Fig. S3). Taken together, these data indicated that avirulent Toxoplasma prefer to infect macrophages (potentially all phagocytes) over other cell types, and PTVI pathway is superior to active penetration pathway in terms of phagocytes infection efficiency.
Fig. 4.
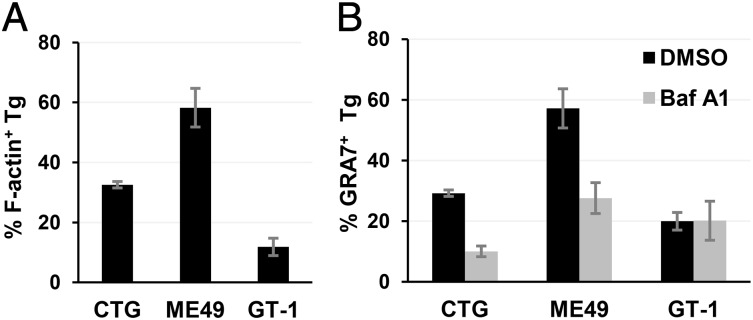
PTVI pathway is preferentially used by avirulent Toxoplasma strains. (A) Comparison of F-actin induction by adherent type III (CTG), type II (ME49), and type I (GT-1) Toxoplasma tachyzoites at resting RAW264.7 cell surface 8 min following synchronized infection by temperature switch. (B) Baf A1 sensitivity of PVM formation by intracellular CTG, ME49, and GT-1 parasites 40 min after infection of resting RAW264.7 cells. Immunostaining and data analysis are same as in Fig. 2C. Data are presented as mean ± SD in triplicates.
Fig. 5.

Avirulent T. gondii strains exhibit enhanced macrophage infection tropism. (A) Representative FACS plots showing infection rate of resting RAW264.7 cells by GFP-PTG and mCherry-RH. For dual infection, RAW264.7 cells were infected with a mixture of GFP-PTG and mCherry-RH at moi = 0.5 for each strain for 40 min at 37 °C. The frequency of GFP+ and mCherry+ among CD11b+ cells is shown on the right. Data are presented as mean ± SD. (B) Immunofluorescent assay of PVM formation by intracellular PTG and RH during single or dual infections at 40 min after infection. PVMs were stained with α-GRA7 Ab. Intracellular and extracellular parasites were distinguished by surface staining with anti-Toxoplasma Ab before cell permeabilization. The number of PVMs per field from 12 randomly selected fields was counted per coverslip in triplicates. On average, there were 48 RAW264.7 cells and 8 MEF cells per field (0.0166 mm2 with approximately 80% confluence of cells). Infections of RAW264.7 are shown on left, and infections of MEF shown on right. (C and D) PECs were harvest from the peritoneum of naïve C57BL/6 mice and infected ex vivo by GFP-PTG or mCherry-RH alone (single infection, s.i) or in combination (dual infection, d.i) at moi = 0.5 per strain for 50 min at 37 °C. (C) Representative FACS plots showing infection rate in macrophages (CD11b+ F4/80+ Ly6G−) (Left) and mean infection rates from triplicate cultures is summarized at Right. (D) The infection rate in B cells (B220+ CD11b−TCRβ−). The ratio of PTG and RH parasites in mixture was examined by fluorescence microscopy. This experiment was repeated four times with similar results. (E and F) Naïve mice were i.p. infected by either 1 million GFP-PTG or 1 million mCherry-RH per mouse during single infection (Single, s.i, n = 2), or by 1 million GFP-PTG and 1 million mCherry-RH per mouse during dual infection (Dual, d.i, n = 4) for 60 min. In vivo infected PECs were harvested and analyzed by using FACSAria. (E) Representative FACS plots showing macrophages infection (CD11b+ F4/80+) (Left) and summarized infection rate (Right). (F) Infection rate in B cells (CD19+ TCRβ−) is shown. Statistical significance of macrophage infection by PTG and RH in dually inoculated mice was analyzed by using two-tailed and paired Student t test. ***P = 0.0009. The dual infection experiments were repeated three times with similar results.
Discussion
In summary, unlike virulent Toxoplasma strains that actively invade host cells at the plasma membrane, their avirulent counterparts mainly use the PTVI pathway—a typical Trojan horse strategy to invade macrophages from within the phagosome. Assuming that the parasite machinery for invasion at the cell surface versus from the phagosome is identical, we think that the key difference between the avirulent and virulent strain use of the PTVI pathway is at the initial phagocytic step. Similar to how other intracellular pathogens are internalized through phagocytosis (21), the uptake of avirulent T. gondii by macrophages likely involves the recognition of parasite ligands by host surface receptors. The parasite and host determinants involved in the surface interaction leading to the binding and phagocytosis of avirulent T. gondii by macrophages are yet to be identified. The absence of this putative phagocytic ligand, coupled with their known hypermotility phenotype, could account for the inefficient phagocytosis of virulent strains by macrophages.
After internalization, engagement of the tachyzoite with the host endocytic system may play a critical role for Toxoplasma to establish the chronic infection, a survival strategy implemented successfully only by the avirulent Toxoplasma strains. The persistence of infection requires not only the acute induction of immune mechanisms that control parasite hyperproliferation but also efficient dissemination of parasites into deep tissues, where they establish a chronic niche. A concept that is now gaining acceptance is that induction of proinflammatory cytokines essential for in vivo control of acute Toxoplasma infection involves intracellular signaling from endosomal and cytoplasmic pattern recognition receptors (PRRs) (22–26). Interestingly, the inflammatory cytokines IL-12 and IL-1β are both induced differentially by avirulent, but not virulent, Toxoplasma strains (9, 27, 28), presumably through intracellular activation of TLRs and other PRRs. However, how these intracellular PRRs are activated during live Toxoplasma infection is still poorly understood. The close association of the internalized avirulent tachyzoites with the endocytic system of professional phagocytes, as reported here, could present opportunities for host PRRs and their respective parasite ligands to interact with each other. This interplay could then underlie a more robust innate and adaptive immune activation, leading to effective control of avirulent parasites. Additionally, avirulent T. gondii strains preferentially infect mononuclear cells, including naive resident macrophages (this report), monocytes (29, 30) and neutrophils (31), and also differentially induce hypermigratory phenotype in infected macrophages and dendritic cells (10, 32). It is tempting to speculate that these maneuvers together represent a constellation of adaptive responses characteristic of avirulent T. gondii. The establishment of chronic infections could then be a consequence of the orchestrated actions and immune-activated phagocytes mediating late parasitic control, both being potentially influenced or significantly regulated by the initial phagocytic route of infection used by avirulent T. gondii.
Avirulent Toxoplasma strains, particularly the type II archetype and its ancestral derivatives such as haplogroup 12 (33), are dominant in infections of humans and their domesticated animals in Europe and North America (34–37). However, the infections caused by virulent type I strains are rare (19). Thus, the adaptive interactions of avirulent Toxoplasma with phagocytic cells (including use of PTVI) discussed above may be linked to their widespread distribution in nature. Considering the recent genetic evidence suggesting that virulent type I strains were derived from an atypical type II ancestor following a sexual recombination event approximately 10,000 years ago (38–40), type I strains may have lost the ability to efficiently induce phagocytosis and, therefore, cannot initiate PTVI. This event, coupled with the acquisition of virulence factors that evade innate immunity (41), may have ultimately led to hypervirulence in type I strains of Toxoplasma.
Materials and Methods
Experimental Animals and Cell Lines.
C57BL/6 wild-type (WT) mice were procured from the Jackson Laboratory. Mice were maintained under specific pathogen-free conditions at the University of Medicine and Dentistry of New Jersey (now Rutgers University) animal care facility. This research work was carried out in accordance with the guidelines of the University of Medicine and Dentistry of New Jersey (now Rutgers University) Institutional Animal Care and Use Committee. Mouse macrophage-like cell line RAW264.7 was purchased from American Type Culture Collection (ATCC) and maintained in COSTAR ultralow cluster cell culture plate (Corning) in RPMI 1640 medium supplemented with 5% (vol/vol) FBS (HyClone), 0.45% d-(+)-glucose, 1 mM sodium pyruvate, and 10 mM Hepes (Invitrogen). Human foreskin fibroblast (HFF) and mouse embryo fibroblast (MEF) cells line were obtained from ATCC and maintained in high-glucose DMEM in the presence of 10% (vol/vol) FBS, 10 U/mL penicillin-streptomycin, and 29.2 µg/mL l-glutamine (Invitrogen).
Parasites.
The uracil auxotrophic carbamoyl phosphate synthase CPS (42) mutant strain of T. gondii was developed in the laboratory of David Bzik (Dartmouth Medical School, Hanover, NH). The strains of GT-1and CTG were generous gifts from David Sibley (Washington University, St. Louis, MO). mCherry-RH was obtained from Marc-Jan Gubbels (Boston College, Boston). GFP-PTG, GFP-RH, and ME49 were purchased from ATCC. All strains were maintained and propagated in confluent HFF. Upon 60–80% lysis of the HFF monolayer, T. gondii tachyzoites were released by passing through 25G needle twice and spun down by centrifugation at 2,000 × g for 10 min. For the CPS strain, the pellet was resuspended in PBS and gamma (γ) irradiated at 15,000 rads before priming of mouse. All T. gondii strains were routinely monitored with MycoSensor PCR assay Kit (Agilent Technologies) and were maintained free of Mycoplasma.
Antibodies, chemical reagents, and procedures for immunofluorescence and ultrastructural microscopy assay can be found in SI Materials and Methods.
Opsonization of T. gondii.
GFP-RH strain was incubated with 1 µg/mL mouse α-SAG1 mAb for 30 min on ice. Opsonized RH were washed twice with invasion medium to remove unbound Abs and used in macrophage infection. Infected cells were fixed and permeabilized 40 min after infection. To visualize opsonized Ab on the surface of internalized RH, donkey α-mouse AF568 were used. The PVMs were visualized with either rabbit α-ROP1 or rabbit α-GRA7 antisera.
Competitive Infection Assay by Flow Cytometry and Immunofluorescent Microscopy.
For in vitro competitive infections between PTG and RH, 1 × 106 resting RAW264.7 cells were either singly or dually infected with 0.5 × 106 GFP-transgenic PTG and/or 0.5 × 106 mCherry-transgenic RH in suspension for 40 min at 37 °C. For ex vivo competitive infection, 1 × 106 PECs harvested from naive mice peritoneum were either singly or dually infected with 0.5 × 106 GFP-transgenic PTG and/or 0.5 × 106 mCherry-transgenic RH in suspension for 50 min at 37 °C. For in vivo competitive infections, 1 × 106 GFP-PTG or 1 × 106 mCherry-RH parasites were i.p. injected into naive mouse peritoneum, and 1 h later PECs were harvested by peritoneal lavage. To make sure equal number of tachyzoites was used during competitive infections, the GFP-PTG and mCherry-RH parasites in mixture were examined by using fluorescent microscope, and the ratio was compensated to 1 before infection. The infected macrophages were fixed with BD Cytofix and surface stained with CD11b-APC. The samples were acquired on a FACSAria (BD), and the frequency of GFP+ and mCherry+ cells were analyzed by using Flowjo (Tree Star). To compare the efficiency of PVM formation by PTG and RH in macrophage (RAW264.7) and MEF, 0.3 × 106 RAW264.7 cells and 0.05 × 106 MEF cells were plated onto coverslips, and 24 h later, both cell types (approximately 80% confluent) were singly or dually infected with 1 × 106 GFP-PTG and/or 1 × 106 mCherry-RH at 37 °C for 40 or 60 min as indicated in the legend to Fig. 5. To exclude extracellular parasites, α-SAG1 Ab was performed before cell permeabilization, and only SAG1 negative parasites were included during PVM scoring. The number of GRA7+ PVMs was enumerated from 12 randomly selected fields (0.016675 mm2 per field, 60× objective) across a coverslip. The average frequency of GRA7+ PVMs per field was calculated from triplicate coverslips.
Statistical Analysis.
Data are represented as mean ± SD. Statistical analysis was carried out by using Microsoft Excel. Statistical significance was analyzed by unpaired two-tails Student t test.
Supplementary Material
Acknowledgments
We thank David Sibley (Washington University in St. Louis) for generously providing parasites, antibodies, and discussions; Ryan Weiss for manuscript proofreading; and numerous colleagues who provided reagents and critical review of the manuscript. This work is supported by NIH Grant R01 AI083405 (to G.S.Y.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316841111/-/DCSupplemental.
References
- 1.Sarantis H, Grinstein S. Subversion of phagocytosis for pathogen survival. Cell Host Microbe. 2012;12(4):419–431. doi: 10.1016/j.chom.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Mordue DG, Desai N, Dustin M, Sibley LD. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med. 1999;190(12):1783–1792. doi: 10.1084/jem.190.12.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobrowolski JM, Sibley LD. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 1996;84(6):933–939. doi: 10.1016/s0092-8674(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 4.Joiner KA, Fuhrman SA, Miettinen HM, Kasper LH, Mellman I. Toxoplasma gondii: Fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 1990;249(4969):641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 5.Mordue DG, Håkansson S, Niesman I, Sibley LD. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp Parasitol. 1999;92(2):87–99. doi: 10.1006/expr.1999.4412. [DOI] [PubMed] [Google Scholar]
- 6.Sibley LD, Weidner E, Krahenbuhl JL. Phagosome acidification blocked by intracellular Toxoplasma gondii. Nature. 1985;315(6018):416–419. doi: 10.1038/315416a0. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez V, et al. Host cell entry by apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe. 2009;5(3):259–272. doi: 10.1016/j.chom.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 8.Andenmatten N, et al. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat Methods. 2013;10(2):125–127. doi: 10.1038/nmeth.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robben PM, et al. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J Immunol. 2004;172(6):3686–3694. doi: 10.4049/jimmunol.172.6.3686. [DOI] [PubMed] [Google Scholar]
- 10.Lambert H, Vutova PP, Adams WC, Loré K, Barragan A. The Toxoplasma gondii-shuttling function of dendritic cells is linked to the parasite genotype. Infect Immun. 2009;77(4):1679–1688. doi: 10.1128/IAI.01289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Courret N, et al. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. 2006;107(1):309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol. 2006;8(10):1611–1623. doi: 10.1111/j.1462-5822.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 13.Morisaki JH, Heuser JE, Sibley LD. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J Cell Sci. 1995;108(Pt 6):2457–2464. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- 14.Conte MP, et al. The effects of inhibitors of vacuolar acidification on the release of Listeria monocytogenes from phagosomes of Caco-2 cells. J Med Microbiol. 1996;44(6):418–424. doi: 10.1099/00222615-44-6-418. [DOI] [PubMed] [Google Scholar]
- 15.Santic M, Asare R, Skrobonja I, Jones S, Abu Kwaik Y. Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect Immun. 2008;76(6):2671–2677. doi: 10.1128/IAI.00185-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, et al. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J Immunol. 2009;182(6):3775–3781. doi: 10.4049/jimmunol.0804190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martens S, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005;1(3):e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling YM, et al. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203(9):2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: Correlation of parasite genotype with human disease. J Infect Dis. 1995;172(6):1561–1566. doi: 10.1093/infdis/172.6.1561. [DOI] [PubMed] [Google Scholar]
- 20.Sibley LD, Boothroyd JC. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature. 1992;359(6390):82–85. doi: 10.1038/359082a0. [DOI] [PubMed] [Google Scholar]
- 21.Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011;11(3):187–200. doi: 10.1038/nri2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raetz M, et al. Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J Immunol. 2013;191(9):4818–4827. doi: 10.4049/jimmunol.1301301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. 2011;286(5):3307–3314. doi: 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrade WA, et al. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe. 2013;13(1):42–53. doi: 10.1016/j.chom.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun. 2014;82(1):460–468. doi: 10.1128/IAI.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witola WH, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun. 2011;79(2):756–766. doi: 10.1128/IAI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosowski EE, et al. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med. 2011;208(1):195–212. doi: 10.1084/jem.20100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gov L, Karimzadeh A, Ueno N, Lodoen MB. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. MBio. 2013;4(4):e00255-13. doi: 10.1128/mBio.00255-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Unno A, et al. Toxoplasma gondii tachyzoite-infected peripheral blood mononuclear cells are enriched in mouse lungs and liver. Exp Parasitol. 2013;134(2):160–164. doi: 10.1016/j.exppara.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Channon JY, Seguin RM, Kasper LH. Differential infectivity and division of Toxoplasma gondii in human peripheral blood leukocytes. Infect Immun. 2000;68(8):4822–4826. doi: 10.1128/iai.68.8.4822-4826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coombes JL, et al. Motile invaded neutrophils in the small intestine of Toxoplasma gondii-infected mice reveal a potential mechanism for parasite spread. Proc Natl Acad Sci USA. 2013;110(21):E1913–E1922. doi: 10.1073/pnas.1220272110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lambert H, Dellacasa-Lindberg I, Barragan A. Migratory responses of leukocytes infected with Toxoplasma gondii. Microbes Infect. 2011;13(1):96–102. doi: 10.1016/j.micinf.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Khan A, et al. Genetic analyses of atypical Toxoplasma gondii strains reveal a fourth clonal lineage in North America. Int J Parasitol. 2011;41(6):645–655. doi: 10.1016/j.ijpara.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ajzenberg D, et al. Genotype of 88 Toxoplasma gondii isolates associated with toxoplasmosis in immunocompromised patients and correlation with clinical findings. J Infect Dis. 2009;199(8):1155–1167. doi: 10.1086/597477. [DOI] [PubMed] [Google Scholar]
- 35.Dubey JP, et al. Endemic toxoplasmosis in pigs on a farm in Maryland: Isolation and genetic characterization of Toxoplasma gondii. J Parasitol. 2008;94(1):36–41. doi: 10.1645/GE-1312.1. [DOI] [PubMed] [Google Scholar]
- 36.Dubey JP, et al. High prevalence and abundant atypical genotypes of Toxoplasma gondii isolated from lambs destined for human consumption in the USA. Int J Parasitol. 2008;38(8-9):999–1006. doi: 10.1016/j.ijpara.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 37.Maksimov P, et al. Serotyping of Toxoplasma gondii in cats (Felis domesticus) reveals predominance of type II infections in Germany. PLoS ONE. 2013;8(11):e80213. doi: 10.1371/journal.pone.0080213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su C, et al. Recent expansion of Toxoplasma through enhanced oral transmission. Science. 2003;299(5605):414–416. doi: 10.1126/science.1078035. [DOI] [PubMed] [Google Scholar]
- 39.Boyle JP, et al. Just one cross appears capable of dramatically altering the population biology of a eukaryotic pathogen like Toxoplasma gondii. Proc Natl Acad Sci USA. 2006;103(27):10514–10519. doi: 10.1073/pnas.0510319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan A, et al. 2011 A monomorphic haplotype of chromosome Ia is associated with widespread success in clonal and nonclonal populations of Toxoplasma gondii. mBio 2(6):e00228-00211. [Google Scholar]
- 41.Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol. 2012;10(11):766–778. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature. 2002;415(6874):926–929. doi: 10.1038/415926a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.