

Background: The functional importance of apolipoprotein A-I (apoA-I) nitration at tyrosine 166 (Tyr166) in vivo is controversial.
Results: Nitrotyrosine 166-apoA-I accounts for 8% of apoA-I within human atheroma, is not HDL-associated, and is functionally impaired.
Conclusion: Buoyant density ultracentrifugation of HDL can lead to erroneous results, particularly with modified apoA-I forms.
Significance: Detection and quantification of nitrotyrosine 166-apoA-I may provide insights into a pathophysiological process within the artery wall.
Keywords: Aminoacyl-tRNA Synthetase, Apolipoproteins, Atherosclerosis, Peroxidase, Post-translational Modification, Dysfunctional HDL, Nitrotyrosine, Orthogonal Amino Acid, Protein Nitration, Unnatural Amino Acid (uAA)
Abstract
We reported previously that apolipoprotein A-I (apoA-I) is oxidatively modified in the artery wall at tyrosine 166 (Tyr166), serving as a preferred site for post-translational modification through nitration. Recent studies, however, question the extent and functional importance of apoA-I Tyr166 nitration based upon studies of HDL-like particles recovered from atherosclerotic lesions. We developed a monoclonal antibody (mAb 4G11.2) that recognizes, in both free and HDL-bound forms, apoA-I harboring a 3-nitrotyrosine at position 166 apoA-I (NO2-Tyr166-apoA-I) to investigate the presence, distribution, and function of this modified apoA-I form in atherosclerotic and normal artery wall. We also developed recombinant apoA-I with site-specific 3-nitrotyrosine incorporation only at position 166 using an evolved orthogonal nitro-Tyr-aminoacyl-tRNA synthetase/tRNACUA pair for functional studies. Studies with mAb 4G11.2 showed that NO2-Tyr166-apoA-I was easily detected in atherosclerotic human coronary arteries and accounted for ∼8% of total apoA-I within the artery wall but was nearly undetectable (>100-fold less) in normal coronary arteries. Buoyant density ultracentrifugation analyses showed that NO2-Tyr166-apoA-I existed as a lipid-poor lipoprotein with <3% recovered within the HDL-like fraction (d = 1.063–1.21). NO2-Tyr166-apoA-I in plasma showed a similar distribution. Recovery of NO2-Tyr166-apoA-I using immobilized mAb 4G11.2 showed an apoA-I form with 88.1 ± 8.5% reduction in lecithin-cholesterol acyltransferase activity, a finding corroborated using a recombinant apoA-I specifically designed to include the unnatural amino acid exclusively at position 166. Thus, site-specific nitration of apoA-I at Tyr166 is an abundant modification within the artery wall that results in selective functional impairments. Plasma levels of this modified apoA-I form may provide insights into a pathophysiological process within the diseased artery wall.
Introduction
Post-translational modification (PTM)4 of proteins through multiple distinct oxidative processes is a common occurrence at sites of inflammation and cardiovascular disease (1–11). Inflammation and oxidative stress are widely touted as playing a role in the pathogenesis of cardiovascular disease; however, studies aimed at understanding the functional consequences of modifications of proteins through these processes have proven difficult. One major reason for this is that exposure of a protein to an oxidative source resulting in PTM of the targeted amino acid typically also oxidizes many other susceptible amino acids, making conclusions of the functional impact of PTM of a specific residue difficult. To circumvent this problem, we and other investigators have used oxidative modification in combination with site-specific mutagenesis studies to provide data in support of the involvement of specific residues and types of PTMs with alterations in protein function (12, 13). Although instructive, this course of action is not ideal for a number of reasons, including the fact that mutagenesis of a targeted residue to a non-oxidizable residue may have unknown effects on other biological interactions or functions of the protein that are not immediately apparent. Additionally, the problems of oxidative modification on other residues within the protein occurring concurrently cannot be avoided and remain a limitation of this approach.
Recent advances in the use of evolved orthogonal amino acid synthetases and their paired tRNACUA has enabled the development of technology allowing for site-specific incorporation of non-physiologic amino acids into recombinant proteins (14–16). This capability has the potential to allow one to study the functional consequences of individual PTM to residues that have previously been implicated as playing a functional role based on their detection by proteomics studies in the material recovered from diseased tissue (16, 17).
We demonstrated previously that apoA-I recovered from human atherosclerotic lesions is heavily oxidized (7–9, 18), a finding that others have replicated (6, 19, 20). Proteomic identification of oxidatively modified sites within apoA-I isolated from atherosclerotic lesion by immunoprecipitation with apoA-I-specific antibodies identified a number of preferred residues for MPO- and NO-derived oxidant PTMs (8, 9). In addition to apoA-I tyrosine 192, for which mutagenesis studies by our group failed to demonstrate changes in biological activity, tyrosine 166 of apoA-I was reported as being of particular interest because this site was also shown to be within a region we identified as a “solar flare,” a solvent-exposed region on nascent HDL that we predicted interacts with lecithin-cholesterol acyltransferase (LCAT) (12). Based upon mutagenesis of this residue to phenylalanine and functional analyses, we predicted that nitration of the endogenous tyrosine at this site may impair LCAT activity (12).
Other investigators have studied oxidative PTMs on apoA-I present in atherosclerotic lesions not by immunoprecipitating total apoA-I but rather exclusively by “floating” HDL-like particles (d = 1.063–1.21 g/ml) using buoyant density ultracentrifugation (6, 7, 19, 21, 22). Proteomics analysis of buoyant density-isolated HDL-like fractions confirms tyrosine 192 as a preferred oxidation site on apoA-I through MPO-catalyzed chlorination in both human plasma- and lesion-derived apoA-I (21, 22). The major site of nitration in plasma-derived apoA-I in HDL was also reported to be tyrosine 192, whereas tyrosine 18 was found to be a major site reported from lesion “HDL-like” particle-localized apoA-I (22). In these latter studies where apoA-I was recovered from plasma or lesions by floating HDL, little nitration at tyrosine 166 was observed, and the involvement of this site in modification was suggested to have little biological relevance (22). Thus, there exists a discrepancy of the importance of modification within apoA-I with regard to the abundance and functional significance of tyrosine 166 oxidative modification.
The quantitative significance and functional consequences of apoA-I nitration at tyrosine 166 are of potential clinical and therapeutic importance. ApoA-I-targeted therapies such as direct delivery of the protein intravenously for promotion of atherosclerotic plaque regression is currently under investigation. A better understanding of the true sites of oxidative modification of apoA-I and their functional significance may thus be relevant to the generation of oxidation-resistant mutant forms of apoA-I with the potential to provide enhanced cardioprotective action compared with that observed with the native form (13). A better understanding would also be useful for development of potential diagnostic tools to monitor processes ongoing within the diseased artery wall or with which to potentially titrate response to therapies.
Herein we show, through use of a novel monoclonal antibody (mAb) that specifically recognizes apoA-I harboring a 3-nitrotyrosine at position 166 (NO2-Tyr166-apoA-I), that this modified form of apoA-I is abundant within the artery wall in a lipid-poor form rather than on an HDL particle. Furthermore, by generating recombinant apoA-I that selectively incorporates a lone 3-nitrotyrosine at 166 in apoA-I, we show that this PTM rendered apoA-I severely impaired in stimulating LCAT activity as a nascent HDL particle but had no effect on cholesterol efflux activity. Given the abundance of NO2-Tyr166-apoA-I observed within arterial tissues, detection of NO2-Tyr166-apoA-I within the circulation may thus serve as a means to monitor a pathophysiologically relevant process occurring in the artery wall during atherosclerosis. Moreover, the present studies showed that methods used to examine apoA-I within the artery wall are important for gauging the quantity and functional significance of modified apoA-I forms in vivo.
EXPERIMENTAL PROCEDURES
Materials
D2O was purchased from Cambridge Isotopes, Inc. (Andover, MA). Chelex-100 resin, fatty acid-free bovine serum albumin (BSA), and crystalline catalase (from bovine liver; thymol-free) were purchased from Roche Applied Science. Human MPO was isolated and characterized as described (1). [3H]Cholesterol was obtained from Amersham Biosciences. The immunoglobulin class of the mouse mAb kit was obtained from Invitrogen. Trypsin was from Promega (Madison, WI), and the RAW264.7 cell line was from the American Type Culture Collection (Manassas, VA). Sodium phosphate, H2O2, and NaOCl were purchased from Fisher. Peroxynitrite (ONOO−) was purchased from Cayman Chemicals (Ann Arbor, MI). All other materials were purchased from Sigma except where indicated.
General Methods
Human apoA-I was purified from circulating HDL obtained from healthy volunteer donors using established methods (12, 23). All donors gave written informed consent, and the Institutional Review Board of the Cleveland Clinic approved all study protocols. Mouse studies involving monoclonal antibody generation were performed under protocols approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic. Lipoproteins, including HDL and HDL-like particles (d = 1.063–1.21) from plasma and tissue homogenates, respectively, were isolated by sequential buoyant density ultracentrifugation at low salt concentrations using D2O/sucrose (24). Protein concentrations were determined by the Markwell modified protein assay (25) with bovine serum albumin as the standard. Cholesterol efflux and LCAT activity assays were performed as described (23, 26, 27). Reconstituted HDL (rHDL) was prepared from isolated apoA-I by the cholate dialysis method (28) using a molar ratio of apoA-I:1-palmitoyl-2-oleoylphosphatidylcholine:cholesterol of 1:100:10. HDL particles were further purified by gel filtration chromatography using a Sephacryl S300 column (GE Healthcare) on a Bio-Rad Biologics DuoFlo FPLC. Reconstituted nascent HDL particle preparations were further characterized by native polyacrylamide gel electrophoresis (PAGE), and lipid composition analyses were determined (23, 12). MPO (donor, hydrogen peroxide; myeloperoxidase, EC 1.11.2.2) was isolated (final A430/A280 ratio of 0.6) as described (29). Enzyme concentration was determined spectrophotometrically (ϵ430 = 170 mm−1 cm−1) (30). H2O2 and −OCl concentrations were each determined spectrophotometrically (ϵ240 = 39.4 m−1 cm−1) (31) and ϵ292 = 350 m−1 cm−1 (32), respectively) immediately prior to use. ONOO− was quantified spectrophotometrically prior to use (ϵ302 = 1.36 mm−1 cm−1) (33). All buffers used were passed through a Chelex-100 column and supplemented with 100 μm diethylenetriaminepentaacetic acid (DTPA) to remove any trace levels of redox-active transition metals. All glassware used was rinsed with 100 μm DTPA, pH 7.4 and then Chelex-100-treated distilled deionized H2O and then baked at 500 °C prior to use. Sodium dodecyl sulfate (SDS)-PAGE was performed as described by Laemmli (34).
Bacterial Cells
Escherichia coli TOP10 cells (Invitrogen) were used for cloning purposes and for expression of His8-C3-apoA-I (referred to as wild type (WT) apoA-I) as described below. NO2-Tyr166-apoA-I protein was expressed in an E. coli TOP10 strain harboring the evolved orthogonal Methanococcus jannaschii (Mj) nitro-Tyr-tRNA synthetase/tRNACUA pair that specifically incorporates 3-nitrotyrosine into amber codons (16).
Plasmids
Plasmid (pUC57-apoA-I) was created by blunt end cloning of an E. coli codon-optimized human apoA-I DNA sequence encoding the mature apoA-I polypeptide, amino acids 25–267 (amino acids preceded by an in-frame amino-terminal His8-C3 protease site tag; gene synthesized by Genescript, Piscataway, NJ) into the EcoRV site of pUC57. The His8-C3-apoA-I DNA fragment was excised with digestion by NcoI and KpnI restriction enzymes, and the gene fragment was cloned into arabinose-inducible pBAD/myc/His6 backbone (Invitrogen) at those same restriction sites, eliminating the vector-encoded His6 tag to create pBAD apoA-I-3C using T4 DNA ligase. The Y166F mutant in apoA-I tyrosine at position 166 was constructed by site-directed mutagenesis changing the TAT (Tyr) to TTT (Phe) using a QuikChangeTM II mutagenesis kit (Stratagene, La Jolla, CA) to create pBAD apoA-I 166F. The His8-C3-apoA-I allele expressing apoA-I with a nitrotyrosine to be incorporated at position 166 was constructed and denoted as pBAD apoA-I-3C-166TAG. Briefly, the codon for the tyrosine 166 residue in the human apoA-I sequence was replaced by an amber stop codon (TAG) by site-directed mutagenesis. Site-directed mutagenesis was carried out using QuikChangeTM II (Stratagene). After mutagenesis, clones were isolated and sequenced to confirm the altered sequence. The amber codon is suppressed by the orthogonal nitrotyrosine Mj-tRNACUA/Mj-tRNA synthetase engineered to incorporate 3-nitrotyrosine only at amber codons (pDULE 5B) (16). The modified protein was expressed and purified as described below for recombinant human apoA-I.
Protein Expression
Plasmids containing the apoA-I wild type and apoA-I Tyr166 TAG stop codon were transformed into E. coli TOP10 cells and TOP10 cells harboring the evolved orthogonal Mj-nitro-Tyr-tRNA synthetase/tRNACUA pair, respectively, and grown under antibiotic selection (100 mg/ml ampicillin and 15 μg/ml tetracycline). Protein expression was carried out using a modified M9 medium. M9 salts were supplemented with 0.375% glycerol, 0.05% glucose, 1 mm MgSO4, 0.1 mm CaCl2, 0.03% casamino acids, 1× basal medium Eagle vitamin solution (Sigma), and 0.05% arabinose. Additionally, with NO2-Tyr166-apoA-I expressed proteins, 1 mm free 3-nitrotyrosine was added. Overnight cultures of E. coli were grown in 50 ml of LB containing the appropriate antibiotics with shaking at 37 °C until the A600 reached 0.8. These cultures were then spun down and resuspended in 10 ml of the M9 supplemented medium described above. The suspension (2 ml) was then added to 500 ml of the M9 supplemented medium and grown at 37 °C with shaking to an A600 of 0.8–1. At this point, cells were left shaking overnight at room temperature. Cells were then spun down, and the pellets were frozen at −80 °C until required for purification as described (23).
After purification of proteins on nickel affinity matrices, proteins were extensively dialyzed in the buffer of choice prior to use. Incorporation of 3-nitrotyrosine into the recombinant apoA-I was detected by scanning spectrophotometry with maximal absorbance at 428 nm. The extent of 3-nitrotyrosine incorporation at the desired position (166) of apoA-I was also confirmed by proteomics analysis quantifying both wild-type and 3-nitrotyrosine-containing tryptic peptides (see below).
Tissue Collection
Surgical specimens of freshly isolated human aortic tissue were obtained as discarded material both at time of organ harvest from transplant donors and during valve/aortic arch (“elephant trunk”) replacement surgery. Aortic tissue was immediately rinsed in ice-cold normal saline until free of visible blood, submerged in argon-sparged phosphate buffer (65 mm sodium phosphate, pH 7.4) supplemented with 100 μm DTPA and 100 μm butylated hydroxytoluene, and stored at −80 °C in threaded cap specimen containers in which headspace was purged with argon prior to freezing. Butylated hydroxytoluene was omitted from buffer in specimens where apoA-I was isolated for functional activity assays.
mAb Generation, Specificity, and Labeling
mAb 10G1.5 (anti-total apoA-I) was used for isolation and quantification of total apoA-I. This mAb was recently developed and extensively characterized in our laboratory and demonstrates equal recognition of lipid-free and lipidated (in rHDL) apoA-I under native conditions as well as following oxidation by exposure to multiple different oxidation systems, including MPO/H2O2/Cl−, MPO/H2O2/NO2−, and CuSO4 (oxidized as outlined below for mAb 4G11.2). For quantitative Western blot analyses, mAb 10G1.5 was IRDye-labeled (Li-COR Biosciences, Lincoln, NE) using a dye:protein ratio of 4:1, and proteins were visualized by infrared imaging. Coupling of dye to the antibody was performed according to the manufacturer's instructions.
A monoclonal antibody specific for 3-nitrotyrosine within apoA-I specifically at residue 166 (mAb 4G11.2) was prepared and characterized as follows. Hybridoma cell lines were generated by immunizing apoA-I−/− mice with purified delipidated human apoA-I isolated from HDL recovered from healthy donors that had been nitrated in vitro in chloride-free phosphate buffer using the complete MPO/H2O2/NO2− system at an apoA-I:H2O2 mol:mol ratio of 1:5 (NO2-A1) using 19 nm MPO, 100 μm DTPA, 40 μm H2O2, and 1 mm NaNO2 as described (12). Several positive clones were further subcloned and screened until an mAb with the desired binding specificity for nitrated apoA-I forms with appropriate site-specific recognition for an epitope containing NO2-Tyr at apoA-I position 166 (see below) was identified. The subclone, mAb 4G11.2, was selected from among these by further screening for equal recognition of lipid-free and lipidated (in rHDL) nitrated apoA-I under native conditions as well as following oxidation by only nitrative oxidation systems but not exposure to multiple other different systems, including MPO/H2O2/Cl− and CuSO4 (oxidized as outlined below).
The specificity of mAb 4G11.2 was tested using apoA-I or rHDL either in native form or following incubation at 37 °C in 60 mm sodium phosphate buffer, pH 7.4 with multiple different oxidation systems. The different MPO systems consisted of 19 nm MPO, 100 μm DTPA, 40 μm H2O2, and either 100 mm NaCl, 1 mm KBr, or 1 mm NaNO2 as indicated. Horseradish peroxidase (HRP; 19 nm) was used with 40 μm H2O2. ApoA-I and rHDL were exposed to MPO or HRP for 90 min at 37 °C, and the reactions were stop by addition of 2 mm methionine and 300 nm catalase. All other oxidation reactions were carried out for 24 h at 37 °C. Final concentrations of oxidants used were: H2O2, 40 μm; ONOO−, 40 μm; ONOO−/HCO3− (40 μm each); CuSO4, 10 μm; CuSO4/H2O2, 10 and 40 μm, respectively; FeCl3, 10 μm; and FeCl3/H2O2, 10 and 40 μm, respectively. ApoA-I or rHDL (prepared from apoA-I or various oxidized versions of apoA-I) were coated at 0.5 μg/ml into enzyme immunoassay plates and probed with 10 ng/ml anti-nitrotyrosine apoA-I monoclonal antibody 4G11.2 at room temperature for 1 h.
Epitope mapping of the recognition region for mAb 4G11.2 was performed by coating enzyme immunoassay plates with peptides (10 μm) of human apoA-I that contained a tyrosine centrally located within the peptide or identical versions of the peptides with 3-nitrotyrosine incorporated as follows: Tyr18 (DLATVYVDVLK), NO2-Tyr18 (DLATV[NO2-Y] VDVLK), Tyr166 (THLAPYSDELR), NO2-Tyr166 (THLAP[NO2-Y]SDELR), Tyr192 (LAEYHAK), and NO2-Tyr192 (LAE[NO2-Y]HAK). Peptides were synthesized by Ohio Peptide (Powell, OH). For quantitative Western blot analyses, purified mAb 4G11.2 was IRDye-labeled (Li-COR Biosciences) by using a LI-COR IRDye 680RD high molecular kit at a dye:protein ratio of 4:1 and visualized by infrared imaging. Coupling was performed according to the manufacturer's instructions.
To produce sufficient levels of 4G11.2 for immunoaffinity purification of apoA-I from arterial tissues, hybridoma clones were injected into pristane-treated male BALB/c mice for ascites tumor production. Ascites fluid was collected, and proteins were precipitated with ammonium sulfate and then chromatographed on a protein A/G column (Pierce) to purify mouse monoclonal antibodies. Isotypes of the mAbs were determined with the mouse mAb isotyping kit (catalogue number 26179, Pierce Rapid Antibody Isotyping Strips plus κ and λ-Mouse, Thermo Scientific Pierce).
Human ApoA-I Quantification
Human apoA-I was quantified by an Food and Drug Administration-approved apoA-I immunoassay using the Abbott ARCHITECT ci8200 Integrated Analyzer System (Abbott Laboratories, Abbott Park, IL). All other apoA-I was quantified by quantitative immunoblot analysis using mAb 10G1.5 as the detecting antibody as determined against a standard curve of known amounts of purified apoA-I standard. Immunoreactive bands were quantified using Image Studio software (version 2; LI-COR Biosciences) or NIH ImageJ (version 1.46).
Aortic Tissue Homogenization
Atherosclerotic lesions from human aortic tissues were obtained from subjects (n > 20) with an average age of 81 ± 6 years. Normal human aortic tissues were obtained at the time of organ harvest from transplant donors (n = 5) with an average age of 27 ± 4 years. Frozen tissue pieces (submerged in argon-sparged 65 mm sodium phosphate buffer, pH 7.4 with headspace purged with argon within screw cap containers) were thawed in an ice/water bath. Immediately after thawing, ice-cold Ca2+- and Mg2+-free Chelex-100-treated PBS that had been supplemented with 100 μm DTPA, pH 7.4 was used to rinse the tissue at least five times to remove residual blood from the tissue. Residual fat was also removed from the aorta segments. The aorta segments were immediately placed into Petri dishes containing ice-cold PBS that had been supplemented with 100 μm DTPA, pH 7.4. The adventitial layer was dissected away, the aorta was liberally rinsed with ice-cold PBS supplemented with 100 μm DTPA at least three additional times, and then the wet weight of the aorta was determined. Aortic tissue was then cut into small pieces in preparation for mincing. Prior to mincing the tissue, a protease inhibitor mixture (catalogue number P8340, Sigma-Aldrich) was added. This mixture was also included in all subsequent solutions used for homogenization and lipoprotein isolation.
Homogenization of all aortic tissues was performed in an ice/water slush bath with a motorized homogenizer using 30-s interval bursts five times, incorporating a 2-min rest between homogenization cycles. Crude homogenates were centrifuged at low speed at 15,000 × g for 30 min at 0 °C, and the insoluble pellet was discarded. This low speed supernatant (designated as lesion homogenate) was then used for sequential buoyant density isolation of LDL/VLDL (d < 1.063), HDL (d = 1.063–1.21), and lipoprotein-depleted (LPD) fractions (d > 1.21). Ultracentrifugation fractions were recovered and then dialyzed at 0–4 °C four times against 4 liters of 5 mm ammonium bicarbonate with addition of antioxidants 50 μm DTPA, pH 7.4 and 25 μm butylated hydroxytoluene changed every 4 h. Finally, buffer was changed against 4 liters of ice-cold Chelex-100-treated 1× PBS, pH 7.4. Butylated hydroxytoluene was omitted from the apoA-I preparations isolated for use in LCAT or cholesterol efflux activity measurements. To further prevent artificial oxidation, all dialyses were performed in large glass air-tight jars with argon-sparged buffer and an argon overlay to purge headspace gases.
Immunoaffinity Isolation of ApoA-I from Aortic Tissue Homogenate
Anti-NO2-Tyr166-apoA-I immunoaffinity resin was prepared by covalently coupling mAb 4G11.2 to AminoLink Plus (Pierce) resin at an antibody density of 1.5 mg of mAb/ml of resin in an amine-free buffer (PBS, pH 7.4) according to the manufacturer's instructions. Reactive non-antibody-bound sites on the resin were blocked with addition of excess ethanolamine. The affinity gel was then drained, and the antibody concentration in the flow-through fractions was determined. The resulting cross-linking efficiency was calculated to be greater than 90%. The gel was then extensively rinsed with 1 m Tris, pH 7.4, 1 m NaCl and then equilibrated in 1× PBS, pH 7.4 prior to use or storage (0.002% sodium azide was added for storage). Individual one-time use affinity columns (1 ml; drained resin) were prepared with immobilized 4G11.2, and the artery wall apoA-I was purified from individual samples of aortic homogenates using conditions that quantitatively recovered apoA-I as confirmed using Western blot analyses of column fractions. Total apoA-I was similarly immunoprecipitated using immobilized mAb 10G1.5 similarly coupled to AminoLink Plus (Pierce) resin.
Immunochemistry Staining of Atherosclerotic Coronary Arteries
Atherosclerotic coronary artery segments (fresh frozen tissue blocks) were obtained from the explanted hearts of patients undergoing cardiac transplantation at the Cleveland Clinic. Fresh frozen tissue blocks from normal coronary arteries were obtained from transplantation donor hearts that had been removed but were subsequently not used. Immunohistochemistry on atherosclerotic and normal coronary arteries was performed using anti-human NO2-Tyr166-apoA-I antibody 4G11.2 or polyclonal rabbit anti-human MPO (catalogue number A0398, Dako North America, Inc., Carpinteria, CA), each at a dilution of 1:400. Mouse normal IgG1 (catalogue number X0931, Dako North America, Inc.) was used as an isotype control for the antibody staining. The staining procedure followed the protocol of the DakoCytomation Envision+ system HRP (3-amino-9-ethylcarbazole) (Dako North America, Inc.).
Modification of ApoA-I by MPO-generated Reactive Nitrogen Species
The MPO-mediated modification reactions were carried out in a 50 mm sodium phosphate buffer, pH 7.0 containing 100 μm DTPA, 1 mg/ml protein (apoA-I), 57 nm purified human MPO (donor, hydrogen peroxide; myeloperoxidase, EC 1.11.2.2; A430/A280 ratio of 0.79), and 100 μm NaNO2. The MPO reactions were initiated by adding hydrogen peroxide at varying concentrations as indicated in Fig. 1 (typically 0–200 μm as needed) and carried out at 37 °C for 1 h. These reaction conditions included physiologically relevant amounts of MPO, chloride, and nitrite and hydrogen peroxide concentrations that ranged from physiologic to pathophysiological.
FIGURE 1.

A monoclonal antibody that recognizes apoA-I nitrated at tyrosine 166. A, specificity and epitope mapping of mAb 4G11.2 to nitrated apoA-I was performed using ELISAs as described under “Experimental Procedures.” The indicated protein (apoA-I or oxidized apoA-I forms) or indicated peptides and nitrated peptides were coated onto enzyme immunoassay plates at 10 μm in triplicate and incubated with mAb4G11.2, and ELISAs were performed. B, 4G11.2 fails to recognize any other oxidized or nitrated residues within apoA-I other than nitrated apoA-I containing an oxidizable tyrosine at position 166. Wild-type or Y166F-mutated apoA-I were oxidized in vitro with increasing molar ratios of oxidant as indicated and then coated onto enzyme immunoassay plates, and ELISAs were performed as described in A. Values represent the average of three reactions; error bars indicate S.D.
Mass Spectrometry Analyses
To confirm that the recombinant apoA-I was nitrated only at position 166 and at complete saturation, after SDS-PAGE, the major gel bands at molecular mass 25 kDa were excised for wild-type apoA-I and for NO2-Tyr166-apoA-I. Samples were first treated with dithiothreitol/iodoacetamide (Sigma), and then proteins were digested using mass spectrometry grade trypsin (Promega) at 37 °C overnight. Tryptic peptides were loaded onto an IntegraFrit sample trap (ProteoPep C18, 300 Å, 150 μm × 2.5 cm; New Objective, Woburn MA) at 1 μl/min with 5% acetonitrile and 0.1% formic acid to desalt the samples. The peptides were subsequently eluted through a column (75 μm × 15 cm) packed in-house with XperTek 218TP (C18, 300-Å pore size, 150-μm particle size; Cobert Associates, St. Louis, MO) at 200 nl/min using a Proxeon Easy-nLC II system (Thermo Scientific, Waltham, MA) with a gradient of 5–65% acetonitrile, 0.1% formic acid over 120 min into an OrbitrapVelos mass spectrometer (Thermo Scientific).
Peak lists were generated using Proteome Discoverer 1.1 (Thermo Scientific, Waltham, MA). The resulting unified search files (*.srf) were searched against the UniProt FASTA database of all apolipoproteins and against a human protein database downloaded from the European Bioinformatics Institute (release 2013_02). The amino acid modifications used for searches included carbamidomethylated cysteine (fixed), oxidized methionine and tryptophan (variable), and 3-chloro- and 3-nitrotyrosine (variable). Only strictly tryptic peptides with a maximum of two missed cleavage sites were allowed in the database searches. Monoisotopic precursor ions were searched with a tolerance of 100 ppm with 0.8 Da for the fragment ions on the data obtained from the hybrid LTQ-Orbitrap mass spectrometer. Unidentified fragment ions in all fragmentation spectra were manually validated using Protein Prospector (University of California, San Francisco). Incorporation of 3-nitrotyrosine only at position 166 with complete saturation was confirmed.
Reconstituted HDL composition was analyzed as described previously using microphosphate for phospholipid quantification and stable isotope dilution liquid chromatography with on-line liquid chromatography (LC) electrospray ionization tandem mass spectrometry (MS/MS) analysis to quantify both amino acid (Phe, Val, and Leu) and cholesterol content on an API 5000 triple quadrupole mass spectrometer (AB SCIEX) (12). Protein content was calculated assuming a protein sequence of human apoA-I and demonstrated comparable results using any of the three amino acids monitored. As a control, the content of cholate in reconstituted HDL particle preparations was also quantified by LC/electrospray ionization/MS/MS on the API 5000 instrument and confirmed to be <0.1 mol % (12).
Statistical Analysis
The statistical significance of differences was determined by Student's t test or analysis of variance where more than two comparisons were made. p values for statistical significance are reported for p < 0.05.
RESULTS
Monoclonal Antibody 4G11.2 Recognizes ApoA-I Selectively Nitrated at Position 166
We first reported that apoA-I was a preferential target of protein nitration within atherosclerotic lesions, and in our later studies, NO2-Tyr166-apoA-I was identified as a preferred site of nitration using proteomics studies (7, 8). We initially sought to develop an mAb that specifically recognized this site-specific nitrated apoA-I to better study its abundance and particle distribution. We further wished to use this mAb as a tool to immunoaffinity purify and probe the function of in vivo apoA-I harboring this modification. As antigen, purified delipidated human apoA-I (isolated from plasma HDL) was nitrated in vitro using the complete MPO/H2O2/NO2− system at a 5:1 molar ratio of oxidant to apoA-I (protein nitration conditions) and injected into several apoA-I−/− mice. After a robust antibody response was generated, splenocytes from the animals were used to generate hybridomas. After screening several thousand hybridoma clones for their ability to recognize apoA-I exposed to the MPO/H2O2/NO2− system but not other oxidized (non-nitrative) forms of apoA-I or native apoA-I, several clones were identified.
At this stage, we next turned our attention to characterizing these hybridoma clones to determine whether any expressed an antibody that selectively recognized apoA-I nitrated specifically at tyrosine 166. One mAb, named 4G11.2, was selected based upon its specific activity of recognition by ELISA, immunoblot analysis, and finally, its ability to immunoprecipitate NO2-Tyr166-apoA-I. Fig. 1, A and B, collectively demonstrate the recognition selectivity of mAb 4G11.2 to detect only nitrated apoA-I at position 166. In Fig. 1A, mAb 4G11.2 only recognizes apoA-I exposed to the MPO/H2O2/NO2− system (NO2-A1) but not unoxidized plasma-purified human apoA-I. Our prior in vitro and in vivo proteomics studies with apoA-I revealed that of the seven potential tyrosine residues only three are nitrated to any significant degree, Tyr18, Tyr166, and Tyr192 (8). Selectivity for mAb 4G11.2 to recognize nitration at tyrosine 166 of apoA-I was then demonstrated by examining mAb binding to synthetic peptides spanning Tyr18, Tyr166, or Tyr192 in either native form or with site-specific incorporation of a nitrotyrosine at the indicated residue (see “Experimental Procedures” for peptide sequences). Of note, mAb 4G11.2 only recognized the NO2-Tyr166-containing peptide but not the non-nitrated version of the Tyr166-containing peptide or other NO2-Tyr-containing peptides (e.g. NO2-Tyr18 and NO2-Tyr192). Selectivity of mAb 4G11.2 was further demonstrated by its lack of recognition of other possibly nitrated tyrosine residues within apoA-I using the apoA-I Tyr166Phe mutant in comparison with wild-type apoA-I after oxidation with increasing levels of oxidant with the MPO/H2O2/NO2− system (Fig. 1B). Although wild-type apoA-I was readily detected by 4G11.2 upon exposure to increasing levels of oxidant, apoA-I Tyr166Phe mutant where only tyrosine 166 is mutated to the relatively non-oxidizable (and non-nitratable) phenylalanine was not (Fig. 1B).
The specificity of mAb 4G11.2 for recognition of either apoA-I or rHDL only following exposure to nitrating oxidant systems and not in native form or following exposure to alternative oxidant systems is shown in supplemental Fig. 1. ApoA-I and rHDL were only recognized following exposure to the MPO/H2O2/NO2− system, ONOO−, or ONOO−/HCO3− but not following exposure to multiple alternative oxidation systems (as described under “Experimental Procedures”). Further studies as outlined below showed that mAb 4G11.2 only recognized protein bands within human plasma and tissue homogenates on Western analyses at the molecular weight of the apoA-I monomer and higher molecular weight forms consistent with dimer, trimer, and higher cross-linked apoA-I forms. Proteomics analyses of immunoaffinity-isolated recovered protein from these tissue matrices revealed apoA-I in each band. Collectively, these results show that mAb 4G11.2 is highly specific for recognition of NO2-Tyr166-apoA-I. Moreover, importantly, 4G11.2 did not recognize either native apoA-I or apoA-I following exposure to multiple alternative oxidative pathways.
NO2-Tyr166-apoA-I Is Abundant within Atherosclerotic Lesions but Present as a Lipid-poor Form Rather than within HDL-like Particles
As noted above, there is some controversy over whether NO2-Tyr166-apoA-I is present within human atherosclerotic lesions and if so to what level. Therefore, we initially sought to use mAb 4G11.2 to determine whether NO2-Tyr166-apoA-I was present within human lesions. Should this be the case, this finding would be an important preface to determining abundance, particle distribution, and function (if present in high enough levels to be recoverable for biological activity assays) within the atherosclerotic artery wall and the normal artery wall. Thus, immunohistochemical staining of human coronary arteries was performed. Both atherosclerotic lesions and normal human coronary artery tissue were examined. NO2-Tyr166-apoA-I was readily observed throughout the atherosclerotic plaque, but essentially no staining was evident in normal human coronary artery tissue (Fig. 2, left). No staining was observed with isotype control antibody, corroborating the specificity of 4G11.2 staining (Fig. 2, right). Staining of parallel sections with rabbit anti-human MPO polyclonal antibodies also revealed an intense staining pattern in human atherosclerotic plaque. Only mild MPO staining was observed in normal human coronary arteries that was localized to the endothelial surface and subendothelial space (Fig. 2, middle).
FIGURE 2.
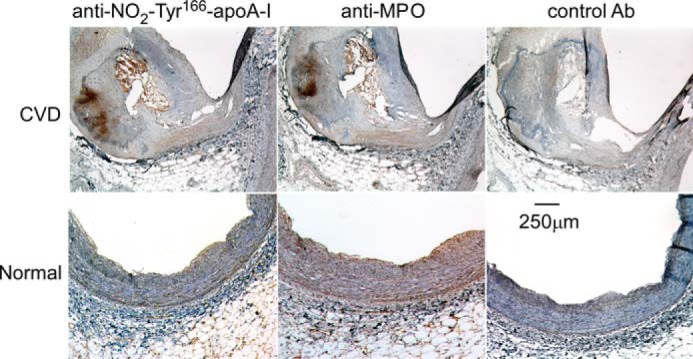
ApoA-I nitrated on tyrosine 166 is located in atherosclerosis-laden human coronary artery. Fresh frozen sections of human coronary aortic tissue segments with (top row; cardiovascular disease (CVD)) and without (bottom row; Normal) atherosclerotic plaque were stained using either mAb 4G11.2 (anti-NO2-Tyr166-apoA-I), anti-MPO-specific antibody, or isotype control IgG (Ab) as described under “Experimental Procedures.” Sections were counterstained with hematoxylin (blue), and MPO-positive staining is red. Magnification, 5×; scale bar, 250 μm.
We next sought to examine both the particle distribution and amount of NO2-Tyr166-apoA-I in human atherosclerotic lesions. Human aortic plaque material recovered fresh at time of surgery was obtained as outlined under “Experimental Procedures.” Tissues were rinsed free of blood with PBS and placed in buffer containing antioxidant mixture, stored in gas-tight containers after sparging with argon to remove air, sealed, and stored at −80 °C until time of isolation (see “Experimental Procedures”). Tissues from multiple subjects were homogenized in antioxidant buffer and centrifuged at low speed to remove gross particulate material. This supernatant (called “homogenate” henceforward) was then subjected to sequential buoyant density gradient ultracentrifugation using the D2O/sucrose method (24) to avoid high salt concentrations present with typical KBr-dependent lipoprotein fractionation methods. Initially, VLDL/LDL-like fractions (d < 1.063) were removed. Then, the HDL-like fraction (d = 1.063–1.21) and LPD fraction (d > 1.21) were recovered as described under “Experimental Procedures.”
Homogenate, HDL-like particle, and LPD fractions were separated by gradient (5–15%) reducing SDS-PAGE, stained for protein with SYPRO Ruby Red, and imaged. Visual inspection of the gel (Fig. 3A) reveals a complex protein mixture with minimal variation in sample to sample protein banding patterns. A notable protein band migrating at ∼27 kDa is also observed, consistent with the molecular mass of apoA-I (Fig. 3A). Western blot analysis of a membrane containing transferred proteins from a parallel-run duplicate gel was probed with anti-NO2-Tyr166-apoA-I antibody (mAb 4G11.2). Of note, NO2-Tyr166-apoA-I within the homogenate and LPD fractions migrating at ∼27 kDa, the size of an apoA-I monomer, along with much lighter staining bands migrating at just over 50 and 75 kDa, consistent with dimeric and trimeric apoA-I forms, were observed (Fig. 3B, molecular masses of apoA-I monomer, dimer, and trimer are marked by arrowheads). Remarkably, the HDL-like particle fraction had very light to no detectable NO2-Tyr166-apoA-I present. In fact, the blot had to be highly overexposed to observe the presence of this modified form of apoA-I in the HDL-like fraction (Fig. 3B, bottom panel).
FIGURE 3.

ApoA-I nitrated on tyrosine 166 present in human atherosclerotic lesions is not located on HDL-like particles. Proteins in atherosclerotic lesion homogenate or after sequential D2O/sucrose buoyant density ultracentrifugation fractionation into HDL-like particle and lipoprotein-depleted fractions from the indicated density ranges were separated by gradient (5–15%) reducing SDS-PAGE. A, SYPRO Ruby-stained gel of the indicated protein samples (5 μg) from homogenate and the indicated density ranges obtained from different atherosclerotic lesion tissue samples (n = 5). B, top panel, Western blot membrane of a duplicate-run gel as in A probed with anti-NO2-Tyr166-apoA-I mAb 4G11.2. B, bottom panel, overexposure of Western blot to show NO2-Tyr166-apoA-I in the HDL-like fraction. Monomers, dimers, and trimers (multimers) of apoA-I-immunoreactive NO2-Tyr166-apoA-I bands are indicated by arrowheads. Molecular mass markers are indicated.
Quantification of NO2-Tyr166-apoA-I within human atherosclerotic lesions showed that a large preponderance of this modified form of apoA-I within lesion tissues is in a lipid-poor form and not in an HDL-like particle with the vast majority recovered within the LPD fraction (Fig. 4A). Remarkably, NO2-Tyr166-apoA-I accounted for ∼8% of the total apoA-I recovered in human atherosclerotic plaque with this almost exclusively being found in the LPD fraction (Fig. 4B). Further quantitative analysis of NO2-Tyr166-apoA-I/total apoA-I found within each fraction is shown in Fig. 4C.
FIGURE 4.
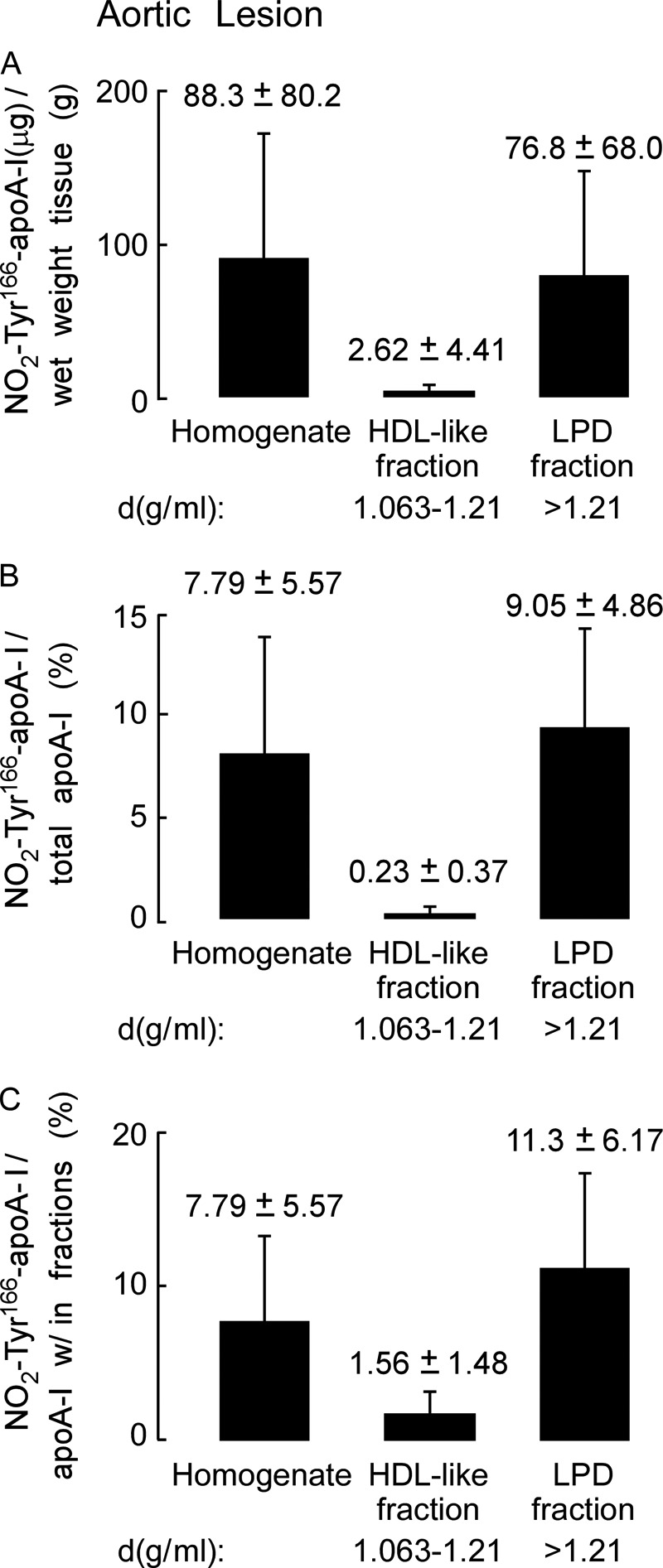
Quantitation of nitrated NO2-Tyr166-apoA-I present in human atherosclerotic lesions. A, recovery of apoA-I (μg) per gram of lesion tissue (wet weight) from the atherosclerotic lesion tissue homogenate and in the HDL-like and LPD fractions was calculated from quantitative Western blot analysis of apoA-I-immunoreactive bands, and total apoA-I content as described under “Experimental Procedures” is qualitatively presented in B. B, percentage of NO2-Tyr166-apoA-I to total apoA-I from atherosclerotic lesion present in the starting homogenate and HDL-like and LPD fractions was calculated as in A. C, percentage of NO2-Tyr166-apoA-I to total apoA-I within each fraction was determined by quantitative Western blot analysis of apoA-I-immunoreactive bands. Values were determined from n = 5 samples; error bars represent ±S.D.
Only Trace Levels of NO2-Tyr166-apoA-I Are Observed in Plasma and as a Lipid-poor Form Not within an HDL Particle
We next examined the distribution and level of NO2-Tyr166-apoA-I within plasma from normal healthy consenting donors (n = 5). Plasma was fractionated by sequential buoyant density ultracentrifugation. The starting plasma, HDL-rich fraction, and lipoprotein-depleted fraction were then separated by gradient (5–15%) reducing SDS-PAGE and either stained with Coomassie Blue (Fig. 5A) or transferred to membranes and probed with either anti-total apoA-I (mAb 10G1.5) (Fig. 5B) or mAb 4G11.2 to visualize NO2-Tyr166-apoA-I (Fig. 5C). As expected, a major band staining at ∼27 kDa in the HDL fraction of the Coomassie Blue-stained gel was noted (Fig. 5A) that corresponds with immunoreactive bands observed after probing with anti-total apoA-I (mAb 10G1.5) (Fig. 5B). For the Western analyses using mAb 10G1.5, which recognizes total apoA-I, unequal (less) protein loading of the HDL fraction was necessary compared with total protein used in the plasma and LPD fractions to avoid grossly overloading apoA-I signal intensities in the HDL fraction (Fig. 5B). Remarkably, as was observed in the atherosclerotic lesion, NO2-Tyr166-apoA-I was detected (with mAb 4G11.2) only in the starting material (plasma) and in the lipid-poor LPD fraction (Fig. 5C) but not within the HDL fraction, despite readily apparent apoA-I within the HDL fraction as visualized with the anti-total apoA-I mAb 10G1.5 (Fig. 5B). As can be seen, mAb 4G11.2 failed to recognize recombinant unmodified apoA-I (Fig. 5C, control sample (C)), whereas mAb 10G1.5 (anti-total apoA-I) easily recognized it (Fig. 5B). Quantification of NO2-Tyr166-apoA-I in plasma within both the HDL and LPD fractions is shown in Fig. 6. Of note, NO2-Tyr166-apoA-I in plasma was only present at 0.14% of total apoA-I. Moreover, even following long overexposure of the blot, immunoreactive bands were not detected in the HDL-rich fraction. Similarly, FPLC fractionation of plasma and subsequent Western blot analyses confirmed no immunodetectable monomeric NO2-Tyr166-apoA-I within fractions where HDL particles elute and detection of the modified protein with lower molecular weight lipid-poor forms of apoA-I (data not shown). Thus, we were not able to detect any NO2-Tyr166-apoA-I in the HDL fraction with all of the NO2-Tyr166-apoA-I partitioned into the LPD fraction (d > 1.21) as a lipid-poor form.
FIGURE 5.

ApoA-I nitrated on tyrosine 166 present in plasma is not on HDL particles, is lipid-poor, and is only found in the lipoprotein-depleted fraction. Plasma from normal healthy human volunteers (n = 5) was fractionated by sequential D2O/sucrose buoyant density gradient centrifugation. A, Coomassie Blue-stained gel after gradient (5–15%) SDS-PAGE of the plasma, HDL, and LPD fraction proteins (10 μg of protein/lane). B, Western blot analysis of proteins on a membrane transferred from a duplicate fractionated protein gel in A with 10 μg of protein loaded per lane from the plasma and LPD fractions and 0.8 μg in the HDL fractions (not to overload the immunoreactive signal) probed with anti-total apoA-I mAb 10G1.5 shows that apoA-I is predominately in the HDL fraction. Recombinant apoA-I (0.8 μg) served as a positive control (C). C, Western blot analysis of the same amounts of proteins on membrane from a duplicate fractionated protein gel as in A probed with anti-NO2-Tyr166-apoA-I mAb 4G11.2 shows that apoA-I is not present in the HDL fraction but is present only in the lipid-poor LPD fraction (d > 1.21 mg/ml) and that mAb 4G11.2 does not recognize non-nitrated apoA-I (C; 0.8 μg). Values were determined from (n = 5 samples. Overexposure of C reveals that there is no NO2-Tyr166-apoA in plasma HDL fractions (not shown).
FIGURE 6.

Quantitation of apoA-I nitrated on tyrosine 166 present in plasma. A, recovery of NO2-Tyr166-apoA-I (μg) per ml of plasma present in the starting plasma and in the HDL-like and LPD fractions after sequential D2O/sucrose buoyant density gradient ultracentrifugation was determined from quantitative Western blot analysis of apoA-I-immunoreactive bands and total apoA-I content as described under “Experimental Procedures” and is qualitatively presented in B and C. B, percentage of NO2-Tyr166-apoA-I to total apoA-I from plasma present in the starting material and HDL-like and LPD fractions was calculated from quantitative Western blot analysis of apoA-I-immunoreactive bands as in A. C, percentage of NO2-Tyr166-apoA-I to total apoA-I within each fraction was determined by quantitative Western blot analysis of apoA-I-immunoreactive bands. Values were determined from n = 5 samples; error bars represent ±S.D. N.D., not able to be detected.
NO2-Tyr166-apoA-I Is Nearly 100-fold Enriched in Lesions Relative to Normal Aortic Tissue and Is Not Found on an HDL-like Particle in Normal Aortic Tissues
Given the stark difference in abundance of NO2-Tyr166-apoA-I observed in the atherosclerotic artery wall versus that found in plasma (i.e. one in 12 apoA-I molecules in the atherosclerotic artery wall versus one in 750 apoA-I molecules in plasma is nitrated on tyrosine 166), we wanted to examine whether NO2-Tyr166-apoA-I could be detected in the normal artery wall. Furthermore, we wanted to determine its abundance and particle distribution. Normal human aortic tissue (from n = 5 subjects) was obtained at time of organ harvest from transplant donors, and homogenates were prepared and fractionated by sequential D2O/sucrose buoyant density ultracentrifugation as described under “Experimental Procedures.” Direct comparison of normal versus lesion aortic homogenates by 5–15% reducing SDS-PAGE following protein staining (Fig. 7A) and following Western blot analysis from parallel-run gels probed with mAb 4G11.2 (Fig. 7B) showed that there was significantly less (nearly 100-fold) immunoreactive apoA-I in normal artery homogenates than in those from atherosclerotic lesions (Fig. 7, C and D).
FIGURE 7.

ApoA-I nitrated on tyrosine 166 obtained from the normal aortic tissue is not on an HDL-like particle and behaves similarly to that isolated from atherosclerotic lesions. Aortic tissue that was histopathologically determined to appear normal was used to prepare a tissue homogenate as described under “Experimental Procedures.” A, aortic tissue homogenates from normal and atherosclerotic human artery wall tissue (n = 5 each) with the indicated amounts of protein were fractionated by gradient (5–15%) SDS-PAGE and stained with SYPRO Ruby Red. B, Western blot of duplicate gel as in A with the indicated amount of protein (15 μg) per lane (n = 5) probed with anti-NO2-Tyr166-apoA-I mAb 4G11.2. C, recovery of apoA-I (μg) per gram of tissue (wet weight) from the normal aortic tissue and from atherosclerotic lesion tissue was determined from quantitative Western blot analysis of apoA-I-immunoreactive bands and total apoA-I content as described under “Experimental Procedures” and is qualitatively presented in Figs. 8B and 4B. D, percentage of NO2-Tyr166-apoA-I to total apoA-I present in the starting material as indicated was quantified as in C. Values were determined from n = 5 samples; error bars represent ±S.D.
We next examined the particle distribution of NO2-Tyr166-apoA-I recovered from normal human artery wall. Western blot analyses with anti-total apoA-I (mAb 10G1.5) demonstrated low but detectable levels of apoA-I in HDL-like particles recovered from normal human aorta (Fig. 8A). Although apoA-I is present on the blot in relatively equal abundance, the amount of protein required to generate an equivalent immunoreactive signal was nearly 20% of the entire HDL-like fraction protein material, whereas the amount of protein loaded in the homogenate or the LPD fraction was only ∼0.2% of the total protein present in those fractions. Identically prepared membranes from SDS-PAGE probed instead with anti-NO2-Tyr166-apoA-I antibody (mAb 4G11.2) revealed no detectable bands within the HDL-like fraction, and all immunoreactive species within the LPD fraction (Fig. 8B). Interestingly, the immunoreactive bands migrated not only at ∼27 kDa, the size of an apoA-I monomer, but also at higher molecular weights as well (corresponding to molecular weights of the apoA-I dimer, trimer, etc.), consistent with appreciable oxidative cross-linking present in this material (Fig. 8B). Quantification of NO2-Tyr166-apoA-I in homogenate and LPD fractions from normal aortic tissues showed that there is only ∼1 μg of NO2-Tyr166-apoA-I/g wet weight of aortic tissue (Fig. 8C), accounting for only ∼0.12–0.14% of total apoA-I within artery wall homogenate and the LPD fraction (Fig. 8D).
FIGURE 8.

ApoA-I nitrated on tyrosine 166 obtained from the normal aortic tissue is not on an HDL-like particle. A, aortic tissue that appears histologically normal was used to prepare a tissue homogenate as described under “Experimental Procedures.” The homogenate was fractionated on sequential D2O/sucrose buoyant density gradients by ultracentrifugation, the indicated amounts of protein were separated by gradient (5–15%) reducing SDS-PAGE, and proteins were transferred to membranes. A, Western blot of normal artery wall proteins (15 μg) from homogenate and HDL-like and LPD fractions probed with anti-total apoA-I antibody mAb 10G1.5 to detect total apoA-I in homogenate and HDL-like and LPD fractions. B, duplicate Western blot as in A probed with anti-NO2-Tyr166-apoA-I mAb 4G11.25 to show anti-NO2-Tyr166-apoA-I only in the starting material and in the LPD fraction. C, recovery of NO2-Tyr166-apoA-I (μg) per gram of normal aortic tissue (wet weight) that was found to be present in the homogenate (Homog.) and in the HDL-like and LPD fractions was determined from quantitative Western blot analysis of apoA-I-immunoreactive bands and total apoA-I content as described under “Experimental Procedures” and is qualitatively presented in Fig. 9. D, percentage of NO2-Tyr166-apoA-I to total apoA-I from normal aortic tissue present in the starting homogenate and HDL-like and LPD fractions was calculated from quantitative Western blot analysis of apoA-I-immunoreactive bands as calculated in C. Values were determined from n = 5 samples; error bars represent ±S.D.
ApoA-I Harboring Site-specific 3-Nitrotyrosine at Position 166 in Vivo Is Functionally Impaired
We did not have access to sufficient quantities of human aortic tissue to recover enough pure NO2-Tyr166-apoA-I for biological characterization. Therefore, to interrogate the functional properties of apoA-I containing a nitrotyrosine at position 166 formed in vivo, we collected units of plasma from 11 different healthy volunteers and immunopurified NO2-Tyr166-apoA-I as described under “Experimental Procedures.” In parallel, bulk total apoA-I was affinity-purified from the same plasma samples using an anti-total apoA-I antibody. Recovery of the NO2-Tyr166-apoA-I and total apoA-I by their respective immunoaffinity resins was quantitative under the conditions used as assessed by Western blot analyses (data not shown). Both NO2-Tyr166-apoA-I and total apoA-I from the individual samples were delipidated, then reconstituted by a cholate dialysis method into rHDL, and further purified by polishing analytical gel filtration FPLC as described under “Experimental Procedures.” Then comparable amounts (1 μg of apoA-I mass) of rHDL formed from each subject's NO2-Tyr166-apoA-I versus total apoA-I were examined for LCAT activity as described under “Experimental Procedures.” Remarkably, rHDLs formed using immunoaffinity-isolated NO2-Tyr166-apoA-I were highly defective in their ability to stimulate LCAT activity, collectively showing a 88.1 ± 8.5% reduction in activity (p < 0.001 compared with rHDLs prepared from total apoA-I; Fig. 9). These results for the first time directly demonstrate that apoA-I harboring a nitrotyrosine at 166 in vivo has impaired LCAT-activating function. They also demonstrate that the apoA-I detected by 4G11.2 in plasma is “dysfunctional.”
FIGURE 9.
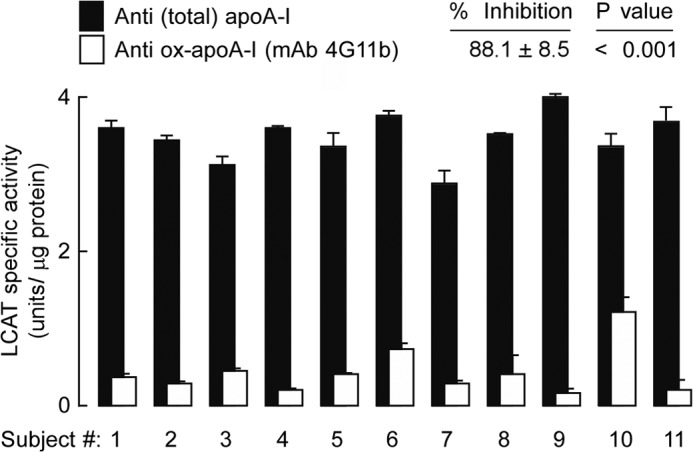
NO2-Tyr166-apoA-I immunopurified from biological material using mAb 4G11.2 is dysfunctional for LCAT activity. LCAT activity was measured in NO2-Tyr166-apoA-I immunoaffinity-purified from normal healthy human plasma (n = 11 different subjects) and reconstituted into rHDL particles as described under “Experimental Procedures.” ApoA-I isolated from plasma HDL recovered from these same healthy donors (n = 11) using an anti-total apoA-I chicken IgY antibody (Sigma) and rHDL formed from these apoA-Is served as individual internal controls for LCAT activity. Bars represent triplicate determinations; error bars represent ±S.D. Percent inhibition was calculated, and p values less than 0.001 were significant.
Functional Interrogation of Site-specific 3-Nitrotyrosine Incorporated into ApoA-I Using an Evolved Orthogonal tRNA Synthetase
Although the above immunopurification studies with mAb 4G11.2 are consistent with results observed with in vitro exposure of rHDL to MPO-generated oxidants (8), it is impossible to determine what the exact contribution of tyrosine 166 nitration in apoA-I alone is toward LCAT activity or other functional assays such as cholesterol efflux because the immunoaffinity-isolated NO2-Tyr166-apoA-I material has many other confounding sites of oxidative modification. To specifically address this problem, we produced wild-type and mutant apoA-I protein that contained 3-nitrotyrosine regiospecifically incorporated only at position 166 through engineering an orthogonal tRNATyr/tyrosyl-tRNA synthetase originally from M. jannaschii (35). By evolving this system to exclusively use nitrotyrosine and not tyrosine (pDULE 5B) and then using it for recombinant expression from an apoA-I amber codon-interrupted gene, we were able to site-specifically insert exclusively 3-nitrotyrosine with Mj-nitro-Tyr-tRNA synthetase/tRNACUA (16). A schematic illustrating the system for incorporating this unnatural amino acid into a protein at a specific site(s) of interest is presented in Fig. 10.
FIGURE 10.
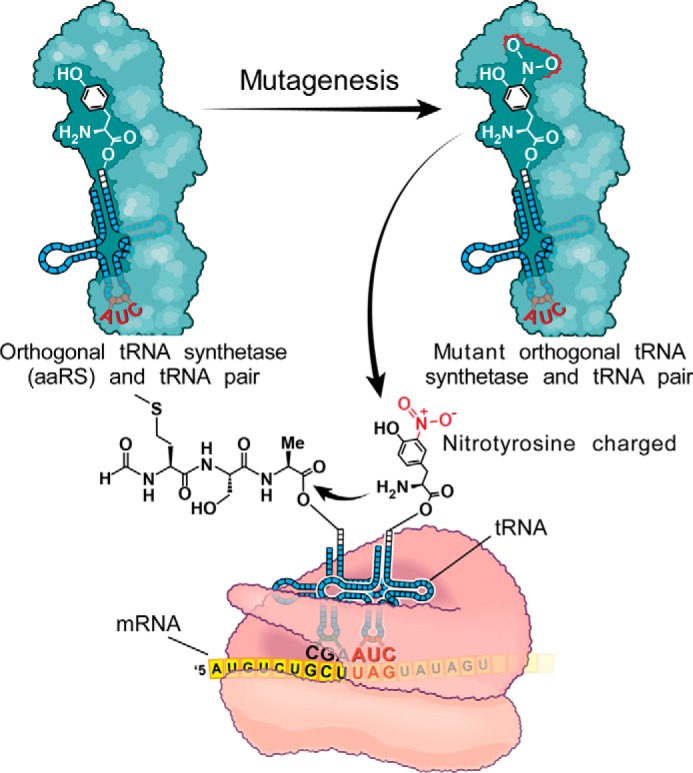
Strategy for incorporation of a nitrotyrosine into a protein of interest using an orthogonal tRNAUAG/nitro-Tyr-tRNA synthetase. The schematic depicts how the orthogonal incorporation of 3-nitrotyrosine site-specifically at amber codons (TAG) into proteins occurs. The tyrosyl-tRNA synthetase from M. jannaschii is mutated at a number of sites based upon structural information, and a library of these mutant tRNA synthetases is screened for their ability to incorporate the 3-nitrotyrosine but not any other natural amino acid. The selected mutated nitro-Tyr-tRNA synthetase then charges the orthogonal tRNACUA with nitrotyrosine, which is then translationally incorporated into the protein of interest site-specifically with high fidelity at the TAG codon. aaRS, aminoacyl-tRNA synthetase.
We prepared an E. coli codon-optimized His8-tagged human apoA-I allele and an Amber166 mutant version of this allele (Y166TAG-apoA-I) for recombinant protein expression (see “Experimental Procedures” for details). Both wild-type and mutant apoA-I alleles were transformed into bacteria harboring the evolved orthogonal Mj-nitro-Tyr-tRNA synthetase/tRNACUA, and expression was induced in minimal medium with supplements either lacking (for the wild-type apoA-I) or supplemented with 1 mm free 3-nitrotyrosine (Y166TAG-apoA-I mutant). Recombinant wild-type and mutant proteins were purified as described under “Experimental Procedures,” and equivalent amounts of each protein were fractionated by gradient (5–15%) reducing SDS-PAGE and stained with Coomassie Blue. The stained gel revealed that the 3-nitrotyrosine-containing protein was purified to the same degree and migrated equivalently to the wild-type His8-apoA-I (Fig. 11A). We then used NO2-Tyr166-apoA-I to reaffirm the specificity and high affinity of mAb 4G11.2 (note that we did not have this site-specific apoA-I form when first generating or characterizing mAb 4G11.2). Western analyses with mAb 4G11.2 failed to recognize recombinant wild-type apoA-I (1.5 μg) but readily recognized the site-specifically nitrated apoA-I even at 1000-fold lower levels of protein (e.g. 1.5 ng) (Fig. 11B). Visual inspection of the purified proteins showed that only the mutant protein is yellow in appearance, consistent with the incorporation of 3-nitrotyrosine (see Fig. 11C, inset). Spectroscopic analysis of the isolated recombinant NO2-Tyr166-apoA-I confirmed the presence of the anticipated visible spectral absorbency maximum peak at 428 nm (Fig. 11C). Moreover, proteomics analyses of the mutant apoA-I protein revealed that 3-nitrotyrosine was indeed incorporated at position 166 (supplemental Fig. 2, A and B). Furthermore, 3-nitrotyrosine is site-specifically incorporated at site 166 with a fidelity that matches that of the natural amino acid incorporation, showing a mass increase of 45.17 m/z on all b and y ions present after the tyrosine in the nitrotyrosine 166 peptide spectra compared with those same b and y ions in wild type apoA-I (supplemental Fig. 2A). This verification of our ability to site-specifically incorporate 3-nitrotyrosine within apoA-I confidently allowed us to probe the functional consequences of nitration of only tyrosine 166 in apoA-I. Additionally, the NO2-Tyr166-His8-apoA-I protein also served as an internal calibrator in quantifying NO2-Tyr166-apoA-I and total apoA-I when using mAb 4G11.2 and anti-total apoA-I antibodies.
FIGURE 11.
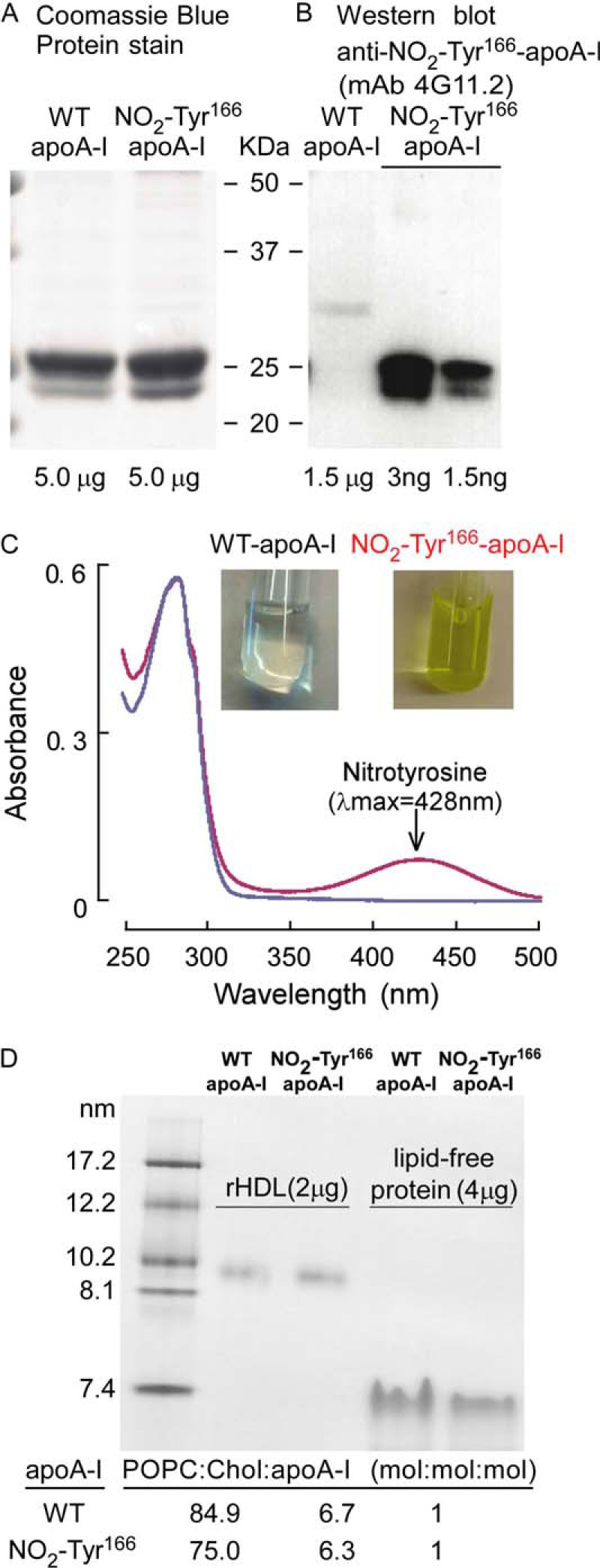
Site-specific incorporation of a nitrotyrosine into position 166 of apoA-I. 3-Nitrotyrosine was site-specifically incorporated into human apoA-I at position 166 in place of tyrosine using an evolved orthogonal Mj-tRNAUAG/nitro-Tyr-tRNA synthetase (16). A, purified WT and NO2-Tyr166-apoA-I proteins (5 μg each) were fractionated by gradient (5–15%) SDS-PAGE, and the gel was stained with Coomassie Blue. B, WT apoA-I protein (1.5 μg) with the indicated amounts of NO2-Tyr166-apoA-I proteins was fractionated by SDS-PAGE, and the proteins were transferred to membrane and probed with anti-NO2-Tyr166-apoA-I mAb 4G11.2 to demonstrate that 4G11.2 fails to recognize non-nitrated apoA-I but does specifically recognize NO2-Tyr166-apoA-I. C, scanning wavelength spectrophotometric absorbance profile for both wild-type and NO2-Tyr166-apoA-I proteins shows that only NO2-Tyr166-apoA-I protein absorbs at 428 nm, the absorbance maximum for nitrotyrosine. The inset shows the yellow tinge in color of the recombinant apoA-I that indicates incorporation of 3-nitrotyrosine; the wild-type apoA-I is colorless. D, native PAGE analyses of lipid-poor WT and NO2-Tyr166-apoA-I proteins and reconstituted HDL particles prepared from each as described under “Experimental Procedures.” Size standards are indicated in nm. Particle compositions were determined as described under “Experimental Procedures” and are listed below the gel as 1-palmitoyl-2-oleoylphosphatidylcholine (POPC):cholesterol (Chol):apoA-I (mol:mol:mol).
In initial studies, we incorporated either wild-type apoA-I or NO2-Tyr166-apoA-I into reconstituted HDL particles and examined the particle preparations for both size distribution and composition. Of note, both reconstituted HDL particle preparations were monodispersed and demonstrated comparable particle size (∼9.6 nm) (Fig. 11D). Compositional analyses of the HDL preparations also demonstrated comparable results for the HDL formed using either wild type or NO2-Tyr166-apoA-I (Fig. 11D). Thus, apoA-I site-specifically nitrated only at tyrosine 166 can generate in vitro reconstituted HDL particles that appear similar in terms of size and lipid composition to those formed by wild type apoA-I.
Thus, we first evaluated the functional effect of selective tyrosine 166 nitration of apoA-I on ABCA1-specific cholesterol acceptor function. Recombinant wild-type apoA-I was used as a control for comparison with recombinant NO2-Tyr166-apoA-I mutant protein. Increasing concentrations of each protein were examined in cholesterol efflux assays using cholesterol-loaded RAW264.7 macrophage cells in the absence and presence of 8-Br-cAMP pretreatment to induce ABCA1 expression, and ABCA1-specific cholesterol efflux activity was quantified. As observed, both the wild-type and mutant apoA-I proteins promoted comparable levels of ABCA1-dependent cholesterol efflux over a range of protein concentrations (Fig. 12A). We next examined reconstituted nascent HDL particles prepared from either wild-type apoA-I or NO2-Tyr166-apoA-I. Comparable amounts (5 μg of apoA-I mass) of each rHDL were incubated with increasing amounts of human LCAT as indicated, and LCAT activity was determined. Importantly, site-specific nitration of only tyrosine 166 in apoA-I was shown to reduce LCAT activity by 58.1 ± 5.5% across multiple LCAT concentrations (Fig. 12B). Further kinetic analyses of LCAT activity in rHDL formed by wild type versus NO2-Tyr166-apoA-I showed reduction in both apparent Km and Vmax for LCAT activity (Fig. 12C). The overall reduction in catalytic efficiency (Vmax/Km) for NO2-Tyr166-apoA-I was ∼45%. Collectively, these results show that site-specific incorporation of 3-nitrotyrosine into apoA-I at only position 166, a solvent-exposed residue and region comprising the solar flare we previously identified as interacting with LCAT (12), results in significant selective impairment in apoA-I LCAT activity with no effect on ABCA1-specific cholesterol acceptor function.
FIGURE 12.

Site-specific incorporation of a nitrotyrosine into position 166 of apoA-I is dysfunctional for LCAT activity when reconstituted into an HDL particle. A, functional interrogation of cholesterol efflux activity by wild-type and NO2-Tyr166-apoA-I proteins to observe whether NO2-Tyr166-apoA-I is dysfunctional as an ABCA1 cholesterol acceptor. ABCA1-dependent cholesterol efflux activity of purified recombinant wild-type apoA-I and NO2-Tyr166-apoA-I proteins was examined in cholesterol-loaded RAW264.7 macrophages incubated with increasing levels of apoA-I protein (wild type and NO2-Tyr166-apoA-I) as indicated. Unfilled bars represent apoA-I wild-type protein; filled bars represent NO2-Tyr166-apoA-I. ABCA1-dependent cholesterol efflux assays were performed as described under “Experimental Procedures.” B, purified wild-type and NO2-Tyr166-apoA-I proteins were used in LCAT activity assays with increasing LCAT added as indicated (see “Experimental Procedures” for details) to examine whether NO2-Tyr166-apoA-I was dysfunctional for LCAT activity. Percent inhibition and p values shown are averages across the different concentrations examined. p values that were significant (less than 0.001) and are indicated by an asterisk. C, double reciprocal plots (Lineweaver-Burk plots) of wild-type and NO2-Tyr166-apoA-I proteins used in LCAT activity assays to determine Km, Vmax, and catalytic efficiency (Cat. Eff.) (calculated values are listed in the inset). Values represent the average of three reactions; error bars indicate S.D. Diff, difference.
DISCUSSION
Although multiple investigators have affirmed that apoA-I is a selective target for oxidative modification within the artery wall (6–9, 13, 18, 19) and in vitro studies corroborate the functional impairment of activity of the lipoprotein following oxidation (7, 8, 12, 13, 19, 21), a debate exists as to relative abundance of different site-specific oxidative modifications in vivo and hence their potential functional and biological importance. In this regard, nitration of tyrosine 166 stands out as a prime example in this debate. Following immunoprecipitation of total apoA-I from homogenates prepared from the atherosclerotic artery wall, it was reported that NO2-Tyr166 modification of apoA-I was an abundant PTM in vivo through proteomics studies (8). In contrast, in studies that followed using buoyant density centrifugation to isolate HDL-like particles, it was concluded that nitration of tyrosine at this site on apoA-I is an exceedingly low abundance product and therefore presumably of little physiologically relevance (22). The present studies unambiguously showed that nitration of apoA-I at tyrosine 166 is an exceptionally abundant modification in apoA-I in the diseased artery wall where ∼1 of every 12 apoA-I molecules recovered harbors this PTM. Furthermore, the present studies showed that apoA-I that harbored this PTM, whether in the human atherosclerotic lesion or the circulation, exhibited marked impairment in function as monitored through loss of LCAT activity. Finally, through use of orthogonal amino acid substitution and incorporation of 3-nitrotyrosine site-specifically at only position 166 of apoA-I, a methodology used for the first time in apoA-I, we were able to directly demonstrate that the simple addition of the NO2− moiety at the meta-position of tyrosine 166 of apoA-I results in generation of a “dysfunctional” protein. ApoA-I nitrated at tyrosine 166, a position previously predicted to be highly solvent-exposed (located in the solar flare domain) and a putative docking site for LCAT interaction (12), is thus affirmed as an abundant dysfunctional form of apoA-I in the atherosclerotic disease-laden artery wall. In fact, NO2-Tyr166-apoA-I appears to be one of the more abundant modified forms of apoA-I recovered from human atherosclerotic plaque.
The reasons for the discrepant results obtained with respect to this site-specific modification are also elaborated with the present studies. Our work suggests that studies of buoyant density-isolated HDL-like particles would not detect this modification as it is primarily associated with lipid-poor forms of apoA-I. Interestingly, this distribution of NO2-Tyr166-apoA-I was observed within both atherosclerotic lesions and the plasma compartment.
There is much interest in understanding the functional role of apoA-I modifications in vivo. The present studies highlight that dramatic differences can be observed whether one uses ultracentrifugation or uses more quantitative apoA-I recovery approaches through the use of a high affinity antibody(s). In these studies, an mAb that is highly selective and specific for nitration of tyrosine 166 in apoA-I exclusively and not for other sites of nitration within the protein or other nitrated proteins was developed and used for quantification.
The role of nitrative and oxidative processes in contributing to the inflammatory milieu within the artery wall has grown in appreciation, as has the involvement of inflammatory processes in the pathogenesis of cardiovascular disease (10, 36–42). The present study reaffirms the concept that MPO and nitric oxide-derived oxidants are abundantly formed within the diseased artery wall and suggests that detection and quantification of circulating levels of this site-specific oxidized form of apoA-I may serve as a barometer for monitoring a potential pathophysiological process occurring in the artery wall. Further studies are needed to evaluate the potential clinical utility of quantifying circulating levels of this specific modified apoA-I form as it relates to cardiovascular disease risks and therapeutic responses.
Because epidemiological studies have long shown an inverse association between circulating HDL-cholesterol (HDLc) or apoA-I levels and both the prevalence of coronary artery disease and its major adverse events (43), a number of strategies have recently been tried as a means of raising HDLc levels for potential therapeutic benefit (43–48). Unfortunately, recent clinical trials targeting raising HDLc levels have met with failure, calling into question the notion of elevating HDLc levels as a means of promoting cardioprotective effects (43, 46–48). One possible explanation for the failures may be that HDLc is simply a “marker” and does not faithfully reflect a process in the disease pathogenesis pathway. Indeed, there is an overwhelming body of evidence using animal models indicating that apoA-I is atheroprotective (44, 45, 49–51). Recent evidence supporting an atheroprotective effect of “functional” as opposed to MPO-modified and dysfunctional apoA-I in vivo has been reported (52). When either native (unoxidized) apoA-I or MPO-oxidized apoA-I was injected subcutaneously into apoE−/− atherosclerosis-laden mice under conditions that showed comparable levels of apoA-I in the plasma compartment, cholesterol efflux, reverse cholesterol transport, atherosclerotic plaque regression, and favorable phenotypic changes in arterial tissue macrophages were only observed with native apoA-I but not MPO-oxidized apoA-I (52). Additional evidence for the atheroprotection that increased levels of apoA-I or functional HDL particles can provide is found in human clinical studies of either direct infusion of apoA-I, HDL, or extracorporeal delipidated forms of apoA-I (53–56). The conflicting clinical trial results related to HDLc and/or apoA-I levels thus emphasize the complexity and incomplete knowledge of the relationship between circulating levels of HDL forms and coronary artery disease pathogenesis (57). The present study suggests that not only might it be the functionality of the HDL particle or apoA-I rather than HDLc mass that may be more important but that development of tools that can help probe the relevant biology of apoA-I that is occurring within the artery wall is also going to be of considerable value. Fully one in every 12 apoA-I molecules in the atherosclerotic artery wall contains site-specific nitration at position 166, whereas only ∼1 in 750–1000 apoA-I molecules in the circulation harbors this modification. These results further underscore that what is occurring in the artery wall is quite distinct from what is happening in the plasma compartment. Consistent with this notion, recent studies using a monoclonal antibody specifically developed to recognize all forms of apoA-I equally well (lipidated and non-lipidated forms as either native or various oxidized forms alike) have shown that the function and distribution of apoA-I in the artery wall are markedly distinct from those in plasma (58).
In recent studies, the discovery of an alternative abundant modified form of apoA-I in human atheroma was reported using mAb developed to specifically recognize apoA-I modified by the MPO/H2O2/Cl− system (59). A 2-oxindolyl alanine moiety at Trp72 of apoA-I (oxTrp72-apoA-I) was shown to be specifically generated by MPO-catalyzed oxidation and to represent ∼20% of the apoA-I present in plaque, nearly twice the abundance of NO2-Tyr166-apoA-I. Like NO2-Tyr166-apoA-I, oxTrp72-apoA-I exists predominantly as a lipid-poor form and is not recovered in the HDL fraction (59). Interestingly, oxTrp72-apoA-I immunoaffinity purified from either human atherosclerotic aortic tissues or plasma was shown to lack ABCA1-dependent cholesterol efflux activity and in vivo HDL biogenesis activity when injected into apoA-I knock-out mice as well as to promote proinflammatory NF-κB-activating activity (59).
Recent studies reveal that apoA-I within human atherosclerotic plaque is 100-fold enriched compared with normal artery wall, predominantly lipid-poor, not associated with HDL, extensively oxidatively cross-linked, and functionally impaired with respect to both cholesterol efflux and LCAT activities (58). The present studies extend these observations by looking at a specific oxidized form of apoA-I that is abundant within the atherosclerotic plaque-laden artery wall. Taken together, the present results and recent findings (52, 59) suggest that additional tools need to be developed to identify trace levels of structurally distinct, functionally impaired modified forms of apoA-I in the circulation that like NO2-Tyr166-apoA-I are abundant in the diseased artery wall. Potential clinical utility of monitoring such modified apoA-I forms was recently demonstrated for oxTrp72-apoA-I in the circulation where elevated levels among stable subjects presenting to an outpatient cardiology clinic were associated with increased cardiovascular disease risk (59). Development of mAbs to distinct dysfunctional apoA-I forms may prove to be a valuable tool in the future as a means of monitoring distinct pathophysiological processes in a diseased artery wall and for monitoring antiatherosclerotic biological activities of HDL-targeting therapies.
Acknowledgment
Mass spectrometry studies were performed within the Lerner Research Institute mass spectrometry facility, which is partially supported through a Center of Innovation award by AB SCIEX.
This work was supported, in whole or in part, by National Institutes of Health Grants P01HL098055, P01HL076491, and HL17964. This work was also supported by a grant from the LeDucq Fondation. Dr. Tang has previously received research grant support from Abbott Laboratories. Drs. Hazen and Smith report being listed as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics. Dr. Hazen reports having been paid as a consultant for the following companies: AstraZeneca Pharmaceuticals LP, Cleveland Heart Lab, Esperion, Lilly, Liposcience Inc., Merck and Co., Inc., Pfizer Inc., Procter and Gamble, and Takeda. Dr. Hazen reports receiving research funds from Cleveland Heart Lab, Liposcience Inc., Procter and Gamble, Roche, and Takeda. Dr. Smith reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics from Cleveland Heart Lab and being paid as a consultant for Esperion. Dr. Hazen reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics and the following companies: Cleveland Heart Lab, Frantz Biomarkers, LLC, Liposcience Inc., and Siemens.

This article contains supplemental Figs. 1 and 2.
- PTM
- post-translational modification
- MPO
- myeloperoxidase
- DTPA
- diethylenetriaminepentaacetic acid
- apoA-I
- apolipoprotein A-I
- LCAT
- lecithin-cholesterol acyltransferase
- 8-Br-cAMP
- 8-bromo-cyclic AMP
- NO2-Tyr166-apoA-I
- apoA-I harboring a 3-nitrotyrosine at position 166
- rHDL
- reconstituted HDL
- Mj
- Methanococcus jannaschii
- LPD
- lipoprotein-depleted
- HDLc
- HDL-cholesterol
- oxTrp72-apoA-I
- 2-oxindolyl alanine moiety at Trp72 of apoA-I.
REFERENCES
- 1. Hazen S. L., Heinecke J. W. (1997) 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 99, 2075–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Souza J. M., Daikhin E., Yudkoff M., Raman C. S., Ischiropoulos H. (1999) Factors determining the selectivity of protein tyrosine nitration. Arch. Biochem. Biophys. 371, 169–178 [DOI] [PubMed] [Google Scholar]
- 3. Viera L., Ye Y. Z., Estévez A. G., Beckman J. S. (1999) Immunohistochemical methods to detect nitrotyrosine. Methods Enzymol. 301, 373–381 [DOI] [PubMed] [Google Scholar]
- 4. Wu W., Samoszuk M. K., Comhair S. A., Thomassen M. J., Farver C. F., Dweik R. A., Kavuru M. S., Erzurum S. C., Hazen S. L. (2000) Eosinophils generate brominating oxidants in allergen-induced asthma. J. Clin. Investig. 105, 1455–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brennan M. L., Wu W., Fu X., Shen Z., Song W., Frost H., Vadseth C., Narine L., Lenkiewicz E., Borchers M. T., Lusis A. J., Lee J. J., Lee N. A., Abu-Soud H. M., Ischiropoulos H., Hazen S. L. (2002) A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 277, 17415–17427 [DOI] [PubMed] [Google Scholar]
- 6. Pennathur S., Bergt C., Shao B., Byun J., Kassim S. Y., Singh P., Green P. S., McDonald T. O., Brunzell J., Chait A., Oram J. F., O'brien K., Geary R. L., Heinecke J. W. (2004) Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J. Biol. Chem. 279, 42977–42983 [DOI] [PubMed] [Google Scholar]
- 7. Zheng L., Nukuna B., Brennan M. L., Sun M., Goormastic M., Settle M., Schmitt D., Fu X., Thomson L., Fox P. L., Ischiropoulos H., Smith J. D., Kinter M., Hazen S. L. (2004) Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 114, 529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zheng L., Settle M., Brubaker G., Schmitt D., Hazen S. L., Smith J. D., Kinter M. (2005) Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J. Biol. Chem. 280, 38–47 [DOI] [PubMed] [Google Scholar]
- 9. Peng D.-Q., Wu Z., Brubaker G., Zheng L., Settle M., Gross E., Kinter M., Hazen S. L., Smith J. D. (2005) Tyrosine modification is not required for myeloperoxidase-induced loss of apolipoprotein A-I functional activities. J. Biol. Chem. 280, 33775–33784 [DOI] [PubMed] [Google Scholar]
- 10. Wang Z., Nicholls S. J., Rodriguez E. R., Kummu O., Hörkkö S., Barnard J., Reynolds W. F., Topol E. J., DiDonato J. A., Hazen S. L. (2007) Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 13, 1176–1184 [DOI] [PubMed] [Google Scholar]
- 11. Vaisar T., Shao B., Green P. S., Oda M. N., Oram J. F., Heinecke J. W. (2007) Myeloperoxidase and inflammatory proteins: pathways for generating dysfunctional high-density lipoprotein in humans. Curr. Atheroscler. Rep. 9, 417–424 [DOI] [PubMed] [Google Scholar]
- 12. Wu Z., Wagner M. A., Zheng L., Parks J. S., Shy J. M., 3rd, Smith J. D., Gogonea V., Hazen S. L. (2007) The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat. Struct. Mol. Biol. 14, 861–868 [DOI] [PubMed] [Google Scholar]
- 13. Peng D.-Q., Brubaker G., Wu Z., Zheng L., Willard B., Kinter M., Hazen S. L., Smith J. D. (2008) Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler. Thromb. Vasc. Biol. 28, 2063–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Young T. S., Schultz P. G. (2010) Beyond the canonical 20 amino acids: expanding the genetic lexicon. J. Biol. Chem. 285, 11039–11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samuelson J. C., Singh-Blom A., Hughes R., Ellington A. (2013) in Enzyme Engineering, pp. 93–114, Humana Press, Totowa, NJ [Google Scholar]
- 16. Franco M. C., Ye Y., Refakis C. A., Feldman J. L., Stokes A. L., Basso M., Melero Fernández de Mera R. M., Sparrow N. A., Calingasan N. Y., Kiaei M., Rhoads T. W., Ma T. C., Grumet M., Barnes S., Beal M. F., Beckman J. S., Mehl R., Estévez A. G. (2013) Nitration of Hsp90 induces cell death. Proc. Natl. Acad. Sci. U.S.A. 110, E1102–E1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neumann H., Hazen J. L., Weinstein J., Mehl R. A., Chin J. W. (2008) Genetically encoding protein oxidative damage. J. Am. Chem. Soc. 130, 4028–4033 [DOI] [PubMed] [Google Scholar]
- 18. Undurti A., Huang Y., Lupica J. A., Smith J. D., DiDonato J. A., Hazen S. L. (2009) Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 284, 30825–30835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bergt C., Pennathur S., Fu X., Byun J., O'Brien K., McDonald T. O., Singh P., Anantharamaiah G. M., Chait A., Brunzell J., Geary R. L., Oram J. F., Heinecke J. W. (2004) The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. U.S.A. 101, 13032–13037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hadfield K. A., Pattison D. I., Brown B. E., Hou L., Rye K. A., Davies M. J., Hawkins C. L. (2013) Myeloperoxidase-derived oxidants modify apolipoprotein A-I and generate dysfunctional high-density lipoproteins: comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCl). Biochem. J. 449, 531–542 [DOI] [PubMed] [Google Scholar]
- 21. Shao B., Bergt C., Fu X., Green P., Voss J. C., Oda M. N., Oram J. F., Heinecke J. W. (2005) Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J. Biol. Chem. 280, 5983–5993 [DOI] [PubMed] [Google Scholar]
- 22. Shao B., Pennathur S., Heinecke J. W. (2012) Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J. Biol. Chem. 287, 6375–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Z., Gogonea V., Lee X., Wagner M. A., Li X. M., Huang Y., Undurti A., May R. P., Haertlein M., Moulin M., Gutsche I., Zaccai G., Didonato J. A., Hazen S. L. (2009) Double superhelix model of high density lipoprotein. J. Biol. Chem. 284, 36605–36619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ståhlman M., Davidsson P., Kanmert I., Rosengren B., Borén J., Fagerberg B., Camejo G. (2008) Proteomics and lipids of lipoproteins isolated at low salt concentrations in D2O/sucrose or in KBr. J. Lipid Res. 49, 481–490 [DOI] [PubMed] [Google Scholar]
- 25. Markwell M. A., Haas S. M., Bieber L. L., Tolbert N. E. (1978) A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 87, 206–210 [DOI] [PubMed] [Google Scholar]
- 26. Parks J. S., Gebre A. K., Furbee J. W. (1999) Lecithin-cholesterol acyltransferase. Assay of cholesterol esterification and phospholipase A2 activities. Methods Mol. Biol. 109, 123–131 [DOI] [PubMed] [Google Scholar]
- 27. Li X. M., Tang W. H., Mosior M. K., Huang Y., Wu Y., Matter W., Gao V., Schmitt D., Didonato J. A., Fisher E. A., Smith J. D., Hazen S. L. (2013) Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler. Thromb. Vasc. Biol. 33, 1696–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matz C. E., Jonas A. (1982) Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J. Biol. Chem. 257, 4535–4540 [PubMed] [Google Scholar]
- 29. Rakita R. M., Michel B. R., Rosen H. (1990) Differential inactivation of Escherichia coli membrane dehydrogenases by a myeloperoxidase-mediated antimicrobial system. Biochemistry 29, 1075–1080 [DOI] [PubMed] [Google Scholar]
- 30. Morita Y., Iwamoto H., Aibara S., Kobayashi T., Hasegawa E. (1986) Crystallization and properties of myeloperoxidase from normal human leukocytes. J. Biochem. 99, 761–770 [DOI] [PubMed] [Google Scholar]
- 31. Nelson D. P., Kiesow L. A. (1972) Enthalpy of decomposition of hydrogen peroxide by catalase at 25 degrees C (with molar extinction coefficients of H2O2 solutions in the UV). Anal. Biochem. 49, 474–478 [DOI] [PubMed] [Google Scholar]
- 32. Morris J. C. (1966) The acid ionization constant of HOCl from 5 to 35°. J. Phys. Chem. 70, 3798–3805 [Google Scholar]
- 33. Reiter C. D., Teng R. J., Beckman J. S. (2000) Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J. Biol. Chem. 275, 32460–32466 [DOI] [PubMed] [Google Scholar]
- 34. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 35. Wang L., Brock A., Herberich B., Schultz P. G. (2001) Expanding the genetic code of Escherichia coli. Science 292, 498–500 [DOI] [PubMed] [Google Scholar]
- 36. Beckman J. S., Koppenol W. H. (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 271, C1424–C1437 [DOI] [PubMed] [Google Scholar]
- 37. Boullier A., Bird D. A., Chang M. K., Dennis E. A., Friedman P., Gillotre-Taylor K., Hörkkö S., Palinski W., Quehenberger O., Shaw P., Steinberg D., Terpstra V., Witztum J. L. (2001) Scavenger receptors, oxidized LDL, and atherosclerosis. Ann. N.Y. Acad. Sci. 947, 214–222; discussion 222–223 [DOI] [PubMed] [Google Scholar]
- 38. Lusis A. J. (2000) Atherosclerosis. Nature 407, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Navab M., Hama S. Y., Reddy S. T., Ng C. J., Van Lenten B. J., Laks H., Fogelman A. M. (2002) Oxidized lipids as mediators of coronary heart disease. Curr. Opin. Lipidol. 13, 363–372 [DOI] [PubMed] [Google Scholar]
- 40. Shishehbor M. H., Aviles R. J., Brennan M. L., Fu X., Goormastic M., Pearce G. L., Gokce N., Keaney J. F., Jr., Penn M. S., Sprecher D. L., Vita J. A., Hazen S. L. (2003) Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 289, 1675–1680 [DOI] [PubMed] [Google Scholar]
- 41. Brennan M. L., Penn M. S., Van Lente F., Nambi V., Shishehbor M. H., Aviles R. J., Goormastic M., Pepoy M. L., McErlean E. S., Topol E. J., Nissen S. E., Hazen S. L. (2003) Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 349, 1595–1604 [DOI] [PubMed] [Google Scholar]
- 42. Libby P. (2012) Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 2045–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gordon T., Castelli W. P., Hjortland M. C., Kannel W. B., Dawber T. R. (1977) High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 62, 707–714 [DOI] [PubMed] [Google Scholar]
- 44. Badimon J. J., Badimon L., Galvez A., Dische R., Fuster V. (1989) High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab. Invest. 60, 455–461 [PubMed] [Google Scholar]
- 45. Badimon J. J., Badimon L., Fuster V. (1990) Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J. Clin. Investig. 85, 1234–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nissen S. E., Tardif J. C., Nicholls S. J., Revkin J. H., Shear C. L., Duggan W. T., Ruzyllo W., Bachinsky W. B., Lasala G. P., Tuzcu E. M., and ILLUSTRATE Investigators (2007) Effect of torcetrapib on the progression of coronary atherosclerosis. N. Engl. J. Med. 356, 1304–1316 [DOI] [PubMed] [Google Scholar]
- 47. Barter P. J., Caulfield M., Eriksson M., Grundy S. M., Kastelein J. J., Komajda M., Lopez-Sendon J., Mosca L., Tardif J.-C., Waters D. D., Shear C. L., Revkin J. H., Buhr K. A., Fisher M. R., Tall A. R., Brewer B., and ILLUMINATE Investigators (2007) Effects of torcetrapib in patients at high risk for coronary events. New Engl. J. Med. 357, 2109–2122 [DOI] [PubMed] [Google Scholar]
- 48. AIM-HIGH Investigators, Boden W. E., Probstfield J. L., Anderson T., Chaitman B. R., Desvignes-Nickens P., Koprowicz K., McBride R., Teo K., Weintraub W. (2011) Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 365, 2255–2267 [DOI] [PubMed] [Google Scholar]
- 49. Rubin E. M., Krauss R. M., Spangler E. A., Verstuyft J. G., Clift S. M. (1991) Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 353, 265–267 [DOI] [PubMed] [Google Scholar]
- 50. Plump A. S., Scott C. J., Breslow J. L. (1994) Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc. Natl. Acad. Sci. U.S.A. 91, 9607–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hughes S. D., Verstuyft J., Rubin E. M. (1997) HDL deficiency in genetically engineered mice requires elevated LDL to accelerate atherogenesis. Arterioscler. Thromb. Vasc. Biol. 17, 1725–1729 [DOI] [PubMed] [Google Scholar]
- 52. Hewing B., Parathath S., Barrett T., Kellie Chung W. K., Astudillo Y. M., Hamada T., Ramkhelawon B., Tallant T. C., Yusufishaq M. S., Didonato J. A., Huang Y., Buffa J., Berisha S. Z., Smith J. D., Hazen S. L., Fisher E. A. (2014) Effects of native and myeloperoxidase-modified apolipoprotein A-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 10.1161/ATVBAHA.113.303044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kinlay S., Libby P., Ganz P. (2001) Endothelial function and coronary artery disease. Curr. Opin. Lipidol 12, 383–389 [DOI] [PubMed] [Google Scholar]
- 54. Nissen S. E., Tsunoda T., Tuzcu E. M., Schoenhagen P., Cooper C. J., Yasin M., Eaton G. M., Lauer M. A., Sheldon W. S., Grines C. L., Halpern S., Crowe T., Blankenship J. C., Kerensky R. (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290, 2292–2300 [DOI] [PubMed] [Google Scholar]
- 55. Tardif J. C., Grégoire J., L'Allier P. L., Ibrahim R., Lespérance J., Heinonen T. M., Kouz S., Berry C., Basser R., Lavoie M. A., Guertin M. C., Rodés-Cabau J., and Effect of rHDL on Atherosclerosis-Safety and Efficacy (ERASE) Investigators (2007) Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA 297, 1675–1682 [DOI] [PubMed] [Google Scholar]
- 56. Sacks F. M., Rudel L. L., Conner A., Akeefe H., Kostner G., Baki T., Rothblat G., de la Llera-Moya M., Asztalos B., Perlman T., Zheng C., Alaupovic P., Maltais J. A., Brewer H. B. (2009) Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J. Lipid Res. 50, 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Feig J. E., Hewing B., Smith J. D., Hazen S. L., Fisher E. A. (2014) High-density lipoprotein and atherosclerosis regression: evidence from preclinical and clinical studies. Circ. Res. 114, 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DiDonato J. A., Huang Y., Aulak K. S., Even-Or O., Gerstenecker G., Gogonea V., Wu Y., Fox P. L., Tang W. H., Plow E. F., Smith J. D., Fisher E. A., Hazen S. L. (2013) Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 128, 1644–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang Y., Didonato J. A., Levison B. S., Schmitt D., Li L., Wu Y., Buffa J., Kim T., Gerstenecker G. S., Gu X., Kadiyala C. S., Wang Z., Culley M. K., Hazen J. E., Didonato A. J., Fu X., Berisha S. Z., Peng D., Nguyen T. T., Liang S., Chuang C. C., Cho L., Plow E. F., Fox P. L., Gogonea V., Tang W. H., Parks J. S., Fisher E. A., Smith J. D., Hazen S. L. (2014) An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 20, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]