

Background: Macrophage polarization regulates human inflammatory disorders.
Results: KLF6 is a novel transcriptional regulator of macrophage polarization.
Conclusion: KLF6 regulates macrophage inflammatory gene expression by modulating functions of NF-κB and PPARγ.
Significance: Pharmacological agents that modulate KLF6 signaling may allow for therapeutic gain in the treatment of inflammatory disorders.
Keywords: Cellular Immune Response, Gene Regulation, Inflammation, Macrophages, Transcription Regulation, Kruppel-like Transcription Factor 6
Abstract
Accumulating evidence supports the importance of macrophage plasticity in a broad spectrum of biological processes operative in health and disease. A major locus of control regulating macrophage polarization is at the transcriptional level, and several major pathways have been elucidated in recent years. In this study, we identify the Kruppel-like transcription factor 6 (KLF6) as a molecular toggle controlling macrophage speciation. KLF6 expression was robustly induced by pro-inflammatory M1 stimuli (e.g. LPS and IFN-γ) and strongly suppressed by M2 stimuli (e.g. IL4 and IL-13) in human and murine macrophages. Gain- and loss-of-function studies suggest that KLF6 is required for optimal LPS-induced pro-inflammatory gene expression, acting cooperatively with NF-κB. Furthermore, KLF6 inhibits anti-inflammatory gene expression by negatively regulating peroxisome proliferator-activated receptor γ expression in macrophages. Collectively, these observations identify KLF6 as a novel transcriptional regulator of macrophage polarization.
Introduction
Clinical, pathological, and experimental studies highlight a central role for the macrophages in a broad spectrum of acute and chronic inflammatory disease conditions (1, 2). Macrophages are strategically located throughout the body, where they play crucial role in host defense, tissue repair, and the recruitment of additional inflammatory cells to the local microenvironment (3). During the inflammatory response, monocytes emigrate from the bloodstream and develop into macrophages within tissues (4). In response to both pathogenic and tissue-derived cues, macrophages undergo additional phenotypic changes, which help them to adapt and tailor an appropriate response (5). These adaptive changes are carefully orchestrated, at least in part, at the transcriptional levels through activation of key pathways that regulate target gene expression and cellular function (6).
Recent refinements to our understanding of macrophage plasticity have revealed the importance of distinct subsets in physiology and disease (7). Macrophage phenotypes exist across an M1 and M2 spectrum in which M1 cells are defined as “classically activated” pro-inflammatory macrophages, whereas M2 cells are “alternatively activated” anti-inflammatory macrophages (8). The balance between the classically activated M1-type and alternatively activated M2-type macrophages at the site of inflammation regulates the physiological inflammatory response, and an alteration in this balance can contribute to various inflammatory disorders (9). The initial inflammatory response is orchestrated by macrophages activated by pro-inflammatory stimuli and are generally referred as M1 macrophages (10). These classically activated macrophages are characterized by the production of high levels of pro-inflammatory factors (e.g. IL-1α, IFN-γ, IL-6, and TNF-α) and promote a robust Th1 immune response (1). At the transcriptional level, factors such as NF-κB, AP-1, STAT-1, and IRF5 have been identified as important for this pro-inflammatory M1 phenotypic response (1). By contrast, the resolution phase of the inflammatory process is orchestrated by alternatively activated macrophages. These M2 macrophages are less-sensitive to pro-inflammatory stimuli and are actively involved in debris scavenging, angiogenesis, tissue remodeling, and secretion of anti-inflammatory cytokines (11). At the transcriptional level, factors, including STAT6, IRF4, and PPARγ,4 have been identified as important in the regulation of characteristic M2 target genes such as Arg1, Mrc1, and Chi3l3 (11). Furthermore, it has been shown that PPARγ and STAT6 can cooperate to regulate many M2 targets establishing the STAT6/PPARγ pathway as essential for alternative macrophage polarization (12).
Kruppel-like factor 6 (KLF6) is a member of the zinc finger family of transcription factors that regulate key cellular processes such as development, differentiation, proliferation, and programmed cell death (13). Recent studies have linked Kruppel-like factors (KLFs) to the biology of macrophage activation and polarization (14, 15). The initial link of this gene family to myeloid biology was the identification of KLF2 as a tonic repressor of macrophage pro-inflammatory activation (14). Furthermore, KLF4 was found to be essential for the macrophage M2 genetic program in vitro and in vivo (15). However, the role of robustly expressed KLF6 in regulation of inflammatory gene expression and macrophage polarization is completely unknown. Here, we provide the evidence that KLF6 promotes an M1 phenotype through cooperation with NF-κB. Furthermore, KLF6 was also found to inhibit the M2 targets by suppressing PPAR-γ expression. Collectively, these findings identify KLF6 as a novel and critical molecular switch regulating macrophage polarization.
EXPERIMENTAL PROCEDURES
Materials
Lipopolysaccharide, phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), and thioglycollate broth were purchased from Sigma. Anti-KLF6, anti-NF-κBp65, anti-p300, anti-PPAR-γ, anti-actin antibodies, and rabbit IgG were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Phycoerythrin-Texas Red-labeled anti-ITGAM (CD11b) antibody (clone M1/70) was obtained from Invitrogen. Phycoerythrin-labeled anti-SIGLEC5 (SiglecF) antibody (E50-2440) and Fc block were obtained from BD Biosciences. CRecombinant mouse IL-4 and MCSF were obtained by R&D Systems (Minneapolis, MN). RAW264.7, THP-1, U937, and J774.1 cell lines were obtained from American Type Culture Collection (Manassas, VA). All the tissue culture supplies were obtained from Corning Inc. (Lowell, MA). Amaxa® mouse macrophage Nucleofector® kit was obtained from Lonza. All other chemicals and reagents used were of analytical grade and were obtained from commercial sources.
Cell Culture
RAW264.7, THP-1, U937, and J774.1 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm glutamine in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Mouse primary macrophages were obtained from the peritoneal cavity by inducing peritonitis with 3% thioglycollate broth in 8–12-week-old mice as described previously (14). The peritoneal lavage and adherent macrophage cell population were examined for eosinophil contamination by FACS analysis using fluorescently labeled anti-CD11b (macrophage marker) and anti-SiglecF (eosinophil marker) antibodies. Bone marrow-derived macrophages were generated as described previously (16). Briefly, bone marrow cells from wild-type, Lyz2cre, and Lyz2cre:Klf6fl/fl mice were harvested from femur and tibia. These bone marrow cells were cultured in cell-culture media supplemented with mouse recombinant macrophage colony-stimulating factor 1 for 7 days. These cells were harvested and utilized for experiments. To generate human macrophages for these studies, peripheral blood mononuclear cells were obtained from healthy blood donors (approved by the Case Western Reserve University Institutional Review Board). These cells were allowed to adhere onto plastic tissue culture surfaces and differentiate into macrophages. Nonadherent cells were removed, and adherent macrophages were utilized for indicated experiments.
Generation of Myeloid-specific Klf6 Null Mice and Cutaneous Inflammation Model
All mouse colonies were maintained in a clean animal facility, and all animal experimentation was approved by the IACUC Committee, Case Western Reserve University. Mouse line expressing lysozyme M promoter-driven Cre recombinase (Lyz2cre) was obtained from The Jackson Laboratory (Bar Harbor, ME). Klf6 floxed (Klf6fl/fl) mice were obtained from Genentech (San Francisco, CA) (17). Klf6 floxed mice were crossed with Lyz2cre mice to generate a myeloid-specific deletion of Klf6. These mice were further backcrossed to Lyz2cre mice to generate male and female offspring expressing two Cre and floxed Klf6 alleles. These mice with two Klf6 floxed and Cre were used as the Klf6 myeloid-specific null group. Mice with only two Cre alleles were used as the control group. Lyz2cre genotyping was performed as described previously (14). Klf6 floxed allele genotyping was performed using site-specific primers (forward primer, 5′-GTCTCTTGACACCTTGACTATCTCTCC-3′, and reverse primer, 5′-CAAGAAGCCTTCAGA GAACACC-3′). To examine the Klf6 genomic excision, total RNA from control and Klf6 myeloid-specific knock-out macrophages were isolated and analyzed by PCR using primer pairs indicated below (forward primer, 5′-TTGCAGTCAGTCCGCTGTTTG-3′, and reverse primer, 5′-CTGCTCCTTCAGAGGTGC-3′). T-cell receptor δ chain was used as loading control and was amplified using forward primer 5′-CAAATGTTGCTTGTCTGGTG-3′ and reverse primer 5′-GTCAGT CGAGTGCACAGTTT-3′. The TPA-induced cutaneous inflammation analyses were performed as described before (18). The right ear in Lyz2cre and Lyz2cre:Klf6fl/fl mice was exposed twice with 2.5 μg of TPA in 20 μl of acetone. The left ear was treated similarly with acetone alone and served as a vehicle control. The mice were sacrificed 24 h after the second TPA application, and ear weight was recorded.
Real Time Quantitative RT-PCR Assay and Western Blot Analysis
Total RNA was isolated from indicated cell types or tissue following designated treatment using TRIzol® reagent (Invitrogen). One microgram of total RNA was reverse-transcribed using M-MuLV reverse transcriptase in the presence of random hexamers and oligo(dT) primer mixtures. Real time quantitative PCR was performed using Universal SYBR Green PCR Master Mix on Applied Biosystems Step One plus real time PCR system using gene-specific primers.
Primary mouse peritoneal macrophages, bone marrow-derived macrophages or RAW264.7, THP-1, U937, and J774.1 cells were lysed using radioimmunoprecipitation lysis buffer (Sigma) supplemented with a protease and phosphatase inhibitor mixture tablet (Roche Applied Science) following the indicated treatment. Equal quantities of total protein were separated by SDS-PAGE and detected by indicated antibody by immunoblotting analysis.
Chromatin Immunoprecipitation, Transient Transfection, and Luciferase Assay Studies
Chromatin immunoprecipitation analyses were performed using the EZ-Magna ChIP G kit (Millipore Corp., Billerica, MA) according to the manufacturer's instruction. Briefly, wild-type bone marrow-derived macrophages were stimulated with LPS or IL-4. Chromatin immunoprecipitations were performed using anti-KLF6 antibody. Chromatin samples from these experiments were analyzed by real time quantitative RT-PCR. Primer pairs flanking the KLF-binding site were targeted to amplify the mouse IL-1α (−976 to −980), IL-1β (−601 to −605), Arg-1 (−1009 to −1013), and Mrc1 (−1637 to −1642) promoters. Chromatin immunoprecipitation performed using IgG was used as negative control.
Transfection of mouse BMDBs was performed utilizing the Amaxa® mouse macrophage Nucleofector® kit according to the manufacturer's instruction. Luciferase reporter plasmids driven by the NF-κB concatemer were transfected alone or were co-transfected with NF-κB-p65 or Klf6 plasmids in RAW264.7 cells using Lipofectamine® transfection reagent (Invitrogen). These cells were exposed to 100 ng/ml LPS for 6 h. Luciferase reporter activity was measured and normalized according to the manufacturer's instructions. Results are presented as relative luciferase activity over the control group.
Statistical Analysis
All data are presented as the means ± S.D. unless indicated. The statistical significance of differences between the two groups was analyzed with Student's t test. Values of p < 0.05 were considered statistically significant.
RESULTS
KLF6 mRNA and Protein Expression in Human and Murine Macrophages
Recent studies have linked several members of the KLF family (e.g. KLF1, KLF2, KLF3, and KLF4) to monocyte/macrophage differentiation and activation (19). However, the expression and function of KLF6 in macrophages are unknown. Therefore, we examined the expression of KLF6 in human and mouse primary macrophages and cell lines. Analysis of human peripheral blood mononuclear cells and macrophages revealed that KLF6 protein and mRNA are abundantly expressed in both cell types with the highest expression in the macrophages (Fig. 1A). Furthermore, analysis of human (U937 and THP-1) and mouse (RAW264.7 and J774.1) monocytic/macrophage cell lines suggested that KLF6 mRNA and protein are also abundantly expressed in these cell lines (Fig. 1B).
FIGURE 1.

KLF6 expression in human and mouse macrophages. A, human peripheral blood mononuclear cells (PBMC) and primary macrophages were analyzed for KLF6 protein (upper panel) and mRNA (lower panel) expression by Western blot analysis and quantitative PCR analysis, respectively. B, human (U937 and THP-1) and mouse (RAW264.7 and J774.1) myeloid cell lines examined for KLF6 protein expression by Western blot (upper panel) and mRNA (lower panel) expression by quantitative PCR analysis. C, mouse thioglycollate-induced peritoneal macrophages and bone marrow-derived macrophages were analyzed for KLF6 protein (upper panel) and mRNA (lower panel) expression by Western blot and quantitative PCR analysis respectively. D, mouse thioglycollate-induced peritoneal macrophages and bone marrow-derived macrophages were analyzed for relative expression of macrophage Klfs by quantitative PCR analysis. Klf6 expressions in these cells were assigned a value of 1, and relative fold increases are indicated. E, Klf6 expression analyzed in mouse liver, kidney, testis, spleen, adipose tissue, lung, heart and bone marrow-derived macrophages was analyzed for relative expression of Klf6 by quantitative PCR analysis. Klf6 levels in liver assigned a value of 1 and relative fold increases are indicated. Actin and 36B4 were used as housekeeping gene for Western blot and quantitative PCR analysis, respectively. Each experiment was performed a minimum of three times. Data represent mean ± S.D., and p values less than 0.05 between indicated groups are considered significant.
We next evaluated KLF6 protein and mRNA expression in mouse thioglycollate-elicited peritoneal macrophages (PMs) and BMDMs. KLF6 protein and mRNA expression were detected in both with higher expression in PMs (Fig. 1C). To determine the expression of Klf6 relative to other KLFs expressed in mouse macrophages, quantitative PCR analysis was performed (Fig. 1D). Klf5 expression in mouse PMs and BMDMs was defined as one, and relative fold expression of individual Klf was assessed. Our results indicate that Klf6 is an abundantly expressed member of the Klf family in murine PMs and BMDMs. Finally, our survey of Klf6 expression analysis across multiple murine tissues indicated that Klf6 is most abundantly expressed in macrophages (Fig. 1E). Collectively, these results demonstrate that KLF6 mRNA and protein are robustly expressed in human and mouse primary macrophages and cell lines.
Pro- and Anti-inflammatory Stimuli Differentially Regulate KLF6 Expression in Macrophages
To gain initial insights into the role of KLF6 in macrophages, we examined the effect of various pro- and anti-inflammatory stimuli on KLF6 mRNA and protein expression. Exposure of human macrophages to IFN-γ or LPS for 6 h induced KLF6 mRNA and protein expression (Fig. 2A). Similar results were seen in murine BMDMs (Fig. 2B). Next, time course studies were undertaken to examine the kinetics of IFN-γ- and LPS-induced KLF6 mRNA and protein expression in mouse BMDMs. IFN-γ induced KLF6 protein and mRNA expression as early as 4 h after treatment in a sustained manner (Fig. 2C). Interestingly, the kinetics in response to LPS was distinct. LPS strongly induced KLF6 mRNA and protein expression at 2 h, peaked at ∼6 h, and then returned to baseline levels by 24 h after exposure (Fig. 2D). Next we assessed the effect of anti-inflammatory M2 stimuli on KLF6 mRNA and protein expression. As shown in Fig. 2, E and F, IL-4 or IL-13 exposure strongly diminished KLF6 mRNA and protein expression in human primary macrophages and murine BMDMs. Furthermore, kinetic studies revealed a gradual reduction of KLF6 mRNA and protein levels in response to IL-4 or IL-13 exposure (Fig. 2, G and H). Collectively, these results indicate that M1 and M2 stimuli differentially regulate KLF6 expression in human and murine primary macrophages.
FIGURE 2.

Pro-inflammatory stimuli induce and anti-inflammatory stimuli suppress KLF6 expression in human and mouse macrophages. A and B, human primary macrophages (A) and mouse bone marrow-derived macrophages (B) were induced with 10 ng/ml IFN-γ and 100 ng/ml LPS separately for 6 h. These cells were analyzed for KLF6 protein (upper panels) and mRNA (lower panels) expression by Western blot and quantitative PCR analysis, respectively. C and D, mouse bone marrow-derived macrophages were induced with 10 ng/ml IFN-γ (C) and 100 ng/ml LPS (D) separately for 0–24 h. These cells were examined for KLF6 protein (upper panels) and mRNA (lower panels) expression kinetics by Western blot and quantitative PCR analysis, respectively. E and F, human primary macrophages (E) and mouse bone marrow-derived macrophages (F) were induced with 10 ng/ml IL-4 and 10 ng/ml IL-13 separately for 18 h. These cells were analyzed for KLF6 protein expression (upper panels) by Western blot and mRNA (lower panels) expression by quantitative PCR analysis. G and H, mouse bone marrow-derived macrophages were induced with 10 ng/ml IL-4 (G) and 10 ng/ml IL-13 (H) separately for 0–24 h. These cells were examined for KLF6 protein (upper panels) and mRNA (lower panels) expression kinetics by Western blot and quantitative PCR analysis, respectively. Actin and 36B4 were used as housekeeping genes for Western blot and quantitative PCR analysis, respectively. Each experiment was performed for a minimum of three times. Data represent mean ± S.D., and p values less than 0.05 between indicated groups are considered significant.
Generation and Characterization of a Myeloid-specific Klf6-deficient Murine Line
Previous studies show that homozygous deletion of Klf6 is embryonically lethal and associated with diminished hematopoiesis (20). To generate myeloid-specific Klf6 null mice, we crossed Klf6 conditional mutant mice containing loxP sites flanking exons 2 and 3 with Lyz2cre mice. These mice were genotyped with site-specific primers as described under “Experimental Procedures” and further backcrossed to Lyz2cre mice to generate offspring expressing two Cre and floxed Klf6 alleles. To examine the deletion of KLF6 in myeloid cells, total RNA from wild type, Lyz2cre, Klf6fl/fl, and Lyz2cre:Klf6fl/fl BMDMs was analyzed for exon 2 and 3 excision by reverse transcription-PCR. Our result confirmed that the Klf6 exon 2- and 3-excised product was only detected in Lyz2cre:Klf6fl/fl mouse BMDMs (Fig. 3A). Consistent with this, qPCR analysis of mRNA confirmed robust reduction of Klf6 levels in Lyz2cre:Klf6fl/fl thioglycollate-elicited peritoneal macrophages and BMDMs compared with Lyz2cre group (Fig. 3, B and C). To corroborate this observation at the protein level, Lyz2cre and Lyz2cre:Klf6fl/fl thioglycollate-elicited peritoneal macrophages and BMDMs (Fig. 3D) were stimulated with 100 ng/LPS for 4 h. Cell lysates were analyzed for KLF6 protein expression. Our results suggest that KLF6 protein is absent in Lyz2cre:Klf6fl/fl thioglycollate-elicited peritoneal macrophages and BMDMs. LPS stimulation only induced KLF6 protein expression in Lyz2cre thioglycollate-elicited peritoneal macrophages and BMDMs. Eosinophil contamination analysis of peritoneal lavage and post-adherent macrophage population (24–72 h) by ITGAM (CD11b) and SIGLEC5 (Siglec F) staining illustrated no significant difference between Lyz2cre and Lyz2cre:Klf6fl/fl groups (Fig. 3, E and F).
FIGURE 3.

Generation and characterization of a myeloid-specific KLF6-deficient mice line. A, wild-type, Lyz2cre, Klf6fl/fl, and Lyz2cre:Klf6fl/fl bone marrow-derived macrophage total RNA was subjected to reverse transcription-PCR analysis using a site-specific primer. Presence of lower band (∼325 bp) indicates genomic excision of a Klf6 floxed site. T-cell receptor δ chain (TCRD) gene was used as loading control. B and C, Lyz2cre and Lyz2cre:Klf6fl/fl mouse thioglycollate-induced peritoneal macrophages and bone marrow-derived macrophages were analyzed for Klf6 mRNA expression by quantitative PCR analysis. 36B4 was used as the housekeeping gene. D, Lyz2cre and Lyz2cre:Klf6fl/fl mouse thioglycollate-induced peritoneal macrophages and bone marrow-derived macrophages were stimulated with 100 ng/ml LPS for 4 h. Cell lysates were analyzed for KLF6 protein expression by Western blot analysis using anti-KLF6 antibody. Actin was used as a loading control. E and F, thioglycollate-elicited peritoneal lavage and post-adherent macrophages (days 1–3) derived from Lyz2cre and Lyz2cre:Klf6fl/fl mice were collected. These cell populations were examined for eosinophil and macrophage levels by SIGLEC5 (FiglecF) and ITGAM (CD11b) staining by FACS analysis (E). Percentage of eosinophils in peritoneal lavage and post-adherent macrophages are represented (F). G and H, age- and sex- matched Lyz2cre and Lyz2cre:Klf6fl/fl mouse total blood was collected be venipuncture in heparin/EDTA-coated tubes. Samples were analyzed by Hemavet 950TM hematology profiling unit. Data represent mean ± S.D., and a p value less than 0.05 between the indicated groups is considered significant.
Next, we examined the effect of myeloid Klf6 deficiency on adult mouse hematopoietic cell compartment. Our results revealed no significant difference in lymphocyte, neutrophil, eosinophil, and basophil populations between Lyz2cre and Lyz2cre:Klf6fl/fl groups (Fig. 3G). However, Lyz2cre:Klf6fl/fl mice exhibited a modest increase in the monocyte populations compared with the Lyz2cre group (Fig. 3G). Further analysis of additional hematological parameters, including RBC and platelet count, showed no significant difference between Lyz2cre and Lyz2cre:Klf6fl/fl groups (Fig. 3H). Collectively, our results demonstrate that myeloid-specific Klf6 deficiency did not significantly alter the hematopoietic cell compartment in adult mice.
KLF6 Regulates Inducible Pro- and Anti-inflammatory Gene Expression in Macrophages
As KLF6 was induced by LPS, we hypothesized that KLF6 may play a role in pro-inflammatory macrophage gene expression. As a first step, RAW264.7 cells were transfected with pCI-neo-Klf6 or pCI-neo plasmid and stimulated with vehicle or 100 ng/ml LPS for 6 h, and pro-inflammatory gene expression was analyzed by qPCR. Klf6 overexpression augmented the LPS-induced expression of IL-1α, IL-1β, TNF-α, MCP-1, COX2, and MIP-1α (Fig. 4A). Next, we sought to examine the effect of Klf6 deficiency on LPS-induced pro-inflammatory gene expression in primary macrophages. Accordingly, Lyz2cre and Lyz2cre:Klf6fl/fl BMDMs were stimulated with LPS, and pro-inflammatory gene expression was analyzed by qPCR. Interestingly, KLF6 deficiency strongly attenuated LPS-induced pro-inflammatory gene (IL-1α, IL-1β, TNF-α, MCP-1, COX2, and MIP-1α) expression in macrophages (Fig. 4B).
FIGURE 4.

KLF6 regulates inducible pro- and anti-inflammatory gene expression in macrophages. A, RAW264.7 cells were transfected with either pCI-neo or pCI-neo-Klf6 using Lipofectamine® transfection reagent. These cells were stimulated with 100 ng/ml LPS for 6 h. Total RNA from these experiments were analyzed for IL-1α, IL-1β, TNF-α, MCP-1, COX-2,and MIP-1α, by quantitative PCR analysis. B, Lyz2cre and Lyz2cre:Klf6fl/fl mouse bone marrow-derived macrophages were stimulated with 100 ng/ml LPS for 6 h. Total mRNA from these experiments was analyzed for IL-1α, IL-1β, TNF-α, MCP-1, COX-2, and MIP-1α expression by quantitative PCR analysis. C, RAW264.7 cells were transiently transfected with either pCI-neo or pCI-neo-Klf6 using Lipofectamine® transfection reagent. These cells were exposed to 10 ng/ml IL-4 for 18 h. Total RNA from these experiments was analyzed for Arg1, Pdcd1lg2, Mrc1, Chi3l3, and Retnla expression by quantitative PCR analysis. D, Lyz2cre and Lyz2cre:Klf6fl/fl mouse bone marrow-derived macrophages were induced with 10 ng/ml IL-4 for 18 h. Total mRNA from these experiments was analyzed for Arg1, Pdcd1lg2, Mrc1, Chi3l3, and Retnla expression by quantitative PCR analysis. 36B4 was used as housekeeping gene for all the experiments. Each experiment was performed a minimum of three times. Data represent mean ± S.D., and a p value of less than 0.05 between indicated groups is considered significant.
Next, we examined whether KLF6 affects IL-4-induced anti-inflammatory gene expression in macrophages. Accordingly, RAW264.7 cells were transfected with control or KLF6 plasmid and induced with 10 ng/ml IL-4, and anti-inflammatory genes were analyzed by qPCR. As expected, IL-4 significantly induced expression of Arg1, Pdcd1lg2, Mrc1, Chi3l3, and Retnla compared with the control group (Fig. 4C). Intriguingly, overexpression of KLF6 in RAW264.7 cells significantly attenuated IL-4-induced anti-inflammatory gene targets. Interestingly, studies utilizing KLF6-deficient BMDMs revealed enhanced expression of Arg1, Pdcd1lg2, Mrc1, Chi3l3, and Retnla following IL-4 stimulation (Fig. 4D). Taken together, our results indicate that KLF6 promotes LPS-induced M1 gene expression but abrogates IL-4 induced M2 gene expression in macrophages.
Myeloid Klf6 Deficiency Results in Impaired Cutaneous Inflammation in Vivo
Next, we sought to determine whether myeloid KLF6 deficiency affects an acute inflammatory response in vivo. We employed the TPA-induced cutaneous inflammation model that is characterized by myeloid infiltration and tissue edema secondary to interstitial fluid accumulation. Accordingly, the right ear of Lyz2cre and Lyz2cre:Klf6fl/fl mice was treated with phorbol ester TPA, and the left ear served as vehicle control. Our results indicate that myeloid deficiency of KLF6 significantly attenuated TPA-induced inflammatory edema in Lyz2cre:Klf6fl/fl mice (Fig. 5A). To examine TPA-induced myeloid cell recruitment, ear tissue extracts from Lyz2cre and Lyz2cre:Klf6fl/fl mice were evaluated for myeloperoxidase activity. As shown in Fig. 5B, myeloid-specific deficiency of Klf6 significantly impaired myeloid cell recruitment in Lyz2cre:Klf6fl/fl mice. Next, we examined the role of myeloid KLF6 in balancing pro- and anti-inflammatory gene expression following TPA treatment. Accordingly, total RNA was obtained from Lyz2cre and Lyz2cre:Klf6fl/fl mouse ears following TPA or vehicle control treatment. Quantitative analysis of major pro-inflammatory cytokines, such as IL-1β, IL-6, MCP-1, and TNF-α, indicated that myeloid deficiency of KLF6 significantly attenuated expression of these cytokines following TPA exposure (Fig. 5, C–F). Concordant with this observation, deficiency of myeloid KLF6 significantly enhanced expression of anti-inflammatory genes, including Mrc1, Arg1, and Retnla. These results clearly indicate that myeloid deficiency of Klf6 significantly alters inflammatory status at the site of inflammation and modulates TPA-induced in vivo inflammation, including edema, myeloid cell infiltration, and major pro-inflammatory cytokine and chemokine expression.
FIGURE 5.

Myeloid Klf6 deficiency reduce cutaneous inflammation in vivo. A, Lyz2cre and Lyz2cre:Klf6fl/fl mice were subjected to TPA-induced cutaneous inflammation model. Ear weight from control and experimental groups was documented utilizing an analytical balance. The percentage increase in ear weight compared with the control is indicated. B, total protein extracts of ear tissue from vehicle and TPA-treated groups from Lyz2cre and Lyz2cre:Klf6fl/fl mice were subjected to myeloperoxidase activity assay. The data are indicated as MPO activity units/micrograms of total protein. C–I, total RNA from Lyz2cre and Lyz2cre:Klf6fl/fl mouse ear tissue exposed to TPA or vehicle control was analyzed for IL-1β, IL-6, MCP-1, TNF-α, MRC1, Arg1, and Retnla expression by quantitative PCR analysis. 36B4 was used as the housekeeping gene. The combined data from three experiments are shown. Data represent mean ± S.D., and a p value less than 0.05 between the indicated groups is considered significant.
KLF6 Regulates Macrophage Gene Expression by Modulating Functions of NF-κB and PPARγ
We next sought to develop a greater understanding of the molecular basis for KLF6's ability to differentially regulate M1/M2 targets. As a first step, we examined if KLF6 directly occupies target gene promoters following macrophage stimulation. Accordingly, wild-type BMDMs were stimulated with LPS, and chromatin immunoprecipitation was performed using anti-KLF6 antibody. Our analysis reveals low level KLF6 occupancy at the IL-1α (−976 to −980) and IL-1β (−601 to −605) promoters under unstimulated conditions was strongly enhanced following LPS stimulation (Fig. 6A). These results support the notion that KLF6 directly binds to inflammatory gene promoter and regulates their expression. In addition, we also noticed that pro-inflammatory genes that are induced by KLF6 (Fig. 4A) are also classical target genes of NF-κB (21). Therefore, we hypothesized that KLF6 may cooperate with NF-κB to regulate its transcriptional activity. To test this notion, RAW264.7 cells were co-transfected with NF-κB luciferase reporter plasmid with NF-κB p65 or pCI-neo-Klf6 plasmid and stimulated with LPS, and luciferase activity was measured. As expected, LPS induced NF-κB transcriptional activity (Fig. 6B). Overexpression of Klf6 alone strongly induced NF-κB transcriptional activity even in the absence of inflammatory stimuli such as LPS. Exposure of these cells to LPS significantly enhanced NF-κB transcriptional activity over cells treated with LPS alone. Overexpression of NF-κB p65 significantly enhanced NF-κB transcriptional activity in the absence or presence of LPS stimulation. Interestingly, overexpression of Klf6 and NF-κB-p65 together significantly enhanced NF-κB transcriptional activity compared with cells with Klf6 or NF-κB-p65 alone. These results suggest that KLF6 cooperates with NF-κB to modulate its transcriptional activity. Next, we investigated whether KLF6 regulates NF-κB recruitment to the pro-inflammatory gene promoter following LPS stimulation. Our results indicate that neither overexpression of Klf6 nor deficiency of Klf6 altered NF-κB p65 recruitment to the IL-1α promoter following LPS stimulation (Fig. 6, C and D). Therefore, we hypothesized that KLF6 may regulate the critical co-activator component required for optimal NF-κB activity. Indeed, our analysis indicated that overexpression of KLF6 promoted and deficiency of KLF6 reduced recruitment of the critical NF-κB transcriptional co-activators p300 to the IL-1α promoter (Fig. 6, C and D). Next, we examined whether KLF6 regulates LPS-induced pro-inflammatory gene expression through cooperating with NF-κB signaling. Accordingly, RAW264.7 cells were co-transfected with KLF6 or NF-κB p65 and stimulated with LPS, and total RNA was analyzed for IL-1α expression (Fig. 6F). Our results suggest that overexpression of Klf6 or NF-κB p65 alone significantly enhanced LPS-induced IL-1α expression. However, combined overexpression of KLF6 and NF-κB p65 dramatically enhanced LPS-induced IL-1α expression in macrophages. To further confirm these observations, RAW264.7 cells were transfected with Klf6 and treated with the NF-κB peptide inhibitor SN-50. These cells were stimulated with LPS, and total RNA was analyzed for IL-1α expression. As shown in Fig. 6E, SN-50 treatment significantly diminished LPS-induced and KLF6-mediated IL-1α expression in macrophages. These results indicate that KLF6 cooperates with NF-κB to regulate LPS-induced pro-inflammatory gene expression in macrophages.
FIGURE 6.

KLF6 regulates macrophage polarization by modulating functions of NF-κB. A, wild-type BMDMs were stimulated with 10 ng/ml LPS, and KLF6 ChIP analysis was performed on IL-1α (−976 to −980) and IL-1β (−601 to −605) promoters. Fold changes in KLF6 enrichment over control are indicated. ChIP analysis performed using IgG was used as negative control. B, RAW264.7 cells were co-transfected with NF-κB concatemer luciferase reporter plasmid in the presence of KLF6 or NF-κB-p65 plasmid. These cells were stimulated with LPS, and induction in luciferase was documented and is indicated as relative luciferase activity over the control group. C and D, RAW264.7 cells overexpressed with Klf6 and Lyz2cre, Lyz2cre:Klf6fl/fl mouse BMDMs were induced with 10 ng/ml LPS. ChIP analysis was performed on IL-1α promoter (−1787 to −1796) utilizing anti-p65 and anti-p300 antibody. ChIP analysis performed using IgG was used as negative control. E, RAW264.7 cells were co-transfected with Klf6 or NF-κB-p65 plasmid. These cells were stimulated with LPS, and total RNA from these cells was isolated. IL-1α mRNA expression was analyzed by quantitative PCR and normalized to 36B4. F, RAW264.7 cells were transfected with Klf6 plasmid. These cells were stimulated with LPS in the presence of NF-κB peptide inhibitor SN-50, and total RNA from these cells was isolated. IL-1α mRNA expression was analyzed by quantitative PCR and normalized to 36B4.
Next, we investigated how KLF6 may inhibit M2 gene expression. As a first step, we examined whether KLF6 directly occupies the anti-inflammatory gene promoter by ChIP analyses in wild-type BMDMs stimulated with vehicle or IL-4 (Fig. 7A). KLF6 was enriched at both the Arg1 (−1009 to −1013) and Mrc1 (−1637 to −1642) promoters under unstimulated conditions; occupancy was strongly reduced following IL-4 stimulation. Furthermore, as these targets are also known to be regulated by PPARγ, we examined whether KLF6 affected PPARγ expression in macrophages. Our results revealed that deficiency of KLF6 caused a significant increase in PPARγ expression under unstimulated condition as well as following IL-4 stimulation (Fig. 7B). Concordant with this result, overexpression of KLF6 in RAW264.7 cells significantly attenuated IL-4-induced PPARγ expression (Fig. 7C). Our analysis at the protein level indicated that deficiency of KLF6 induced and overexpression of KLF6 attenuated IL-4-induced PPARγ expression in macrophages (Fig. 7D). Next, we investigated whether KLF6 regulates PPARγ recruitment to anti-inflammatory genes such as the arginase1 promoter following IL-4 stimulation. Our results demonstrate that overexpression of KLF6 reduced and deficiency of KLF6 induced PPARγ recruitment to the arginase1 promoter following IL-4 stimulation (Fig. 7, E and F).
FIGURE 7.

KLF6 regulates macrophage polarization by modulating functions of PPARγ. A, wild-type BMDMs were stimulated with 10 ng/ml IL-4, and KLF6 ChIP analysis was performed on Arg1 (−1009 to −1013) and Mrc1 (−1637 to −1642) promoters. Fold changes in KLF6 enrichment over control are indicated. ChIP analysis performed using IgG was used as negative control. B, Lyz2cre and Lyz2cre:Klf6fl/fl mouse BMDMs were induced with 10 ng/ml IL-4 for 18 h. Total mRNA was analyzed for PPARγ expression by quantitative PCR analysis. C, RAW264.7 cells were overexpressed with KLF6 and stimulated with 10 ng/ml of IL-4. These cells are analyzed for PPARγ expression by quantitative PCR analysis. D and E, RAW264.7 cells were overexpressed with Klf6, and Lyz2cre/ Lyz2cre:Klf6fl/fl mouse BMDMs were induced with 10 ng/ml IL-4 for 18 h. Total cell lysate was analyzed for PPARγ protein expression by Western blot analysis. Actin was used as loading control. E and F, RAW264.7 cells overexpressed with Klf6 and Lyz2cre, Lyz2cre:Klf6fl/fl mouse BMDMs were induced with 10 ng/ml IL-4. ChIP analysis was performed on arginase1 promoter (−986 to −1003) utilizing anti-PPARγ antibody. ChIP analysis performed using IgG was used as negative control. G, Lyz2cre and Lyz2cre:Klf6fl/fl mouse BMDMs were exposed to GW9662 (2.5 μm) or rosiglitazone (5 μm). These cells were induced with 10 ng/ml IL-4 for 18 h, and Arg1 mRNA expression was analyzed by quantitative PCR analysis. H, Lyz2cre and Lyz2cre:Klf6fl/fl mouse BMDMs were transfected with control siRNA or siRNA targeting PPARγ by nucleofection. These cells were induced with IL-4 for 18 h, and Arg1 mRNA expression was analyzed by quantitative PCR analysis. - was used as a housekeeping gene. Each experiment was performed a minimum of three times. Data represent mean ± S.D., and a p value less than 0.05 between indicated groups are considered significant.
Next, we examined whether the PPARγ agonist/antagonist can modulate IL-4-induced anti-inflammatory gene expression in KLF6-deficient macrophages. Accordingly, Lyz2cre and Lyz2cre:Klf6fl/fl BMDMs were stimulated with IL-4 in the presence or absence of the PPARγ agonist rosiglitazone or antagonist GW9662 (Fig. 6H). The Arg1 expression levels were analyzed by quantitative PCR analysis. Our results indicate that IL-4-induced Arg1 expression was significantly attenuated following GW9662 treatment. Interestingly, GW9662 treatment abolished elevated levels of Arg1 level in Lyz2cre:Klf6fl/fl BMDMs. By contrast, rosiglitazone treatment enhanced IL-4-induced Arg1 expression of Lyz2cre:Klf6fl/fl BMDMs compared with Lyz2cre BMDMs. Similar results were observed with PPARγ-specific antagonist T0070907 or agonist GW1929 (data not shown). Next, we examined whether genetically altering the PPARγ level in KLF6-deficient macrophages could alter IL-4-induced Arg1 expression in these macrophages. Accordingly, Lyz2cre Lyz2cre:Klf6fl/fl BMDMs were nucleofected with PPARγ-specific siRNA or control siRNA. These cells were stimulated with IL-4, and macrophage Arg1 expression was evaluated by quantitative PCR analysis. As shown in Fig. 7H, IL-4-induced Arg1 expression was significantly diminished in Lyz2cre:Klf6fl/fl BMDMs nucleofected with PPARγ-specific siRNA. Taken together, these results suggest that KLF6 serves as a repressor of M2 target genes through direct binding and regulation of PPARγ.
DISCUSSION
The central findings of this study are as follows: 1) KLF6 mRNA and protein are abundantly expressed in human and murine macrophages; 2) pro-inflammatory stimuli such as IFN-γ and LPS induce KLF6 mRNA/protein expression in human and murine macrophages; 3) anti-inflammatory stimuli such as IL-4 and IL-13 suppress KLF6 mRNA/protein expression in human and murine macrophages; 4) KLF6 promotes LPS-induced pro-inflammatory gene expression in macrophages; 5) KLF6 attenuates IL-4-induced anti-inflammatory gene expression in macrophages; 6) myeloid-specific deficiency of KLF6 significantly reduced TPA-induced cutaneous inflammation; and 7) KLF6 regulates macrophage inflammatory gene expression by modulating functions of NF-κB and PPARγ. Collectively, these observations identify KLF6 as a molecular toggle that promotes macrophage pro-inflammatory gene expression while suppressing anti-inflammatory gene expression (Fig. 8).
FIGURE 8.
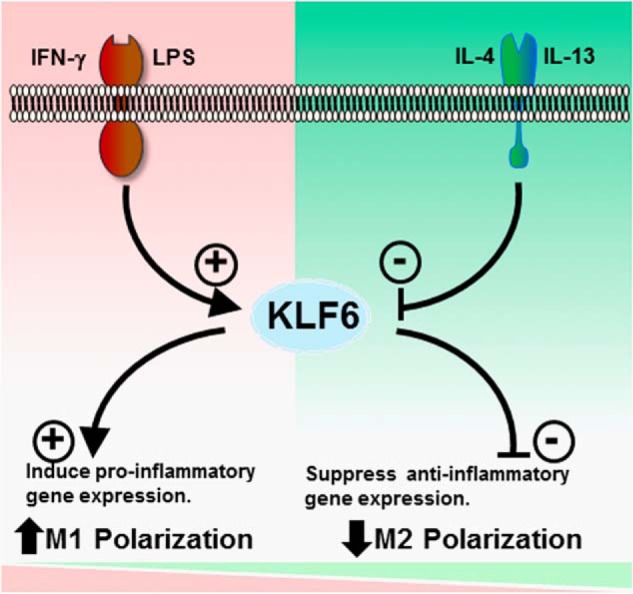
KLF6 promotes Th1 stimulus-induced M1 gene expression and suppresses Th2 stimulus-induced M2 gene expression in macrophages.
KLF6 is a broadly expressed member of the KLF family that has been implicated in a number of key cellular processes such as development, differentiation, proliferation, and programmed cell death in diverse cell types (22). Alterations in KLF6 expression or function have been implicated in the pathogenesis of numerous human diseases, including cancer, hepatic steatosis, and hepatic fibrosis (13). Previous studies have also linked this factor to hematopoietic biology (20). For example, although homozygous mutation of Klf6 was embryonically lethal at E12.5, mutants were noted to exhibit severe reductions in yolk sac hematopoiesis. Consistent with this finding, embryoid body differentiation studies demonstrated defects in erythrocyte, macrophage, and mixed colony formation (20). However, the role of KLF6 in adult hematopoietic cell function has not been investigated. Our approach to delete the myeloid compartment employed lysozyme Mcre. Although this cre is expressed at low levels during myeloid development, its activity is most significant after activation in the mature state (23). Thus, it is not surprising that development of the myeloid and other hematopoietic lineages was unaffected. However, our findings do support a key role for KLF6 in toggling the two inflammatory phenotypic states of a macrophage. These findings do provide the impetus for future investigations focused on the role of KLF6 in the function of other hematopoietic lineages.
Studies from multiple laboratories highlight the importance of transcriptional control in macrophage polarization and function (24). The activation of stimulus-specific transcription factors allows expression of a subset of genes that confer the functional properties of the polarized state (8). A number of major transcription factor families have been linked to macrophage subset specification such as NF-κB, STATs, HIFs, IRFs, and PPARs. In some cases, distinct members of the same family have been identified with a particular phenotypic state. For example, Stat1 regulates M1, and Stat6 promotes the M2 phenotype (25). Similar effects have been reported for HIF-1α/HIF-2α and IRF5/IRF4 (7, 26). In this regard, the findings in this study coupled with recent reports are particularly relevant. Previous studies have indicated that that KLF2 and KLF4 promote the anti-inflammatory phenotype (14, 15). In contrast to KLF6, KLF4 was induced by IL-4/IL-13 and reduced by LPS/INFγ. Furthermore, KLF2 and KLF4 induced anti-inflammatory targets and inhibited pro-inflammatory targets findings that are essentially the diametric opposite of those observed in this study for KLF6. Finally, KLF4 was found to augment PPARγ expression/activity while reducing NF-κB function (15). Here, KLF6 was found to reduce PPARγ expression/activity while cooperating with NF-κB to augment pro-inflammatory gene expression (Figs. 6 and 7). Collectively, these findings raise the intriguing possibility that in response to external cues the differential regulation of macrophage KLFs is an important event required to exact characteristic gene programs. Furthermore, these effects are likely coordinated through intersection with other major regulatory families such as NF-κB and PPARγ.
In summary, the in vitro, ex vivo, and in vivo observations presented here highlight the importance of KLF6 in macrophage polarization (Fig. 8). As polarization has been shown to be important in various macrophage functions in acute (e.g. infections and sepsis) and chronic inflammatory conditions (e.g. obesity, insulin resistance, and atherosclerosis), future studies assessing the effect of myeloid Klf6 deletion on these biological processes are warranted. Confirmation would provide the requisite stimulus for efforts targeting KLF6 for therapeutic gain in the treatment of numerous inflammatory disease states.
This work was supported, in whole or in part, by National Institutes of Health Grants HL097023 (to G. H. M.), HL57506 MERIT Award (to D. I. S.), HL72952, HL75427, HL76754, and HL086548 (to M. K. J.), and R00 phase of K99/R00 Award from NHLBI Pathway to Independence Award (to the G. H. M. laboratory). This work was also supported by American Heart Association Scientist Development Grant 11SDG7390041 (to R. D.).
- PPARγ
- peroxisome proliferator-activated receptor γ
- KLF
- Kruppel-like transcription factor
- BMDM
- bone marrow-derived macrophage
- HIF-1α
- hypoxia-inducible factor-1α
- NF-κB
- nuclear factor-κB
- TPA
- phorbol ester 12-O-tetradecanoylphorbol-13-acetate
- PM
- peritoneal macrophage
- qPCR
- quantitative PCR.
REFERENCES
- 1. Olefsky J. M., Glass C. K. (2010) Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246 [DOI] [PubMed] [Google Scholar]
- 2. Serbina N. V., Jia T., Hohl T. M., Pamer E. G. (2008) Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26, 421–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murray P. J., Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muller W. A. (2011) Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. 6, 323–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Auffray C., Sieweke M. H., Geissmann F. (2009) Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27, 669–692 [DOI] [PubMed] [Google Scholar]
- 6. Medzhitov R., Horng T. (2009) Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 [DOI] [PubMed] [Google Scholar]
- 7. Sica A., Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lawrence T., Natoli G. (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 [DOI] [PubMed] [Google Scholar]
- 9. Biswas S. K., Chittezhath M., Shalova I. N., Lim J. Y. (2012) Macrophage polarization and plasticity in health and disease. Immunol. Res. 53, 11–24 [DOI] [PubMed] [Google Scholar]
- 10. Szabo S. J., Sullivan B. M., Peng S. L., Glimcher L. H. (2003) Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 21, 713–758 [DOI] [PubMed] [Google Scholar]
- 11. Odegaard J. I., Chawla A. (2011) Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 6, 275–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szanto A., Balint B. L., Nagy Z. S., Barta E., Dezso B., Pap A., Szeles L., Poliska S., Oros M., Evans R. M., Barak Y., Schwabe J., Nagy L. (2010) STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 33, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andreoli V., Gehrau R. C., Bocco J. L. (2010) Biology of Kruppel-like factor 6 transcriptional regulator in cell life and death. IUBMB Life 62, 896–905 [DOI] [PubMed] [Google Scholar]
- 14. Mahabeleshwar G. H., Kawanami D., Sharma N., Takami Y., Zhou G., Shi H., Nayak L., Jeyaraj D., Grealy R., White M., McManus R., Ryan T., Leahy P., Lin Z., Haldar S. M., Atkins G. B., Wong H. R., Lingrel J. B., Jain M. K. (2011) The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity 34, 715–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liao X., Sharma N., Kapadia F., Zhou G., Lu Y., Hong H., Paruchuri K., Mahabeleshwar G. H., Dalmas E., Venteclef N., Flask C. A., Kim J., Doreian B. W., Lu K. Q., Kaestner K. H., Hamik A., Clément K., Jain M. K. (2011) Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 121, 2736–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis B. K. (2013) Isolation, culture, and functional evaluation of bone marrow-derived macrophages. Methods Mol. Biol. 1031, 27–35 [DOI] [PubMed] [Google Scholar]
- 17. Leow C. C., Wang B. E., Ross J., Chan S. M., Zha J., Carano R. A., Frantz G., Shen M. M., de Sauvage F. J., Gao W. Q. (2009) Prostate-specific Klf6 inactivation impairs anterior prostate branching morphogenesis through increased activation of the Shh pathway. J. Biol. Chem. 284, 21057–21065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nayak L., Goduni L., Takami Y., Sharma N., Kapil P., Jain M. K., Mahabeleshwar G. H. (2013) Kruppel-like factor 2 is a transcriptional regulator of chronic and acute inflammation. Am. J. Pathol. 182, 1696–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao Z., Sun X., Icli B., Wara A. K., Feinberg M. W. (2010) Role of Kruppel-like factors in leukocyte development, function, and disease. Blood 116, 4404–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsumoto N., Kubo A., Liu H., Akita K., Laub F., Ramirez F., Keller G., Friedman S. L. (2006) Developmental regulation of yolk sac hematopoiesis by Kruppel-like factor 6. Blood 107, 1357–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lawrence T., Bebien M., Liu G. Y., Nizet V., Karin M. (2005) IKKα limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 434, 1138–1143 [DOI] [PubMed] [Google Scholar]
- 22. McConnell B. B., Yang V. W. (2010) Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 90, 1337–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ye M., Iwasaki H., Laiosa C. V., Stadtfeld M., Xie H., Heck S., Clausen B., Akashi K., Graf T. (2003) Hematopoietic stem cells expressing the myeloid lysozyme gene retain long-term, multilineage repopulation potential. Immunity 19, 689–699 [DOI] [PubMed] [Google Scholar]
- 24. Ostuni R., Natoli G. (2011) Transcriptional control of macrophage diversity and specialization. Eur. J. Immunol. 41, 2486–2490 [DOI] [PubMed] [Google Scholar]
- 25. Hawiger J. (2001) Innate immunity and inflammation: a transcriptional paradigm. Immunol. Res. 23, 99–109 [DOI] [PubMed] [Google Scholar]
- 26. Martinez F. O., Helming L., Gordon S. (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483 [DOI] [PubMed] [Google Scholar]