

Background: Muscle-specific deficiency of iron-sulfur (Fe-S) cluster scaffold protein (ISCU) leads to myopathy.
Results: Cells carrying the myopathy-associated G50E ISCU mutation demonstrate impaired Fe-S cluster biogenesis and mitochondrial dysfunction.
Conclusion: Reduced mitochondrial respiration as a result of diminished Fe-S cluster synthesis results in muscle weakness in myopathy patients.
Significance: The molecular mechanism behind disease progression should provide invaluable information to combat ISCU myopathy.
Keywords: Electron Transport System (ETS), Iron-Sulfur Protein, Mitochondrial Diseases, Molecular Chaperone, Reactive Oxygen Species (ROS), Hsp70, J-protein, Iron-Sulfur Cluster, Myopathy
Abstract
Iron-sulfur (Fe-S) clusters are versatile cofactors involved in regulating multiple physiological activities, including energy generation through cellular respiration. Initially, the Fe-S clusters are assembled on a conserved scaffold protein, iron-sulfur cluster scaffold protein (ISCU), in coordination with iron and sulfur donor proteins in human mitochondria. Loss of ISCU function leads to myopathy, characterized by muscle wasting and cardiac hypertrophy. In addition to the homozygous ISCU mutation (g.7044G→C), compound heterozygous patients with severe myopathy have been identified to carry the c.149G→A missense mutation converting the glycine 50 residue to glutamate. However, the physiological defects and molecular mechanism associated with G50E mutation have not been elucidated. In this report, we uncover mechanistic insights concerning how the G50E ISCU mutation in humans leads to the development of severe ISCU myopathy, using a human cell line and yeast as the model systems. The biochemical results highlight that the G50E mutation results in compromised interaction with the sulfur donor NFS1 and the J-protein HSCB, thus impairing the rate of Fe-S cluster synthesis. As a result, electron transport chain complexes show significant reduction in their redox properties, leading to loss of cellular respiration. Furthermore, the G50E mutant mitochondria display enhancement in iron level and reactive oxygen species, thereby causing oxidative stress leading to impairment in the mitochondrial functions. Thus, our findings provide compelling evidence that the respiration defect due to impaired biogenesis of Fe-S clusters in myopathy patients leads to manifestation of complex clinical symptoms.
Introduction
Iron-sulfur (Fe-S) clusters are indispensable and ubiquitous cofactors that are involved in a variety of regulatory processes, including catalysis and electron carrier activity (1). Although the Fe-S clusters are relatively simple inorganic cofactors, their synthesis, assembly, and successive incorporation into apoproteins are highly intricate processes in living cells (2, 3). Fe-S clusters are usually integrated into proteins through coordination of the iron atoms by cysteine or histidine residues, although in more complex Fe-S clusters alternative ligands like Asp, Arg, and Ser and functional groups like CO and CN have been reported (4). Coordination of Fe-S clusters to electron transport chain complexes is indispensable for respiratory function within the cell. For example, complex I of bacteria contains nine Fe-S clusters, whereas eukaryotic complex I harbors eight Fe-S clusters, which are anchored to domains exposed to the cytosol or mitochondrial matrix, respectively (5, 6). On the other hand, mammalian complex II possesses three distinct Fe-S clusters of the [2Fe-2S], [3Fe-4S], and [4Fe-4S] type (7). Besides their importance in electron transfer, Fe-S proteins also play a pivotal role in enzyme-substrate reactions. Eukaryotic Fe-S cluster-containing enzymes, such as succinate dehydrogenase and aconitase, play a critical role in TCA6 cycle metabolism (8, 9). Therefore, the biogenesis of these functionally important Fe-S clusters in mitochondria is an indispensable process for mitochondrial function as well as cell survival.
Mitochondria are the major cellular compartment for Fe-S cluster biogenesis in eukaryotes, including the mammalian system. The central part of Fe-S protein biogenesis in human mitochondria is the de novo synthesis of the Fe-S cluster on a highly conserved scaffold protein, ISCU, before its transfer to apoproteins (10). Mammalian ISCU is a nuclear encoded protein, predominantly localized in the mitochondrial matrix compartment, and comprises 167 amino acids with an N-terminal targeting signal. However, the presence of cytosolic ISCU has also been reported in humans (11). In Saccharomyces cerevisiae, there are two orthologs of human ISCU, namely Isu1 and Isu2, which are localized in the matrix compartment (12). In yeast, the ISU1 and ISU2 double deletion mutant is inviable, thus signifying its central importance in the Fe-S cluster biogenesis (12). The overall biogenesis process can be broadly categorized into two critical events: (a) the de novo assembly of an Fe-S cluster on a scaffold protein and (b) the transfer of the Fe-S cluster from the scaffold to target apoproteins (13). A cysteine desulfurase, Nfs1, assists in the sulfur transfer process to the scaffold protein (14). This reaction is aided by direct interaction between Nfs1 and the scaffold protein (11, 15). Upon transfer of iron and sulfur to the scaffold, the Fe-S cluster is formed by an unknown mechanism (13, 16). The iron-binding protein frataxin (Yfh1 in yeast and CyaY in bacteria) is believed to function as an iron donor (17). The terminal transfer process is assisted by a dedicated chaperone system comprising the mtHsp70 (Ssq1 in yeast) and the DnaJ-like cochaperone, HSCB (Jac1 in yeast) (18, 19). The interaction of a J-protein cochaperone, such as Jac1 (HSCB in humans), with Fe-S scaffold Isu1 is conserved in evolution and indispensable in vivo (15, 20, 21).
Because Fe-S proteins play a critical role in a wide range of cellular activities, a mutation in different components of the synthesis machinery disrupts the process of Fe-S cluster biogenesis and is thus associated with multiple pathological conditions in humans. For instance, one mutation identified in the human mitochondrial iron-sulfur assembly enzyme, ISCU, is known to cause severe myopathy (ISCU myopathy; OMIM *611911). ISCU myopathy is a recessively inherited disorder characterized by lifelong exercise intolerance, where minor exertion causes pain of active muscles, shortness of breath, fatigue, and tachycardia (22, 23). The disease is non-progressive, but in certain cases, metabolic acidosis, rhabdomyolysis, and myoglobinuria have also been reported (24, 25). Myopathy as a result of ISCU deficiency was found to have high incidence rates in individuals of Northern European ancestry with a carrier rate of 1:188 in the Northern Swedish population (23). Most affected individuals are homozygous for a mutation in intron 4 (g.7044G→C) of ISCU that results in synthesis of aberrantly spliced ISCU mRNA, successively causing accumulation of truncated non-functional ISCU protein (22, 26, 27). Recently, a progressive myopathy associated with early onset of severe muscle weakness, extreme exercise intolerance, and cardiomyopathy has been reported in some patients. Interestingly, these patients were compound heterozygous for the common intronic splice mutation (g.7044G→C) on one allele, leading to truncated protein and a novel (c.149G→A) missense mutation in exon 3 on the other allele. The missense mutation in exon 3 changes a completely conserved glycine residue to a glutamate at the 50th position (G50E) in the amino acid sequence (28). The transmission of the G50E mutation alone was found to be recessive because the carrier population did not show significant symptoms of the disease. However, the exact molecular mechanisms of disease development as a result of G50E mutation in ISCU in conjunction with the g.7044G→C allele in compound heterozygous patients have not been elucidated.
Due to the critical function played by ISCU scaffold protein in the Fe-S cluster biogenesis process in humans, the G50E mutant is expected to contribute significantly toward ISCU myopathy. In this report, we delineate the impact of the G50E mutation on mitochondrial function by utilizing the HeLa cell line and yeast as a model system. Our findings highlight that the G50E mutation leads to severe growth defects, compromised Fe-S cluster-containing enzyme activity, sensitivity to oxidative stress, increased cellular reactive oxygen species (ROS), elevated iron level, and reduced interaction of scaffold protein with its interacting partners, thus contributing significantly toward mitochondrial myopathy. Moreover, at the protein level, the G50E mutation was found to form a higher order oligomeric structure that probably reduces the functionality of the protein.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HeLa cells were transfected with pCI-neo-ISCU and pCI-neo-G50E ISCU using Lipofectamine 2000 for expression of wild type ISCU and G50E ISCU. Cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Sigma). The cells were incubated at 37 °C in 5% CO2 for 48 h prior to experiments. The adherent cultures of HeLa cells were grown to ∼90% confluence. The cells were trypsinized, washed with ice-cold 1× phosphate-buffered saline (PBS), pH 7.4, and utilized for different experiments and mitochondria isolation.
Yeast Strains, Genetic Analysis, Plasmid Construction, and Mutagenesis
For genetic analysis in yeast, full-length human WT ISCU and yeast WT ISU1 were amplified from a HeLa cell cDNA library (Stratagene) and W303 yeast genomic DNA, respectively. ISCU and ISU1 with a C-terminal FLAG tag were cloned in pRS414 yeast expression vector under the TEF or GPD promoter containing a Trp marker for selection. The Δisu1/isu2 double deleted yeast haploid strain, W303 (trp1-1 ura3-1 leu2-3, 112 his3-11, 15 ade2-1 can1–100 GAL2+ met2-Δ1 lys2-Δ2 isu2::HIS3 isu1::LEU2), contained the plasmid pRS316-ISU1 for the maintenance of viability. For in vivo phenotype analysis, the haploid Δisu1/isu2 strain was transformed with pRS414-ISCU, pRS414-G50E ISCU, pRS414-ISU1, and pRS414-isu1G50E. The transformants were selected on tryptophan omission plates at 30 °C. The transformants (Trp+) were then subjected to spot test analysis using serial dilutions of the cells on different media like Trp−, Trp− containing H2O2, and medium containing 5-fluoroorotic acid (U.S. Biological) to eliminate the WT ISU1 containing the plasmid pRS316.
For purification of ISCU and Isu1 using a bacterial expression system, the ORFs without N-terminal signal sequences of ISCU (aa 35–167) and ISU1 (aa 37–165) were inserted in between the BamHI-SalI restriction sites of the pRSFDuet-1 vector, carrying an N-terminal vector backbone His6 tag. The glutathione S-transferase (GST) fusion constructs of hNFS1 (aa 56–457 into BamHI-SalI), yNFS1 (aa 37–497 into BamHI-XhoI), HSCB (aa 22–235 into BamHI-SalI), and JAC1 (aa 1–184 into BamHI-XhoI) were generated by introducing the respective coding sequences downstream of the GST tag in the pGEX-KG vector, respectively. For mammalian transfection experiments, the full-length human ISCU and G50E ISCU were cloned downstream to a CMV-based promoter of pCI-neo vector (Promega) with a FLAG tag at the 3′-end of the open reading frame. The G50E point mutation was created through QuikChange site-directed mutagenesis, using high fidelity Pfu Turbo DNA polymerase (Stratagene). All of the clones were verified by DNA sequencing reactions carried out at Eurofins Inc. and Macrogen Inc.
Protein Expression and Purification
For purification of N-terminal His6-tagged human WT ISCU and G50E ISCU or yeast WT Isu1 and Isu1G50E, the proteins were expressed in Escherichia coli BL21(DE3) strain by allowing growth at 30 °C to an A600 of 0.6, followed by induction using 0.5 mm IPTG for 6 h. Cells were harvested by centrifugation and then lysed in buffer A (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1 mm dithiothreitol, 20 mm imidazole, 1 mm PMSF, and 10% glycerol) along with 0.2 mg/ml lysozyme, followed by incubation at 4 °C for 1 h. The samples were gently treated with 0.2% deoxycholate, followed by DNase I (10 μg/ml) treatment for 15 min at 4 °C. The cells were further lysed by sonicating three times (for 15 s each) at 25% amplitude using an Ultrasonic processor with 2-min intervals in ice. The cell lysates were clarified by centrifuging at 22,000 × g for 30 min at 4 °C. The supernatant was incubated with nickel-nitrilotriacetic acid-Sepharose (GE Healthcare) for 2 h at 4 °C. Unbound proteins and nonspecific contaminants were removed by multiple washes of buffer A alone followed by sequential single washes of buffer B (buffer A along with 0.05% Triton X-100), buffer C (buffer A along with 1 mm ATP, 10 mm MgCl2), buffer D (buffer A along with 1 m NaCl), and buffer E (buffer C along with 40 mm imidazole). Finally, the bound proteins were eluted with buffer A containing 250 mm imidazole. Purification of other proteins was also achieved by the same strategy as mentioned above with minor modifications. For GST-tagged proteins, the purification was performed in a conventional manner using the GST-Sepharose beads (GE Healthcare). The bound proteins were stored in buffer containing 50 mm Tris-Cl, pH 7.5, and 100 mm NaCl at 4 °C.
Measurement of Mitochondrial ROS Levels by FACS
In order to analyze the extent of superoxide generation by the mitochondria, we utilized MitoSOX Red dye (Molecular Probes). This dye is specifically targeted to the mitochondria, where it undergoes oxidation by the mitochondrial superoxide radicals and fluoresces upon binding to mitochondrial DNA, with an emission maximum of 580 nm. Transfected HeLa cells cultured for 48 h and 0.1 OD (A600) of yeast cells from the early log phase were harvested and incubated with the dye (0.5 μm for HeLa and 2.5 μm for yeast) for 10 min, following which they were washed with 1× PBS and subjected to FACS analysis using a 488-nm argon laser for excitation (BD FACS Canto II). The respiratory inhibitor rotenone (1 mm) was used as a positive control for generating higher superoxide levels. The mean fluorescence intensity values of 10,000 events were recorded per sample and plotted to compare the relative ROS levels using the WinMDI version 2.9 software (29).
Fluorescence Imaging in Cell Lines
HeLa cells harboring WT ISCU and G50E ISCU were seeded on a coverslip (5000 cells/well in 12-well plates) and cultured for 48 h. Medium was removed, cells were washed with 1× PBS, and cells were stained with 0.1 μm MitoSOX Red and 250 ng/ml Hoechst 33342 for 15 min in 300 μl of 1× PBS at 37 °C in a 5% CO2 incubator. Prior to analysis, cells were twice washed with 1× PBS. Images were acquired using a ×63 objective lens on a Zeiss apotome fluorescence microscope. For MitoSOX Red fluorescence imaging of yeast, 0.1 OD (A600) of cells from mid-log phase grown at 37 °C were incubated with 5 μm dye for 15 min. The cells were washed twice with 1× PBS before analysis. Images were acquired using a ×100 objective lens on a Leica fluorescence microscope. A similar strategy was used for H2DCF-DA and calcein blue staining. HeLa cells were treated with 1 μm H2DCF-DA for 15 min, and yeast cells were treated with 5 μm H2DCF-DA for 20 min, and for calcein blue staining, HeLa cells were treated with 3 μm calcein blue for 20 min. To test the specificity of calcein blue staining, the mutant cells were treated with a 1 mm concentration of the iron-specific chelator deferoxamine (DFO) (30). Images of HeLa cells were taken using a ×63 objective lens of the Zeiss apotome fluorescence microscope, and images of the yeast cell were taken with a ×100 objective lens on a Leica fluorescence microscope.
In Vitro GST Pull-down Analysis
Purified GST-hNFS1 (1.5 μm)/GST-yNfs1 (1.5 μm) and GST-HSCB (1 μm)/GST-Jac1 (2.5 μm) were incubated with a 10-μl bed volume of glutathione-agarose beads in 200 μl of GST buffer A (50 mm Tris-Cl, pH 7.5, 100 mm NaCl, 40 mm imidazole, 50 μm pyridoxal phosphate, 10% glycerol) and GST buffer B (50 mm Tris-Cl, pH 7.5, 100 mm NaCl, 40 mm imidazole, 0.2% Triton X-100, 5% glycerol) for an interaction study with either WT or mutant ISCU/Isu1, respectively. Unbound proteins were removed by washing the beads three times with the respective GST buffers. After removing unbound proteins, the samples were blocked with 0.1% BSA for 20 min at 20 °C, followed by washing two times with GST buffer to remove excess unbound BSA. The beads were resuspended in 200 μl of GST binding buffer and incubated with increasing concentrations of either WT or mutant ISCU/Isu1 for 30 min at 20 °C. The GST beads were washed three times with GST buffer and resolved on SDS-PAGE followed by Coomassie dye staining.
Enzymatic Activity Assay and Mitochondrial ATP Level Quantification
Enzymatic assays of complex I, complex II, and complex IV were performed according to a protocol described previously (31). Briefly, 15 μg of protein corresponding to mitochondria isolated from HeLa cells were subjected to three cycles of freeze-thawing before complex I activity was assayed. Mitochondria were resuspended in 50 mm of potassium phosphate buffer (pH 7.5) containing 100 μm NADH, 3 mg/ml fatty acid-free BSA, 300 μm sodium azide (NaN3). The reaction was started by adding 60 μm ubiquinone, and the decrease of absorbance at 340 nm was monitored for 2 min. To check for specificity of the reaction, the experiment was performed both in the presence and absence of the specific inhibitor rotenone (10 μm). For complex II activity analysis, 2 μg of protein corresponding to HeLa cell mitochondria and 10 μg of protein corresponding to yeast cell mitochondria were resuspended in 25 mm potassium phosphate buffer (pH 7.5) containing 20 mm succinate, 1 mg/ml fatty acid-free BSA, 300 μm NaN3, 10 μm rotenone, 10 μg/ml antimycin A, and 80 μm 2,6-dichlorophenolindophenol, followed by incubation at 37 °C for 10 min. The reaction was started by adding 60 μm ubiquinone, and the decrease in the absorbance at 600 nm was recorded for 3 min. For complex IV activity, 2 μg of protein corresponding to HeLa cell mitochondria and 10 μg of protein corresponding to yeast cell mitochondria were resuspended in 50 mm potassium phosphate buffer (pH 7). The reaction was started by adding 60 μm reduced cytochrome c, and the decrease in absorbance at 550 nm for 3 min was observed. To check the specificity of complex IV activity, this experiment was performed in the presence and in the absence of complex IV inhibitor NaN3 (300 μm). Aconitase activity was also measured following a similar protocol as described earlier (32). Briefly, 25 μg of protein corresponding to HeLa cell mitochondria and 50 μg of protein corresponding to yeast cell mitochondria were dissolved in buffer A (50 mm Tris-Cl, pH 8.0, 50 mm NaCl) containing 1% deoxycholic acid and incubated for 10 min. The reaction was started by the addition of sodium citrate dihydrate, and the absorbance increase at 235 nm for 2 min was followed in a quartz cuvette.
Mitochondrial ATP was quantified using the mitochondrial ToxGloTM assay kit (Promega). Briefly, 1–2 μg of mitochondria were resuspended in 80 μl of SE buffer (250 mm sucrose, 1 mm EDTA, 10 mm MOPS-KOH, pH 7.2) and mixed with 20 μl of 5× cytotoxicity reagent (bis-AAF-R110) for 1 min by orbital shaking (700 rpm). The reaction mixture was incubated at 37 °C for 30 min followed by equilibration of the assay plate at room temperature (25 °C) for 5 min. Subsequently, 100 μl of ATP detection reagent was added to each well and mixed by orbital shaking (700 rpm) for 5 min. The luminescence was measured using a microplate counter (Model 2450, PerkinElmer Life Sciences).
Analysis of Mitochondrial Mass and Membrane Potential
The total mitochondrial masses of the transfected HeLa cells and transformed yeast strains were determined using 10-N-nonyl acridine orange (NAO) (Molecular Probes). Briefly, 0.2 OD (A600) of log phase yeast cells were harvested and washed with 1× PBS followed by incubation with 10 mm NAO for 30 min at room temperature. Subsequently, the cells were washed and resuspended in 1× PBS, and FACS analysis was performed using the BD FACS Canto II flow cytometer. An argon laser was used for the excitation at wavelength 488 nm, whereas the emission was recorded at 520 nm. For each analysis, 10,000 events were recorded, and the data were analyzed based on three independent experiments, using the WinMDI version 2.9 software. In order to detect changes in mitochondrial membrane potential, isolated mitochondria (50 μg) from HeLa cells and yeast strains were incubated with JC-1 dye (5 mg/ml) for 5 min in the dark. Subsequently, they were excited at 490 nm and subjected to an emission scan between 500 and 620 nm in a JASCO FP-6300 spectrofluorometer. The ratios of the peaks at 590 (aggregate form of the JC-1 dye) and 530 nm (monomeric form) were used as an indicator of membrane polarization. The WT mitochondria were incubated in 100 μm valinomycin for 15 min prior to dye staining as a control for complete depolarization.
Coimmunoprecipitation (Co-IP) in Mitochondrial Lysate
Mitochondria isolated from HeLa cells or yeast (1 mg of protein) were lysed for 3 min on ice in 1 ml of buffer A (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 0.2% Tween 20, 10 μm leupeptin, 10 μm pepstatin, 2 mm phenylmethylsulfonyl fluoride (PMSF), 50 μm pyridoxal phosphate, and 1 mm ascorbic acid). Membrane debris was removed by centrifugation for 10 min at 18,000 × g, and the supernatant was incubated with 5 μl of anti-FLAG antibody (0.5 μg/μl) for 2 h on a rotary shaker at 4 °C. This supernatant was added to 30 μl of protein G beads pre-equilibrated with buffer A and kept for 2 h on a rotary shaker at 4 °C. Beads were collected by centrifugation for 5 min at 800 × g and washed three times with 800 μl of buffer A. Bound proteins were subjected to SDS-PAGE and identified by immunostaining.
Iron Quantification Analysis
Assessment of total cellular iron levels by atomic absorption spectroscopy (AAS) was performed using 1 million HeLa cells and 0.2 OD (A600) of yeast cells. Mitochondrial iron estimation by AAS was performed using 50 μg of protein corresponding to HeLa cell and yeast mitochondria. Samples were digested with concentrated HCl, diluted, and subjected to AAS (atomic absorption spectrometer AA 200) according to previously described procedures with minor modifications (12). For colorimetric cellular iron quantification, HeLa cells (∼1 million) and 0.2 OD (A600) of yeast cells were sonicated prior to analysis. For colorimetric mitochondrial iron estimation, 0.2–0.4 mg of mitochondrial protein from HeLa cells and yeast were determined following methods described previously (33). Protein determinations were performed by using the Bio-Rad protein assay kit with BSA as a standard.
Gel Filtration Chromatography
For oligomerization analysis, 500 μl of protein samples (2 mg/ml) were filtered through a 0.22-μm filter and subjected to gel filtration analysis using a Superdex 200 10/300 GL column (GE Healthcare) in buffer G (50 mm Tris-Cl, pH 7.5, 50 mm NaCl, 50 mm imidazole). The column was calibrated prior to the experiment using standard molecular weight marker from Bio-Rad (thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.35 kDa)), and the void volume (V0) was calculated using blue dextran (2000 kDa). The elution volume (Ve) of standard markers, WT, and mutant ISCU proteins was determined under similar buffer and pH conditions. The molecular mass was calculated from the standard plot of Ve/V0 versus the log of the molecular mass of standard molecular weight markers.
Miscellaneous
Cell proliferation was assessed by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay according to the manufacturer's recommendations (Invitrogen). MTT was added directly to the 72-h grown culture medium and was reduced by metabolically active cells to insoluble purple formazan dye crystals. The absorbance of the sample was read directly in the wells at an optimal wavelength of 570 nm. For calcein blue FACS analysis, HeLa cells were cultured for 72 h and treated with 1 μm calcein blue dye for 5 min prior to FACS analysis. Oligomerization of ISCU in the mitochondrial lysate was analyzed by blue native PAGE (BN-PAGE) similar to the protocols published previously (34). Briefly, mitochondrial lysate was prepared in buffer containing 0.5% Tween 20 to disrupt the ISCU-bound subcomplexes with interacting partner proteins. The supernatant was subjected to BN-PAGE separation followed by immunostaining using anti-ISCU antibodies. One-way (non-parametric) ANOVA was performed using GraphPad Prism. A p value of <0.05 was defined as significant, and asterisks were used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RESULTS
Myopathy-associated Glycine 50 Residue of ISCU Is Critical for Cell Viability
In humans, the synthesis of the Fe-S cluster utilizes the mitochondrial scaffold protein ISCU as a platform for the complex interactions between various intervening proteins (Fig. 1A). ISCU is among the most conserved proteins of Fe-S cluster biogenesis machinery throughout eukaryotic evolution (33). Recent studies have reported that a missense mutation changes the conserved glycine 50 residue to glutamate (G50E) and contributes significantly toward the development of myopathy (28). Multiple-protein sequence alignment of predicted ISCU orthologs suggested significant conservation of the glycine 50 residue across different species, highlighting its importance in the protein function. Additionally, ISCU orthologs also showed a significant sequence homology throughout the length of the protein across genera. Human ISCU revealed a 71% sequence identity at the amino acid level with S. cerevisiae Isu1 and 75% sequence identity with E. coli IscU (Fig. 1B). In the three-dimensional modeled structure of ISCU (generated using a Protein Data Bank entry 1wfz template), glycine 50 is located at the N-terminal exposed random coil region, which forms a lid over the core β-strands of the protein (Fig. 1C).
FIGURE 1.

Comparative analysis of ISCU proteins and measurement of cellular viability. A, a proposed model of Fe-S cluster synthesis highlighting ISCU as a central scaffold in the biogenesis process. Assembly of the Fe-S cluster includes transfer of the sulfur atoms from the NFS1-ISD11 complex and iron from the putative iron donor protein frataxin. The transfer and incorporation into recipient apoproteins are facilitated by the ATP-dependent mtHsp70 chaperone GRP75 and the DnaJ-like cochaperone HSCB. B, predicted orthologs of ISCU from different species (Homo sapiens (Hs), Mus musculus (Mm), S. cerevisiae (Sc), Arabidopsis thaliana (At), and E. coli (Ec)) are aligned using ClustalW software. Identical, conserved, and semiconserved residues are highlighted in yellow, green, and cyan, respectively. The amino acid positions corresponding to the myopathy mutation (glycine 50) are boxed. C, the structure of human ISCU (aa 44–162) is modeled based on M. musculus Iscu (Protein Data Bank code 1wfz) using the SWISS-MODEL program. The N-terminal side chain depicting the conserved residue glycine 50 is highlighted. N and C, N and C terminus of ISCU, respectively. D, ISCU protein levels in the mitochondrial lysate of the UT HeLa cells and cells carrying WT ISCU and G50E ISCU in a pCI-neo plasmid after 72 h of transfection. The mitochondrial protein Tim23 was used as a loading control. E, MTT assay for the measurement of cellular viability (represented as bars) of UT, WT, and G50E mutant in HeLa cells. p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
Although the G50E mutation is associated with clinical cases of myopathy patients, the cellular defects and the molecular mechanisms that lead to the disease condition are still elusive. The importance of the glycine 50 residue in the maintenance of cell viability was analyzed by an MTT assay. Wild type (WT) or G50E mutant constructs of ISCU were transfected into HeLa cells and were allowed to be expressed for 72 h (Fig. 1D). The relative cell viability was determined by an MTT assay based on the conversion of MTT into formazan crystals in living cells. We observed a more than 2-fold reduction in cell viability in the case of cells overexpressing a mutant copy of ISCU as compared with that of cells expressing a WT copy of protein or untransfected (UT) controls (Fig. 1E), thus highlighting the critical role of glycine 50 in normal protein function required for the maintenance of cell growth.
Human G50E ISCU Mutant Exhibits Decreased Fe-S Cluster-containing Enzyme Activity and Reduced Cellular Respiration
Mitochondria are the major site for the synthesis of Fe-S clusters in eukaryotic cells (11). Because ISCU forms the key component for Fe-S assembly, we investigated whether the reduced cell viability in the G50E mutant is due to impaired Fe-S cluster biogenesis. To assess the biogenesis of functional Fe-S cluster, we monitored the enzyme activity of Fe-S cluster-containing proteins using purified mitochondria from HeLa cells cultured for 48 h. Before enzymatic analysis, the purity of isolated mitochondria was assessed using immunodecoration with ISCU-specific antibody for the enriched mitochondrial fraction. As a positive control, components of the mitochondrial inner membrane translocon machinery, such as Tim23 and Tim44 proteins, were probed. To detect the other organellar contamination, immunoblotting was performed for other markers, such as cathepsin D (lysosomes), catalase (peroxisomes), and superoxide dismutase (nuclear), as negative controls. Negligible amounts of non-mitochondrial proteins were detected in the enriched mitochondrial fraction, thus confirming the purity of the mitochondria utilized for the enzymatic analysis (Fig. 2A).
FIGURE 2.

Measurement of Fe-S cluster enzyme activity, cellular respiration, mitochondrial mass, and membrane potential in HeLa cells. A, the purity and enrichment of mitochondria obtained from HeLa cells were analyzed by immunodecoration using antibodies against mitochondrial specific markers as positive controls (Tim23, Tim44, and ISCU) and other organellar specific antibodies, such as catalase (peroxisomes), cathepsin (lysosomal), and SOD (cytosolic/nuclear) as negative controls. Equivalent amounts of cell and mitochondrial lysates (100 μg of protein) were loaded for the comparison. B, activity of mitochondrial complex I of the ETC in UT HeLa cells overexpressing WT ISCU and G50E ISCU in the presence or absence of the complex I inhibitor rotenone. A.U., arbitrary units. C, activity of ETC complex II in the presence of inhibitors of complex I, III, and IV represented in bar charts. D, assessment of the activity of mitochondrial matrix enzyme aconitase in HeLa cells denoted in bar charts. E, activity of complex IV of the ETC measured in the presence or absence of complex IV inhibitor sodium azide (NaN3). F, relative ATP levels measured in mitochondria isolated from UT, WT, and mutant protein-expressing HeLa cells using the mitochondrial ToxGlo assay kit. G and H, purified mitochondria isolated from HeLa cells were stained with the JC-1 dye and subjected to an excitation at 490 nm followed by an emission wavelength scan ranging from 500 to 620 nm. The fluorescence intensity values obtained were plotted against the wavelengths to calculate the relative distribution of polarized versus depolarized mitochondria (G). A relative fluorescence intensity obtained from the JC-1 dye (G) was quantified and plotted as a ratio of multimer (590 nm) to monomer (530 nm) (H). I and J, the mean fluorescence intensity histogram for UT, WT, and G50E ISCU HeLa cells stained with NAO followed by flow cytometric analysis for the measurement of overall mitochondrial mass. The values were plotted based on three independent experiments (I). The total mitochondrial mass for each HeLa cell line obtained from I was quantitated, and normalized values were plotted on a bar chart (J). p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
We chose three model iron-sulfur proteins, namely membrane-associated complex I and complex II of the electron transport chain and matrix-localized TCA cycle enzyme aconitase. The activity of complex I (NADH:ubiquinone oxidoreductase) containing eight Fe-S clusters was found to be significantly reduced in the case of the G50E mutant as compared with mitochondria isolated from WT ISCU and untransfected HeLa cells. The specificity of the reaction was further verified by inhibiting complex I with its specific inhibitor, rotenone. As expected, rotenone-treated mitochondria showed considerably reduced enzymatic activity as compared with the untreated controls. A cumulative reduction in complex I function was observed in the case of rotenone-treated G50E mitochondria, thus highlighting the importance of the residue for normal protein function (Fig. 2B). Similarly, a marked reduction in the activity of complex II (succinate dehydrogenase) composed of three Fe-S centers was observed in G50E mutant mitochondria (Fig. 2C). We further validated our results by measuring the activity of a mitochondrial matrix Fe-S cluster enzyme, aconitase, that reversibly converts citrate to isocitrate in the second step of the TCA cycle. Supporting the above observation, the activity of aconitase was found substantially lessened in the G50E mutant mitochondria, suggesting impairment in the Fe-S cluster biogenesis (Fig. 2D). In order to address the specificity in the impairment of Fe-S biogenesis by the loss of function of ISCU as a result of the mutation, we checked the activity of complex IV or cytochrome c oxidase that contains two heme centers instead of Fe-S centers. Notably, complex IV showed a marginal declined activity in mutant mitochondria as a consequence of decreased activity of Fe-S cluster-containing ferrochelatase, which is involved in the heme synthesis (Fig. 2E). In light of the loss of electron transport chain complex activity, we checked for the mitochondrial respiratory activity in HeLa cells. A defect in mitochondrial respiration results in diminished ATP synthesis (35, 36). In agreement with the above data, we observed that the G50E mutant mitochondria harbored a substantially lesser amount of ATP as compared with mitochondria isolated from WT ISCU and untransfected HeLa cells (Fig. 2F). Together, these results highlight that the G50E ISCU mutation impairs Fe-S cluster biogenesis, leading to decreased activity of Fe-S cluster-containing electron transport chain (ETC) enzymes, thereby causing a defect in the cellular respiration in myopathy patients.
G50E Mutation in ISCU Results in Loss of Mitochondrial Membrane Potential and Mitochondrial Mass
Previous reports have suggested that impairment of Fe-S cluster biogenesis often leads to mitochondrial dysfunction (37). Therefore, we embarked upon analyzing whether the loss of Fe-S cluster biogenesis as a result of G50E mutation results in impairment of mitochondrial function. First, we assessed the maintenance of mitochondrial inner membrane potential, using the membrane potential-sensitive cationic dye JC-1. In the presence of an intact membrane potential, the JC-1 dye accumulates inside the mitochondrial matrix to form J-aggregates, which fluoresces at the red region with an emission maximum of 590 nm. However, upon dissipation of membrane potential, JC-1 remains in the cytoplasm as a green fluorescent monomeric form with an emission maximum at 530 nm. The ratio of red to green fluorescence serves as an indicator for functional mitochondria membrane potential (38). Indeed, WT mitochondria showed decreased fluorescence at 530 nm, indicating the presence of intact membrane potential. In contrast, the G50E mutant showed a significant increment in the fluorescence intensity at 530 nm and reduction at 590 nm as compared with mitochondria from untransfected HeLa cells (Fig. 2G, compare peaks represented in red with those in green and black). Upon quantification of the red/green ratio, a 21% reduction in mitochondrial membrane potential was estimated for the G50E mutant (Fig. 2H). Valinomycin, which is known to disrupt the membrane potential of intact mitochondria, was used as a positive control (39). Second, we assessed the overall mitochondrial mass using a fluorescent dye, 10-N-nonyl acridine orange (NAO), which binds to the cardiolipin of mitochondrial membrane, thus giving an estimate of the total mitochondrial mass present inside the cell (40). Interestingly, the G50E mutant showed a significant decrease in mitochondrial mass as the fluorescence signal peak was shifted toward the lower region as compared with WT ISCU and untransfected HeLa cells (Fig. 2I, compare peaks represented in red with green and black ones). Upon quantification of mean fluorescence intensity values, a 52% reduction in the overall mitochondrial mass was observed in the G50E mutant in comparison with the WT ISCU (Fig. 2J). These findings indicate that the G50E mutation in ISCU results in the reduced biogenesis of mitochondria along with loss of membrane potential.
G50E Mutation in ISCU Results in Increased Iron Load and ROS Level in Mitochondria
A defect in Fe-S cluster biogenesis is often associated with impairment of mitochondrial iron homeostasis. It has been reported earlier in a neurodegenerative disorder, Friedreich ataxia, that deprivation of Fe-S biogenesis results in accumulation of iron inside mitochondria (41). Considering this fact, we investigated the cellular and mitochondrial iron levels in WT ISCU HeLa cells and G50E mutant cells. The overall cellular free iron was measured using an iron-sensitive probe, calcein blue, through flow cytometry and fluorescence imaging. Calcein blue is a cell-permeable metallo-fluorochromic indicator dye, which upon binding with free iron results in reduction in fluorescence intensity (42, 43). Flow cytometric analysis of G50E mutant cells stained with calcein blue showed a reduction in fluorescence intensity (shift in the fluorescence peak toward a lower region) as compared with WT ISCU and untransfected HeLa cells, suggesting the presence of higher free iron content in mutant cells (Fig. 3, A and B; compare peaks indicated in red with green and black ones). The flow cytometric results were further validated by fluorescence imaging of WT and mutant HeLa cells, which revealed the diminished fluorescence intensity in the G50E mutant, thus confirming an overall enhanced cellular unbound/free iron level. To demonstrate the specific quenching of calcein by iron, the G50E HeLa cells were treated with 1 mm iron chelator, deferoxamine (DFO). Upon DFO treatment, an elevated fluorescence in G50E HeLa cells was observed, suggesting that the quenching of calcein fluorescence observed in G50E HeLa cells was the consequence of enhanced unbound/free iron levels and not due to interference from other metal ions (Fig. 3, C and D). The increment in the cellular iron load as observed in G50E mutant HeLa cells through fluorescence-based assays was additionally validated by an iron-specific colorimetric assay and atomic absorption spectroscopy analysis using whole cell or mitochondrial lysates. Both the colorimetric assay and AAS analysis showed a substantial increment in the mitochondrial iron load in G50E mutant as compared with WT ISCU mitochondria, indicating that impaired Fe-S biogenesis leads to accumulation of iron levels in the organelle (Fig. 3, E to H).
FIGURE 3.

Estimation of total cellular and mitochondrial iron content of G50E ISCU in HeLa cells. A, flow cytometric estimation of overall cellular free iron levels using calcein blue dye in UT HeLa cells and cells harboring the WT ISCU and G50E ISCU. A total of 10,000 events were analyzed for each case, and the fluorescence intensity values were plotted as a mean of three independent experiments. B, the free iron content was quantitated from the FACS experiment in A and represented in a bar chart. A.U., arbitrary units. C, calcein blue fluorescence images of UT, WT ISCU, G50E ISCU, and G50E ISCU HeLa cells treated with 1 mm iron chelator, deferoxamine (DFO). D, the calcein blue fluorescence intensity obtained from the images in C was quantitated using ImageJ software and represented in a bar chart. The total cellular iron (E) and mitochondrial iron levels (F) in HeLa cells were determined by colorimetric analysis and represented in a bar chart after normalization of values obtained for G50E and WT ISCU against UT. The total cellular iron (G) and mitochondrial iron levels (H) in each HeLa cell type were determined using AAS. The data are represented in a bar chart after normalization of values obtained for UT and WT against G50E ISCU. p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
Increased cellular iron load and loss of respiratory complex activity is well known to promote ROS production (44). In retrospect, higher ROS levels also promote accumulation of iron inside cells (45–47). Based on our observations of enhanced mitochondrial iron load and respiratory defect in G50E mutant, we further assessed the maintenance of mitochondrial ROS levels in HeLa cells. The accumulation of mitochondrial ROS was determined by measuring the superoxide levels with MitoSOX Red dye, which is specifically targeted to mitochondria in live cells. Oxidation of MitoSOX Red reagent by superoxide produces red fluorescence, which is quantified by flow cytometric analysis and correlated with the amount of ROS present in the mitochondria. We observed that ISCU G50E mutation led to an elevation in the mitochondrial superoxide levels indicated by a significant shift in the fluorescence signal peak toward higher ROS levels as compared with WT HeLa cells (Fig. 4A, compare peaks represented in red with green and black ones; blue represents rotenone, a control). Upon quantification of flow cytometric data, a greater than 6-fold increase in MitoSOX staining was observed in G50E HeLa cells (Fig. 4B). As a positive control, cells were treated with 1 mm rotenone, which elevates the mitochondrial superoxide levels by inhibiting respiratory complex I. In support of the flow cytometric data, live cell imaging of G50E mutant cells treated with MitoSOX Red dye revealed robust enhancement in the fluorescence intensity as compared with WT (Fig. 4, C and D), suggesting elevation in the mitochondrial superoxide levels.
FIGURE 4.
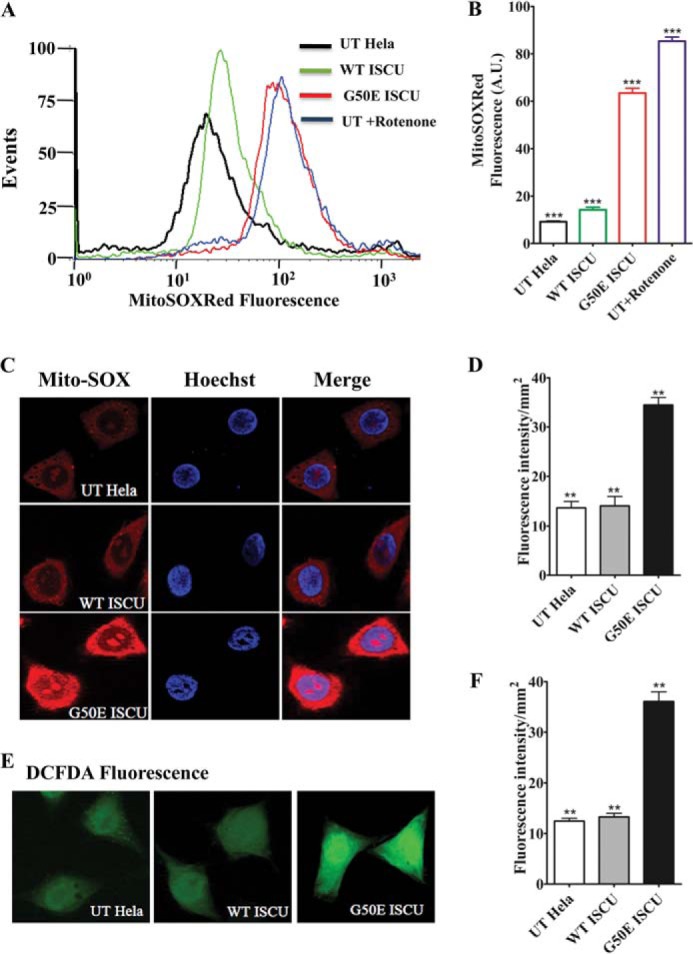
Estimation of mitochondrial and total cellular ROS of G50E ISCU in HeLa cells. A, the mitochondrial superoxide level in UT HeLa cells and cells harboring the WT ISCU and G50E ISCU were estimated flow cytometrically using fluorescent MitoSOX Red dye. B, the total superoxide levels obtained from the aforementioned experiment (A) were quantitated and represented in a bar chart. The UT cells treated with 1 mm rotenone were used as a positive control. For flow cytometry experiments, 10,000 events were analyzed for each case, and the values were plotted based on three independent experiments. C, untransfected HeLa cells and cells transfected with WT ISCU and G50E ISCU were stained with the mitochondrial superoxide indicator MitoSOX Red (red fluorescence) and nuclear counterstain Hoechst 33342 (blue fluorescence), and the images were recorded in a Zeiss Apotome fluorescence microscope using a ×63 objective lens. D, the total MitoSOX Red fluorescence intensity of images from the aforementioned experiments was quantitated using ImageJ software. E, fluorescence images of UT HeLa cells and cells harboring WT ISCU and G50E ISCU stained with H2DCF-DA dye to detect enhancement in overall cellular ROS levels (green fluorescence). The images were recorded in a Leica fluorescence microscope using a ×63 objective lens. F, the total H2DCF-DA dye fluorescence intensity from the images (E) was quantitated using ImageJ software. p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
To address whether elevation of mitochondrial superoxide levels alters the overall cellular ROS levels, the WT and mutant cells were stained with H2DCF-DA dye followed by fluorescence microscopic analysis. H2DCF-DA is a chemically reduced form of fluorescein and, upon modification by peroxide species present in the cells, produces a green fluorescence signal. Consistent with the MitoSOX data, G50E HeLa cells showed significantly increased green fluorescence as compared with WT, further validating that the G50E mutation results in enhancement of the overall cellular ROS levels (Fig. 4, E and F). In summary, the above findings indicate that an enormous increment in the superoxide and overall ROS levels in G50E ISCU mutant induces a severe oxidative stress, thus inflicting multiple cellular dysfunctions, including impairment in the mitochondrial functions.
Analogous G50E Mutation in Yeast Isu1 Protein Leads to Inviability and Mitochondrial Dysfunction
Our observations in the mammalian system highlighted that the glycine 50 residue is crucial for proper functioning of ISCU. The G50E mutation in ISCU results in mitochondrial dysfunction that facilitates the development of myopathy in humans. Therefore, to understand whether the glycine 50 residue plays a similar conserved role in other ISCU orthologs across kingdoms, we performed analogous amino acid substitution (G50E) in the S. cerevisiae ISU1 gene. Yeast mitochondrial Isu1 is an ortholog of human ISCU and is essential for the biogenesis of Fe-S clusters in mitochondria. In yeast mitochondria, ISU1 has a second paralog, ISU2, believed to be a result of whole genome duplication. However, Isu1 is the predominant form that is expressed under all growth conditions, and the Δisu1/isu2 double deletion mutant is inviable (12).
We first assessed whether WT human ISCU could rescue the inviability of the Δisu1/isu2 strain. To test this possibility, a centromeric plasmid pRS414 TEF carrying a WT copy of human ISCU was transformed into a Δisu1/isu2 strain harboring a WT copy of ISU1 on the pRS316 plasmid having URA3 as a selectable marker. The transformed strains were selected on tryptophan-omitted medium. The resultant yeast transformants were scored for viability, by drop test analysis on medium containing 5-fluoroorotic acid, which selects for cells that have lost the pRS316 plasmid, thereby losing the WT copy of ISU1 in this case. Interestingly, human WT ISCU rescued the inviability of the Δisu1/isu2 strain (Fig. 5A). Importantly, on the other hand, both human G50E ISCU and yeast isu1G50E resulted in failure to rescue inviability of the Δisu1/isu2 strain on 5-fluoroorotic acid at all temperatures tested, thus highlighting the critical nature of amino acid glycine 50 of Isu1 for in vivo function in yeast.
FIGURE 5.

Growth and functional defects associated with analogous G50E mutation in yeast Isu1. A, Δisu1/isu2 yeast strain carrying a WT copy of ISU1 in pRS316 plasmid transformed with WT ISU1 or isu1G50E mutant and subjected to serial drop dilution analysis on 5-fluoroorotic acid medium (5-FOA) and incubated at the indicated temperatures for 96 h (top). Similarly, human WT ISCU and G50E ISCU mutant in pRS414 TEF vector were transformed in the Δisu1/isu2 yeast strain and subjected to serial drop dilution analysis on 5-fluoroorotic acid medium as described in the aforementioned analysis (bottom). B, Δisu1/isu2 yeast strain harboring a WT copy of ISU1 in pRS316 plasmid was transformed with WT ISU1 and mutant isu1G50E in a centromeric plasmid pRS414 TEF (top) or pRS414 GPD (bottom). Cells were spotted on Trp− plates followed by incubation for 72 h at the indicated temperatures. C, immunoblot analysis for Isu1 protein levels in the mitochondrial lysates of overexpressed strains (under pRS414 GPD vector as indicated in B) prepared from WT and mutant yeast cells using anti-FLAG antibodies. The mitochondrial protein Mge1 was used as a loading control. D and E, the estimation of mitochondrial mass was performed by flow cytometric analysis using NAO dye. For each flow cytometric analysis, 10,000 events were analyzed, and the relative fluorescence intensity values obtained from three independent experiments were plotted (D). Relative mitochondrial mass in each yeast strain determined from the aforementioned FACS analysis was quantitated (E). F, purified mitochondria from yeast strains were stained with JC-1 dye to determine the mitochondrial membrane potential by scanning the emission wavelength ranging from 500 to 620 nm, and the fluorescence intensity (absorbance units (A.U.)) was plotted against the wavelength. Mitochondria treated with the valinomycin were used as a positive control. G, the ratio of fluorescence intensities between multimer (590 nm) and monomer (530 nm) was quantitated and represented in a bar chart. p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
The isu1G50E mutant is inviable in the haploid state and hence found to be unsuitable for the in vivo functional analysis. Therefore, to analyze the effect of the G50E Isu1 mutation on Fe-S biogenesis, the isu1G50E mutant was first overexpressed under the mild TEF promoter in yeast strains harboring a WT copy of the ISU1 gene on the pRS316 plasmid. However, at similar expression levels, the isu1G50E mutant did not exhibit any growth phenotype at all temperature conditions tested (Fig. 5B, top). On the other hand, when the isu1G50E mutant was expressed under the control of the strong GPD promoter, the cells were temperature-sensitive at 37 °C as compared with WT (Fig. 5B, bottom). Moreover, both WT Isu1- and isu1G50E-overexpressing strains showed equal levels of protein expression in the mitochondrial lysate upon immunoblot analysis using anti-FLAG antibodies (Fig. 5C). This implies that higher expression levels of the isu1G50E mutant in the presence of a WT copy of the ISU1 gene lead to a semidominant negative phenotype at 37 °C. We utilized isu1G50E overexpressing under the GPD promoter in yeast strains harboring a WT copy of ISU1 for further in vivo experiments.
To address whether the deleterious effect of the analogous G50E mutation in Isu1 is due to functional defects in yeast mitochondria, we first measured the mitochondrial mass of isu1G50E yeast cells using NAO staining. Similar to the G50E ISCU HeLa cell mutant, isu1G50E yeast strains also showed a significantly reduced mitochondrial mass as compared with WT (Fig. 5, D and E). Likewise, assessment of mitochondrial membrane potential using JC-1 dye staining exhibited 25% lower membrane potential in isu1G50E yeast as compared with that of WT, suggesting impairment in the biogenesis as well as mitochondrial functions (Fig. 5, F and G).
G50E Mutation in Yeast Isu1 Leads to a Similar Reduction in Fe-S Cluster-containing Enzyme Activity, ATP Levels, and Elevation in Mitochondrial Iron Level and Mitochondrial ROS
The indispensability of the glycine 50 residue of ISCU for cell viability and mitochondrial function has already been established using HeLa cells. In order to ascertain the conservation of function of the glycine 50 residue across genera, we performed a similar cellular analysis in isu1G50E yeast cells pre-exposed to temperature stress at 37 °C. To address whether the inviability of isu1G50E mutant yeast is due to an impaired Fe-S biogenesis process, we measured the Fe-S cluster enzyme activity. Supporting our results observed in human cell lines, isu1G50E yeast strains showed 2.6-fold reduced complex II activity (Fig. 6A) and 3-fold lower activity of aconitase (Fig. 6C) as compared with WT Isu1-harboring yeast cells. In contrast, complex IV that does not possess Fe-S clusters demonstrates a marginal decrease in the activity in mutant yeast cells (Fig. 6B). These results provide evidence to indicate that the G50E mutation in Isu1 leads to a similar impairment in the Fe-S cluster biogenesis, hence decreasing the activity of Fe-S cluster-containing enzymes. As a result of the loss of ETC enzyme complex activity, the isu1G50E-overexpressing yeast mitochondria showed 1.8-fold lower respiratory activity in terms of total mitochondrial ATP content as compared with WT Isu1 yeast mitochondria (Fig. 6D). These observations provide compelling evidence to prove that, similar to the mammalian system, the G50E mutation in yeast Isu1 also leads to impairment in Fe-S cluster biogenesis and reduced cellular respiration.
FIGURE 6.

Measurement of enzymatic activity, respiration, and iron levels in isu1G50E mutant yeast strains. A, activity of mitochondrial complex II of the ETC in yeast strains carrying either vector alone control (pRS414), or WT ISU1, or isu1G50E in pRS414 GPD plasmid in the presence or absence of inhibitors of complex I, III, and IV. B, bar chart indicating the activity of complex IV of the ETC in yeast cells in the presence or absence of complex IV inhibitor sodium azide (NaN3). C, activity of mitochondrial matrix enzyme aconitase in aforementioned yeast strains. D, quantification of ATP levels in the mitochondria isolated from WT, and mutant yeast strains were measured using the mitochondrial ToxGlo assay. E and F, estimation of total cellular iron content (E) and mitochondrial iron levels (F) in WT and mutant strains were determined by colorimetric analysis. The normalized values against the mutant strain are plotted on a bar chart. G and H, measurement of total cellular iron content (G) and mitochondrial iron levels (H) in the aforementioned yeast strains obtained using AAS analysis. The normalized values against the mutant strain are plotted on a bar chart. p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. A.U., absorbance units. Error bars, S.E.
To check alterations in the cellular iron content due to defective Fe-S cluster biogenesis, we checked for the iron accumulation in mutant isu1G50E yeast cells by a colorimetry-based assay and AAS analysis. The colorimetric assay showed 17% higher cellular iron and 34% higher mitochondrial iron levels in the G50E mutant (Fig. 6, E and F). Similarly, AAS analysis revealed 22% higher cellular iron and 32% elevated mitochondrial iron levels in the mutant as compared with WT Isu1 yeast strains, thus confirming our previous observations using HeLa cells (Fig. 6, G and H).
Previous reports and our observations from the human cell lines have indicated that mitochondrial respiratory defects and iron overload results in increased ROS levels in the mitochondrial compartment. To check the ROS levels in the mitochondria of isu1G50E mutant yeast cells, flow cytometric analysis was performed using MitoSOX Red dye. As indicated in Fig. 7, A (compare the peak represented in red with the green and blue ones) and B, isu1G50E yeast strains exhibited substantially higher mitochondrial ROS as compared with WT Isu1 yeast strains. The enhanced ROS levels in yeast were usually associated with the enhanced sensitivity toward the extraneous stress. To probe the sensitivity of the isu1G50E mutant strain to extraneous oxidative stress, cells were subjected to hydrogen peroxide (H2O2) treatment. As shown in Fig. 7C, the isu1G50E strain was responsive to oxidative stress at 37 °C, exemplified by a compromised growth phenotype in minimal medium containing 1 mm H2O2. On the other hand, WT Isu1 grew normally in the presence of 1 mm H2O2-containing medium. The sensitivity of the isu1G50E strain to extraneous oxidative stress was further demonstrated by fluorescence live cell imaging. The isu1G50E strain stained with MitoSOX Red dye showed an enhanced basal level of fluorescence in untreated conditions as compared with the WT Isu1 strain, consistent with the flow cytometric analysis (Fig. 7D, top left panels, compare Untreated). Importantly, isu1G50E cells showed a robust increment in red fluorescence when treated with H2O2 as compared with the WT Isu1 yeast strain, indicating its susceptibility to cellular redox alterations (Fig. 7D, right panels, compare Treated). A similar analysis was performed to measure the overall cellular ROS production using H2DCF-DA dye in response to oxidative stress. As shown in Fig. 7E, isu1G50E yeast cells displayed higher basal peroxide levels as compared with the WT Isu1 strain, in untreated conditions (Fig. 7E, bottom left, compare Untreated). However, a significant enhancement in green fluorescence was observed for the isu1G50E yeast strain as compared with WT Isu1 yeast cells after H2O2 treatment (Fig. 7E, right panels, compare Treated). These findings conclusively establish that the isu1G50E strain harbors enhanced mitochondrial and overall cellular ROS levels due to defective Fe-S cluster biogenesis.
FIGURE 7.

Estimation of cellular ROS levels and oxidative stress sensitivity of isu1G50E. A, flow cytometric analysis of mitochondrial superoxide levels in yeast strains carrying WT ISU1 and isu1G50E in pRS414 GPD plasmid using MitoSOX Red dye. Relative superoxide level obtained from the aforementioned analysis was quantitated and represented in a bar chart (B). WT cells treated with 1 mm rotenone were set as a positive control. For flow cytometry experiments, 10,000 events were analyzed for each case, and the values were plotted based on three independent experiments. C, assessment of oxidative stress sensitivity of WT and mutant strains performed by drop test analysis on selective medium with and without treatment of 1 mm H2O2 for 2 h. Equivalent numbers of yeast cells from the WT Isu1 and isu1G50E strains were spotted by serial dilution and allowed to grow at 37 °C for 72 h. D, fluorescence imaging analysis of WT Isu1 and isu1G50E yeast strains stained with mitochondrial superoxide indicator MitoSOX Red (red fluorescence). The images obtained for untreated strains are represented in the left-hand panels (bright field (BF), MitoSOX (middle), and Merge column). The images acquired for the strains after treatment with 1 mm H2O2 for 2 h are indicated in the right-hand panels. E, fluorescence images of WT Isu1 and isu1G50E yeast strains stained with cellular peroxide indicator H2DCF-DA (green fluorescence). The images obtained for untreated strains are represented in the left-hand panels, and images after treatment with 1 mm H2O2 for 2 h are indicated in the right-hand panels. The images were recorded in a Leica fluorescence microscope using a ×100 objective lens. p < 0.05 was defined as significant, and asterisks are used to denote significance as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
Human G50E ISCU and Yeast isu1G50E Mutant Proteins Are Defective in Interaction with Sulfur Donor hNFS1/yNfs1 and J-protein, HSCB/Jac1
ISCU serves as a platform for the de novo synthesis of an Fe-S cluster in a highly coordinated manner along with other key partner proteins of the machinery. Previously, it has been demonstrated that ISCU protein specifically associates with hNFS1, a cysteine desulfurase that is involved in the transfer of inorganic sulfur to Fe-S cluster assembly (11). On the other hand, biochemical studies in yeast have demonstrated that the mitochondrial DnaJ-like cochaperone Jac1 interacts with Isu1 to enhance the specific transfer of a scaffold-bound Fe-S cluster to bona fide Fe-S apoproteins (20, 48). In addition to that, recently, it has been reported in yeast that binding of yNfs1 and cochaperone Jac1 to the iron-sulfur cluster scaffold Isu1 is mutually exclusive (15). Therefore, we asked whether the G50E mutation in ISCU results in compromised interaction with the sulfur donor NFS1 or with the J-protein HSCB, leading to defective Fe-S cluster biogenesis. To evaluate whether the G50E ISCU mutant protein is capable of forming a subcomplex with hNFS1 or with HSCB, we performed in vitro GST pull-down analysis using purified proteins. The interaction with hNFS1 was investigated by incubating immobilized GST-bound hNFS1 with increasing concentrations of WT ISCU or G50E ISCU mutant. The GST pull-down analysis showed an efficient interaction of hNFS1 with WT ISCU (Fig. 8, A and B). Interestingly, however, its interaction with the G50E mutant was significantly compromised (Fig. 8, A and B). As a control, GST alone did not interact with either WT or mutant ISCU protein (Fig. 8A, middle). In addition to that, we assessed the interaction of mutant with the J-protein cochaperone by using a similar GST pull-down analysis. Interestingly, the G50E ISCU mutant exhibits significantly reduced interaction with HSCB as compared with WT ISCU (Fig. 8, C and D).
FIGURE 8.

Interaction of G50E mutant (ISCU/Isu1) with hNFS1/yNfs1 and HSCB/Jac1 by GST pull-down analysis. Prebound 1.5 μm GST-hNFS1 (A) and 1.0 μm GST-HSCB (C) were incubated with increasing concentrations of purified WT and mutant human ISCU protein, as indicated. The unbound proteins were washed with buffer and analyzed by SDS-PAGE followed by Coomassie dye staining. GST alone (2.5 μm) was used as a negative control, and 25% input of ISCU served as a loading control. The band intensities were densitometrically quantitated using ImageJ software for hNFS1 (B) and HSCB (D). E–H, prebound 1.5 μm GST-yNfs1 (E) and 2.5 μm of GST-Jac1 (G) were incubated with increasing concentrations of purified WT and mutant yeast Isu1 protein as indicated. The unbound proteins were washed with buffer and analyzed by SDS-PAGE followed by Coomassie dye staining. The band intensities were densitometrically quantitated using ImageJ software for yNfs1 (F) and Jac1 (H). I, mitochondria lysates were prepared from HeLa cells expressing either FLAG-tagged WT or G50E ISCU in buffer A containing 0.2% Tween 20 and subjected to immunoprecipitation using anti-FLAG antibodies. Fractions were analyzed for the presence of hNFS1 and HSCB by immunostaining. 10% of total mitochondrial extract was used as input co-IP control (right). J, FLAG-tagged WT Isu1 or isu1G50E mutant mitochondrial lysates were subjected to immunoprecipitation using anti-FLAG antibodies and analyzed for the presence of yNfs1 and Jac1 by immunostaining. 10% of total mitochondrial extract was used as input co-IP control (right). K, surface representation of the (IscS-IscU)2 complex showing the two IscS molecules (green and cyan) and the two IscU molecules (purple and yellow). Close-up view, IscU surface highlights the glycine position in red, located adjacent to the interface of IscS and IscU.
Based on these findings, we further investigated the presence of similar defective interactions between the yeast isu1G50E mutant and orthologous protein partners, such as Nfs1 and Jac1, using GST pull-down analysis. Similar to our observations in humans, yNfs1 showed diminished interaction with isu1G50E, whereas WT Isu1 exhibited robust association with yNfs1 (Fig. 8, E and F). Likewise, Jac1 also showed a compromised interaction with isu1G50E as compared with WT, similar to HSCB in humans (Fig. 8, G and H).
To further confirm our in vitro observations, we assessed the subcomplex formation between G50E ISCU and hNFS1 or HSCB in HeLa cell mitochondrial lysates using in organelle co-IP analysis. For this purpose, we isolated mitochondria from HeLa cells expressing FLAG-tagged WT ISCU or G50E ISCU. The respective mitochondrial extracts were prepared in 0.2% Tween 20-containing buffer and then subjected to co-IP analysis using anti-FLAG antibodies. Both hNFS1 and HSCB were coprecipitated along with ISCU in extracts of WT mitochondria, indicating that WT ISCU can form a stable subcomplex with hNFS1 and HSCB. However, co-IP analysis with HeLa cell mitochondria expressing G50E ISCU showed significantly reduced immunoprecipitation of hNFS1 and HSCB, highlighting the importance of the glycine 50 residue for subcomplex formation in vivo (Fig. 8I). A similar co-IP analysis in isu1G50E yeast mitochondrial lysates also showed impaired interaction of Isu1 with yeast yNfs1 and Jac1 (Fig. 8J). Interestingly, in the crystal structure, the position of the glycine residue is located adjacent to the interaction interface of IscS and IscU (ortholog of ISCU and NFS1 from Archaeoglobus fulgidus), suggesting that the glycine residue plays a critical role to maintain complex structure (Fig. 8K). In summary, our results suggest that the glycine 50 residue is critical for ISCU function, and the G50E mutation in ISCU/Isu1 results in compromised interaction with its bona fide partner proteins, which are essential for the assembly process as well as transfer of the Fe-S cluster into apoproteins.
G50E Mutant ISCU Exhibits a Semidominant Phenotype by Forming Oligomeric Complex with WT Protein
It has been reported earlier that, in different organisms, ISCU homologs exist in variable oligomeric states. For example, E. coli apo-IscU was reported to exist as a monomer or an S–S bridged dimer (49, 50). However, IscU from Azotobacter vinelandii and Schizosaccharomyces pombe exists as a dimer, retaining either one or two [2Fe-2S] centers or a single [4Fe-4S] cluster (51, 52). In the case of Aquifex aeolicus, the IscU holoprotein forms a trimer containing a substoichiometric [2Fe-2S] cluster, whereas its apo-form gets dissociated into a smaller species, which is a mixture of the monomeric and predominant dimeric forms (53). Therefore, to investigate whether there is any alteration in the stoichiometry of G50E ISCU mutant protein, we purified recombinant WT apo-ISCU and G50E apo-ISCU from E. coli under aerobic conditions. The purified mutant protein retained structural integrity similar to WT as confirmed by circular dichroism (Fig. 9A) and partial trypsin and chymotrypsin digestion analysis (Fig. 9, B and C). The oligomeric state of WT and mutant proteins was analyzed by subjecting them to gel filtration chromatography using a Superdex 200 10/300 GL column. In the case of WT ISCU, a single elution peak was obtained, corresponding to a molecular mass of 30–33 kDa (Fig. 9, D and E). This suggests that apo-ISCU possibly exists as a dimer, based on the calculated molecular mass of 14.5 kDa, from the ISCU primary sequence. Interestingly, gel filtration analysis of the G50E ISCU mutant resulted in the appearance of two elution peaks of different molecular weights, a smaller peak with a molecular mass of 30–33 kDa corresponding to the dimeric form. On the other hand, the second elution peak with a molecular mass of 180–185 kDa probably corresponds to the higher order oligomeric forms of the G50E ISCU (Fig. 9, F and G). Upon quantitation, we observed that the G50E ISCU showed an increased propensity to form larger species with a ratio of 30% dimer to 70% oligomeric form. To test the formation of oligomeric species in G50E ISCU protein under in vivo conditions, the mitochondrial lysate was subjected to BN-PAGE analysis followed by immunostaining with ISCU-specific antibodies. Besides dimeric forms, the G50E ISCU protein showed a distinct oligomeric species as compared with wild type controls, thus further supporting the findings of the in vitro purified system (Fig. 9H).
FIGURE 9.

Secondary structure analysis and oligomerization study of G50E ISCU. A, UV CD spectra of WT ISCU (solid line) and G50E ISCU (dashed line) in 20 mm phosphate buffer (pH 8) were recorded at 10 °C. B and C, 5 μg of WT ISCU (left) or G50E ISCU (right) protein was preincubated in cleavage buffer for 20 min at 10 °C. The proteolysis was initiated by the addition of 1 μl (1:50 dilution of 1 μg/μl stock) of trypsin (B) and/or chymotrypsin (C) as indicated. The reaction was stopped at the indicated time intervals using 2 mm PMSF. The samples were boiled in SDS sample buffer and analyzed using SDS-PAGE followed by Coomassie dye staining. D, gel filtration chromatography elution profile of WT ISCU protein separated on a Superdex 200 column with a single peak corresponding to the molecular size of ∼30–33 kDa indicated. E, elution fractions from the gel filtration of WT ISCU were analyzed on 15% SDS-PAGE and stained with Coomassie dye. F and G, gel filtration elution profile of G50E ISCU mutant separated under similar conditions. Two major peaks corresponding to molecular sizes ∼30–33 kDa and ∼180–185 kDa are highlighted (F). The eluted fractions from the gel filtration of G50E ISCU mutant were separated on 15% SDS-PAGE and stained with Coomassie Blue dye (G). H, mitochondrial lysate (500 μg of the protein) prepared from HeLa cells expressing either WT or G50E ISCU mutant was separated on BN-PAGE followed by immunostaining with anti-ISCU-specific antibody. The positions of dimer and oligomers are highlighted with reference to BN-PAGE markers. I, mitochondria lysates were prepared from HeLa cells expressing either FLAG-tagged WT or G50E ISCU in buffer A containing 0.2% Tween 20 and subjected to immunoprecipitation using anti-FLAG antibodies. The presence of endogenous WT ISCU and HSCB was detected by immunostaining using either anti-ISCU- or anti-HSCB-specific antibodies (left). 10% of total mitochondrial extract was used as input loading control for co-IP (right). J, co-IP was performed using mitochondrial lysate prepared from yeast cells expressing either FLAG-tagged WT Isu1 or isu1G50E and subjected to immunodetection. The blot was detected for the presence of His6-tagged WT Isu1 and Jac1 using anti-His, Jac1-specific antibodies (left). 10% of total mitochondrial extract was used as input co-IP control (right).
To unravel the mechanistic insights into how overexpression of G50E ISCU or isu1G50E mutant leads to the semidominant phenotype, we tested for their ability to form subcomplexes with WT protein by co-IP analysis. For the immunodetection, the wild type or G50E ISCU was overexpressed as FLAG-tagged in HeLa cells and subjected to co-IP in the mitochondrial lysate using anti-FLAG antibodies followed by immunostaining with the ISCU-specific antibody. Both FLAG-tagged WT and G50E ISCU displayed the ability to form the subcomplex with endogenous WT ISCU (Fig. 9I, bottom). As a co-IP control, the blot was further probed for the HSCB, which showed a diminished interaction (Fig. 9I, top) with the G50E ISCU mutant as compared with WT, consistent with the previous results (Fig. 8I). A similar co-IP analysis was performed in yeast mitochondrial lysate prepared from either overexpressed FLAG-tagged WT Isu1 or isu1G50E using FLAG-specific antibodies. The endogenous Isu1 protein was expressed as His6-tagged and detected using an anti-His-specific antibody. Consistent with the findings in HeLa cells, similar subcomplexes were detected between FLAG-tagged WT and isu1G50E protein with endogenous His6-tagged WT Isu1 (Fig. 9J, bottom and middle). As a co-IP control, the isu1G50E protein showed a diminished interaction with Jac1 (Fig. 9J, top) as compared with WT Isu1, in agreement with the previous results (Fig. 8J). In conclusion, these findings highlight that the semidominant phenotype associated with the G50E ISCU mutation is attributable to its tendency to form an oligomeric complex with endogenous wild type protein, thus probably contributing toward the impairment in the scaffolding function by altering the stoichiometry of the protein association during the assembly process.
DISCUSSION
Following Friedreich ataxia, ISCU myopathy is the second most prevalent pathophysiological condition associated with impairment of Fe-S cluster biogenesis in human mitochondria. However, the mechanism of how mutations in the scaffold protein ISCU generate complex cellular traits that contribute to the disease progression is poorly understood. Our experimental data provide the first compelling evidence to highlight a mitochondrial specific etiology associated with the ISCU myopathy mutation, G50E. The glycine 50 residue is highly conserved in the ISCU class of proteins, across phylogenetic boundaries. Due to its conserved nature, its importance in the Fe-S cluster scaffolding function is unanimous across species. The G50E mutation in the mammalian system causes marked reduction in the cell viability, whereas the analogous mutation in yeast displays a lethal phenotype, highlighting its critical nature for the protein function.
The Fe-S clusters are built in the ISCU/Isu1 platform through precise coordinated events involving the iron and sulfur transfer process. Therefore, it is reasonable to predict that the mutations in the ISCU scaffold protein can directly influence the initial assembly process as well as the rate of Fe-S cluster synthesis in the mitochondrial matrix. In support of this hypothesis, we observed that the G50E mutation in ISCU showed a significantly attenuated enzyme activity of proteins containing Fe-S clusters, both in the mammalian and in the yeast system. Most notably, the activities of multienzyme complexes (I and II) of the ETC was considerably reduced due to compromised Fe-S cluster biogenesis in G50E ISCU mitochondria. This is in agreement with clinical symptoms of ISCU myopathy patients showing marked impairment in the succinate oxidation to fumarate, due to decreased activity of succinate dehydrogenase and aconitase, in muscle mitochondria (54). In addition, our experimental evidence also suggests that the compromised activity of the Fe-S cluster-containing multienzyme complex of the ETC results in diminished pumping of protons across the inner membrane, leading to decreased membrane potential and ATP synthesis and further resulting in overall reduced respiratory activity causing impaired mitochondrial function. Consequently, a significant decrease in the overall mitochondria mass was observed in the G50E ISCU mutant, probably due to enhanced mitophagy. This is one of the most important cellular etiologies that contributes toward disease outcome, particularly in skeletal muscles and cardiomyocytes that demand the highest energy requirement for their constitutive contractile functions. Therefore, the reduced respiratory activity associated with ISCU mutation may directly lead to the manifestation of disease symptoms, such as severe muscle weakness, muscle wasting, and mild cardiac hypertrophy, as observed in affected individuals (28).
Mitochondrial Fe-S cluster biogenesis impairment invariably associates with loss of iron homeostasis inside cells (55). In a well characterized mitochondrial disease, such as Friedreich ataxia, a defect in the function of frataxin leads to decreased Fe-S cluster synthesis. In addition, frataxin deficiency also results in an increase in iron levels in the mitochondrial matrix compartment (41). Our results highlight that the reduction in Fe-S cluster synthesis associated with the myopathy mutation in the ISCU/Isu1 scaffold protein leads to accumulation of iron levels in the mitochondrial compartment similar to frataxin deficiency. Thus, our study reconciles well with the results of histochemical analysis obtained from skeletal muscle biopsy of myopathy patients that showed an intracellular iron overload (22).
The ROS, such as superoxide and peroxide radicals, are primarily generated as a byproduct of oxidative phosphorylation, due to incomplete reduction of free electrons from the ETC complexes (56–58). However, the cellular redox balance is critically regulated through multiple well orchestrated pathways involving members of several antioxidant systems (59). The up-regulation of ROS levels has been implicated in multiple pathological conditions, including cardiovascular diseases, hypertension, atherosclerosis, and neurodegenerative diseases (60, 61). Importantly, our analyses demonstrate a significant up-regulation of ROS levels in the G50E mutant mitochondria of the mammalian system as well as in the yeast system. This is presumably due to a loss in ETC complex activity in response to decreased synthesis of Fe-S clusters, thus enhancing the leakage of free electrons and leading to the production of superoxide radicals. Alternatively, the mitochondrial overloading of a reduced form of iron can also serve as a source for the production of potent hydroxyl radicals via Fenton's reaction (62). These hydroxyl radicals are extremely deleterious to the cellular components because they can cause further damage to the pre-existing Fe-S clusters of the ETC complexes as well as other biomolecules, leading to severe oxidative stress and mitochondrial dysfunction. Based on our findings, we hypothesize that the free radical-mediated damage may therefore significantly contribute to the development of complex secondary traits in myopathy patients.
The initial Fe-S cluster assembly process is aided by a precise coordinated interaction of the scaffold protein with putative iron donors, such as frataxin/Yfh1 and cysteine desulfurase, hNFS1/yNfs1, which is in complex with the Isd11 protein. On the other hand, the molecular chaperone J-protein HSCB (Jac1 in yeast) and its partner mtHsp70 (Ssq1 in yeast) play a critical role in the transfer of Fe-S clusters from the ISCU/Isu1 scaffold to recipient proteins (63). Both are indispensable processes for the homeostasis of iron metabolism in the mitochondrial compartment. Our in vitro as well as ex vivo experiments reveal that the G50E mutation in ISCU/Isu1 results in significantly compromised interaction with the sulfur donor, hNFS1/yNfs1, and the J-protein cochaperone, HSCB/Jac1. As a result, the myopathy-associated ISCU mutant may have severe impairment in the initial synthesis of the cluster as well as in the rate of transfer into the apoproteins, thereby affecting the overall rate of Fe-S cluster biogenesis. Studies from the yeast model system have earlier indicated that formation of the Jac1-Isu1 complex as well as its targeting to the Hsp70s is an essential process in Fe-S cluster biogenesis (15, 20). Most notably, recent analysis has also revealed that yNfs1 and Jac1 both have a mutually exclusive binding surface on Isu1 and that amino acid residues from the Isu1 core β-strands (Leu-63, Val-72, and Phe-94) are involved in the interaction with Jac1 and yNfs1 in yeast (15). Strikingly, the G50E mutant also exhibits a similarly compromised interaction with both HSCB and hNFS1, indicating that they possess a mutually exclusive interaction surface on the ISCU protein, thus confirming the previous observation. Based on the severity of the phenotype observed with the myopathic mutation in humans as well as in the yeast system, it is reasonable to predict that glycine 50 is one of the critical residues required for mediating the hNFS1 and HSCB interaction.
Although the functional oligomeric state of the human ISCU is not very clear, the monomeric form probably is not a functional entity in terms of coordinating geometry to form Fe-S clusters. For example, in A. vinelandii and S. pombe, IscU exists as a dimer in which one or two [2Fe-2S] or [4Fe-4S] clusters are constituted (53). In the case of A. aeolicus, IscU exists in a trimeric form, the [2Fe-2S] cluster is positioned at the interface between two subunits, and the third subunit provides additional stability for the interaction (64). Results from gel filtration analysis demonstrate that WT apo-ISCU mostly exists in a dimeric form, whereas apo-G50E ISCU showed a higher tendency to form a larger oligomeric species. Most notably, apo-G50E ISCU displayed a tendency to form oligomeric subcomplexes with wild type protein when overexpressed, thus imparting a semidominant negative phenotype. The formation of such higher molecular weight oligomers in the G50E mutant in compound heterozygous conditions may therefore impair the precise coordination geometry required to assemble the Fe-S cluster on ISCU. On the other hand, the oligomeric forms of the G50E mutant may also hamper the physical interaction surface of ISCU with its interacting partners hNFS1 and HSCB, resulting in their compromised interaction, thereby abrogating the overall Fe-S cluster synthesis and diminishing mitochondrial function in myopathy patients.
In conclusion, our study elucidates insights into the complex protein-protein interactions involving the ISCU scaffold protein, in the process of Fe-S cluster biogenesis, in the mammalian system. Additionally, our findings delineate the cellular mechanisms behind the impairment of mitochondrial function associated with the G50E mutation, leading to manifestation of pathological symptoms observed in ISCU myopathy. Although our basic understanding of Fe-S cluster biogenesis is primarily obtained from a yeast model system, a relevant complex process in the mammalian system is poorly understood. Following Friedreich ataxia, ISCU myopathy is the second most prevalent mitochondrial disorder identified with a high penetrance rate in the European lineage and is connected with the impairment in Fe-S cluster biogenesis. Thus, our findings at the cellular as well as in molecular level should provide invaluable information to understand the disease progression in both homozygous and compound heterozygous affected individuals.
Acknowledgments
We thank Dr. Elizabeth A. Craig for providing the Δisu1/isu2 yeast strain and anti-Jac1 antibody. We thank Dr. Roland Lill and Dr. Wing-Hang Tong for anti-yeast Nfs1 and anti-human NFS1 antibodies, respectively. We also thank the Flow Cytometry Facility of the Indian Institute of Science (Bangalore, India) for FACS experiments and the Solid State and Structural Chemistry Unit of the Indian Institute of Science (Bangalore, India) for atomic absorption spectroscopic analysis.
This work was supported by Council of Scientific and Industrial Research, India, Grant 37(1534)/12/EMR-II (to P. D. S).
- TCA
- tricarboxylic acid
- ROS
- reactive oxygen species
- aa
- amino acids
- NAO
- 10-N-nonyl acridine orange
- IP
- immunoprecipitation
- AAS
- atomic absorption spectroscopy
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BN-PAGE
- blue native PAGE
- UT
- untransfected
- ETC
- electron transport chain.
REFERENCES
- 1. Craig E. A., Marszalek J. (2002) A specialized mitochondrial molecular chaperone system: a role in formation of Fe/S centers. Cell. Mol. Life Sci. 59, 1658–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Craig E. A., Voisine C., Schilke B. (1999) Mitochondrial iron metabolism in the yeast Saccharomyces cerevisiae. Biol. Chem. 380, 1167–1173 [DOI] [PubMed] [Google Scholar]
- 3. Lill R. (2009) Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 [DOI] [PubMed] [Google Scholar]
- 4. Meyer J. (2008) Iron-sulfur protein folds, iron-sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 13, 157–170 [DOI] [PubMed] [Google Scholar]
- 5. Hinchliffe P., Sazanov L. A. (2005) Organization of iron-sulfur clusters in respiratory complex I. Science 309, 771–774 [DOI] [PubMed] [Google Scholar]
- 6. Sazanov L. A. (2007) Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry 46, 2275–2288 [DOI] [PubMed] [Google Scholar]
- 7. Sun F., Huo X., Zhai Y., Wang A., Xu J., Su D., Bartlam M., Rao Z. (2005) Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057 [DOI] [PubMed] [Google Scholar]
- 8. Beinert H., Kennedy M. C., Stout C. D. (1996) Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 96, 2335–2374 [DOI] [PubMed] [Google Scholar]
- 9. King A., Selak M. A., Gottlieb E. (2006) Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682 [DOI] [PubMed] [Google Scholar]
- 10. Lill R., Dutkiewicz R., Elsässer H. P., Hausmann A., Netz D. J., Pierik A. J., Stehling O., Urzica E., Mühlenhoff U. (2006) Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim. Biophys. Acta 1763, 652–667 [DOI] [PubMed] [Google Scholar]
- 11. Tong W. H., Rouault T. (2000) Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 19, 5692–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garland S. A., Hoff K., Vickery L. E., Culotta V. C. (1999) Saccharomyces cerevisiae ISU1 and ISU2: members of a well-conserved gene family for iron-sulfur cluster assembly. J. Mol. Biol. 294, 897–907 [DOI] [PubMed] [Google Scholar]
- 13. Fontecave M., Ollagnier-de-Choudens S. (2008) Iron-sulfur cluster biosynthesis in bacteria: mechanisms of cluster assembly and transfer. Arch. Biochem. Biophys. 474, 226–237 [DOI] [PubMed] [Google Scholar]
- 14. Biederbick A., Stehling O., Rösser R., Niggemeyer B., Nakai Y., Elsässer H. P., Lill R. (2006) Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation. Mol. Cell. Biol. 26, 5675–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Majewska J., Ciesielski S. J., Schilke B., Kominek J., Blenska A., Delewski W., Song J. Y., Marszalek J., Craig E. A., Dutkiewicz R. (2013) Binding of the chaperone Jac1 protein and cysteine desulfurase Nfs1 to the iron-sulfur cluster scaffold Isu protein is mutually exclusive. J. Biol. Chem. 288, 29134–29142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson D. C., Dean D. R., Smith A. D., Johnson M. K. (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74, 247–281 [DOI] [PubMed] [Google Scholar]
- 17. Bencze K. Z., Kondapalli K. C., Cook J. D., McMahon S., Millán-Pacheco C., Pastor N., Stemmler T. L. (2006) The structure and function of frataxin. Crit. Rev. Biochem. Mol. Biol. 41, 269–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Craig E. A., Huang P., Aron R., Andrew A. (2006) The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev. Physiol. Biochem. Pharmacol. 156, 1–21 [DOI] [PubMed] [Google Scholar]
- 19. Schilke B., Williams B., Knieszner H., Pukszta S., D'Silva P., Craig E. A., Marszalek J. (2006) Evolution of mitochondrial chaperones utilized in Fe-S cluster biogenesis. Curr. Biol. 16, 1660–1665 [DOI] [PubMed] [Google Scholar]
- 20. Ciesielski S. J., Schilke B. A., Osipiuk J., Bigelow L., Mulligan R., Majewska J., Joachimiak A., Marszalek J., Craig E. A., Dutkiewicz R. (2012) Interaction of J-protein co-chaperone Jac1 with Fe-S scaffold Isu is indispensable in vivo and conserved in evolution. J. Mol. Biol. 417, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andrew A. J., Dutkiewicz R., Knieszner H., Craig E. A., Marszalek J. (2006) Characterization of the interaction between the J-protein Jac1p and the scaffold for Fe-S cluster biogenesis, Isu1p. J. Biol. Chem. 281, 14580–14587 [DOI] [PubMed] [Google Scholar]
- 22. Mochel F., Knight M. A., Tong W. H., Hernandez D., Ayyad K., Taivassalo T., Andersen P. M., Singleton A., Rouault T. A., Fischbeck K. H., Haller R. G. (2008) Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 82, 652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olsson A., Lind L., Thornell L. E., Holmberg M. (2008) Myopathy with lactic acidosis is linked to chromosome 12q23.3–24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum. Mol. Genet. 17, 1666–1672 [DOI] [PubMed] [Google Scholar]
- 24. Kollberg G., Melberg A., Holme E., Oldfors A. (2011) Transient restoration of succinate dehydrogenase activity after rhabdomyolysis in iron-sulphur cluster deficiency myopathy. Neuromuscul. Disord. 21, 115–120 [DOI] [PubMed] [Google Scholar]
- 25. Larsson L. E., Linderholm H., Mueller R., Ringqvist T., Soernaes R. (1964) Hereditary metabolic myopathy with paroxysmal myoglobinuria due to abnormal glycolysis. J. Neurol. Neurosurg. Psychiatry 27, 361–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kollberg G., Holme E. (2009) Antisense oligonucleotide therapeutics for iron-sulphur cluster deficiency myopathy. Neuromuscul. Disord. 19, 833–836 [DOI] [PubMed] [Google Scholar]
- 27. Sanaker P. S., Toompuu M., Hogan V. E., He L., Tzoulis C., Chrzanowska-Lightowlers Z. M., Taylor R. W., Bindoff L. A. (2010) Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy. Biochim. Biophys. Acta 1802, 539–544 [DOI] [PubMed] [Google Scholar]
- 28. Kollberg G., Tulinius M., Melberg A., Darin N., Andersen O., Holmgren D., Oldfors A., Holme E. (2009) Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 132, 2170–2179 [DOI] [PubMed] [Google Scholar]
- 29. Goswami A. V., Samaddar M., Sinha D., Purushotham J., D'Silva P. (2012) Enhanced J-protein interaction and compromised protein stability of mtHsp70 variants lead to mitochondrial dysfunction in Parkinson's disease. Hum. Mol. Genet. 21, 3317–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uchiyama A., Kim J. S., Kon K., Jaeschke H., Ikejima K., Watanabe S., Lemasters J. J. (2008) Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology 48, 1644–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. (2012) Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 7, 1235–1246 [DOI] [PubMed] [Google Scholar]
- 32. Pierik A. J., Netz D. J., Lill R. (2009) Analysis of iron-sulfur protein maturation in eukaryotes. Nat. Protoc. 4, 753–766 [DOI] [PubMed] [Google Scholar]
- 33. Schilke B., Voisine C., Beinert H., Craig E. (1999) Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 96, 10206–10211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meisinger C., Ryan M. T., Hill K., Model K., Lim J. H., Sickmann A., Müller H., Meyer H. E., Wagner R., Pfanner N. (2001) Protein import channel of the outer mitochondrial membrane: a highly stable Tom40-Tom22 core structure differentially interacts with preproteins, small tom proteins, and import receptors. Mol. Cell. Biol. 21, 2337–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee S., Sheck L., Crowston J. G., Van Bergen N. J., O'Neill E. C., O'Hare F., Kong Y. X., Chrysostomou V., Vincent A. L., Trounce I. A. (2012) Impaired complex-I-linked respiration and ATP synthesis in primary open-angle glaucoma patient lymphoblasts. Invest. Ophthalmol Vis. Sci. 53, 2431–2437 [DOI] [PubMed] [Google Scholar]
- 36. Wen J. J., Garg N. J. (2010) Mitochondrial complex III defects contribute to inefficient respiration and ATP synthesis in the myocardium of Trypanosoma cruzi-infected mice. Antioxid. Redox Signal. 12, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sheftel A., Stehling O., Lill R. (2010) Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab. 21, 302–314 [DOI] [PubMed] [Google Scholar]
- 38. Nowikovsky K., Reipert S., Devenish R. J., Schweyen R. J. (2007) Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 14, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 39. Johnson L. V., Walsh M. L., Bockus B. J., Chen L. B. (1981) Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 88, 526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gallet P. F., Maftah A., Petit J. M., Denis-Gay M., Julien R. (1995) Direct cardiolipin assay in yeast using the red fluorescence emission of 10-N-nonyl acridine orange. Eur. J. Biochem. 228, 113–119 [DOI] [PubMed] [Google Scholar]
- 41. Delatycki M. B., Camakaris J., Brooks H., Evans-Whipp T., Thorburn D. R., Williamson R., Forrest S. M. (1999) Direct evidence that mitochondrial iron accumulation occurs in Friedreich ataxia. Ann. Neurol. 45, 673–675 [PubMed] [Google Scholar]
- 42. Kakhlon O., Cabantchik Z. I. (2002) The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic. Biol. Med. 33, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 43. Devireddy L. R., Hart D. O., Goetz D. H., Green M. R. (2010) A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 141, 1006–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gutteridge J. M., Maidt L., Poyer L. (1990) Superoxide dismutase and Fenton chemistry. Reaction of ferric-EDTA complex and ferric-bipyridyl complex with hydrogen peroxide without the apparent formation of iron(II). Biochem. J. 269, 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Py B., Barras F. (2010) Building Fe-S proteins: bacterial strategies. Nat. Rev. Microbiol. 8, 436–446 [DOI] [PubMed] [Google Scholar]
- 46. Jang S., Imlay J. A. (2007) Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem. 282, 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dixon S. J., Stockwell B. R. (2014) The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 [DOI] [PubMed] [Google Scholar]
- 48. Uhrigshardt H., Singh A., Kovtunovych G., Ghosh M., Rouault T. A. (2010) Characterization of the human HSC20, an unusual DnaJ type III protein, involved in iron-sulfur cluster biogenesis. Hum. Mol. Genet. 19, 3816–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adinolfi S., Rizzo F., Masino L., Nair M., Martin S. R., Pastore A., Temussi P. A. (2004) Bacterial IscU is a well folded and functional single domain protein. Eur. J. Biochem. 271, 2093–2100 [DOI] [PubMed] [Google Scholar]
- 50. Kato S., Mihara H., Kurihara T., Takahashi Y., Tokumoto U., Yoshimura T., Esaki N. (2002) Cys-328 of IscS and Cys-63 of IscU are the sites of disulfide bridge formation in a covalently bound IscS/IscU complex: implications for the mechanism of iron-sulfur cluster assembly. Proc. Natl. Acad. Sci. U.S.A. 99, 5948–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agar J. N., Krebs C., Frazzon J., Huynh B. H., Dean D. R., Johnson M. K. (2000) IscU as a scaffold for iron-sulfur cluster biosynthesis: sequential assembly of [2Fe-2S] and [4Fe-4S] clusters in IscU. Biochemistry 39, 7856–7862 [DOI] [PubMed] [Google Scholar]
- 52. Wu G., Mansy S. S., Wu Sp S. P., Surerus K. K., Foster M. W., Cowan J. A. (2002) Characterization of an iron-sulfur cluster assembly protein (ISU1) from Schizosaccharomyces pombe. Biochemistry 41, 5024–5032 [DOI] [PubMed] [Google Scholar]
- 53. Shimomura Y., Kamikubo H., Nishi Y., Masako T., Kataoka M., Kobayashi Y., Fukuyama K., Takahashi Y. (2007) Characterization and crystallization of an IscU-type scaffold protein with bound [2Fe-2S] cluster from the hyperthermophile, Aquifex aeolicus. J. Biochem. 142, 577–586 [DOI] [PubMed] [Google Scholar]
- 54. Hall R. E., Henriksson K. G., Lewis S. F., Haller R. G., Kennaway N. G. (1993) Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Invest. 92, 2660–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Richardson D. R., Lane D. J., Becker E. M., Huang M. L., Whitnall M., Suryo Rahmanto Y., Sheftel A. D., Ponka P. (2010) Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. U.S.A. 107, 10775–10782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Batandier C., Fontaine E., Kériel C., Leverve X. M. (2002) Determination of mitochondrial reactive oxygen species: methodological aspects. J. Cell Mol. Med. 6, 175–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kudin A. P., Bimpong-Buta N. Y., Vielhaber S., Elger C. E., Kunz W. S. (2004) Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 279, 4127–4135 [DOI] [PubMed] [Google Scholar]
- 58. Liu Y., Fiskum G., Schubert D. (2002) Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80, 780–787 [DOI] [PubMed] [Google Scholar]
- 59. Trachootham D., Lu W., Ogasawara M. A., Nilsa R. D., Huang P. (2008) Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. de Moura M. B., dos Santos L. S., Van Houten B. (2010) Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ. Mol. Mutagen. 51, 391–405 [DOI] [PubMed] [Google Scholar]
- 61. Kirkinezos I. G., Moraes C. T. (2001) Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol. 12, 449–457 [DOI] [PubMed] [Google Scholar]
- 62. Levi S., Rovida E. (2009) The role of iron in mitochondrial function. Biochim. Biophys. Acta 1790, 629–636 [DOI] [PubMed] [Google Scholar]
- 63. Dutkiewicz R., Schilke B., Cheng S., Knieszner H., Craig E. A., Marszalek J. (2004) Sequence-specific interaction between mitochondrial Fe-S scaffold protein Isu and Hsp70 Ssq1 is essential for their in vivo function. J. Biol. Chem. 279, 29167–29174 [DOI] [PubMed] [Google Scholar]
- 64. Shimomura Y., Wada K., Fukuyama K., Takahashi Y. (2008) The asymmetric trimeric architecture of [2Fe-2S] IscU: implications for its scaffolding during iron-sulfur cluster biosynthesis. J. Mol. Biol. 383, 133–143 [DOI] [PubMed] [Google Scholar]