

Background: Membrane recruitment of the calcium channel β2e-subunit remains elusive.
Results: Charge neutralization of β2e N-terminal residues abolishes its lipid-binding and surface-targeting abilities and its slow inactivation-conferring phenotype.
Conclusion: The β2e-subunit anchors to the membrane via electrostatic interactions likely involving an N-terminal in-plane α-helix and a tryptophan side-chain penetration.
Significance: A novel mechanism for β-subunit membrane-anchoring supporting slow inactivation is presented.
Keywords: Calcium Channels, Electrophysiology, Phospholipid, Plasma Membrane, Tryptophan
Abstract
The β-subunit associates with the α1 pore-forming subunit of high voltage-activated calcium channels and modulates several aspects of ion conduction. Four β-subunits are encoded by four different genes with multiple splice variants. Only two members of this family, β2a and β2e, associate with the plasma membrane in the absence of the α1-subunit. Palmitoylation on a di-cysteine motif located at the N terminus of β2a promotes membrane targeting and correlates with the unique ability of this protein to slow down inactivation. In contrast, the mechanism by which β2e anchors to the plasma membrane remains elusive. Here, we identified an N-terminal segment in β2e encompassing a cluster of positively charged residues, which is strictly required for membrane anchoring, and when transferred to the cytoplasmic β1b isoform it confers membrane localization to the latter. In the presence of negatively charged phospholipid vesicles, this segment binds to acidic liposomes dependently on the ionic strength, and the intrinsic fluorescence emission maxima of its single tryptophan blue shifts considerably. Simultaneous substitution of more than two basic residues impairs membrane targeting. Coexpression of the fast inactivating R-type calcium channels with wild-type β2e, but not with a β2e membrane association-deficient mutant, slows down inactivation. We propose that a predicted α-helix within this domain orienting parallel to the membrane tethers the β2e-subunit to the lipid bilayer via electrostatic interactions. Penetration of the tryptophan side chain into the lipidic core stabilizes the membrane-bound conformation. This constitutes a new mechanism for membrane anchoring among the β-subunit family that also sustains slowed inactivation.
Introduction
The α1-subunit (CaVα1) of HVA2 calcium channels encompasses the ion conduction pathway, the voltage sensor, and the multiple intracellular domains that provide sites of interaction for regulatory proteins (1). One of these proteins is the β-subunit (CaVβ) that binds to a highly conserved site among HVA channels and modulates several aspects of channel function, including surface expression and biophysical properties (2, 3). CaVβs are special members of the membrane-associated guanylate kinase family of proteins containing only two of the three canonical domains shared by this group as follows: an Src homology 3 (SH3) and a guanylate kinase domain (4–6). In contrast to these highly conserved domains, the linker region between them as well as the N- and C-terminal flanking segments (HOOK, NT, and CT, respectively, Fig. 1) vary in sequence and length among the four β-subunit isoforms (β1 to β4) identified until now. Each β subtype is encoded by a different gene that generates multiple splice variants giving rise to functionally different proteins. An extreme case occurs among the β2 splice variants (β2a to β2e) that, despite their sequence similarity, have different subcellular localizations and even have opposing regulatory effects on calcium currents (7, 8). β2 variants are between 604 and 655 amino acids long and they differ only in their most proximal N-terminal segment, hereafter referred to as NTv (the subscript v stands for variable, Fig. 1). The subtypes 2a and 2e are the only members of the CaVβ family that are targeted to the plasma membrane in the absence of the α1 pore-forming subunit, and when associated with it, they inhibit the voltage-dependent inactivation of the channel complex (7, 9, 10). Coexpression of the fast-inactivating R-type calcium channel with β2a in Xenopus laevis oocytes or mammalian cell lines increases considerably the time needed for the current to return to 50% of its peak amplitude and shifts the steady-state inactivation curve toward more positive potentials as compared with channels composed of cytosolic, nonmembrane-associated β-isoforms (10–13). These unique modulatory properties of β2a rely on two continuous N-terminal cysteine residues that undergo palmitoylation and target the protein to the plasma membrane (9, 14, 15). Substitution of these two residues disrupts membrane localization as well as the slow inactivation-conferring phenotype of β2a (16). The prevalent view is that membrane anchoring restricts the free movement of the channel inactivation gate, preventing it from closing. Colecraft and co-workers (7) showed that CaV1.2 L-type calcium channels also inactivate more slowly in the presence of the two membrane-anchored β2 variants as compared with their fully cytosolic distributed counterparts, β2b, β2c, and β2d. In contrast to β2a, the mechanism by which the β2e-subunit is targeted to the plasma membrane remains to be elucidated. Here, we exchanged the variable N-terminal region between the β2e and the β1b isoform, which is distributed entirely in the cytoplasm, and we showed that this maneuver is sufficient to target the β1b chimera to the plasma membrane. Moreover, the same N-terminal segment binds in vitro to negatively charged liposomes in an ionic strength-dependent manner. β2eNTv includes seven positively charged residues and a net charge of +4 at physiological pH. We studied the role of these basic amino acids in membrane association and found that substitution of these residues by alanine inhibits the targeting of the protein to the plasma membrane and the in vitro interaction with anionic lipids. Coexpression of wild-type β2e with the fast inactivating CaVα1 encoding the CaV2.3 R-type calcium channel results in channel complexes that inactivate significantly more slowly than CaV2.3 expressed with a β2e mutant in which all positively charged residues within NTv were replaced by alanine. Our results demonstrate that the positively charged N-terminal variable segment of β2e is responsible for tethering the protein to the plasma membrane via electrostatic interactions and support the model suggesting that membrane anchoring underlies the slow inactivation-conferring phenotype.
FIGURE 1.
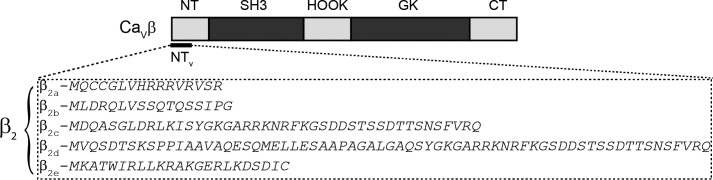
Scheme of the domain organization of CaVβ and amino acid sequences of the N-terminal variable segments of β2 splice variants. The two highly conserved domains among the CaVβ family, SH3 and guanylate kinase (GK), shown in dark gray, are flanked by three nonconserved regions: N-terminal (NT), linker region joining the SH3 and GK domains (HOOK), and C-terminal (CT). The amino acid sequences corresponding to the variable segment (NTV) among all β2 splice variants (β2a to β2e) from rat are shown.
EXPERIMENTAL PROCEDURES
cDNA Constructs
Full-length rat β2e (UniProtKB accession number Q8VGC3-4) and rat β1b (UniProtKB accession number P54283) cDNAs were subcloned into pcDNA 3.1 vector and fused at the C terminus to YFP (for confocal imaging) or monomeric red fluorescence protein (for electrophysiology) using standard overlapping PCR methods. β2e mutants were generated by PCR using a common antisense and specific sense primers bearing the corresponding mutation. The chimeric β2eNTv/β1b was also generated using overlapping PCR methods by replacing residues 1–57 of β1b with residues 1–23 of β2e. The cDNAs encoding wild-type β2eNTv (amino acids 1–23) or mutant versions were subcloned into PGEX 6P-1 vector using standard PCR methods to produce GST-β2eNTv domain fusion constructs. YFP was fused to the N terminus of CaV2.3-coding region (UniProtKB accession number Q15878) using standard PCR methods and subcloned into pRcCMV vector.
Transfection of tsA201 Cells
For confocal microscopy, cells were grown on plastic μ-dishes (Ibidi) in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and l-glutamine (2 mm) and incubated in a 5% CO2-humidified atmosphere. Cells were transiently transfected using Lipofectamine 2000TM (Invitrogen) according to the manufacturer's instructions with the corresponding plasmids encoding the different β2e derivatives. For whole-cell patch clamp recordings, cells were cotransfected with YFP-CaV2.3 and the corresponding β2e fused to monomeric red fluorescence protein-encoding plasmids.
Confocal Microscopy and Colocalization Analysis
Confocal images were acquired 24–30 h after transfection on living cells using a 63× oil immersion objective on a Leica inverted confocal microscope. For colocalization analysis, prior to imaging the cells were stained with the plasma membrane marker CellMaskTM (Invitrogen) according to the manufacturer's instructions. YFP fusion proteins were excited with a 514-nm laser, and emission was monitored at 520–550 nm, and the plasma membrane stain was excited at 633 nm and monitored at 640–700 nm. Quantification of the degree of colocalization between fluorescently labeled β-subunit and the plasma membrane marker was done by calculating the Manders coefficient (MC) using the JACoP plugin (17) embedded in ImageJ 1.44p software (National Institutes of Health, Bethesda) (18). At least five different fields of view (in total 100–200 transfected cells) for each β2e derivative from at least two different transfections were analyzed. The Manders coefficient represents the fraction of pixels in the YFP channel that overlaps with some signal in the CellMaskTM (red) detection channel. For comparison of samples, a two-tailed Student's t test was used. Values are expressed as mean ± S.E.
Liposome Preparation
Phospholipid unilamellar vesicles were prepared by extrusion of multilamellar vesicles through 100-nm pore size polycarbonate filters using a handheld extruder (LiposoFast, Avestin, Inc). 1-Palmitoyl-2-oleoyl-sn-glycerophosphocholine (PC, Avanti 840034) either alone or combined with 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (PS, Avanti 840034) in a 1:3 (PS/PC) molar ratio was dried under a stream of nitrogen. Any residual solvent was removed under vacuum. The resulting lipid film was rehydrated to reach a final concentration of 1 mm with a buffer containing 50 mm HEPES pH 7.5 supplemented with the indicated NaCl concentration and then subjected to three-to five freeze-thaw cycles using liquid nitrogen and a water bath at 37 °C. The lipid suspension was either extruded to obtain uniformly sized (about 100 nm diameter) large unilamellar vesicles (LUVs) for protein-liposome cosedimentation assays or sonicated at RT using a bath sonicator to obtain small unilamellar vesicles (SUVs) for tryptophan fluorescence studies (Morrissey Lab Protocols, University of Illinois) (19, 20).
Control PS/PC liposomes were prepared by adding 0.1% fluorescently labeled 7-nitro-2–1,3-benzoxadiazol-4-yl-PS (Avanti 810198). The fluorescence intensity was monitored using a microplate reader (FLUOstar Omega (BMG Labtec).
Protein-Liposome Cosedimentation Assays
Cosedimentation assays were performed as described previously (21). Briefly, 2.0 μg of protein were incubated with LUVs in 50 mm HEPES pH 7.5 buffer supplemented with the indicated NaCl concentration (total volume 100 μl) for 15 min at room temperature. Samples were ultracentrifuged at 150,000 × g for 40 min at 4 °C using a TLA 120.2 rotor (Beckman Instruments). Pellets were resuspended in the same volume as the supernatant, and equal volumes of each fraction from each sample were resolved by SDS-PAGE (12%). The quantification of proteins present in the different fractions was performed by densitometry of Coomassie Blue-stained gels using ImageJ software (18). The fraction of the protein bound to liposomes (protein-bound) was calculated from the quotient between the fraction of nonaggregated protein in the pellet and the pool of nonaggregated protein as shown in Equation 1,
 |
where PP(Lip) is the fraction of protein in the pellet in the presence of lipids, and PP(Ø Lip) is the fraction of protein in the pellet in the absence of liposomes (corresponding to the aggregated protein).
Tryptophan Fluorescence Measurements
Measurements were performed on a FluoroLog 3 spectrofluorometer (HORIBA Jobin Yvon) using 1–2 μm of a synthetic peptide (Eurogentec) containing the N-terminal region of β2e (1MKATWIRLLKRAKGERLKDSD21) dissolved in different concentrations of SUVs in 50 mm HEPES pH 7.5 buffer supplemented with the indicated NaCl concentration. After combining the reagents in a 1-cm cuvette, the sample was mixed by pipetting and allowed to settle for ∼5 min. Upon excitation of the samples at 280 nm, the emission spectrum was recorded in front face excitation mode from 290 to 440 or 300 to 440 nm. At each wavelength, the signal was integrated over 0.2 s. The bandwidth was 6–8 nm on the excitation side and 1–2.5 nm on the emission side to optimize the signal levels. Data analysis was performed using Origin (OriginLab) and SigmaPlot (Systat Software). To infer the wavelength of maximal emission, a log-normal distribution was fitted to the spectra (22). For the quantification of binding of vesicles to the peptide, it was assumed that the fluorescence emission results from a two-state equilibrium between unbound and bound peptide. It follows that every measured spectrum is a linear combination of two spectra. Accordingly, the fraction of peptide that is bound to lipids was determined by a deconvolution of the spectral data into the spectra resulting from experiments with 0 and 1.500 μm lipid concentration, corresponding to free and fully lipid-bound peptide, respectively (23). The resulting data were fitted by a Hill function.
Protein Purification
Wild-type β2e and β2eNTv(7+ less), both fused to a hexahistidine tag, and the GST fusion proteins were expressed in bacteria and purified using affinity and size-exclusion chromatography as described previously (11).
Protein-Protein Binding Assays
The highly conserved β-binding site among all α1 pore-forming subunits of high voltage gated calcium channels known the as α1 interaction domain (AID) was fused to GST (GST-AID). Either 4 μg of GST-AID or of GST alone (in control experiments) was pre-coupled to glutathione beads (11). Coupled beads were then incubated with 4 μg of either β2e or β2eNTv(7+ less) for 1 h at room temperature. After several washes, the bound proteins were eluted with SDS-loading buffer and resolved on SDS-PAGE. The gel was stained with Coomassie Blue.
Electrophysiological Recordings
Whole-cell patch clamp recordings were performed with an EPC-10 amplifier implemented with the PatchMaster software (HEKA, Elektronik). Borosilicate pipettes (Harvard Apparatus) with resistances of 0.9–2 megohms were pulled on a Sutter P-1000 puller (Harvard Apparatus) and fire-polished using a Narishige MF-830 microforge. Series resistance compensation was applied, resulting in less than 5-mV voltage error. The external recording solutions contained the following (in mm): 140 tetraethylammonium-MeSO3, 10 BaCl2, 10 HEPES buffer pH 7.3 adjusted with tetraethylammonium hydroxide although the internal solution contained 135 cesium-MeSO3, 10 EGTA, 5 CsCl, 1 MgCl2, 4 MgATP, 10 HEPES, and the pH was adjusted to 7.3 with CsOH. The data analysis was performed using a combination of FitMaster (HEKA), Origin (OriginLab), SigmaPlot (Systat Software), and Excel (Microsoft) software. All data are presented as mean ± S.E.
RESULTS
N Terminus of β2e Is Responsible for Targeting the Protein to the Plasma Membrane
Because β2 variants differ only in the most proximal amino acid sequence within the N-terminal region, it is likely that the differences in their subcellular location are caused by signals within this part of the protein. To assess the impact of β2eNTv on membrane anchoring, we deleted this segment (residues 2–23) from the rat β2e. Wild-type β2e, the deletion mutant, and the entire cytosolic β1b isoform were expressed in tsA201 cells as YFP fusion proteins for confocal imaging analysis. Plasma membrane localization of the YFP fusion proteins was analyzed by staining the transfected cells with the CellMaskTM plasma membrane marker and quantifying their degree of colocalization using the MC as described under “Experimental Procedures.” For the plasma membrane-associated β2e-YFP (7), we obtained an MC of 0.48 ± 0.01, for YFP alone, which distributes homogeneously within the cell, the value was 0.10 ± 0.01 (t test; p < 0.01, Fig. 2). After deletion of the NTv segment, the degree of colocalization decreased significantly (MC = 0.14 ± 0.01, t test; p < 0.01, Fig. 2). We also exchanged the N-terminal variable regions between β2e and the entire cytosolic β1b-subunit to generate a chimeric protein encompassing β2eNTv in the β1b background (β2eNTv/β1b). This maneuver increases MC from 0.12 ± 0.01 for β1b to 0.35 ± 0.03 for the chimeric construct (t test; p < 0.01, Fig. 2B), a magnitude approaching that of wild-type β2e. This indicates that substitution of the N-terminal segment of β1b by β2eNTv transfers to the former the capability to associate with the plasma membrane.
FIGURE 2.

The Variable N-terminal segment of the β2e-subunit is responsible for targeting the protein to the plasma membrane. A, confocal fluorescence images of tsA201 cells expressing the indicated protein constructs fused to YFP as follows: full-length β2e (β2e); β2e lacking NTV (β2e ΔNTV); full-length β1b (β1b); chimeric protein encompassing NTV of β2e in a β1b background (β2e NTV/β1b). YFP moiety alone was used as control protein. Prior to visualization, cells were stained with the red plasma membrane marker CellMaskTM. Images from β2e fusion proteins, green channel (panels a–e); plasma membranes, red channel (panels f–j); and merge (panels k–o) are shown. Scale bar, 15 μm and is valid for all images. The bottom panels show crude lysates from cells expressing the indicated construct resolved by SDS-PAGE and visualized by fluorescence scanning. Numbers denote the molecular mass of standard proteins. The migration of the fusion constructs is consistent with their predicted molecular mass. B, bar plot of the colocalization analysis between β2e derivatives and the plasma membrane marker according to Manders coefficient. Values are expressed as mean ± S.E.
To exclude the possibility that the alterations in protein sorting were due to expression artifacts or degradation, crude homogenates from cells expressing the different fluorescent proteins were run on SDS-PAGE and visualized by fluorescence scanning (Fig. 2A, bottom panels). The results show that all YFP fusion proteins were expressed in their full length indicating that the changes observed in their subcellular location are associated with the absence or presence of β2eNTv. Together, these results demonstrate that β2eNTv is the molecular determinant for targeting the protein to the plasma membrane.
The Variable N-terminal Segment of β2e Associates in an Ionic Strength-dependent Manner with Phospholipid Vesicles
Peripheral membrane proteins commonly associate with the plasma membrane via covalent lipid modification of specific residues and/or via electrostatic interactions between positively charged amino acid residues and negatively charged phospholipids (24). Substitution of the single cysteine residue in β2eNTv by alanine (β2e C23A) does not impair the relocation of the protein to the plasma membrane (Fig. 3). The degree of colocalization between this mutant and the plasma membrane marker is slightly higher than the one estimated for the wild-type protein (MC = 0.62 ± 0.06, Fig. 3B). We conclude that palmitoylation is not responsible for the membrane association of this subunit, as is the case for β2a (14). β2eNTv contains no predictable additional protein modification sites such as prenylation or myristoylation but is enriched in positively charged residues. Therefore, we investigated the role of electrostatic interactions in the membrane association of β2e using protein-liposome cosedimentation assays. β2eNTv was fused to GST (GST-β2eNTv), expressed in bacteria, purified, and incubated at different ionic strengths with LUVs containing either negatively charged (PS/PC) or neutral (PC) phospholipids. After centrifugation, the supernatant and pellet fractions containing lipid-free and lipid-associated protein, respectively, were analyzed using SDS-PAGE (Fig. 4A). The presence of lipids in the supernatant and in the pellet fractions was confirmed in control experiments where a fluorescently labeled PS analog (7-nitro-2–1,3-benzoxadiazol-4-yl-PS) was added during the preparation of liposomes. The majority of the lipids was found in the pellet fraction at the different NaCl concentrations tested (99 ± 1, 96 ± 2, and 91 ± 4% in 50, 300, and 400 mm NaCl, respectively).
FIGURE 3.
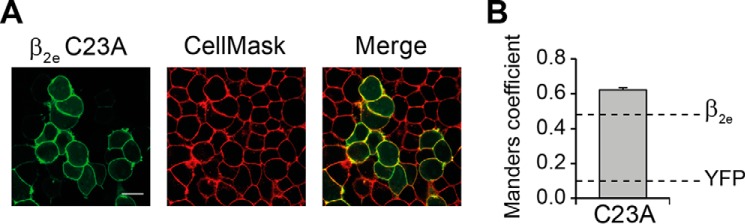
Subcellular localization of β2e C23A mutant protein. A, confocal fluorescence images of tsA201 cells expressing β2e C23A fused to YFP. Prior to visualization, cells were stained with the red plasma membrane marker CellMaskTM. Images from β2e C23A fusion protein (green channel), plasma membranes (red channel), and merge are shown. Scale bar, 15 μm. B, bar plot of the colocalization analysis for β2e C23A mutant. Values are expressed as mean ± S.E. Dashed lines correspond to the average values obtained for β2e and YFP alone from Fig. 2B. The MC value for β2e C23A was significantly different from that measured in cells expressing the wild-type protein (t test; p < 0.05).
FIGURE 4.
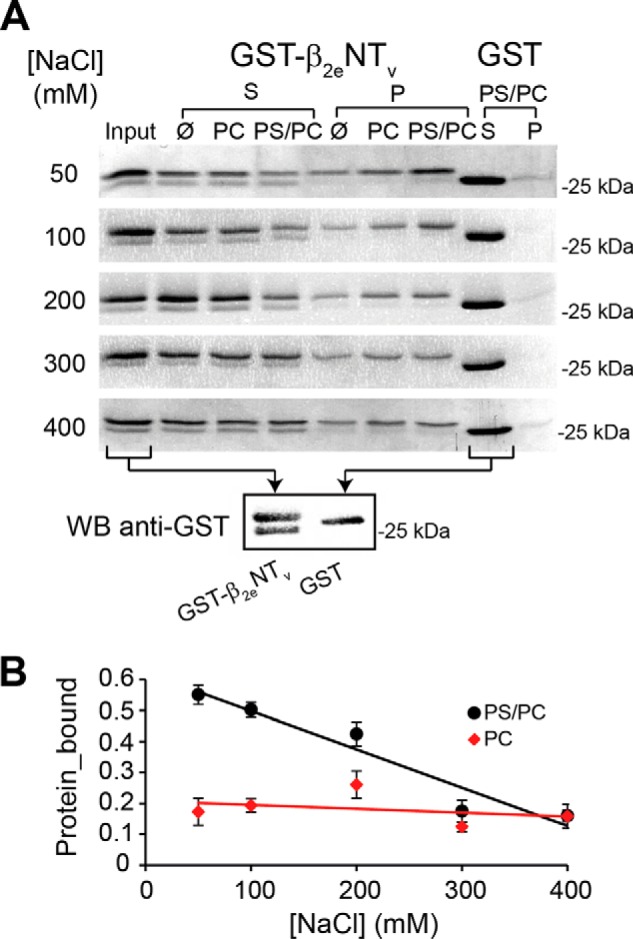
Variable N-terminal segment of the β2e-subunit binds to acidic liposomes in an ionic strength-dependent manner. A, protein-liposome cosedimentation assays using β2eNTv fused to GST (GST-β2eNTV). Representative SDS-PAGE gels from supernatant (S) and pellet (P) fractions after centrifugation of GST-β2eNTV in assay buffer only (Ø indicates absence of liposomes) or preincubated with either neutral (PC) or negatively charged (PS/PC) liposomes at different NaCl concentrations from 50 to 400 mm. Controls assays using the GST moiety alone mixed with PS/PC liposomes were performed in parallel (last two lanes). A reference lane (input) that contains the total amount of protein used in each assay (2 μg) was included. Each assay was repeated at least three times. The bottom panel shows a Western blot (WB) analysis using an anti-GST antibody of the recombinant proteins used, GST-β2eNTV and GST alone. B, plot of the fraction of GST-β2eNTv bound to either PS/PC or PC liposomes at different NaCl concentrations. For details see under “Experimental Procedures.” Values are expressed as mean ± S.E.
To discard the possibility that the protein observed in the pellet arises from an interaction between GST and the lipids, parallel experiments were performed in which GST-β2eNTv was replaced by GST moiety alone. GST is virtually excluded from the pellet in all conditions after centrifugation (Fig. 4A).
A lower molecular band copurifies with GST-β2eNTv and distributes almost evenly in the supernatant fraction (Fig. 4A). In Western blot analysis, this protein was recognized by an anti-GST antibody (Fig. 4A) indicating that it is a degradation product that may lack completely or partially the 23-amino acid residues of β2eNTv. Although unplanned, this protein provided an extra internal control for the assay.
A fraction of the GST-β2eNTv is found in the pellet in the absence of phospholipids, indicating a degree of aggregation of the fusion protein arising from β2eNTv moiety. Despite this, quantification of the protein bound to the liposomes demonstrates that at relatively low ionic strength (50 mm NaCl) a significantly higher amount of protein cosediments with negatively charged LUVs (PS/PC) than with neutral PC liposomes (Fig. 4B). Moreover, association of GST-β2eNTv with PS/PC liposomes shows more acute dependence on the salt concentration (Fig. 4B). Increasing the ionic strength up 400 mm NaCl results in a persistent decrease in the quantity of GST-β2eNTv found in the pellet with PS/PC liposomes that is not observed in the presence of PC liposomes. Altogether, these results indicate that electrostatic interactions play an important role in the binding of β2eNTv to anionic liposomes.
Positively Charged Residues within β2eNTv Are Required for Targeting the Protein to the Plasma Membrane
The binding of β2eNTv to negatively charged phospholipid vesicles implies that positively charged residues within this segment are key players. To test this, we generated several β2e constructs bearing simultaneous substitutions of lysine and arginine residues within β2eNTv, in which alanine replaced either all seven (β2eNTv(7+ less)), four (β2eNTv(4+ less)), or two basic residues at once (β2e R7A,K10A and β2eK2A,R16A). Quantification of the plasma membrane localization of these mutant proteins was done as in Fig. 2. Substitution of all seven positively charged residues decreased dramatically the degree of colocalization as compared with the wild-type protein (MC = 0.13 ± 0.01 for β2eNTv(7+ less), t test; p < 0.01, Fig. 5B). The MC value obtained for β2eNTv(7+ less) is comparable with the one estimated for YFP alone indicating a loss of surface membrane targeting. Neutralization of four or two basic residues also leads to a significant decrease in the plasma membrane localization of the protein (MC = 0.19 ± 0.01 for β2eNTv(4+ less), 0.22 ± 0.02 for β2e R7A,K10A and 0.20 ± 0.02 for β2eK2A,R16A, t test; p < 0.01, Fig. 5, A and B). However, the charge-conserving mutations of the double mutant proteins at the same amino acid positions (β2e R7K,K10R and β2e K2R,R16K, Fig. 5, A and B) preserve plasma membrane localization, demonstrating that the charge of the residue is the determinant factor in targeting the protein to the surface membrane.
FIGURE 5.

Charge-neutralizing mutations of the basic residues within β2eNTv impair the association of the protein with the plasma membrane. A, confocal fluorescence images of tsA201 cells expressing β2e mutants bearing the indicated simultaneous amino acid substitutions of lysine and arginine residues within NTv as follows: NTv(7+ less), seven lysine and arginine residues at NTv were substituted by alanine; NTv(4+ less), four basic residues were replaced with alanine (Arg11, Lys13, Arg16, and Lys18), and R7A,K10A, K2A,R16A, R7K,K10R, and K2R,R16K. β2e mutant proteins (panels a–f), plasma membranes (panels g–l), and merge images (panels m–r) visualized as in Fig. 2A. Scale bar, 15 μm and is valid for all images. B, bar plot of the colocalization analysis for the mutant proteins shown in A done as in Fig. 2B. Dashed lines correspond to the average values obtained for β2e and YFP alone from Fig. 2B. C, protein-liposome cosedimentation assays in 50 mm NaCl (as described in Fig. 4A) for β2eNTv bearing the charge-neutralizing R7A,K10A (GST-β2eNTv R7A,K10A, top panel) and the charge-conserving mutations at the same positions (GST-β2eNTv R7K,K10R, bottom panel). D, plot of the fraction of protein bound to anionic liposomes (PS/PC) normalized to wild-type GST-β2eNTv under the same salt conditions (50 mm NaCl). Values are expressed as mean ± S.E.
We introduced the double mutations R7A,K10A and R7K,K10R into the GST-β2eNTv fusion proteins (GST-β2eNTv R7A,K10A and GST-β2eNTv R7K,K10R, respectively) to investigate the correlation between surface membrane targeting and liposome binding. The charge neutralizing mutations significantly reduced the fraction of protein associated with negatively charged liposomes, whereas the ability of the charge conserving mutant to bind to PS/PC liposomes is preserved to a high extent as compared with wild-type β2eNTv (Fig. 5, C and D). These results suggest that the loss of plasma membrane localization upon charge neutralization is mainly due to the impaired ability of the protein to establish electrostatic interactions with anionic phospholipids.
Single point mutations of all arginine or lysine residues within β2eNTv to alanine moderately decrease the degree of colocalization with the plasma membrane marker compared with the WT protein (Fig. 6, A and B). The values of MC ranged between 0.36 ± 0.01 and 0.42 ± 0.02 for all proteins bearing a single residue mutation except for K2A mutant with an MC equal to 0.28 ± 0.02 (Fig. 6B). This suggests that Lys2 is involved in a more intimate protein-membrane interaction.
FIGURE 6.

Individual substitutions of positively charged residues within β2eNTv differentially alter plasma membrane targeting. A, confocal fluorescence images of tsA201 cells expressing the indicated β2e mutations at single lysine or arginine residues within β2eNTv. β2e single-point mutants (panels a–f), plasma membranes (panels g–l), and merged images (panels m–r) are visualized as in Fig. 2A. Scale bar represents 15 μm and is valid for all images. B, colocalization analysis and dashed lines as in Fig. 5B.
Fluorescence Emission Maxima of the Trp5 Blue Shifts in the Presence of Anionic Liposomes Indicating Penetration of This Residue into the Lipid Bilayer
Tryptophan residues in peripheral membrane proteins are commonly found in association with the membrane's hydrophobic core (25). We took advantage of the presence of a single tryptophan residue at position 5 (Trp5) within β2eNTv to study the association of this segment with lipids using intrinsic fluorescence measurements. At first, we substituted this residue by alanine in β2e-YFP and found that this mutation results in a significant decrease in the degree of colocalization with the plasma membrane marker (MC = 0.16 ± 0.01) compared with WT (Fig. 7, A and B). However, the binding of β2eNTv W5A to negatively charged liposomes under low salt conditions (50 mm NaCl) is only slightly diminished as compared with wild-type β2eNTv, although association with neutral liposomes is significantly impaired (Fig. 7, C and D). This indicates that Trp5 may indeed contribute directly to lipid binding through hydrophobic interactions, which will be more relevant at the relatively higher ionic strength of the cellular environment.
FIGURE 7.

Hydrophobic interactions mediated by the single tryptophan residue within β2eNTv contribute to the plasma membrane targeting of the β2e subunit. A, confocal fluorescence images of tsA201 cells expressing β2e W5A fused to YFP and stained with the CellMaskTM plasma membrane marker, visualized using the green and red channel, respectively. Scale bar, 15 μm. B, colocalization analysis for β2e W5A, and dashed lines are as in Fig. 3B. C, representative protein-liposome cosedimentation assay of GST-β2eNTv W5A. D, plot of the fraction of protein bound to anionic (PS/PC) and neutral (PC) liposomes normalized to wild-type GST-β2eNTv under the same salt conditions (50 mm NaCl). Values are expressed as mean ± S.E. E, representative emission spectra of β2eNTv peptide (residues 1–21) after excitation with 280 nm in the presence of 50 mm NaCl and different concentrations of PC/PS SUVs. The lipid concentrations were (in order of increasing peak intensity, in μm) 0, 80, 150, 300, 600, and 1500. F, plot of emission maxima of spectra as shown in E against the lipid concentrations of PC/PS SUVs at 50 and 400 mm NaCl, as well as PC SUV at 50 mm NaCl. G, plot of the fraction of bound peptide (for details see under “Experimental Procedures”) against the lipid concentration of PC/PS SUVs at 50 mm NaCl. The line represents the fit of a Hill equation to the data, resulting in KD = 182 ± 34 μm and nH = 1.5 ± 0.1. Values are expressed as mean ± S.E.
At a low salt concentration, incubation of a synthetic peptide corresponding to β2eNTv (residues 1–21) with increasing concentrations of negatively charged PS/PC SUVs induced a pronounced blue shift in Trp5 fluorescence emission maximum with a concomitant increase in the fluorescence intensity (Fig. 7E). The observed shift in emission maximum from 346 nm in the absence of vesicles to 323 nm in the presence of saturating PS/PC concentrations (Fig. 7F) indicates that this residue changes from an aqueous into an essentially hydrophobic environment (e.g. the hydrocarbon core of the membrane) (see also “Discussion”) (26). The fit of a Hill function to the lipid concentration dependence of the fraction of bound peptide at low ionic strength yields a KD of 182 ± 34 μm (Fig. 7G).
At higher ionic strength (400 mm NaCl), full binding was not reached with the employed lipid concentrations (Fig. 7F) indicating a decrease of the binding affinity. This finding is consis2eNTv to PS/PC liposomes observed in cosedimentation assays (see Fig. 4). In the presence of neutral PC vesicles, virtually no changes in the intrinsic Trp5 fluorescence were monitored at all salt concentrations tested (Fig. 7F). In contrast, we observed a small fraction of the GST-β2eNTv cosedimenting with PC vesicles (Fig. 4). Although the cause of this difference is unclear, it is worth noting that fluorescence measurements were performed with SUVs, rather than LUVs, to minimize differential light-scattering effects (19). Altogether these results indicate that Trp5 penetrates into the plasma membrane when electrostatic interactions between the positively charged residues and the anionic lipids are allowed.
Plasma Membrane Anchoring of β2e Regulates the Rate of Inactivation of R-type Calcium Channels
Coexpression of Cav2.3 with the membrane-anchored β2a in tsA201 cells and Xenopus oocytes induces a dramatic increase in the time needed for the current to return to 50% of its peak amplitude (hereafter referred to as t0.5) and shifts the steady-state inactivation curve toward more depolarizing voltages with respect to the CaV2.3 coexpressed with cytosolic β-isoforms (β1b, β3, and β4) (10–13). Here, we compared the effects of the wild-type β2e-subunit and the membrane association-deficient mutant containing no basic residues within β2eNTv (β2eNTv(7+ less), see Fig. 5, A and B) on CaV2.3 channel complexes expressed in tsA201 cells.
We tested whether substitution of all positively charged residues in β2eNTv interferes with the ability of the protein to bind to the α1-subunit. For this purpose, the highly conserved binding site in all α1-subunits of HVA, referred as to AID (27), was fused to GST (GST-AID) and used as bait protein in a standard in vitro binding assay. Either wild-type β2e or the β2eNTv(7+ less) were added as prey. Control binding reactions were performed in parallel using GST alone as bait. Both wild-type β2e and the mutant protein bind to the GST-AID to a comparable extent (Fig. 8A).
FIGURE 8.

Membrane association of the β2e-subunit modulates the voltage-dependent inactivation of the R-type calcium channels. A, binding assay of wild-type β2e and β2eNTv(7+ less) membrane association-deficient mutant to the highly conserved α1 interaction domain (AID). Either GST moiety alone or AID site fused to GST (GST-AID) were pre-coupled to glutathione-agarose beads and incubated with either wild-type β2e or β2eNTv(7+ less) proteins. After several washes, the bound proteins were eluted and resolved on SDS-PAGE. Lane 1, molecular mass in kDa; lane 2, binding of wild-type β2e to GST-AID; lane 3, binding of wild-type β2e to GST; lane 4, binding of β2eNTv(7+ less) to GST-AID; lane 5, binding of β2eNTv(7+ less) to GST; lanes 6-9, inputs proteins. B, representative current traces from tsA201 cells coexpressing the R-type CaV2.3 α1-subunit of voltage-activated calcium channels either with wild-type β2e or β2 NTv(7+ less). Currents were elicited during a 10-s pulse to 0 mV from a holding potential of −90 mV. C, average t0.5 for CaV2.3/β2e and CaV2.3/β2eNTv(7+ less) channel complexes. t0.5 significantly decreases from 1.26 ± 0.14 s (n = 6) for CaV2.3/β2e to 0.16 ± 0.01 s (n = 6) for CaV2.3/β2eNTv(7+ less) channel complexes (t test; p < 0.01). D, average steady-state inactivation curves from cells expressing the indicated channel subunit combinations. Inactivation was determined after a 10-s pre-pulse ranging from −120 to +30 mV in 15-mV increments from a holding potential of −90 mV followed by a short deactivation pulse to −90 mV and a 5-s test pulse at 0 mV. The interval between sweeps was of 60 s. Continuous lines correspond to the Boltzmann distribution that best fit the experimental data for the indicated channel complex. Values are expressed as mean ± S.E. V0.5 significantly varies from −49.0 ± 3.1 mV (n = 6) for CaV2.3-β2e to −67.0 ± 0.8 mV (n = 6) for CaV2.3-β2eNTv(7+ less) channel complexes (t test; p < 0.01).
Coexpression of CaV2.3 with the nonmembrane-associated β2eNTv(7+ less) mutant leads to an 8-fold decrease in t0.5 at 0 mV as compared with CaV2.3 coexpressed with wild-type β2e (Fig. 8, B and C). Moreover, the steady-state inactivation curve from cells expressing CaV2.3 and β2eNTv(7+ less) complexes is shifted to more negative potentials with respect to cells transfected with CaV2.3 and β2e (Fig. 8D). Half-inactivation voltages (V0.5) obtained from the fit to a Boltzmann distribution for CaV2.3 coexpressed with β2e WT and mutant were similar to those previously reported for membrane-anchored β2a and the cytosolic β1b, respectively (12).
These results imply that different mechanisms of β-subunit membrane association lead to a functional analogy in the inhibition of the voltage dependence of inactivation.
DISCUSSION
Proteins use diverse mechanisms to associate with lipid membranes, including electrostatic interactions between protein segments enriched in positively charged amino acid residues and anionic lipids as well as hydrophobic interactions between the membrane lipids and nonpolar regions of the protein and/or covalently attached lipid anchors (24). Here, we identified a short domain located at the N-terminal region of β2e that is responsible for tethering the protein to the plasma membrane. This domain consists of several positively charged residues distributed along 23 amino acids and hydrophobic residues intercalated between them. A consensus secondary structure prediction program that integrates several methods based on different principles (28) calculated a helical structure within residues 2–13. Most notably, within this putative helix a short in-plane membrane anchor segment was predicted (Fig. 9A) (29). This region is highly conserved across highly divergent species suggesting a conserved structure-function (Fig. 9A). Alanine scanning mutagenesis revealed that Lys2 has a stronger effect on membrane affinity than the other residues (see Fig. 6). Lys2 maps to the same side of a helical wheel plot together with the nonpolar Trp5 and Leu9 residues (Fig. 9B). The significant blue shift of the fluorescence emission of Trp5 upon addition of negatively charged lipids indicates that this residue moves to a highly hydrophobic environment that is not likely to be provided by structural rearrangement of this short peptide bearing a net charge of +4. The direction and magnitude of the changes in the emission maxima of Trp5 in the presence of anionic phospholipids are consistent with a relatively deep membrane penetration of this residue. Fluorescence studies on model peptides assign an average distance of 7 Å from the lipid bilayer center to a Trp emission fluorescence wavelength maximum comparable with the one we observed with the β2eNTv peptide at the highest lipid concentration (323 nm, Fig. 7, E and F) (30). Considering the width of a monolayer as 15 Å, this would imply that the Trp5 penetrates about 8 Å into the plasma membrane beyond the lipid head groups and, consequently, that the putative in-plane helix is in close contact or interweaved with the headgroups (Fig. 9C). Aside from strengthening the association with the plasma membrane, the Trp5 may be important for the intracellular trafficking of the protein, for example by promoting binding to intracellular membrane vesicles. This would explain why the protein bearing an alanine at that position binds to anionic lipids but is not found at the cell surface. Mutation of Leu9 into alanine also results in a loss of plasma membrane targeting (data not shown), reinforcing the possibility that hydrophobic interactions may be important under physiological conditions for associating the protein with the plasma membrane. Our data are consistent with a mechanism by which β2e anchors to the plasma membrane via an N-terminal helix that orients parallel to the plane of the bilayer (in-plane membrane association) and inserts relatively deeply into the headgroups (Fig. 9C). Association with the plasma membrane would take place in two steps as follows: first, the protein is recruited to the cell surface via electrostatic interactions with its positively charged residues, and second, the membrane-bound conformation is stabilized by the penetration of hydrophobic residues (i.e. Trp5 and Leu9) into the lipidic core. Although we cannot evaluate the precise contribution of electrostatic and hydrophobic interactions to the binding of β2e to the plasma membrane in vivo, our experiments show that hydrophobic interactions become more relevant in the relatively higher ionic strength of the cellular environment.
FIGURE 9.

Model depicting the association of the β2e-subunit with the plasma membrane. A, amino acid sequence and predicted secondary structure for the β2e N-terminal variable region (β2e NTV). The positively charged and the nonpolar residues within β2eNTv are highlighted in blue and yellow, respectively. The green cylinder below the sequence delimits the predicted α-helix (residues 2–13) according to the consensus secondary prediction results (NPS@Web server) (28). Within the helix an in-plane membrane anchor segment (residues 4–7) is predicted (black line) (29). The lower panel shows the amino acid alignment of β2eNTv segment from (in parenthesis, UniProtKB accession numbers) rat (Q8VGC3), human (Q08289), mouse (Q8CC27), dog (E2RCN6), chicken (E1BZF6), Xenopus (F7EDB7), and zebrafish (B2XY77). B, helical wheel diagram of the predicted consensus helix within β2eNTv extending from residues 2 to 13 (Helical Wheel Applet, University of Virginia). The potential location of Arg16 considering a longer helix is also shown (37). The relatively critical residues deduced from alanine scanning mutagenesis, Lys2, Trp5 together with Leu9 (data not shown) lie on one side of the helix. C, proposed model to explain membrane anchoring of β2e. The β2e-subunit includes a helical membrane anchor site in its N-terminal region that associates in plane with the membrane via electrostatic interactions between its positively charged residues and the phospholipids headgroups. After the electrostatic recruitment of the protein to the plasma membrane, the bound conformation is stabilized by a relatively deep penetration of hydrophobic residues, including the aromatic side chain of Trp5 and the aliphatic chain of Leu9 into the lipid core. We suggest that the predicted phosphorylation sites (36), represented as lines, in the flexible segment joining the NTv helix and the core of the protein (labeled β) may induce dissociation of the protein from the plasma membrane, providing additional regulatory input for calcium entry through voltage-gated calcium channels.
The interaction with lipids can alter the subcellular localization, conformation, and activity of proteins. An example is β2a, which switches from a fast to a slow inactivation-conferring phenotype upon palmitoylation and membrane association. Here, we demonstrated that β2e is not an exception; when associated with the plasma membrane, β2e displays very different channel modulatory activity compared with the membrane anchor-deficient mutant. The fact that both β2a and β2e inhibit voltage-dependent inactivation of CaV2.3 channels, although they associate differently with lipids, supports the model suggesting that membrane anchoring is sufficient to slow down channel inactivation by restricting the movement of the inactivation gate (31).
The dynamic nature of β2a palmitoylation provides a further control mechanism regulating in vivo calcium influx through voltage-gated calcium channels (15). However, it remains unknown whether or not the association of β2e with the cell membrane is subject to regulation in vivo. In fluorescence confocal images of cells expressing β2e, the protein distributes mainly in the surface membrane, but a slight diffuse fluorescence is visible within the cell, arguing in favor of a regulated membrane targeting (32). Although additional experiments will be required to test this hypothesis, we speculate that the phosphorylation state of the β2e N-terminal domain may be particularly important for controlling the association of this subunit with the plasma membrane as has been suggested for other proteins (32, 33). Phosphorylation of CaVβ subunits that play a role in channel regulation has been described (34, 35). The N-terminal region of β2e contains several potential phosphorylation sites downstream of the membrane anchor segment identified in this study. Prediction programs reveal a phosphorylation hot spot within a very short protein segment encompassing residues 29–39 (36). Phosphorylation of these sites would decrease the positive charge density of β2eNTv and, consequently, weaken its interaction with membrane lipids.
In summary, in addition to providing a mechanism by which β2e anchors to the plasma membrane, our study suggests a structure for the N-terminal region of the protein for which no data are available. The high resolution structure of three β isoforms (β2, β3, and β4) has been solved, but these do not provide structural data for the variable regions outside the SH3-guanylate kinase domains.
This is the first β-subunit described that associates with the plasma membrane via electrostatic interactions. The fact that single point mutations within this domain appear to alter the protein membrane affinity makes it feasible to use this protein as a model to correlate membrane binding affinities to the degree of inhibition of voltage-dependent inactivation conferred by membrane-anchored β-subunits.
Acknowledgments
We thank Drs. Christoph Fahlke, Ingo Weyand, and Juan Garcia for insightful discussion.
This work was supported by Deutsche Forschungsgemeinschaft Grant Hi 800/3-1 (to P. H.).
- HVA
- high voltage-activated
- SH3
- Src homology 3 domain
- NTv
- N-terminal variable segment
- LUV
- large unilamellar vesicles
- SUV
- small unilamellar vesicles
- MC
- Manders coefficient
- PC
- 1-palmitoyl-2-oleoyl-sn-glycero-phosphocholine
- PS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- AID
- α interaction domain.
REFERENCES
- 1. Catterall W. A. (2011) Voltage-gated calcium channels. Cold Spring Harbor Perspect. Biol. 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dolphin A. C. (2003) β subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 35, 599–620 [DOI] [PubMed] [Google Scholar]
- 3. Hidalgo P., Neely A. (2007) Multiplicity of protein interactions and functions of the voltage-gated calcium channel β-subunit. Cell Calcium 42, 389–396 [DOI] [PubMed] [Google Scholar]
- 4. Chen Y. H., Li M. H., Zhang Y., He L. L., Yamada Y., Fitzmaurice A., Shen Y., Zhang H., Tong L., Yang J. (2004) Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature 429, 675–680 [DOI] [PubMed] [Google Scholar]
- 5. Opatowsky Y., Chen C. C., Campbell K. P., Hirsch J. A. (2004) Structural analysis of the voltage-dependent calcium channel β-subunit functional core and its complex with the α1-interaction domain. Neuron 42, 387–399 [DOI] [PubMed] [Google Scholar]
- 6. Van Petegem F., Clark K. A., Chatelain F. C., Minor D. L., Jr. (2004) Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takahashi S. X., Mittman S., Colecraft H. M. (2003) Distinctive modulatory effects of five human auxiliary β2 subunit splice variants on L-type calcium channel gating. Biophys. J. 84, 3007–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Link S., Meissner M., Held B., Beck A., Weissgerber P., Freichel M., Flockerzi V. (2009) Diversity and developmental expression of L-type calcium channel β2 proteins and their influence on calcium current in murine heart. J. Biol. Chem. 284, 30129–30137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chien A. J., Gao T., Perez-Reyes E., Hosey M. M. (1998) Membrane targeting of L-type calcium channels. Role of palmitoylation in the subcellular localization of the β2a subunit. J. Biol. Chem. 273, 23590–23597 [DOI] [PubMed] [Google Scholar]
- 10. Olcese R., Qin N., Schneider T., Neely A., Wei X., Stefani E., Birnbaumer L. (1994) The amino termini of calcium channel β-subunits set rates of inactivation independently of their effect on activation. Neuron 13, 1433–1438 [DOI] [PubMed] [Google Scholar]
- 11. Hidalgo P., Gonzalez-Gutierrez G., Garcia-Olivares J., Neely A. (2006) The α1-β subunit interaction that modulates calcium channel activity is reversible and requires a competent α-interaction domain. J. Biol. Chem. 281, 24104–24110 [DOI] [PubMed] [Google Scholar]
- 12. Jones L. P., Wei S. K., Yue D. T. (1998) Mechanism of auxiliary subunit modulation of neuronal α1E calcium channels. J. Gen. Physiol. 112, 125–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yasuda T., Chen L., Barr W., McRory J. E., Lewis R. J., Adams D. J., Zamponi G. W. (2004) Auxiliary subunit regulation of HVA calcium channels expressed in mammalian cells. Eur. J. Neurosci. 20, 1–13 [DOI] [PubMed] [Google Scholar]
- 14. Chien A. J., Carr K. M., Shirokov R. E., Rios E., Hosey M. M. (1996) Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. J. Biol. Chem. 271, 26465–26468 [DOI] [PubMed] [Google Scholar]
- 15. Qin N., Platano D., Olcese R., Costantin J. L., Stefani E., Birnbaumer L. (1998) Unique regulatory properties of the type 2a Ca2+ channel β-subunit caused by palmitoylation. Proc. Natl. Acad. Sci. U.S.A. 95, 4690–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez-Gutierrez G., Miranda-Laferte E., Nothmann D., Schmidt S., Neely A., Hidalgo P. (2008) The guanylate kinase domain of the β-subunit of voltage-gated calcium channels suffices to modulate gating. Proc. Natl. Acad. Sci. U.S.A. 105, 14198–14203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolte S., Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 18. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao D., Wallace B. A. (1984) Differential light scattering and absorption flattening optical effects are minimal in the circular dichroism spectra of small unilamellar vesicles. Biochemistry 23, 2667–2673 [DOI] [PubMed] [Google Scholar]
- 20. Mui B., Chow L., Hope M. J. (2003) Extrusion technique to generate liposomes of defined size. Methods Enzymol. 367, 3–14 [DOI] [PubMed] [Google Scholar]
- 21. Hui E., Bai J., Chapman E. R. (2006) Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys. J. 91, 1767–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burstein E. A., Emelyanenko V. I. (1996) Log-normal description of fluorescence spectra of organic fluorophores. Photochem. Photobiol. 64, 316–320 [Google Scholar]
- 23. Keller S., Böthe M., Bienert M., Dathe M., Blume A. (2007) A simple fluorescence-spectroscopic membrane translocation assay. Chembiochem 8, 546–552 [DOI] [PubMed] [Google Scholar]
- 24. Khandelia H., Ipsen J. H., Mouritsen O. G. (2008) The impact of peptides on lipid membranes. Biochim. Biophys. Acta 1778, 1528–1536 [DOI] [PubMed] [Google Scholar]
- 25. Lomize A. L., Pogozheva I. D., Lomize M. A., Mosberg H. I. (2007) The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct. Biol. 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reshetnyak Y. K., Koshevnik Y., Burstein E. A. (2001) Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 81, 1735–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pragnell M., De Waard M., Mori Y., Tanabe T., Snutch T. P., Campbell K. P. (1994) Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature 368, 67–70 [DOI] [PubMed] [Google Scholar]
- 28. Combet C., Blanchet C., Geourjon C., Deléage G. (2000) NPS@: network protein sequence analysis. Trends Biochem. Sci. 25, 147–150 [DOI] [PubMed] [Google Scholar]
- 29. Sapay N., Guermeur Y., Deléage G. (2006) Prediction of amphipathic in-plane membrane anchors in monotopic proteins using a SVM classifier. BMC Bioinformatics 7, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chung L. A., Lear J. D., DeGrado W. F. (1992) Fluorescence studies of the secondary structure and orientation of a model ion channel peptide in phospholipid vesicles. Biochemistry 31, 6608–6616 [DOI] [PubMed] [Google Scholar]
- 31. Stotz S. C., Jarvis S. E., Zamponi G. W. (2004) Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J. Physiol. 554, 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cornell R. B., Taneva S. G. (2006) Amphipathic helices as mediators of the membrane interaction of amphitropic proteins, and as modulators of bilayer physical properties. Curr. Protein Pept. Sci. 7, 539–552 [DOI] [PubMed] [Google Scholar]
- 33. Cho W., Stahelin R. V. (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34, 119–151 [DOI] [PubMed] [Google Scholar]
- 34. Chien A. J., Hosey M. M. (1998) Post-translational modifications of β-subunits of voltage-dependent calcium channels. J. Bioenerg. Biomembr. 30, 377–386 [DOI] [PubMed] [Google Scholar]
- 35. Martin S. W., Butcher A. J., Berrow N. S., Richards M. W., Paddon R. E., Turner D. J., Dolphin A. C., Sihra T. S., Fitzgerald E. M. (2006) Phosphorylation sites on calcium channel α1- and β-subunits regulate ERK-dependent modulation of neuronal N-type calcium channels. Cell Calcium 39, 275–292 [DOI] [PubMed] [Google Scholar]
- 36. Blom N., Gammeltoft S., Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 37. Rost B., Sander C. (1993) Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232, 584–599 [DOI] [PubMed] [Google Scholar]