

Background: BCAR1 (breast cancer antiestrogen resistance protein 1) and BCAR3 promote antiestrogen resistance and malignancy in breast cancer.
Results: Mutations preventing the tight BCAR1-BCAR3 association disrupt downstream signaling events required for antiestrogen resistance, including ERK1/2 activation.
Conclusion: Complex formation is critical for BCAR1-BCAR3 oncogenic activities.
Significance: The BCAR1-BCAR3 complex and associated signaling events represent promising therapeutic targets in breast cancer.
Keywords: Anticancer Drug, Breast Cancer, Cancer, Drug Resistance, MAP Kinases (MAPKs), Protein-Protein Interactions, ERK1/2, NSP3, PEA15, Mesenchymal Phenotype
Abstract
Most breast cancers are estrogen receptor-positive and treated with antiestrogens, but aberrant signaling networks can induce drug resistance. One of these networks involves the scaffolding protein BCAR1/p130CAS, which regulates cell growth and migration/invasion. A less investigated scaffolding protein that also confers antiestrogen resistance is the SH2 domain-containing protein BCAR3. BCAR1 and BCAR3 bind tightly to each other through their C-terminal domains, thus potentially connecting their associated signaling networks. However, recent studies using BCAR1 and BCAR3 interaction mutants concluded that association between the two proteins is not critical for many of their interrelated activities regulating breast cancer malignancy. We report that these previously used BCAR mutations fail to cause adequate loss-of-function of the complex. By using structure-based BCAR1 and BCAR3 mutants that lack the ability to interact, we show that BCAR3-induced antiestrogen resistance in MCF7 breast cancer cells critically depends on its ability to bind BCAR1. Interaction with BCAR3 increases the levels of phosphorylated BCAR1, ultimately potentiating BCAR1-dependent antiestrogen resistance. Furthermore, antiestrogen resistance in cells overexpressing BCAR1/BCAR3 correlates with increased ERK1/2 activity. Inhibiting ERK1/2 through overexpression of the regulatory protein PEA15 negates the resistance, revealing a key role for ERK1/2 in BCAR1/BCAR3-induced antiestrogen resistance. Reverse-phase protein array data show that PEA15 levels in invasive breast cancers correlate with patient survival, suggesting that PEA15 can override ERK1/2 activation by BCAR1/BCAR3 and other upstream regulators. We further uncovered that the BCAR3-related NSP3 can also promote antiestrogen resistance. Thus, strategies to disrupt BCAR1-BCAR3/NSP3 complexes and associated signaling networks could ultimately lead to new breast cancer therapies.
Introduction
In their acquisition of an increasingly more malignant and aggressive phenotype, breast cancer cells rely on signaling networks that deregulate proliferation/survival and migration/invasiveness as well as confer resistance to chemotherapeutic drugs. Intrinsic or acquired drug resistance and metastatic dissemination are the most serious obstacles to breast cancer eradication (1, 2). High expression of the scaffolding protein breast cancer antiestrogen resistance 1 (BCAR1,3 also known as p130CAS) has been implicated in the intrinsic resistance to antiestrogens and other chemotherapeutic agents as well as the acquisition of more mesenchymal/migratory characteristics and basal-like features (3–14). BCAR1 also contributes to the malignant characteristics of breast cancer cells overexpressing the HER2/ErbB2 oncogene (15–17). Furthermore, high BCAR1 protein levels in breast cancer patients have been correlated with the triple-negative phenotype, poor prognosis, rapid disease recurrence, and resistance to therapy with the estrogen receptor antagonist tamoxifen (4, 5, 13, 18, 19). All four members of the CAS protein family regulate signaling assemblies through their multiple conserved domains. These include an N-terminal SH3 domain, a substrate domain containing multiple tyrosine and serine phosphorylation sites, and two focal adhesion targeting domains that in three of the family members are separated by an intervening SRC binding domain (11).
Although they lack enzymatic activity, CAS family proteins play a central role in cell physiology by serving as scaffolds for multiple protein partners to relay signals downstream of integrins and receptor-tyrosine kinases (11, 12, 20). CAS proteins bind non-receptor-tyrosine kinases, such as the focal adhesion kinase FAK through their SH3 domain and SRC family kinases through their SRC binding domain. Motifs phosphorylated by SRC in the BCAR1 substrate domain generate multiple docking sites for the SH2 domains of adaptor proteins such as CRK, which links BCAR1 to the C3G and DOCK180 exchange factors (12, 20–22). C3G activates the Ras family GTPases R-RAS and RAP1, thus promoting integrin-mediated adhesion. DOCK180 activates the RHO family GTPase RAC1, which promotes the formation of membrane ruffles and protrusions as well as cell migration and invasiveness. Notably, among the family members only BCAR1 has been shown to confer antiestrogen resistance (11, 12, 17, 23).
Another multidomain signaling protein that has been associated with antiestrogen resistance and a mesenchymal phenotype in breast cancer is breast cancer antiestrogen resistance 3 (BCAR3, also known as AND-34) (23–27). BCAR3 is a member of the novel SH2-containing protein (NSP) family and was identified together with BCAR1 in a screen for gene products whose increased expression promotes proliferation of estrogen receptor-positive breast cancer cells treated with antiestrogens (3, 24). BCAR3 plays a key role in the regulation of BCAR1 signaling and subcellular localization as well as activates signaling effectors (such as PI3K, RAC1, PAK1, and cyclin D1) that promote antiestrogen resistance and a migratory/invasive phenotype (23, 28–33).
BCAR3 and the two other members of the NSP family, NSP1 (gene name SH2D3A) (25) and NSP3 (also known as SHEP1 and CHAT, gene name SH2D3C) (25, 34, 35), share a common domain structure. This distinctive structure includes an N-terminal segment subject to alternative splicing, a SH2 domain that can bind activated receptor-tyrosine kinases and the PTPα phosphatase (at least in the case of BCAR3), and a C-terminal CDC25-type guanine nucleotide exchange factor (GEF)-like domain (11, 36, 37). Recent x-ray crystallography studies have shown that this GEF-like domain has a unique conformation that is incompatible with the binding of RAS GTPases but is capable of functioning as an adaptor module that mediates extremely tight binding to the C-terminal focal adhesion targeting domain of CAS family proteins. The tight linkage formed by their C-terminal domains allows members of the BCAR1 and NSP families to connect the signaling networks associated with each multidomain scaffolding protein.
This is in line with earlier suggestions that the level of the BCAR1-BCAR3 complex in breast cancer cell lines correlates with malignancy more closely than the levels of each individual protein (23). Therefore, disrupting the complex could sensitize breast cancer cells to chemotherapeutic drugs and favor reversion to a less malignant state. Surprisingly, recent studies of mutations in the C-terminal interaction domains of mouse BCAR1 (L791P) and BCAR3 (R743A) have shown that the mutants retain many biological activities of the wild-type proteins despite their reported decreased ability to interact (30, 38, 39). These findings have cast doubt on the importance of the BCAR1-BCAR3 association for many of their interrelated biological functions regulating breast cancer aggressiveness.
However, our recent binding data using recombinant BCAR1 and NSP3 C-terminal domains suggested that certain single amino acid mutations in the binding interfaces of BCAR1 and NSP proteins may not be sufficient to adequately disrupt the very tight interaction between the two proteins (36). For example, even the drastic change in amino acid charge in the human NSP3 R627E mutant (corresponding to mouse BCAR3 R743E; Table 1) diminished but did not abolish the interaction of the NSP3 C-terminal domain with the BCAR1 C-terminal domain (36). The difficulty in abrogating BCAR1-NSP interactions may be due to the presence of two separate binding sites that cooperate to yield the extremely strong binding between CAS and NSP family proteins (36). Therefore, we sought to shed light on the exact role of the BCAR1-BCAR3 linkage in breast cancer antiestrogen resistance by using structure-guided double mutations to effectively disrupt the interaction between the two proteins. Additionally, we utilized the mutants to unravel signaling events through which the BCAR1-BCAR3 complex promotes antiestrogen resistance. Knowing whether the physical association between BCAR1 and BCAR3 represents a key factor in breast cancer malignancy and drug resistance as well as characterizing the signaling events involved has important implications for breast cancer prognosis and therapy.
TABLE 1.
BCAR1 and BCAR3 interface mutations
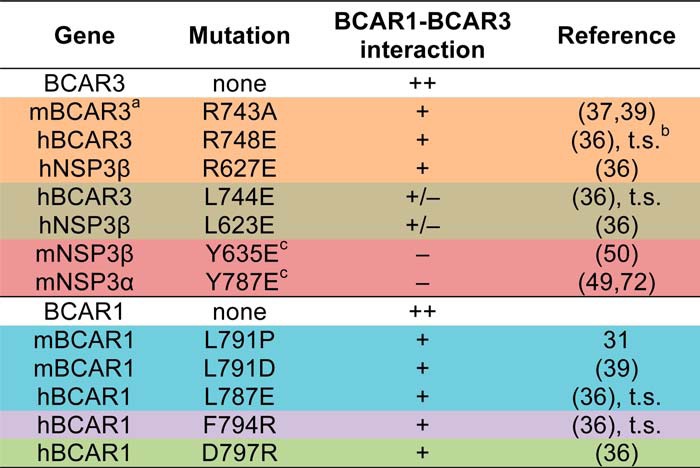
a, mouse; h, human.
b, t.s., this study.
c, allosteric mutation.
EXPERIMENTAL PROCEDURES
Plasmids and Lentiviral Constructs
The human BCAR1 cDNA (nucleotides 2–2614, GenBankTM accession number BC062556.1) was cloned in the XhoI and EcoRI sites of the pEGFP-C2 plasmid (Clontech). The human BCAR3 cDNA (nucleotides 146–2623, GenBankTM accession number BC039895.1) was obtained in the pCMV-SPORT6 vector from Open Biosystems/Thermo Scientific (Huntsville, Alabama; catalog #MHS1010-7508662) and subcloned in the HindIII and EcoRI sites of pcDNA3.1. The human PEA15 cDNAs (nucleotides 1–393, EMBL accession number CR541733.1) was cloned into the KpnI and BamHI sites of the pEGFP-C1 plasmid (Clontech Laboratories, Mountain View, CA).
The BCAR1 cDNA was also inserted between the XhoI and BamHI sites of the pLVX-IRES-Neo lentiviral vector (#632181; Clontech Laboratories), and the BCAR3 cDNA was also inserted between the EcoRI and XbaI sites of the pLVX-IRES-ZsGreen1 lentiviral vector (#632187; Clontech Laboratories). The human NSP3α cDNA (nucleotides 55–2123, GenBankTM accession number BC027962.1) was inserted between the EcoRI and NotI sites of pLVX-IRES-ZsGreen1. The mouse NSP3β cDNA (nucleotides 75–2183, GenBankTM accession number NM_001252547.1) was inserted between the EcoRI and BamHI sites of pLVX-IRES-ZsGreen1. Wild-type and mutant EGFP-PEA15 cDNAs were digested from pEGFP-C1-PEA15 using NheI and BamHI and inserted in the SpeI and BamHI sites of pLVX-IRES-ZsGreen1. The NSP3 shRNA (sequence, CCGGCCTGGACTCATCGCCAGAGAACTCGAGTTCTCTGGCGATGAGTCCAGGTTTTTG) in the pLKO.1-puro lentiviral vector was obtained from Sigma (TRCN0000072865). All mutants were generated by site-directed mutagenesis with the Pfu turbo DNA polymerase from Agilent Technologies (Santa Clara, CA) and verified by sequencing the entire cDNAs.
All infectious lentiviruses, except for those encoding EGFP-PEA15, were prepared in HEK293T-LV cells (GeneTarget) using the pCMV-dR8.74 and pMD2.G second generation packaging system and concentrated by ultracentrifugation in a sucrose gradient by the Sanford-Burnham Medical Research Institute Viral Vector core facility. The EGFP-PEA15 lentiviruses were prepared in Lenti-X 293T cells using the Lenti-XTM HTX packaging system (#631247; Clontech Laboratories) and used without concentration.
Cell Culture, Transfections, and Lentiviral Infections
HEK293T and MCF7 cells were obtained from ATCC (Manassas, VA) and cultured in DMEM supplemented with 10% fetal bovine serum and Pen/Strep. Transient transfections of HEK293T cells were carried out in 6-well plates using Lipofectamine® 2000 Transfection Reagent (Invitrogen) and 1.5 μg of total DNA (0.75 μg BCAR1 constructs and/or 0.75 μg BCAR3 constructs).
For lentiviral infections, MCF7 and HEK293T cells were incubated with lentiviruses overnight. After a first round of amplification, infected cells expressing wild-type or mutant BCAR3 together with ZsGreen from the bicistronic mRNAs were sorted by FACS to isolate cells expressing high levels of ZsGreen (corresponding to 2–12% of the infected cells) and expanded. To generate double-infected cells, the sorted cells expressing BCAR3 were subsequently infected with lentiviruses encoding BCAR1 or EGFP-PEA15 together with the neomycin resistance selection marker and selected using 1 mg/ml G418 (Geneticin, Roche Applied Science).
To measure growth in the presence of ICI182780, stable transduced MCF7 cells were counted 3 times using a hemocytometer and plated in triplicate wells in 6-well plates (50,000 cells/well) in medium containing the estrogen receptor antagonist ICI182780 (100 nm; Tocris Bioscience, Bristol, UK). The culture medium was replaced every 2 days. Over the length of the assay, the cells reaching about 90% confluency were trypsinized, counted, and replated at 50,000 cells/well. At the end of the growth assay, all cells were trypsinized and counted. The doubling times of the cultures were calculated using the algorithm available online at Doubling Time.
Immunoprecipitation
Transfected HEK293T cells were lysed in non-denaturing buffer (50 mm Tris-HCl, pH 7.5, 120 mm NaCl, 1% Triton X-100, 2 mm EDTA, 5% glycerol, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1 mg/ml aprotinin, 1 mg/ml leupeptin, 1 mg/ml pepstatin, and 1 mm phenylmethylsulfonyl fluoride) and centrifuged at 16,000 × g for 15 min at 4 °C to remove insoluble material. Lysates were precleared using 20 μl of GammaBind beads (GE Healthcare) for 30 min. EGFP-BCAR1 and BCAR3 were immunoprecipitated from 400 μg of cell lysate by incubation for 2 h at 4 °C with 1 μl of GFP antibody (#GTX20290, GeneTex, Irvine, CA) or 0.6 μg of BCAR3 antibody (#sc-47811, Santa Cruz Biotechnology, Santa Cruz, CA) immobilized on GammaBind beads.
For the immunoprecipitations, tissue or cell lysates were incubated with antibody for 2 h at 4 °C followed by a 1 h of incubation with GammaBind beads. Beads were washed 3 times with 1 ml of lysis buffer, and immunocomplexes were eluted by boiling for 5 min in SDS-containing sample buffer.
Immunoblotting
Cells were lysed in modified radioimmune precipitation assay buffer and centrifuged at 16,000 × g for 15 min at 4 °C, and SDS-containing sample buffer was added. Cell lysates and immunoprecipitates were resolved by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA) and then probed with antibodies to BCAR1 (#sc-860; Santa Cruz Biotechnology and #610272; BD Biosciences), BCAR1 phospho-Tyr-165 and phospho-Tyr-410 (#4015 and #4011; Cell Signaling Technology, Danvers, MA), BCAR3 (#sc-47811; Santa Cruz Biotechnology), SRC (#05-184; Millipore), SRC phospho-Tyr-416 (#2101; Cell Signaling), AKT (#9272; Cell Signaling Technology), AKT phospho-Ser-473 (#9271; Cell Signaling), ERK1/2 (#9102; Cell Signaling), phospho-ERK1/2 (#9101; Cell Signaling), cyclin D1 (#556470; BD Biosciences), GAPDH (#9484; ABCam, Cambridge, MA), and GFP (#GTX20290; GeneTex). Incubation with primary antibodies was followed by incubation with anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (HRP, Millipore). Immunoblots were developed with ECL chemiluminescence HRP detection reagent (GE Healthcare). Band quantification was carried out using NIH ImageJ.
Immunofluorescence Microscopy
For immunocytochemistry, transduced MCF7 cells were plated at low density on glass coverslips coated with fibronectin (10 μg/ml; Millipore). The cells were then fixed with 4% formaldehyde for 15 min and permeabilized with 0.5% Triton X-100 in PBS for 3 min at room temperature. After a 30-min incubation with 10% normal goat serum in PBS, the cells were incubated overnight at 4 °C with rhodamine phalloidin, anti-BCAR1 antibody (2.5 μg/ml; #610272; BD Biosciences), anti-BCAR3 antibody (4 μg/ml; #sc-47811; Santa Cruz Biotechnology), or anti-NSP3 antibody (40).
RESULTS
The Ability Of BCAR3 to Increase the Levels of Phosphorylated BCAR1 Depends on Their Physical Association
Hallmarks of BCAR1 signaling are BCAR1 tyrosine phosphorylation and the appearance of a BCAR1 upper band with slower electrophoretic mobility in SDS-PAGE gels, which represents a form with increased serine/tyrosine phosphorylation (9, 29, 41, 42). BCAR3 overexpression in MCF7 breast cancer cells was previously shown to increase the proportion of the BCAR1 upper band, which was attributed to increased serine phosphorylation (29). Surprisingly, overexpression of the mouse BCAR3 R743A mutant also caused this effect (39). Because in previous work the R743A mutant showed reduced association with BCAR1 in coimmunoprecipitation experiments using a lysis buffer containing the strong ionic detergent SDS (Table 1), it was proposed that BCAR3 can enhance BCAR1 serine phosphorylation through a mechanism that does not depend on the interaction between the two proteins (39). Consistent with these previous results, we found that even the more drastic R748E mutation in human BCAR3 (corresponding to R743E in mouse BCAR3; Table 1) fails to impair the BCAR3-dependent increase of the BCAR1 upper band in MCF7 cells (Fig. 1a). However, we also found that in transiently transfected HEK293 cells the BCAR3 R748E mutant still avidly interacts with BCAR1 in coimmunoprecipitation experiments using a lysis buffer lacking SDS (Fig. 1c).
FIGURE 1.

Structure-guided mutation of multiple residues in the binding surfaces of BCAR3 or BCAR1 disrupts their interaction and abrogates crucial signaling events. a, stable lentiviral transduction of BCAR3 wild-type (WT) or the L748E single mutant in MCF7 breast cancer cells similarly increase BCAR1 levels, BCAR1 substrate domain tyrosine phosphorylation (detected with an antibody to phospho-Tyr-410), and the proportion of the hyperphosphorylated BCAR1 upper band (top arrow). An enlargement of the BCAR1 blot is also included to more clearly show the upper and lower BCAR1 bands, which are marked by the two arrows. In contrast, the L744E/R748E double mutant has lost these activities. V, empty lentiviral vector control. Cell lysates were probed by immunoblotting with the indicated antibodies. A band recognized non-specifically by the BCAR1 antibody demonstrates equal loading of the lanes (loading control). b, BCAR1-BCAR3 complex modeled by overlaying the crystal structure of the BCAR3 C-terminal domain (PDB:3T6A) onto the complex structure of the BCAR1 and NSP3 C-terminal domains (PDB ID 3T6G). The two domains are shown in schematic representation, and residues in the binding interfaces that were mutated are shown in sphere representation (left, BCAR3, orange; right, BCAR1, green). c, effects of BCAR1 and BCAR3 mutations on the association between the two proteins as determined by coimmunoprecipitation (IP) from HEK293 cells transfected with the indicated combinations of empty vector controls, EGFP-BCAR1, and BCAR3 wild-type or mutant plasmids. To avoid possible interference with the BCAR1-BCAR3 association by the immunoprecipitating antibody, BCAR1 was immunoprecipitated with an antibody to the EGFP tag (which was also used for detection of EGFP-BCAR1 by immunoblotting), and BCAR3 was immunoprecipitated with an antibody to an epitope near the N terminus. Cell lysates were also probed, revealing low expression of the BCAR3 L744E and L744E/R748E mutants, which is likely due to their impaired interaction with both wild-type and transfected BCAR1. GAPDH was probed as a loading control. Mutations: 744, L744E; 748, R748E; 787, L787E; 794, F794R. d, transient transfection of BCAR1 in HEK293 cells increases the levels of cotransfected wild-type BCAR3 and different mutations in the BCAR3-interacting domain of BCAR1 impair this effect to different extents. Cells were transiently transfected with empty vector control (V) or with a BCAR3 WT plasmid together with BCAR1 wild-type, L787E (787) or F794R (794) single mutants, or the BCAR1 L787E/F794R double mutant. Cell lysates were probed by immunoblotting with the indicated antibodies, with the immunoblot for GAPDH demonstrating equal loading of the lanes.
To identify mutations in the BCAR1-BCAR3 binding interface that would most effectively disrupt the interaction allowing elucidation of its mechanistic relevance, we modeled the human BCAR1-BCAR3 complex by overlaying the BCAR3 crystal structure onto the available structure of the BCAR1-NSP3 complex (36) (Fig. 1b). Examination of the binding interface suggested that a L744E mutation would be more effective for disrupting the interaction with BCAR1 than the R748E mutation because Leu-744 is more centrally located within the binding interface (Fig. 1b). This was confirmed in coimmunoprecipitation experiments from transiently transfected HEK293 cells (lysed in a buffer lacking SDS), which revealed that the L744E mutation disrupts the binding to BCAR1 much more effectively than the R748E mutation (Fig. 1c), highlighting the advantage of a structure-guided mutation strategy. Combining the BCAR3 L744E and R748E mutations yielded a double mutant BCAR3 that lost any detectable binding to BCAR1 in coimmunoprecipitation experiments (Fig. 1c) and that did not up-regulate the hyperphosphorylated BCAR1 upper band (Fig. 1a).
BCAR1 substrate domain tyrosine phosphorylation, which is critical for BCAR1 signaling, is also known to be enhanced by BCAR3/NSP proteins (11, 12, 20). It can be detected with antibodies to the Tyr-165 and Tyr-410 substrate domain phosphorylation sites, which also recognize other similar YXXP phosphorylated motifs in the BCAR1 substrate domain (43). Using our BCAR3 mutants, we found that tyrosine-phosphorylated BCAR1 is increased by the R748E BCAR3 mutant similarly to wild-type BCAR3 (Fig. 1a). However, this effect was not observed with the BCAR3 L744E/R748E double mutant, which has lost the ability to interact with BCAR1 (Fig. 1a). Taken together, these results suggest that mutation of Arg-748 to either Ala or Glu is not sufficient to adequately disrupt the strong BCAR1-BCAR3 interaction under the conditions found in the cellular environment and that the BCAR3-induced increase in phosphorylated BCAR1 is in fact highly dependent on the association between the two proteins.
The Physical Association of BCAR1 and BCAR3 Leads to the Stabilization of Both Proteins
The increase in phosphorylated BCAR1 is accompanied by protein stabilization resulting in higher overall BCAR1 levels, which has been reported to be induced by BCAR3 and the related NSP3 (23, 40). Accordingly, we found that both BCAR3 wild-type and the R748E mutant similarly increase BCAR1 protein levels, whereas the L744E/R748E double mutant has lost this ability (Fig. 1a), demonstrating the importance of a physical interaction with BCAR3 for the stabilization of BCAR1. Interestingly, we also found that in a reciprocal fashion BCAR1 expression stabilizes BCAR3, as transient transfection of BCAR1 in HEK293 cells enhanced the levels of cotransfected BCAR3 (Fig. 1d). Thus, association between the two proteins up-regulates the levels and activity of the BCAR1-BCAR3 signaling module.
To further validate our results, we sought to investigate the effects of BCAR1 mutations capable of disrupting the association with BCAR3, again using the available structural information as a guide (36). To this end we identified the F794R mutation in BCAR1, which is central in the BCAR3 binding interface (Fig. 1b), and found that this mutation substantially but not completely disrupts association with BCAR3 in coimmunoprecipitation experiments using lysis buffer without SDS (Fig. 1c and Table 1). Mutation of the more peripheral residue Leu-787 to Glu (L787E) was less effective (Fig. 1b and Table 1), but the combination of F794R and L787E mutations greatly reduced binding to BCAR3, with only a weak residual interaction still detectable (Fig. 1c). We next examined these BCAR1 mutants for their ability to stabilize BCAR3. The effects were consistent with their degree of BCAR3 binding impairment. The L787E and F794R single mutants stabilized BCAR3 less effectively than wild-type BCAR1, whereas the L787E/F794R double mutant exhibited greater functional deficiency (Fig. 1d). Thus, the observed stabilization of BCAR3 by BCAR1 is also inherently linked to their physical association. Furthermore, as in the case of the BCAR3 mutations, these results suggest that the interaction module cannot be easily disrupted and that double mutations in BCAR1 more effectively impair the association with BCAR3 than single mutations.
In summary, the effects of BCAR1/BCAR3 mutants with different degrees of interaction impairment demonstrate that a physical interaction is required for a wide spectrum of signaling events associated with the interplay between BCAR1 and BCAR3. However, a weakened association is still sufficient to support many of these effects. These data also imply that the physiological roles of BCAR1-BCAR3 complexes and most likely other BCAR1-NSP complexes can only be reliably deduced by analyzing the effects of mutations, such as multiple targeted mutations, that drastically impair the very tight binding between the two protein families.
The Mesenchymal Phenotype and Antiestrogen Resistance Induced by BCAR3 Require Complex Formation with BCAR1
Mesenchymal features are a hallmark of cancer malignancy and often accompany drug resistance (1, 44–47). Overexpression of BCAR3 in the epithelial-like, estrogen receptor-expressing MCF7 breast cancer cells is known to lead to a more mesenchymal and migratory phenotype characterized by the formation of peripheral membrane ruffles and lamellipodia that are rich in filamentous actin and also BCAR1 and BCAR3 (23, 30, 31, 33, 39). To investigate the role of the association of BCAR3 with BCAR1 in the actin cytoskeleton rearrangements induced by BCAR3 in MCF7 cells, we used the BCAR3 L744E/R748E double mutant. By labeling actin filaments, we found that wild-type BCAR3 and the R748E single mutant both similarly induce the formation of membrane ruffles (Fig. 2, a and b). These ruffles also contain prominent BCAR1 and BCAR3 immunoreactivity (Fig. 2, a and c), in agreement with previous results obtained by overexpressing the mouse BCAR3 R743A mutant (39). In contrast, the BCAR3 L744E/R748E double mutant (which effectively disrupts BCAR3 binding to BCAR1) lost the ability to induce membrane ruffles (Fig. 2, a–c), supporting the importance of the BCAR1-BCAR3 module in promoting a mesenchymal morphology in breast cancer cells.
FIGURE 2.

BCAR3 and the related NSP3 induce formation of membrane ruffles, which requires complex formation with BCAR1. a, MCF7 cell populations stably transduced with lentiviral vectors encoding ZsGreen alone (V) or together with wild-type BCAR3 (WT), the BCAR3 R748E single mutant (748), or the BCAR3 L744E/R748E double mutant (744/748) were stained with phalloidin to label filamentous actin (top row) or with an anti-BCAR3 antibody (bottom row). The phalloidin-stained cells were also imaged for ZsGreen fluorescence to identify the cells expressing BCAR3 and ZsGreen from the bicistronic transcript (middle row). BCAR3 wild-type and the R748E single mutant both promote the formation of membrane ruffles compared with the vector control cells (arrows point to examples of ruffles), whereas the BCAR3 L744E/R748E double mutant does not. Furthermore, BCAR3 immunoreactivity is evident in the ruffles. Scale bars = 25 μm. b, the histogram shows the percentage of ZsGreen-expressing cells that contain ruffles. **, p < 0.01, and ***, p < 0.001 for the comparison with vector control-infected cells by one-way ANOVA and Dunnett's post hoc test. c, overexpression of BCAR3 and NSP3 in MCF7 cells promotes the formation of membrane ruffles containing BCAR1. Levels and subcellular localization of endogenous BCAR1 are shown by immunolabeling (arrows point to examples of ruffles). ZsGreen fluorescence identifies the cells expressing BCAR3 or NSP3 from the bicistronic transcripts. Scale bar = 40 μm.
We next examined the role of the BCAR1-BCAR3 association in antiestrogen resistance. We lentivirally transduced wild-type or mutant BCAR3 and monitored the growth of MCF7 cells in the presence of the widely used estrogen receptor antagonist ICI182780. The results show that BCAR3 promotes cell growth under these conditions (Fig. 3a) and restores a more healthy cellular appearance (Fig. 3b) compared with control vector-transduced cells, as expected (24, 39). We obtained similar results with the R748E single mutant, which retains substantial interaction with BCAR1, in agreement with previous findings with BCAR3 carrying the less disruptive R743A mutation (39). In contrast, the BCAR3 L744E/R748E double mutant has lost the ability to rescue the cells from the deleterious effects of the antiestrogen (Fig. 3, a and b), demonstrating the importance of the association with BCAR1 for BCAR3-dependent breast cancer antiestrogen resistance.
FIGURE 3.

BCAR3 and the related NSP3 promote antiestrogen resistance, which requires complex formation with BCAR1. a, MCF7 cell populations stably transduced with lentiviral vectors encoding ZsGreen alone (V) or together with wild-type or mutant BCAR3 were grown for 33 days in the presence of the estrogen receptor antagonist ICI182780. The histogram shows the means ± S.E. from triplicate measurements. The doubling time of the cultures (in days) is also shown above the bars. ***, p < 0.001 for the comparison with vector control-infected cells by one way ANOVA and Dunnett's post hoc test. The BCAR3 double mutant, but not the single mutant, has lost the ability to promote antiestrogen resistance. The somewhat higher growth observed in the presence of the BCAR3 R748E single mutant may be due to its slightly higher expression compared with wild-type BCAR3 (see Fig. 1a). b, phase contrast images illustrate the detrimental effects of ICI182780 treatment for 5 days on the morphology of cells transduced with control vector or the BCAR3 double mutant, whereas expression of BCAR3 wild-type and the R748E mutant have a more healthy appearance consistent with their faster proliferation. Scale bar = 100 μm. c, MCF7 breast cancer cell populations stably expressing BCAR3, NSP3α, NSP3β, or vector control were grown for 35 days in the presence of ICI182780. The histogram shows averages from triplicate measurements ± S.E. The doubling time of each cell population, in days, is indicated above each bar. *, p < 0.05, ***, p < 0.001 for the comparisons with control vector-infected cells by one-way ANOVA and Tukey's post hoc test. Other comparisons, indicated by bars, are also shown; ns, not significant. d, lysates of MCF7 breast cancer cell populations stably expressing BCAR3, NSP3α, NSP3β, or vector control were probed by immunoblotting with the indicated antibodies. Although the NSP3 antibody used may recognize better NSP3β than NSP3α (see “Experimental Procedures”), immunoblotting with an NSP3 antibody recognizing the C terminus also indicated that NSP3β was more highly expressed than NSP3α. An enlargement of the BCAR1 blot is also included to more clearly show the upper and lower BCAR1 bands, which are marked by the two arrows. GAPDH was probed as the loading control.
The BCAR3-related NSP3 Can Promote Morphological Changes and Antiestrogen Resistance
Besides BCAR3, we also examined the effects of the NSP family member NSP3, which is also expressed in breast cancer cells albeit at lower levels than BCAR3 (23). Several NSP3 isoforms have been described that differ in the N-terminal region preceding the SH2 domain due to alternative splicing (11, 48). In particular, the NSP3α isoform contains a distinctive ∼200-amino acid N-terminal region that mediates constitutive association with the plasma membrane (11, 49). In contrast, the NSP3β isoform contains a shorter N-terminal region of ∼60 amino acids and, like BCAR3, is not constitutively localized at the plasma membrane. However, both NSP3β and BCAR3 can localize at the membrane through binding of their SH2 domains to tyrosine-phosphorylated motifs of membrane-associated proteins, which in turn leads to increased SRC activity and BCAR1 phosphorylation (11, 25, 31, 32, 35, 37, 40, 50).
By using lentiviruses to introduce the NSP3α or NSP3β isoform in MCF7 cells, we found that both caused morphological changes that were similar but less pronounced than those induced by BCAR3 (Fig. 2c), consistent with previous data in COS and NIH3T3 cells (35, 50). Thus, NSP3 can also promote a more mesenchymal/migratory phenotype in MCF7 cells. In addition, overexpression of NSP3 in these cells revealed that both the α and the β isoforms can significantly promote MCF7 cell growth in the presence of ICI182780, with NSP3α appearing to be more effective than NSP3β (Fig. 3c) as it was expressed at much lower levels (Fig. 3d). The different efficiency of NSPα and NSP3β suggests that the plasma membrane localization of NSP proteins is important for conferring antiestrogen resistance. The membrane localization of NSP3α seems to also be critical for increasing the hyperphosphorylated BCAR1 upper band (Fig. 3d). In contrast, lentivirally transduced or endogenous NSP3β predominantly stabilizes the lower BCAR1 band in MCF7 cells (Fig. 3d) as previously reported (29).
Signaling Pathways That Depend on BCAR1-BCAR3 Association as Candidates for Mediating Antiestrogen Resistance
We next examined signaling pathways associated with overexpression and interaction of BCAR1 and BCAR3 in MCF7 cells grown in the presence of antiestrogen. Concomitant with overcoming the growth inhibition of ICI182780, overexpression of BCAR3 alone enhanced phosphorylation of Tyr-416 in the activation loop of SRC and tyrosine phosphorylation in the substrate domain of endogenously expressed BCAR1 (Fig. 4, first and second lanes). These effects are consistent with BCAR3-dependent increase in SRC activity leading to increased BCAR1 downstream signaling as previously reported in cells not treated with antiestrogens (9, 30, 32, 51). Similar to antiestrogen resistance, SRC and BCAR1 phosphorylation were not induced by the BCAR3 L744E/R748E interaction-deficient mutant (Fig. 4, third lane), highlighting the essential role of BCAR3 complex formation with endogenous BCAR1 for these signaling effects. We also examined AKT, ERK1/2, and cyclin D1, which are major BCAR1 signaling effectors involved in the development of resistance to antiestrogens and other chemotherapeutic drugs through their effects on survival (mainly AKT) and proliferation (all three proteins) (2, 7, 10, 15, 52). Overexpression of BCAR3 wild-type, but not the L744E/R748E interaction-deficient mutant, markedly enhanced ERK1/2 activation (based on Thr-202/Tyr-204 phosphorylation) and cyclin D1 levels but not AKT activation (based on Ser-473 phosphorylation) (Fig. 4, first through third lanes).
FIGURE 4.

Effects of BCAR1-BCAR3 interaction on antiestrogen resistance and signaling networks. Lentivirally transduced MCF7 breast cancer cell populations expressing BCAR1/BCAR3 WT, the BCAR3 L744E/R748E mutant (M), the BCAR1 L787E/F794E/D797R mutant (M), or the appropriate control lentiviral vectors (V) were grown for 35 days in the presence of ICI182780. The histogram shows averages ± S.E. from triplicate measurements. The doubling time of each cell population, in days, is indicated above each bar. The doubling time of the parental cells in the absence of ICI182780 was 2 days. ***, p < 0.001 for the comparisons with control vector-infected cells by one-way ANOVA and Tukey's post hoc test. Other comparisons, indicated by bars, are also shown; ns, not significant. In the immunoblots, lysates from cells treated with ICI182780 for 8 days were probed with the indicated antibodies.
BCAR1 overexpression alone also induced antiestrogen resistance, concomitant with an increase in SRC activation, phosphorylated BCAR1, ERK1/2 activation, cyclin D1 levels, and AKT activation (Fig. 4, first and fourth lanes). However, except for AKT activation, these effects were not as pronounced as with BCAR3 overexpression (Fig. 4, second and fourth lanes). Thus, although BCAR1 is capable of conferring some antiestrogen resistance in MCF7 cells (where BCAR3 expression is very low), BCAR3 is needed for efficient activation of a number of the signaling events linked to BCAR1. Indeed, coexpression of BCAR3 with BCAR1 strongly potentiated cell growth in the presence of antiestrogen as well as all the signaling events monitored, except for AKT activation (Fig. 4, fourth and fifth lanes). In contrast, the BCAR3 L744E/R748E interaction-deficient mutant did not potentiate the effects of BCAR1 overexpression (Fig. 4, fourth and sixth lanes), indicating that the underlying mechanism depends on the physical association between the two proteins.
We also used an interaction-deficient BCAR1 mutant to further examine the importance of the association with BCAR3 in BCAR1 signaling. To ensure a virtually complete disruption of the binding with BCAR3, we generated a BCAR1 mutant encompassing the L787E and F794E mutations as well as a third mutation in the binding interface, D797R (Fig. 1b). We found that the BCAR1 L787E/F794E/D797R triple mutant promotes MCF7 cell growth in the presence of ICI182780 to the same extent as wild-type BCAR1 alone, both when overexpressed alone (Fig. 4, fourth and seventh lanes) or when co-expressed with the BCAR3 L744E/R748E mutant (Fig. 4, sixth and ninth lanes). As in the case of wild-type BCAR1, the increased growth induced by the BCAR1 triple mutant correlated with increased levels of activated SRC, tyrosine-phosphorylated BCAR1, activated AKT and ERK1/2, and cyclin D1 (Fig. 4, first and seventh lanes). Of note, wild-type but not mutant BCAR3 increased the growth in ICI182780 of MCF7 cells overexpressing the interaction-deficient mutant of BCAR1 (Fig. 4, seventh through ninth lanes), which is likely due to increased signaling of its complex with endogenous BCAR1.
Overall, these results suggest that the antiestrogen resistance conferred by BCAR3 depends on its ability to stabilize BCAR1 as well as activate SRC and promote BCAR1 tyrosine phosphorylation, leading to downstream signaling (9, 11, 37). Accordingly, SRC activation was most elevated in the three cell populations expressing wild-type BCAR3, the same ones that grew best in the presence of ICI182780.
Among the various signaling events analyzed, the extent of ERK1/2 activation most closely correlated with the degree of antiestrogen resistance and with BCAR1 tyrosine phosphorylation. Cyclin D1 levels were also generally correlated with antiestrogen resistance, as expected given the ability of the cells to grow in the presence of the antiestrogen (2, 23). Only AKT activation correlated most closely with BCAR1 wild-type or mutant overexpression but not with BCAR1-BCAR3 complex formation, suggesting that AKT activation is mediated by BCAR1 independently of BCAR3. To our knowledge, these results are the first to suggest the involvement of ERK1/2 activation in breast cancer antiestrogen resistance induced by BCAR1/BCAR3.
ERK1/2 Activity Is Required For BCAR1/BCAR3-mediated Antiestrogen Resistance
We next investigated if the ERK1/2 activity connected to the BCAR1-BCAR3 signaling assembly is critical for antiestrogen resistance. Thus, we used the ERK1/2 regulator, phosphoprotein, enriched in astrocytes of 15 kDa (PEA15). PEA15 is a cytoplasmic protein that binds to both phosphorylated and non-phosphorylated ERK, sequestering them in the cytoplasm (53). Through this mechanism, PEA15 potently inhibits ERK1/2 nuclear functions (54, 55). Because PEA15 is expressed at low levels in MCF7 cells (54), we delivered wild-type PEA15 as well as an inactive mutant (PEA-15 R71E) through lentiviral infection.
Because overexpression of BCAR3 alone is sufficient to strongly increase antiestrogen-resistant growth by interacting with endogenous BCAR1 (Fig. 4), we examined whether wild-type PEA15 inhibits the effects of BCAR3 on MCF7 cell growth in the presence of ICI182780. We found that PEA15 potently suppresses the growth of BCAR3-overexpressing cells as well as the already minimal growth of cells transduced with control lentivirus (Fig. 5). In contrast, the PEA15 R71E mutant, which does not sequester ERK1/2 in the cytoplasm, did not inhibit growth (Fig. 5). A peculiar hallmark of ERK1/2 sequestration by PEA15 is a concomitant enrichment in phospho-ERK1/2 (53–55). Indeed, expression of wild-type but not mutant PEA15 in MCF7 cells led to the characteristic increase in phospho-ERK1/2 (Fig. 5), confirming ERK1/2 sequestration by wild-type but not mutant PEA15. Taken together, these results demonstrate a key role of ERK1/2 activity in antiestrogen resistance conferred by BCAR1/BCAR3.
FIGURE 5.

BCAR1/BCAR3-induced antiestrogen resistance requires ERK1/2 activity. MCF7 cell populations with or without stable BCAR3 lentiviral expression were infected with lentiviruses encoding EGFP alone or fused to PEA15 wild-type or the PEA15 R71E mutant. The cells were then grown for 39 days in the presence of ICI182780. The histogram shows average cell numbers from triplicate measurements ± S.E. The doubling time of each cell population, in days, is indicated above each bar. *, p < 0.05; ***, p < 0.001; ns, not significant for the comparison with the EGFP-expressing control cells (first lane) by one-way ANOVA and Tukey's post hoc test. Other comparisons, indicated by bars, are also shown. The immunoblots show lysates from cells treated with ICI182780 for 4 days and probed with the indicated antibodies. PEA15 counteracts estrogen-independent growth induced by BCAR3 overexpression in MCF7 cells.
The inhibitory effects of PEA15 on the growth of breast cancer cells and its ability to counteract the antiestrogen resistance of MCF7 cells overexpressing BCAR3 together with previous data showing a correlation of high PEA15 expression with benign markers and low invasiveness in clinical breast cancer specimens (54, 56) suggest that tumors with high PEA15 levels are less malignant than tumors with low PEA15. Thus, PEA15 protein expression should be considered when evaluating the prognostic significance of BCAR1 and BCAR3 in breast cancer specimens (4, 5, 14, 18, 19). In particular, high BCAR1/BCAR3 protein/phosphorylation levels would be expected to correlate with high ERK1/2 activity and short patient survival only when PEA15 expression is low but should correlate with low ERK1/2 activity and prolonged survival when PEA15 expression is high. Indeed, our analysis of 408 tumors from a breast invasive carcinoma study (TCGA, provisional) using the cBio Cancer Genomics Portal (57, 58) revealed that patients with high PEA15 protein levels in their tumors survived significantly longer than the remaining patients (Fig. 6, left). Conversely, patients with low PEA15 protein levels in their tumors survived a significantly shorter time (Fig. 6, right). We also found that high PEA15 expression significantly correlates with high ERK1/2 phosphorylation and low PEA15 expression with low ERK1/2 phosphorylation (Fig. 6), consistent with PEA15-dependent protection of phospho-ERK1/2 from dephosphorylation but PEA15-mediated inhibition of ERK1/2 nuclear activity (55). This suggests that ERK1/2 phosphorylation is not a reliable prognostic marker for malignancy in breast cancer due to the discrepancy between ERK1/2 activity and ERK1/2 phosphorylation caused by PEA15. However, these data show that PEA15 expression in invasive breast cancer correlates with overall survival and thus could represent a useful new prognostic indicator. The potential usefulness of PEA15 as a critical biomarker is consistent with its ability to override the effects of ERK1/2 activation induced for example by BCAR1/BCAR3 but also other ERK1/2 upstream regulators.
FIGURE 6.
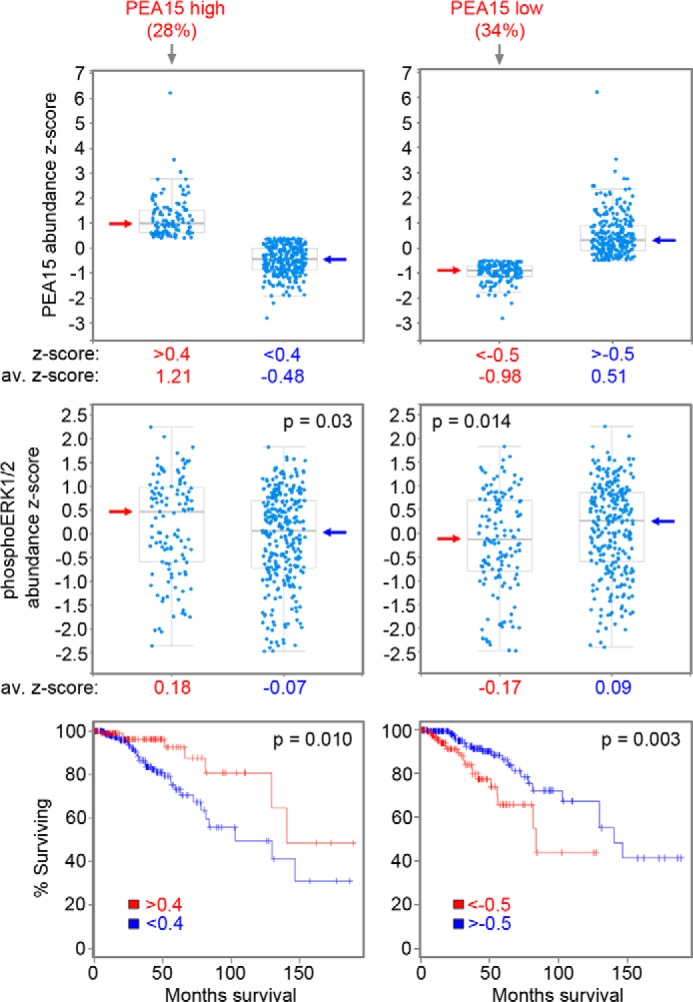
PEA15 expression levels in invasive breast cancers correlate with ERK1/2 phosphorylation and patient overall survival. A cohort of 408 tumors with large scale reverse phase protein array data from a TCGA provisional invasive breast cancer study was analyzed. Top left, Boxplot representation of the group including the 28% of the tumors with highest PEA15 expression (abundance z-score >0.4, average abundance z-score = 1.21) and the group including the remaining 72% of the tumors (abundance z-score <0.4, average z-score = −0.48). Middle left, Boxplot representation of ERK1/2 phospho-Thr-202 levels in the tumors with highest PEA15 expression (average phospho-ERK1/2 abundance z-score = 0.18) versus the remaining tumors (average phospho-ERK1/2 abundance z-score = −0.07, p = 0.03 for the difference in phospho-ERK1/2 levels by two-sided, two-sample Student's t test). Bottom left, patients with tumors expressing the highest levels of PEA15 survived significantly longer than the remaining patients (Kaplan-Meier plot, p = 0.010 by log-rank test) despite their higher phospho-ERK1/2 levels. Top right, Boxplot representation of the group including the 34% of the tumors with lowest PEA15 expression (abundance z-score <-0.5, average z-score = −0.98) and the group including the remaining 66% of the tumors (abundance z-score >-0.5, average z-score = 0.51). Middle right, Boxplot representation of phospho-ERK1/2 levels in the tumors with the lowest PEA15 expression (average ERK1/2 phospho-Thr-202 abundance z-score = −0.17) versus the remaining tumors (average phospho-ERK1/2 abundance z-score = 0.09, p = 0.014 for the difference in phospho-ERK1/2 levels by two-sided, two-sample Student's t test). Bottom right, patients with tumors expressing the lowest levels of PEA15 survived significantly less than the remaining patients (Kaplan-Meier plot, p = 0.003 by log-rank test) despite their lower phospho-ERK1/2 levels. Arrows in the boxplots mark the median values.
DISCUSSION
NSP family proteins have emerged as critical regulators of BCAR1 and other members of the CAS family under physiological as well as pathological conditions (11, 12). Our results show that direct association of the CAS family member BCAR1 and the NSP family member BCAR3 is required to increase BCAR1 abundance, phosphorylation, and downstream signaling, ultimately promoting a motile mesenchymal morphology and antiestrogen resistance in breast cancer cells. The physiological importance of integrating the signaling networks associated with BCAR1 and BCAR3 is consistent with the remarkable adaptation of the highly conserved GEF-like domain of NSP proteins into an efficient module for tight interaction with CAS family proteins (36). We show that the BCAR1-BCAR3 complex can retain many of its cellular functions even when binding is weakened by single amino acid mutations, which is consistent with the very high affinity achieved through two cooperating binding interfaces in the C-terminal domains of CAS and NSP family proteins. Hence, to investigate the role of the BCAR1-BCAR3 complex, we used a structure-guided mutation strategy (55), resulting in a BCAR3 mutant in which both Leu-744 and Arg-748 were replaced by glutamic acid to strongly disrupt BCAR1 binding. In addition, we have complemented the findings obtained with this BCAR3 double mutant by using the BCAR1 L787E/F794E/D797R triple mutant. The consistent and substantial effects of our interaction-deficient mutations in either BCAR1 or BCAR3 suggest that they are due to disruption of the association between the two proteins and not of other possible activities of the BCAR1 or BCAR3 C-terminal domains.
We have shown that previously used single BCAR1/BCAR3 mutants can retain substantial complex formation ability and signaling activities that approximate those of the wild-type proteins, which led to the inaccurate conclusion that linking the BCAR1 and BCAR3 signaling networks are not crucial for their cellular functions. Accordingly, we found that mutation of Arg-748 to glutamic acid in human BCAR3 does not detectably impair the ability of BCAR3 to increase BCAR1 abundance and phosphorylation as well as induce cell morphological changes and antiestrogen resistance, similar to previous data obtained with the less drastic corresponding R743A mutation in mouse BCAR3 (39). Nevertheless, some signaling events do seem to be influenced by the decreased binding affinity of the weaker mutants. Thus, the R743A mutation has been reported to affect some BCAR3 functions. For example, it can inhibit BCAR3 association with the SRC kinase and BCAR3-dependent SRC activation as well as impair BCAR3-dependent fibroblast migration (37, 39, 51).
We show that the L744E mutation in BCAR3 weakens the interaction with BCAR1 much more effectively than the R748E mutation. We previously observed a similar difference for the corresponding mutations in human NSP3β, with the L623E mutation (corresponding to BCAR3 L744E) having a stronger effect on BCAR1 binding than the R627E mutation (corresponding to BCAR3 R748E; Table 1) (36). These similarities highlight the high conservation of the BCAR1 binding interface in different NSP family members.
Mutation of Leu-791 in mouse BCAR1 (corresponding to Leu-787 in human BCAR1; Table 1) also did not result in complete loss of function, consistent with the fact that this residue is peripherally located in the binding interface. In previous studies, the L791P mutation did not affect the ability of BCAR1 to promote SRC activation and COS cell migration (30), whereas the L791D mutation partially reduced the BCAR3-dependent increase in the hyperphosphorylated BCAR1 upper band (39) and BCAR3-dependent SRC activation (51). Furthermore, we have shown that the similar L787E mutation did not drastically impair association with BCAR3 in our coimmunoprecipitation experiments. Thus, mutation of BCAR1 Leu-787-like mutation of Arg-743 in mouse BCAR3 or Arg-748 in human BCAR3 weakens but does not abrogate BCAR1-BCAR3 binding. These examples highlight the importance of carefully evaluating the extent to which amino acid replacements disrupt protein-protein interactions to accurately evaluate the physiological activities of protein complexes.
The critical role of the interaction between CAS and NSP family members is also supported by the impaired activities of BCAR3ΔC, a truncated form of BCAR3 lacking the entire GEF-like domain and thus completely unable to interact with BCAR1. According to a number of studies, BCAR3ΔC cannot promote SRC or RAC1 activation, cell motility, and antiestrogen resistance (28, 31, 39, 59, 60). On the other hand, a few studies using truncated forms of BCAR1 or BCAR3 lacking their interaction domains have suggested that association between the two proteins is dispensable for SRC activation by BCAR3 and for increasing the hyperphosphorylated BCAR1 upper band (29, 59). It will be important to resolve these discrepancies.
A consequence of BCAR1-NSP family complex formation is an increase in the expression levels of both interacting partners, possibly due to reciprocal protection from proteolytic degradation, thus leading to reciprocal enhancement of the activities of both proteins. We also found that increasing expression of the BCAR3-related NSP3 decreases the levels of coexpressed BCAR3 and vice versa,4 consistent with a mechanism involving competition of the two NSP family members for binding to BCAR1 and the importance of a physical association for stabilization. However, precisely how the reciprocal protection from proteolytic degradation may occur remains to be determined.
Interaction with BCAR3 increases not only the abundance of BCAR1 but also its serine/tyrosine phosphorylation, suggesting potentiation of BCAR1 signaling activity. However, the precise role of BCAR1 serine phosphorylation (which does not seem to be important for antiestrogen resistance) and the kinase(s) responsible remain to be identified (39). On the other hand, SRC activation by BCAR3 (30, 32) has been recently shown to promote phosphorylation of the Tyr-789 motif of PTPα, a receptor-type phosphatase that can potentiate SRC activation by dephosphorylating its inhibitory Tyr-527 phosphorylation site (37). Binding of the BCAR3 SH2 domain to phosphorylated PTPα leads to recruitment of the associated BCAR1, positioning it for SRC-mediated phosphorylation (32, 37). Tyrosine-phosphorylated motifs in the BCAR1 substrate domain are well known to mediate downstream signaling by generating binding sites for the SH2 domains of adaptors such as CRK (9, 11, 12, 22).
We observed a close correlation between the levels of tyrosine phosphorylation in the BCAR1 substrate domain and the growth of MCF7 cells overexpressing BCAR1/BCAR3 in culture medium supplemented with the antiestrogen ICI182780. This is consistent with previous studies demonstrating the importance of BCAR1 tyrosine phosphorylation for antiestrogen resistance in estrogen receptor-positive breast cancer cells (10, 52, 61) and for survival and migration/invasion in estrogen receptor-negative breast cancer cells (9). Interestingly, BCAR1 substrate domain phosphorylation and also BCAR3 tyrosine phosphorylation have been reported to be elevated in breast cancer cell lines of the basal-like subtype (that includes a majority of the aggressive triple-negative cells) (9, 14, 62). In these cells BCAR1 and BCAR3 expression levels were found to be similar to those in non-basal-like cell lines, supporting the importance of elevated signaling (rather than mere expression) as part of the oncogenic SRC signature.
Several signaling pathways have been implicated in antiestrogen resistance downstream of BCAR1/BCAR3, although they were investigated in cells grown without antiestrogens, whereas we have examined signaling in cells grown in ICI182780. Increased activity of PI3K, RAC1, and PAK1 was reported to be required but not sufficient for BCAR3-induced antiestrogen resistance in MCF7 and other estrogen-dependent breast cancer cell lines (23, 27, 28). Furthermore, increased cyclin D1-dependent transcription downstream of PAK1 was previously shown to correlate with antiestrogen resistance induced by BCAR3 (23, 28). Here we show that ERK1/2 activity is elevated in cells overexpressing BCAR1/BCAR3 and correlates with their ability to grow in the presence of ICI182780. ERK1/2 signaling was previously found to play a particularly important role in breast cancer malignancy and resistance to hormonal therapy (63, 64). Interestingly, BCAR1 has been reported to promote ERK1/2 activation and cyclin D1 expression by forming a complex with activated SRC and the ERα estrogen receptor, thus mediating the early effects of estrogen (65). However, BCAR1/BCAR3 likely achieve similar effects through a different mechanism in cells grown in medium containing ICI182780 because this pure antiestrogen is known to down-regulate the ERα receptor (2, 27). Notably, ERK1/2 activity can also up-regulate BCAR1 expression, which may result in a positive feedback loop increasing the malignancy of antiestrogen resistant breast cancer cells (66).
Confirming the importance of the ERK1/2 pathway, we show that the wild-type form of the ERK1/2 inhibitor PEA15 inhibits BCAR1-BCAR3-induced growth of MCF7 cells in the presence of antiestrogens, whereas mutant PEA15 deficient in ERK1/2 binding was ineffective. Previous studies reported that PEA15 inhibits breast cancer cell growth and invasiveness concomitant with sequestration of cytoplasmic ERK1/2 but did not demonstrate the importance of ERK1/2 binding in the growth effects of PEA15, a protein capable of multiple activities (54, 56, 67). Interestingly, high PEA15 protein expression in breast cancer clinical samples has been significantly correlated with positive estrogen/progesterone receptor status and a low level of the proliferation marker, proliferating cell nuclear antigen (54). Furthermore, consistent with a tumor suppressor role, PEA15 is poorly expressed in the highly malignant triple negative breast cancers, which lack estrogen and progesterone receptors as well as the receptor-tyrosine kinase HER2 (54).
Here we show that PEA15 can counteract BCAR1-BCAR3-dependent antiestrogen resistance in MCF7 cells and that this activity depends on its ability to bind ERK1/2. These data suggest that promoting PEA15 expression or its ability to sequester ERK1/2 could help overcome the loss of sensitivity to antiestrogens that often occurs in response to hormonal treatment. Unfortunately, the TCGA breast cancer reverse phase protein array dataset analyzed does not include information on BCAR1/BCAR3 protein expression and phosphorylation. Thus, we could not extract additional information on the correlation of BCAR1/BCAR3, PEA15, and ERK1/2 protein and phosphorylation levels from the available TCGA data. Nevertheless, the data show that PEA15 protein levels in invasive breast cancers significantly correlate with overall patient survival despite the fact that tumors with high PEA15 also have significantly increased phospho-ERK1/2 levels. This suggests an overriding effect of the tumor suppressing effects of PEA15, highlighting the potential prognostic usefulness of monitoring PEA15 protein levels in conjunction with BCAR1/BCAR3 and ERK1/2 signaling. It should also be noted that PEA15 phosphorylation on Ser-104 and Ser-116, which results in loss of ERK1/2 inhibition, was shown to promote resistance to proapoptotic cytokines and chemotherapeutic drugs such as paclitaxel in MCF7 cells (68). This suggests a context-dependent regulation of PEA15 tumor suppressive activity (67, 69) that should be further investigated and also underlines the need to examine both PEA15 protein and phosphorylation levels.
Although NSP3 was previously not found to significantly promote antiestrogen resistance (39), our data show that NSP3 can also enable significant MCF7 cell growth in the presence of ICI182780. These results support the potential value of screening breast cancers not only for BCAR1 and BCAR3 but also for NSP3 in order to predict resistance to antiestrogens. The fact that the NSP3α isoform more effectively promotes antiestrogen resistance than the β isoform supports the importance of NSP plasma membrane localization for BCAR1 regulation, consistent with previous data (31, 50). Indeed, we have shown that the NSP3α isoform, but not the β isoform, increases the abundance of the hyperphosphorylated BCAR1 upper band in MCF7 cells. The different effects of BCAR3 and NSP3β also suggest that the SH2 domain of NSP3 interacts with a different partner that may not be highly expressed/phosphorylated in MCF7 cells. Such a partner instead appears to be phosphorylated in the mouse neonatal brain, where loss of endogenous NSP3β reduces BCAR1 tyrosine phosphorylation and the abundance of the hyperphosphorylated BCAR1 upper band (40).
Morphologically, antiestrogen resistance has been associated with a less epithelial and more mesenchymal appearance and thus with migratory/invasive properties (1, 23, 45–47, 52, 70). BCAR1 tyrosine phosphorylation has a well known role in cell migration/invasiveness (6, 9, 11, 71), and we show that BCAR3 and NSP3 can both promote the formation of actin-rich membrane ruffles containing BCAR1 in MCF7 cells, consistent with previous data (30, 31, 33). In agreement with this, besides promoting antiestrogen resistance, BCAR3 overexpression in MCF7 cells was previously shown to promote mesenchymal features such as a decrease in cell-cell junctions containing E-cadherin and increased fibronectin expression (23, 29). These morphological changes have been proposed to underlie the increased migratory/invasive properties of MCF7 cells overexpressing BCAR3 (23, 31, 33, 39).
In conclusion, our results show that linking BCAR1 and NSP signaling networks promotes antiestrogen resistance and a mesenchymal phenotype by enhancing SRC and BCAR1 signaling through a mechanism that requires a direct association between BCAR1 and NSP proteins and leads to increased ERK1/2 activity. Therefore, our findings support the notion that inhibiting the association of BCAR1 with BCAR3 (and possibly NSP3) could help counteract breast cancer resistance to antiestrogens. The fact that the interaction is remarkably tight and modifications that substantially weaken it are not sufficient to impair many of the functional effects of the BCAR1-BCAR3 complex suggests that inhibiting downstream signaling pathways may be a more viable therapeutic approach (10). Finally, our data also suggest that inhibiting ERK1/2 activity (for example through PEA15) may be a useful strategy to counteract BCAR1/BCAR3-dependent breast cancer malignancy and resistance to chemotherapeutic agents.
Acknowledgment
We thank K. Vuori for the gift of the EGFP-BCAR1 plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA160457.
Y. Wallez and E. B. Pasquale, unpublished data.
- BCAR1
- breast cancer antiestrogen resistance 1
- NSP
- novel SH2-containing protein
- GEF
- guanine nucleotide exchange factor
- ANOVA
- analysis of variance.
REFERENCES
- 1. Riggins R. B., Schrecengost R. S., Guerrero M. S., Bouton A. H. (2007) Pathways to tamoxifen resistance. Cancer letters 256, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Musgrove E. A., Sutherland R. L. (2009) Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 9, 631–643 [DOI] [PubMed] [Google Scholar]
- 3. Brinkman A., van der Flier S., Kok E. M., Dorssers L. C. (2000) BCAR1, a human homologue of the adapter protein p130Cas, and antiestrogen resistance in breast cancer cells. J. Natl. Cancer Inst. 92, 112–120 [DOI] [PubMed] [Google Scholar]
- 4. van der Flier S., Chan C. M., Brinkman A., Smid M., Johnston S. R., Dorssers L. C., Dowsett M. (2000) BCAR1/p130Cas expression in untreated and acquired tamoxifen-resistant human breast carcinomas. Int. J. Cancer 89, 465–468 [DOI] [PubMed] [Google Scholar]
- 5. van der Flier S., Brinkman A., Look M. P., Kok E. M., Meijer-van Gelder M. E., Klijn J. G., Dorssers L. C., Foekens J. A. (2000) Bcar1/p130Cas protein and primary breast cancer. Prognosis and response to tamoxifen treatment. J. Natl. Cancer Inst. 92, 120–127 [DOI] [PubMed] [Google Scholar]
- 6. Brábek J., Constancio S. S., Shin N. Y., Pozzi A., Weaver A. M., Hanks S. K. (2004) CAS promotes invasiveness of Src-transformed cells. Oncogene 23, 7406–7415 [DOI] [PubMed] [Google Scholar]
- 7. Ta H. Q., Thomas K. S., Schrecengost R. S., Bouton A. H. (2008) A novel association between p130Cas and resistance to the chemotherapeutic drug adriamycin in human breast cancer cells. Cancer Res. 68, 8796–8804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wendt M. K., Smith J. A., Schiemann W. P. (2009) p130Cas is required for mammary tumor growth and transforming growth factor β-mediated metastasis through regulation of Smad2/3 activity. J. Biol. Chem. 284, 34145–34156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cunningham-Edmondson A. C., Hanks S. K. (2009) p130Cas substrate domain signaling promotes migration, invasion, and survival of estrogen receptor-negative breast cancer cells. Breast Cancer Res. Treat. 2009, 39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soni S., Lin B. T., August A., Nicholson R. I., Kirsch K. H. (2009) Expression of a phosphorylated p130(Cas) substrate domain attenuates the phosphatidylinositol 3-kinase/Akt survival pathway in tamoxifen resistant breast cancer cells. J. Cell Biochem. 107, 364–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wallez Y., Mace P. D., Pasquale E. B., Riedl S. J. (2012) NSP-CAS Protein Complexes. Emerging Signaling Modules in Cancer. Genes Cancer 3, 382–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tikhmyanova N., Little J. L., Golemis E. A. (2010) CAS proteins in normal and pathological cell growth control. Cell Mol. Life Sci. 67, 1025–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tornillo G., Elia A. R., Castellano I., Spadaro M., Bernabei P., Bisaro B., Camacho-Leal Mdel P., Pincini A., Provero P., Sapino A., Turco E., Defilippi P., Cabodi S. (2013) p130Cas alters the differentiation potential of mammary luminal progenitors by deregulating c-Kit activity. Stem Cells 31, 1422–1433 [DOI] [PubMed] [Google Scholar]
- 14. Hochgräfe F., Zhang L., O'Toole S. A., Browne B. C., Pinese M., Porta Cubas A., Lehrbach G. M., Croucher D. R., Rickwood D., Boulghourjian A., Shearer R., Nair R., Swarbrick A., Faratian D., Mullen P., Harrison D. J., Biankin A. V., Sutherland R. L., Raftery M. J., Daly R. J. (2010) Tyrosine phosphorylation profiling reveals the signaling network characteristics of basal breast cancer cells. Cancer Res. 70, 9391–9401 [DOI] [PubMed] [Google Scholar]
- 15. Cabodi S., Tinnirello A., Di Stefano P., Bisarò B., Ambrosino E., Castellano I., Sapino A., Arisio R., Cavallo F., Forni G., Glukhova M., Silengo L., Altruda F., Turco E., Tarone G., Defilippi P. (2006) p130Cas as a new regulator of mammary epithelial cell proliferation, survival, and HER2-neu oncogene-dependent breast tumorigenesis. Cancer Res. 66, 4672–4680 [DOI] [PubMed] [Google Scholar]
- 16. Cabodi S., Tinnirello A., Bisaro B., Tornillo G., del Pilar Camacho-Leal M., Forni G., Cojoca R., Iezzi M., Amici A., Montani M., Eva A., Di Stefano P., Muthuswamy S. K., Tarone G., Turco E., Defilippi P. (2010) p130Cas is an essential transducer element in ErbB2 transformation. FASEB J. 24, 3796–3808 [DOI] [PubMed] [Google Scholar]
- 17. Pylayeva Y., Gillen K. M., Gerald W., Beggs H. E., Reichardt L. F., Giancotti F. G. (2009) Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Invest. 119, 252–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Flier S., van der Kwast T. H., Claassen C. J., Timmermans M., Brinkman A., Henzen-Logmans S. C., Foekens J. A., Dorssers L. C. (2001) Immunohistochemical study of the BCAR1/p130Cas protein in non-malignant and malignant human breast tissue. Int. J. Biol. Markers 16, 172–178 [PubMed] [Google Scholar]
- 19. Dorssers L. C., Grebenchtchikov N., Brinkman A., Look M. P., van Broekhoven S. P., de Jong D., Peters H. A., Portengen H., Meijer-van Gelder M. E., Klijn J. G., van Tienoven D. T., Geurts-Moespot A., Span P. N., Foekens J. A., Sweep F. C. (2004) The prognostic value of BCAR1 in patients with primary breast cancer. Clin. Cancer Res. 10, 6194–6202 [DOI] [PubMed] [Google Scholar]
- 20. Defilippi P., Di Stefano P., Cabodi S. (2006) p130Cas. A versatile scaffold in signaling networks. Trends Cell Biol. 16, 257–263 [DOI] [PubMed] [Google Scholar]
- 21. Feller S. M. (1998) Physiological signals and oncogenesis mediated through Crk family adapter proteins. J. Cell. Physiol. 177, 535–552 [DOI] [PubMed] [Google Scholar]
- 22. Klemke R. L., Leng J., Molander R., Brooks P. C., Vuori K., Cheresh D. A. (1998) CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol. 140, 961–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Near R. I., Zhang Y., Makkinje A., Vanden Borre P., Lerner A. (2007) AND-34/BCAR3 differs from other NSP homologs in induction of anti-estrogen resistance, cyclin D1 promoter activation, and altered breast cancer cell morphology. J. Cell Physiol. 212, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Agthoven T., van Agthoven T. L., Dekker A., van der Spek P. J., Vreede L., Dorssers L. C. (1998) Identification of BCAR3 by a random search for genes involved in antiestrogen resistance of human breast cancer cells. EMBO J. 17, 2799–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu Y., Brush J., Stewart T. A. (1999) NSP1 defines a novel family of adaptor proteins linking integrin and tyrosine kinase receptors to the c-Jun N-terminal kinase/stress-activated protein kinase signaling pathway. J. Biol. Chem. 274, 10047–10052 [DOI] [PubMed] [Google Scholar]
- 26. Cai D., Clayton L. K., Smolyar A., Lerner A. (1999) AND-34, a novel p130Cas-binding thymic stromal cell protein regulated by adhesion and inflammatory cytokines. J. Immunol. 163, 2104–2112 [PubMed] [Google Scholar]
- 27. Felekkis K. N., Narsimhan R. P., Near R., Castro A. F., Zheng Y., Quilliam L. A., Lerner A. (2005) AND-34 activates phosphatidylinositol 3-kinase and induces anti-estrogen resistance in a SH2 and GDP exchange factor-like domain-dependent manner. Mol. Cancer Res. 3, 32–41 [PubMed] [Google Scholar]
- 28. Cai D., Iyer A., Felekkis K. N., Near R. I., Luo Z., Chernoff J., Albanese C., Pestell R. G., Lerner A. (2003) AND-34/BCAR3, a GDP exchange factor whose overexpression confers antiestrogen resistance, activates Rac, PAK1, and the cyclin D1 promoter. Cancer Res. 63, 6802–6808 [PubMed] [Google Scholar]
- 29. Makkinje A., Near R. I., Infusini G., Vanden Borre P., Bloom A., Cai D., Costello C. E., Lerner A. (2009) AND-34/BCAR3 regulates adhesion-dependent p130Cas serine phosphorylation and breast cancer cell growth pattern. Cell. Signal. 21, 1423–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riggins R. B., Quilliam L. A., Bouton A. H. (2003) Synergistic promotion of c-Src activation and cell migration by Cas and AND-34/BCAR3. J. Biol. Chem. 278, 28264–28273 [DOI] [PubMed] [Google Scholar]
- 31. Schrecengost R. S., Riggins R. B., Thomas K. S., Guerrero M. S., Bouton A. H. (2007) Breast cancer antiestrogen resistance-3 expression regulates breast cancer cell migration through promotion of p130Cas membrane localization and membrane ruffling. Cancer Res. 67, 6174–6182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schuh N. R., Guerrero M. S., Schrecengost R. S., Bouton A. H. (2010) BCAR3 regulates Src/p130 Cas association, Src kinase activity, and breast cancer adhesion signaling. J. Biol. Chem. 285, 2309–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilson A. L., Schrecengost R. S., Guerrero M. S., Thomas K. S., Bouton A. H. (2013) Breast cancer antiestrogen resistance 3 (BCAR3) promotes cell motility by regulating actin cytoskeletal and adhesion remodeling in invasive breast cancer cells. PLoS ONE 8, e65678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dodelet V. C., Pazzagli C., Zisch A. H., Hauser C. A., Pasquale E. B. (1999) A novel signaling intermediate, SHEP1, directly couples Eph receptors to R-Ras and Rap1A. J. Biol. Chem. 274, 31941–31946 [DOI] [PubMed] [Google Scholar]
- 35. Sakakibara A., Hattori S. (2000) Chat, a Cas/HEF1-associated adaptor protein that integrates multiple signaling pathways. J. Biol. Chem. 275, 6404–6410 [DOI] [PubMed] [Google Scholar]
- 36. Mace P. D., Wallez Y., Dobaczewska M. K., Lee J. J., Robinson H., Pasquale E. B., Riedl S. J. (2011) NSP-Cas protein structures reveal a promiscuous interaction module in cell signaling. Nat. Struct. Mol. Biol. 18, 1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun G., Cheng S. Y., Chen M., Lim C. J., Pallen C. J. (2012) Protein tyrosine phosphatase α phosphotyrosyl 789 binds BCAR3 to position Cas for activation at integrin-mediated focal adhesions. Mol. Cell. Biol. 32, 3776–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garron M. L., Arsenieva D., Zhong J., Bloom A. B., Lerner A., O'Neill G. M., Arold S. T. (2009) Structural insights into the association between BCAR3 and Cas family members, an atypical complex implicated in anti-oestrogen resistance. J. Mol. Biol. 386, 190–203 [DOI] [PubMed] [Google Scholar]
- 39. Vanden Borre P., Near R. I., Makkinje A., Mostoslavsky G., Lerner A. (2011) BCAR3/AND-34 can signal independent of complex formation with CAS family members or the presence of p130Cas. Cell. Signal. 23, 1030–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roselli S., Wallez Y., Wang L., Vervoort V., Pasquale E. B. (2010) The SH2 domain protein Shep1 regulates the in vivo signaling function of the scaffolding protein Cas. Cell. Signal. 22, 1745–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patwardhan P., Shen Y., Goldberg G. S., Miller W. T. (2006) Individual Cas phosphorylation sites are dispensable for processive phosphorylation by Src and anchorage-independent cell growth. J. Biol. Chem. 281, 20689–20697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sakai R., Iwamatsu A., Hirano N., Ogawa S., Tanaka T., Mano H., Yazaki Y., Hirai H. (1994) A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 13, 3748–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fonseca P. M., Shin N. Y., Brábek J., Ryzhova L., Wu J., Hanks S. K. (2004) Regulation and localization of CAS substrate domain tyrosine phosphorylation. Cell. Signal. 16, 621–629 [DOI] [PubMed] [Google Scholar]
- 44. Yu M., Bardia A., Wittner B. S., Stott S. L., Smas M. E., Ting D. T., Isakoff S. J., Ciciliano J. C., Wells M. N., Shah A. M., Concannon K. F., Donaldson M. C., Sequist L. V., Brachtel E., Sgroi D., Baselga J., Ramaswamy S., Toner M., Haber D. A., Maheswaran S. (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lacroix M., Leclercq G. (2004) Relevance of breast cancer cell lines as models for breast tumours. An update. Breast Cancer Res. Treat. 83, 249–289 [DOI] [PubMed] [Google Scholar]
- 46. Neve R. M., Chin K., Fridlyand J., Yeh J., Baehner F. L., Fevr T., Clark L., Bayani N., Coppe J. P., Tong F., Speed T., Spellman P. T., DeVries S., Lapuk A., Wang N. J., Kuo W. L., Stilwell J. L., Pinkel D., Albertson D. G., Waldman F. M., McCormick F., Dickson R. B., Johnson M. D., Lippman M., Ethier S., Gazdar A., Gray J. W. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hiscox S., Morgan L., Barrow D., Dutkowskil C., Wakeling A., Nicholson R. I. (2004) Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype. Inhibition by gefitinib (“Iressa,” ZD1839). Clin. Exp. Metastasis 21, 201–212 [DOI] [PubMed] [Google Scholar]
- 48. Vervoort V. S., Roselli S., Oshima R. G., Pasquale E. B. (2007) Splice variants and expression patterns of SHEP1, BCAR3 and NSP1, a gene family involved in integrin and receptor tyrosine kinase signaling. Gene 391, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Regelmann A. G., Danzl N. M., Wanjalla C., Alexandropoulos K. (2006) The hematopoietic isoform of Cas-Hef1-associated signal transducer regulates chemokine-induced inside-out signaling and T cell trafficking. Immunity 25, 907–918 [DOI] [PubMed] [Google Scholar]
- 50. Dail M., Kalo M. S., Seddon J. A., Côté J. F., Vuori K., Pasquale E. B. (2004) SHEP1 function in cell migration is impaired by a single amino acid mutation that disrupts association with the scaffolding protein cas but not with Ras GTPases. J. Biol. Chem. 279, 41892–41902 [DOI] [PubMed] [Google Scholar]
- 51. Makkinje A., Vanden Borre P., Near R. I., Patel P. S., Lerner A. (2012) Breast cancer anti-estrogen resistance 3 (BCAR3) protein augments binding of the c-Src SH3 domain to Crk-associated substrate (p130cas). J. Biol. Chem. 287, 27703–27714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cowell L. N., Graham J. D., Bouton A. H., Clarke C. L., O'Neill G. M. (2006) Tamoxifen treatment promotes phosphorylation of the adhesion molecules, p130Cas/BCAR1, FAK and Src, via an adhesion-dependent pathway. Oncogene 25, 7597–7607 [DOI] [PubMed] [Google Scholar]
- 53. Formstecher E., Ramos J. W., Fauquet M., Calderwood D. A., Hsieh J. C., Canton B., Nguyen X. T., Barnier J. V., Camonis J., Ginsberg M. H., Chneiweiss H. (2001) PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 1, 239–250 [DOI] [PubMed] [Google Scholar]
- 54. Bartholomeusz C., Gonzalez-Angulo A. M., Kazansky A., Krishnamurthy S., Liu P., Yuan L. X., Yamasaki F., Liu S., Hayashi N., Zhang D., Esteva F. J., Hortobagyi G. N., Ueno N. T. (2010) PEA-15 inhibits tumorigenesis in an MDA-MB-468 triple-negative breast cancer xenograft model through increased cytoplasmic localization of activated extracellular signal-regulated kinase. Clin. Cancer Res. 16, 1802–1811 [DOI] [PubMed] [Google Scholar]
- 55. Mace P. D., Wallez Y., Egger M. F., Dobaczewska M. K., Robinson H., Pasquale E. B., Riedl S. J. (2013) Structure of ERK2 bound to PEA-15 reveals a mechanism for rapid release of activated MAPK. Nat. Commun. 4, 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Glading A., Koziol J. A., Krueger J., Ginsberg M. H. (2007) PEA-15 inhibits tumor cell invasion by binding to extracellular signal-regulated kinase 1/2. Cancer Res. 67, 1536–1544 [DOI] [PubMed] [Google Scholar]
- 57. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N. (2012) The cBio cancer genomics portal. An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Riggins R. B., Thomas K. S., Ta H. Q., Wen J., Davis R. J., Schuh N. R., Donelan S. S., Owen K. A., Gibson M. A., Shupnik M. A., Silva C. M., Parsons S. J., Clarke R., Bouton A. H. (2006) Physical and functional interactions between Cas and c-Src induce tamoxifen resistance of breast cancer cells through pathways involving epidermal growth factor receptor and signal transducer and activator of transcription 5b. Cancer Res. 66, 7007–7015 [DOI] [PubMed] [Google Scholar]
- 60. Felekkis K., Quilliam L. A., Lerner A. (2006) Characterization of AND-34 function and signaling. Methods Enzymol. 407, 55–63 [DOI] [PubMed] [Google Scholar]
- 61. Brinkman A., de Jong D., Tuinman S., Azaouagh N., van Agthoven T., Dorssers L. C. (2010) The substrate domain of BCAR1 is essential for anti-estrogen-resistant proliferation of human breast cancer cells. Breast Cancer Res. Treat. 120, 401–408 [DOI] [PubMed] [Google Scholar]
- 62. van Agthoven T., Godinho M. F., Wulfkuhle J. D., Petricoin E. F., 3rd, Dorssers L. C. (2012) Protein pathway activation mapping reveals molecular networks associated with antiestrogen resistance in breast cancer cell lines. Int. J. Cancer 131, 1998–2007 [DOI] [PubMed] [Google Scholar]
- 63. Gee J. M., Robertson J. F., Ellis I. O., Nicholson R. I. (2001) Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int. J. Cancer 95, 247–254 [DOI] [PubMed] [Google Scholar]
- 64. Bartholomeusz C., Gonzalez-Angulo A. M., Liu P., Hayashi N., Lluch A., Ferrer-Lozano J., Hortobágyi G. N. (2012) High ERK protein expression levels correlate with shorter survival in triple-negative breast cancer patients. Oncologist 17, 766–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cabodi S., Moro L., Baj G., Smeriglio M., Di Stefano P., Gippone S., Surico N., Silengo L., Turco E., Tarone G., Defilippi P. (2004) p130Cas interacts with estrogen receptor α and modulates non-genomic estrogen signaling in breast cancer cells. J. Cell Sci. 117, 1603–1611 [DOI] [PubMed] [Google Scholar]
- 66. Kumbrink J., Kirsch K. H. (2012) Regulation of p130(Cas)/BCAR1 expression in tamoxifen-sensitive and tamoxifen-resistant breast cancer cells by EGR1 and NAB2. Neoplasia 14, 108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fiory F., Formisano P., Perruolo G., Beguinot F. (2009) Frontiers. PED/PEA-15, a multifunctional protein controlling cell survival and glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 297, E592–E601 [DOI] [PubMed] [Google Scholar]
- 68. Stassi G., Garofalo M., Zerilli M., Ricci-Vitiani L., Zanca C., Todaro M., Aragona F., Limite G., Petrella G., Condorelli G. (2005) PED mediates AKT-dependent chemoresistance in human breast cancer cells. Cancer Res. 65, 6668–6675 [DOI] [PubMed] [Google Scholar]
- 69. Condorelli G., Vigliotta G., Cafieri A., Trencia A., Andalò P., Oriente F., Miele C., Caruso M., Formisano P., Beguinot F. (1999) PED/PEA-15. An anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene 18, 4409–4415 [DOI] [PubMed] [Google Scholar]
- 70. Shah A. N., Gallick G. E. (2007) Src, chemoresistance and epithelial to mesenchymal transition. Are they related? Anticancer Drugs 18, 371–375 [DOI] [PubMed] [Google Scholar]
- 71. Brábek J., Constancio S. S., Siesser P. F., Shin N. Y., Pozzi A., Hanks S. K. (2005) Crk-associated substrate tyrosine phosphorylation sites are critical for invasion and metastasis of SRC-transformed cells. Mol. Cancer Res. 3, 307–315 [DOI] [PubMed] [Google Scholar]
- 72. Browne C. D., Hoefer M. M., Chintalapati S. K., Cato M. H., Wallez Y., Ostertag D. V., Pasquale E. B., Rickert R. C. (2010) SHEP1 partners with CasL to promote marginal zone B-cell maturation. Proc. Natl. Acad. Sci. U.S.A. 107, 18944–18949 [DOI] [PMC free article] [PubMed] [Google Scholar]