

Background: The structural changes in short chain dehydrogenase-like l-threonine dehydrogenase (SDR-like l-ThrDH) that occur during catalysis remain unknown.
Results: Several analyses revealed dynamical changes in SDR-like l-ThrDH.
Conclusion: NAD+ and l-threonine binding induced changes in flexibility and structure.
Significance: This report demonstrates that the dynamical structural changes of SDR-like l-ThrDH are important for the reactivity and specificity of the enzyme.
Keywords: Crystal Structure, Dehydrogenase, Enzyme Kinetics, Molecular Dynamics, NAD
Abstract
Crystal structures of short chain dehydrogenase-like l-threonine dehydrogenase from Cupriavidus necator (CnThrDH) in the apo and holo forms were determined at 2.25 and 2.5 Å, respectively. Structural comparison between the apo and holo forms revealed that four regions of CnThrDH adopted flexible conformations when neither NAD+ nor l-Thr were bound: residues 38–59, residues 77–87, residues 180–186, and the catalytic domain. Molecular dynamics simulations performed at the 50-ns time scale revealed that three of these regions remained flexible when NAD+ was bound to CnThrDH: residues 80–87, residues 180–186, and the catalytic domain. Molecular dynamics simulations also indicated that the structure of CnThrDH changed from a closed form to an open form upon NAD+ binding. The newly formed cleft in the open form may function as a conduit for substrate entry and product exit. These computational results led us to hypothesize that the CnThrDH reaction progresses by switching between the closed and open forms. Enzyme kinetics parameters of the L80G, G184A, and T186N variants also supported this prediction: the kcat/Km, l-Thr value of the variants was >330-fold lower than that of the wild type; this decrease suggested that the variants mostly adopt the open form when l-Thr is bound to the active site. These results are summarized in a schematic model of the stepwise changes in flexibility and structure that occur in CnThrDH upon binding of NAD+ and l-Thr. This demonstrates that the dynamical structural changes of short chain dehydrogenase-like l-threonine dehydrogenase are important for the reactivity and specificity of the enzyme.
Introduction
l-Threonine 3-dehydrogenase (EC 1.1.1.103, l-ThrDH)2 is expressed in a broad range of species from bacteria to mammals (1). l-ThrDH utilizes oxidized NAD+ as a coenzyme to catalyze dehydrogenation of the 3′ carbon atom of l-Thr. The product 2-amino 3-keto butyrate is decomposed rapidly to aminoacetone and CO2 (2) or converted to glycine by 2-amino 3-keto butyrate CoA ligase (3). The structures of two types of l-ThrDH enzymes have been reported. One type is classified within the medium chain dehydrogenase/reductase (MDR) superfamily, and is similar to alcohol dehydrogenase (ADH) (3, 4). MDR has ∼350 amino acid residues and contains a Rossmann fold domain in its C-terminal region (5). The functions of MDR family members are divergent (6), and this type of l-ThrDH is classified as a zinc-dependent MDR as well as an ADH. Several biochemical and structural studies have been performed on this type of l-ThrDH (3, 7–10). The other type is classified within the extended short chain dehydrogenase/reductase (SDR) (4) and is similar to UDP-galactose 4-epimerase (GalE). SDR family members have Rossmann fold domains in their N-terminal regions. Early studies revealed that SDRs can be classified further into “classical” and “extended” subtypes: the former type has ∼250 amino acid residues, and the latter type has ∼100 additional residues in the C-terminal region (4). Many SDRs have a Tyr-based catalytic center (4). In this report, we refer to the former and latter l-ThrDHs as “ADH-like l-ThrDH” and “SDR-like l-ThrDH,” respectively. Here, we describe our analysis of the structural and functional properties of an SDR-like l-ThrDH from Cupriavidus necator (CnThrDH).
SDR-like l-ThrDH was originally identified in Cytophaga sp. strain KUC1 (CyThrDH) by Kazuoka et al. (11), who reported that this enzyme had no sequence similarity with previously described ADH-like l-ThrDHs. More recently, Millerioux et al. (12) demonstrated that SDR-like l-ThrDH is essential for lipid biosynthesis in Trypanosoma brucei, the causative pathogen of human African trypanosomiasis. The reaction catalyzed by SDR-like l-ThrDH proceeds via an ordered Bi-Bi mechanism; NAD+ binds to SDR-like l-ThrDH before binding of l-Thr, and the product 2-amino-3-keto butyrate is released before NADH (11). SDR-like l-ThrDH has higher substrate specificity for l-Thr than ADH-like l-ThrDH. In fact, Ueatrongchit and Asano (13) showed that CnThrDH reacts with only two compounds, l-Thr and dl-α-amino β-hydroxyvalerate, and has no activity toward 36 other compounds, including other amino acids and their derivatives. Because of its high specificity for l-Thr, CnThrDH can be used to quantitate the l-Thr concentration in blood plasma; the plasma level of l-Thr reflects the symptom of certain diseases and can therefore be a criterion for diagnosis of these conditions (13–15). Yoneda et al. (16) were the first to determine the crystal structure of the SDR-like l-ThrDH from Flavobacterium frigidimaris KUC-1 (FfThrDH); they showed that the overall structure of FfThrDH is similar to other GalE structures. In a subsequent study, they also determined structures of l-Thr-bound SDR-like l-ThrDH from Thermoplasma volcanium (TvThrDH (Y137F)) (17) and proposed a catalytic mechanism for this enzyme.
To date, no studies have described the dynamic structural changes that these enzymes undergo during their reactions. In many other NAD+-dependent enzymes that contain Rossmann fold motifs, as SDR-like l-ThrDH does, structural changes occur upon binding of NAD+ and substrates to the enzyme. These changes play significant roles in determining the high substrate specificity of the enzymes. One such enzyme is ADH, in which a structural change is induced by binding of NAD+ and substrate (18). ADH may adopt both open and closed forms; a flexible loop containing residues 293–298 is involved in switching between the two forms (18, 19). This switching has also been confirmed by molecular dynamics (MD) simulation (20). In ADH, the switching is essential for the high substrate specificity of the enzyme (19). Based on these findings, it seems likely that binding of NAD+ and l-Thr should induce structural changes in SDR-like l-ThrDH.
To investigate the structural changes that occur in SDR-like l-ThrDH, we determined two crystal structures: CnThrDH (apo) and CnThrDH (holo). In the apo form, NAD+ and l-Thr did not bind to the active site, whereas in the holo form, both compounds bound to the active site. In the apo form, four regions of the protein adopted a flexible conformation. Furthermore, using the structure of NAD+-bound CnThrDH (CnThrDH (holo, with NAD+)) as a starting model, we performed MD simulations at the 50-ns time scale to predict the structural changes that occur upon NAD+ binding. In the final state of the simulations, CnThrDH adopted an open form. To probe the mechanism underlying this conformational change, we mutated residues predicted to be involved in switching between the open and closed forms and determined the kinetic parameters of the mutant enzymes. The results of the enzyme kinetics analyses revealed that CnThrDH performs its reaction by switching between the open and closed forms. Based on our findings, we propose a model for the stepwise dynamic structural changes induced in SDR-like l-ThrDH upon binding of NAD+ and l-Thr.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of CnThrDH Variants
To generate CnThrDH variants, the CnThrDH-pET-15b plasmid, created in a previous study (13), was used as a template. Site-directed mutagenesis was performed using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). CnThrDH variants were confirmed by sequencing.
Overexpression and Purification of CnThrDH
CnThrDH WT and variant plasmids were transformed into Escherichia coli strain BL21(DE3). Overexpression of CnThrDH WT was performed as previously described (13). Overexpressing cells were collected, suspended in buffer A (20 mm potassium phosphate, pH 7.0, 100 mm NaCl, and 10 mm imidazole), and sonicated. The insoluble fraction was removed by centrifugation (28,000 × g, 30 min), and the supernatant was collected and loaded onto a Ni2+-Sepharose column. The column was washed with 50 ml of buffer A, and then with 50 ml of buffer A containing 70 mm imidazole, after which CnThrDH (WT) was eluted and collected buffer A containing 300 mm imidazole. The eluted CnThrDH was concentrated and applied to a gel filtration column (Superdex 75 pg) that had been equilibrated with buffer B (10 mm potassium phosphate, pH 7.0, 50 mm NaCl). The presence of CnThrDH in eluted fractions was confirmed by SDS-PAGE, and fractions containing CnThrDH were collected and concentrated for crystallization. The same procedure was applied to purify other CnThrDH variants.
Crystallization of apo and holo Forms of CnThrDH
The CnThrDH sample was concentrated to more than 20 mg/ml by Amicon Ultra-15 Centrifugal filter devices (Millipore, Billerica, MA). Protein concentration was calculated by measuring UV-visible absorbance at 280 nm on a Nanodrop UV-visible spectrometer (Thermo Fisher Scientific, Waltham, MA). The concentrated sample was divided between two conditions: part of the sample was used directly for screening of crystallization conditions for CnThrDH (apo), and the remainder of the sample was mixed with buffer C (10 mm potassium phosphate, pH 7.0, 50 mm NaCl, 25 mm NAD+) at a sample:buffer ratio of 9:1 for screening of crystallization conditions for CnThrDH (holo).
Screening of crystallization conditions for CnThrDH (apo) was performed using index screening and PEG/ION screening kits (Hampton Research, Aliso Viejo, CA). A 2-μl sample was mixed with 1.0 μl of each reservoir solution from the screening kit. After a 1-month incubation at 20 °C, block-shaped CnThrDH (apo) crystals appeared in PEG/ION screen conditions 1–8 (20% (w/v) PEG3350 and 0.2 m CaCl2). For CnThrDH (holo), a 2-μl sample was mixed with 0.4 μl of 1.0 m magnesium chloride solution and 1.0 μl or each reservoir solution from the PEG/ION screening kit. After 2 weeks, plate-like crystals appeared in PEG/ION screen conditions 1–29 (20% (w/v) PEG3350 and 0.2 m potassium acetate).
X-ray Data Collection
The CnThrDH (apo) crystal used for diffraction data collection was quickly soaked in reservoir solution containing 10% (w/v) PEG400 before the crystal was mounted. The CnThrDH (holo) crystal was soaked for a few minutes in reservoir containing 10 mm l-Thr and 10% (w/v) PEG400. Because the crystals suffered serious damage when NAD+ was present, NAD+ was omitted from the soaking reservoir. The soaked crystals were mounted and flash-cooled under a nitrogen stream (−173 °C). Diffraction data were collected using a Quantum 315 CCD detector at BL-17A of the Photon Factory (Tsukuba, Japan). These data were integrated and scaled using HKL2000 and SCALEPACK (21). Initial phases were determined by molecular replacement using MOLREP (22), with the crystal structure of TvThrDH (Protein Data Bank code 3A4V) as a template. Model building and structure refinement with local NCS restraints were performed using COOT (23) and REFMAC (24), respectively. Water molecules were automatically placed by ARP/WARP (25). All figures were prepared using PyMOL (26). Crystallographic and refinement parameters are shown in Table 1.
TABLE 1.
Statistics of x-ray diffraction data collection for CnThrDH (apo) and CnThrDH (holo)
|
CnThrDH |
||
|---|---|---|
| apo form | holo form | |
| Space group | P212121 | P21 |
| Unit cell parameters | ||
| a (Å) | 73.01 | 91.60 |
| b (Å) | 83.49 | 88.11 |
| c (Å) | 106.2 | 113.60 |
| α (degree) | 90.00 | 90.00 |
| β (degree) | 90.00 | 105.47 |
| γ (degree) | 90.00 | 90.00 |
| X-ray source | PF, BL-17A | PF, BL-17A |
| Wavelength (Å) | 0.98 | 0.98 |
| Resolution (Å) | 48.8–2.25 (2.31–2.25) | 40.9–2.50 (2.57–2.50) |
| No. of reflectionsa | 439,253 | 446,486 |
| No. of unique reflections | 31,326 | 60,238 |
| Completeness (%) | 99.9 (100) | 100 (100) |
| I/σ(I) | 52.2 (10.7) | 36.8 (8.0) |
| Rmergeb | 0.090 (0.354) | 0.099 (0.398) |
| B of Wilson plot (Å)2 | 29.9 | 37.1 |
| Rc | 0.229 | 0.205 |
| Rfreed | 0.264 | 0.240 |
| RMSD of geometry | ||
| Bond length (Å) | 0.008 | 0.008 |
| Bond angle (degree) | 1.143 | 1.255 |
| Geometry | ||
| Ramachandran outlier (%) | 0.2 | 0.1 |
| Ramachandran favored (%) | 99.8 | 99.9 |
| Average B factor (Å)2 | ||
| Protein atoms | 36.4 | 44.2 |
| Ligand atoms | NDe | 38.1 |
| Solvent atoms | 26.7 | 26.7 |
| Protein Data Bank code | 3WMW | 3WMX |
a Sigma cutoff was set to none (F > 0σF).
b Rmerge = ΣhΣi|Ii(h)−<I(h)>|/ΣhI(h), where Ii(h) is the ith measurement of reflection h, and <I(h)> is the mean value of the symmetry-related reflection intensities. The values in brackets are for the shell of the highest resolution.
c r = Σ||Fo| − |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors used in the refinement, respectively.
d Rfree is the R factor calculated using 5% of the reflections chosen at random and omitted from the refinement.
e ND, not determined.
Molecular Dynamics Simulation
Chains A and C and the NAD+ molecule of the CnThrDH (holo) structure (CnThrDH (holo, with NAD+)) were used as initial structures for the MD simulations. CnThrDH (holo, with NAD+) was loaded into the Molecular Operating Environment 2011 software (27), and protonation of each amino acid residue and NAD+ was performed using the Protonate3D procedure within Molecular Operating Environment (27). An 11 × 10 × 11-nm3 cuboid box was prepared after completion of protonation. Protonated CnThrDH (holo, with NAD+) was moved to the center of the box, and TIP3P water molecules were solvated into the box at a solvent density of 1.0 g/cm3. Energy minimization of the system was performed using the Charmm27 force field as follows: first, energy minimization was performed by constraining the backbone atoms of CnThrDH, and then the constraint was turned off. Before starting the MD simulation, the protonation states of the histidine residues of CnThrDH (holo, with NAD+) at pH 7.0 were re-estimated using the PROPKA web server (28). After the total charge of the system was calculated using a custom-made Python script, sodium (Na+) or chloride (Cl−) ions were added to the system to neutralize the total charge to zero.
All MD simulations were performed using the NAMD 2.9 software (29) with periodic boundary conditions. Electrostatic interactions over long distances were evaluated by the standard particle mesh Ewald method (30). All hydrogens were constrained, and the RATTLE algorithm (31) was employed. The simulation time step was 2 fs, and the isothermal-isobaric (NPT) ensemble was applied to the system. The temperature was set at 300 K, and a Langevin thermostat was applied to the system. Pressure was set at 1.0 bar.
Before commencing collection of trajectories from the MD simulation, we performed energy minimization for 20 ps and confirmed that total energy of the system would be constant. After completion of the minimization, 150 ps of preliminary simulation was performed while gradually raising the temperature of the system from 0 to 300 K in steps of 100 K. This simulated annealing procedure was repeated at least three times, and then productive MD simulation was performed for 50 ns. The structures were saved as trajectories every 1 ps. The trajectory analysis tool Wordom (32) was used to analyze the trajectory data.
Assay of Enzyme Activity toward l-Thr and NAD+
The enzyme activities of WT and variant CnThrDH were measured by quantitating the amount of NADH produced as a product of the l-ThrDH reaction at various concentrations of l-Thr and NAD+. Enzyme activity toward l-Thr and NAD+ was measured using assay buffer 1 (0.1 m Gly-KOH-KCl, pH 10.0, 2.5 mm NAD+, and 5–500 mm l-Thr) and assay buffer 2 (0.1 m Gly-KOH-KCl, pH 10.0, 0.050–0.50 mm NAD+, and 100 mm l-Thr). The assay buffer and enzyme solution diluted with buffer B were prepared separately and incubated for 1 h at 30 °C. To start the reaction, 980 μl of the assay buffer was transferred to a cuvette, and then 20 μl of the enzyme solution was added. The cuvette was installed immediately into a UV-visible spectrometer (UV-1700 PharmaSpec; Shimadzu), and the time-dependent spectrum change of NADH at 340 nm was measured over 2 min. NADH concentration was calculated using the molar extinction coefficient (6300 m−1 cm−1) of the NADH at 340 nm. Initial velocities at different concentrations of l-Thr and NAD+ were calculated using an original program written in Python. The initial velocity was plotted using the ORIGIN program, and the enzyme kinetics parameters kcat, Km, l-Thr, and Km, NAD+ were calculated using the Michaelis-Menten equation and applying the nonlinear least squares method.
RESULTS AND DISCUSSION
Crystal Structures of CnThrDH (apo) and CnThrDH (holo)
The crystal structure of the CnThrDH (apo) form was determined at 2.25 Å resolution (Fig. 1A). The space group is P212121, and there are two molecules in an asymmetric unit. There is no electron density representing NAD+ or l-Thr at the active site.
FIGURE 1.
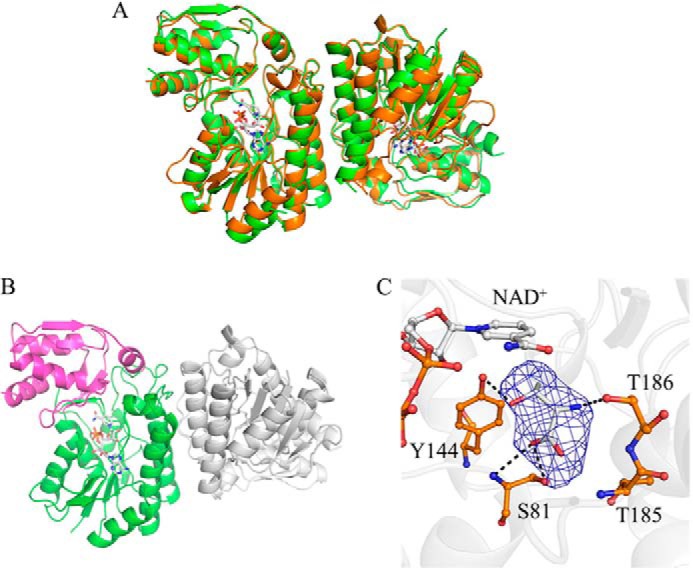
Crystal structures of CnThrDH (apo) and CnThrDH (holo) forms. A, superposed structures of the apo (green) and the holo (orange) forms. NAD+ is shown as stick model (gray). These two structures are similar to each other; the RMSD between these forms was 0.44 Å2. B, structure of catalytic domain and NAD+ binding domain of the CnThrDH (holo). The catalytic domain (amino acids 176–214, 245–279, and 301–313) and the NAD+ binding domain (amino acids 1–175, 215–244, and 280–300) are colored magenta and green, respectively. C, l-Thr binding form of the CnThrDH (holo). Fo-Fc difference Fourier map (2.5 σ, in blue mesh) is superposed on the stick model of l-Thr. Potential hydrogen bonds (broken lines) with lengths less than 3.4 Å are shown.
The crystal structure of the CnThrDH (holo) form, which contains NAD+ and l-Thr bound at the active site, was determined at 2.5 Å resolution (Fig. 1A, orange). The space group is P21, and there are four molecules in the asymmetric unit. The contacts between chains A and C and chains B and D represent native dimer contacts in SDR-like l-ThrDH. These contacts are also present in the previously determined structures of two other SDR-like l-ThrDHs, FfThrDH, and TvThrDH (16, 17). Electron density was observed for amino acid residues 6–313 in chains A, B, and C, and all of these residues could be assigned to the electron density map (Fig. 1A, orange). For chain D, the electron density of the Gly182–Gly184 region was too low to assign these residues on the map.
SDR-like l-ThrDH can be divided into the NAD+-binding and catalytic domains (16, 17). In the CnThrDH structure, the NAD+-binding domain consists of residues 1–175, 215–244, and 280–300 (Fig. 1B, green), and the catalytic domain is formed by residues 176–214, 245–279, and 301–313 (Fig. 1B, magenta).
The overall structure of CnThrDH (holo) was almost the same as those of two other previously determined SDR-like l-ThrDHs, FfThrDH (Protein Data Bank code 2YY7), and TvThrDH (Protein Data Bank code 3A4V). The root mean square distance (RMSD) value for Cα atoms was 0.50 Å between CnThrDH (holo) and FfThrDH and 0.40 Å between CnThrDH (holo) and TvThrDH.
Active Site Structure of CnThrDH (holo)
In the CnThrDH (holo) structure, the electron density map revealed a molecule bound at the active site. This electron density can be observed more clearly for chains A and B than for chains C and D. We predicted that this electron density represents the substrate, l-Thr, because it corresponds to l-Thr in the TvThrDH (Y137F) variant, in which l-Thr forms hydrogen bonds with three residues: Ser81, Tyr144, and Thr186 (Fig. 1C) (17). In the data deposited in the Protein Data Bank, l-Thr is placed in the electron density map of chains A and B.
In the CnThrDH (holo) structure, all four chains have almost the same overall structure; the RMSD values for Cα atoms between each chain were less than 0.55 Å. Therefore, we assumed that all four chains have the same fold and subsequently used the chain A structure as a representative model of CnThrDH (holo).
Four Flexible Regions in CnThrDH (apo)
Comparison of the structures of the apo and the holo forms (Fig. 1) revealed that four regions were more flexible in CnThrDH (apo) than in CnThrDH (holo). Invisible and unclear electron density corresponding to residues 38–59 and 77–87 indicated that these regions were flexible in CnThrDH (apo) (Fig. 2, A and B). Residues 180–186 (Fig. 2A) and the catalytic domain (Fig. 1B, magenta) were also flexible in CnThrDH (apo) (Fig. 2), as judged from their high B-factor values. On the other hand, in the CnThrDH (holo) structure, these four regions were rigid: the electron density map for these four regions was clearly observed, and their B-factor values were low.
FIGURE 2.
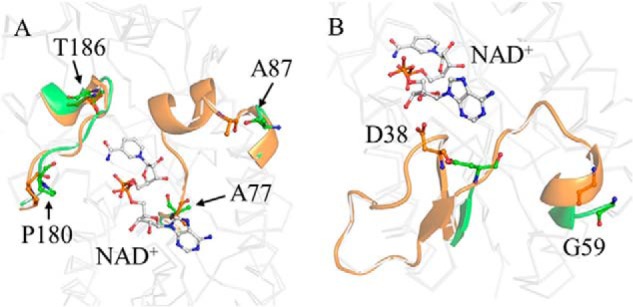
Flexible regions of CnThrDH. A, structures of the apo (green) and the holo (orange) forms. Residues 77–87 and 180–186 are shown in ribbon format. Residues 78–86 region were invisible, and Ala77 and Ala87 had higher B-factor values, in the apo form. Although residues 180–186 region were visible in the electron density map, the B-factor value of this region in the apo form (61 Å2) was ∼1.5 times higher than in the holo form (42 Å2) despite the high resolution of the structure. B, residues 38–59. Residues 39–58 were invisible, and Asp38 and Gly59 had higher B-factor values, in the apo form.
The Rfree value of the CnThrDH (apo) structure (0.264 in Table 1) was higher than that of the CnThrDH (holo) structure (0.240 in Table 1), in contrast to their relative resolutions. In the apo structure, there were many disordered residues that were not assigned into the electron density map, and this might have made the Rfree value of the apo structure higher that of the holo structure. In fact, in the asymmetric unit, the rates of disordered residues in the protein molecule were 17.9% (apo) and 9.0% (holo), respectively. Similar phenomena have been observed in other enzymes (33, 34). To summarize the results, the following four regions were flexible in the CnThrDH (apo) form: residues 38–59, residues 77–87, residues 180–186, and the catalytic domain.
Molecular Dynamics Simulation of CnThrDH (holo, with NAD+)
The comparison between the apo and holo forms suggested that changes in flexibility and structure occurred upon binding of NAD+ and l-Thr. Despite much effort, we could not obtain a crystal structure of CnThrDH with only NAD+ bound at the active site. Therefore, we performed MD simulations to analyze the flexibility of NAD+-bound CnThrDH.
First, we analyzed the time-dependent change of RMSD values for Cα atoms (Fig. 3A). RMSD values increased during the first 20 ns of the simulation to a maximum of 2.8 Å; subsequently, the values equilibrated ∼2.0 Å (Fig. 3A). Radius of gyration values for Cα atoms followed the same general pattern as the RMSD values (Fig. 3B), increasing to a maximum of 20.2 Å and then equilibrating ∼19.7 Å. These results indicate that the simulation reached equilibrium.
FIGURE 3.

A and B, plots of time-dependent change of RMSD values (A) and radius of gyration (RGYR) values (B) for Cα atoms of the CnThrDH (holo, with NAD+) structure. C, plots of RMSF values of the CnThrDH (holo, with NAD+) form. RMSF values were calculated using all trajectory data (total, 50,000 structures) outputted from the 50-ns MD simulation. The values corresponding to residues belonging to the NAD+-binding domain (amino acids 1–175, 215–244, and 280–300) and catalytic domain (amino acids 176–214, 245–279, and 303–313) are colored black and red, respectively.
To evaluate backbone flexibility during the simulations, we calculated the root mean square fluctuation (RMSF) values for all Cα atoms (Fig. 3C). High RMSF values indicate residues that fluctuate dynamically during the simulation. The average RMSF value for all residues was 1.66 Å. High RMSF values (more than 3.0 Å) were confirmed for residues 80–87, 180–186, and 267–273 (Fig. 3C), strongly indicating that these three regions adopted flexible forms in the simulation.
Residues 38–59 and 77–79 regions were flexible in the CnThrDH (apo) form (Fig. 2); however, their RMSF values were almost the same as the average RMSF value: 1.63 and 1.58 Å for residues 38–59 and 77–79, respectively. This observation suggested that these regions adopt rigid forms when NAD+ binds to CnThrDH. These details are discussed in the next section.
The catalytic domain adopted a more flexible form than the NAD+-binding domain. RMSF values for a part of the catalytic domain, residues 267–273, were high, as were those for residues 80–87 and 180–186. Furthermore, the average RMSF value for the catalytic domain (2.22 Å; Fig. 3C, red bars) was ∼1.5 times higher than that of the NAD+-binding domain (1.44 Å; Fig. 3C, black bars). Summarizing these results, the following regions were assigned as flexible regions in CnThrDH (holo, with NAD+): residues 80–87, residues 180–186, and the catalytic domain.
Structural Changes of CnThrDH (holo, with NAD+) Associated with an Increase in Simulation Time
We next analyzed time-dependent structural changes in the CnThrDH (holo, with NAD+) trajectories generated by the simulation. From comparisons between the simulation's initial (0 ns) and final (50 ns) structure, we confirmed that structural changes occurred in residues 80–87 and the catalytic domain, and especially in residues 180–186 and 267–273 (Fig. 4A). During the simulation, these regions moved in the direction indicated by an arrow in Fig. 4A. Surface representations of the initial and final structures are shown in Fig. 4 (B and C, respectively).
FIGURE 4.

Structural comparison of the CnThrDH (holo, with NAD+) between the initial (0 ns) and final (50 ns) states of the MD simulation. A, three flexible regions with high RMSF values. The initial and final structures are colored yellow and magenta, respectively. These regions moved in the direction indicated by the arrow. B and C, surface representations of the initial (B) and final (C) states. The three flexible regions are colored orange. In the initial state (B), there is no cleft for entry of l-Thr; therefore, this is termed the “closed” form). On the other hand, the final state (C) contains a newly formed cleft; this state is termed the “open” form.
In the initial structure, three regions, the regions containing residues 80–87, 180–186, and 267–273, formed stable hydrophobic interactions with each other (Fig. 4B, orange). As a result, there was no cleft through which l-Thr could enter the active site, and the initial structure had a “closed” form. By contrast, in the final structure, these hydrophobic interactions had disappeared (Fig. 4C, orange), resulting in formation of a cleft connected to the active site. Thus, the final state had an “open” form. The open form is the predominant structure in the simulation (supplemental Movie S1). Therefore, CnThrDH (holo, with NAD+) should mostly adopt the open form in solution. The cleft in the open form (Fiure. 4C) should serve as a conduit for entry of the substrate (e.g. l-Thr) and for exit of the product (e.g. 2-amino-3-keto butyrate). No previous report has described such a conduit within an SDR-like l-ThrDH in complex with NAD+; to date, all reported structures (16, 17) are of closed forms, in which no such conduit is present.
In residues 38–59, which was flexible in CnThrDH (apo), the simulation revealed structural change smaller than those of the three flexible regions described above. This observation suggested that residues 38–59 form a rigid conformation in CnThrDH (holo, with NAD+) via interactions between this region and NAD+: in particular, the carboxyl group of Asp38 and the carboxyamide group of Asn54 would form hydrogen bonds with NAD+.
To summarize the structural changes revealed in the simulation, CnThrDH is predicted to mainly adopt the open form when only NAD+ is bound at the active site. Once NAD+ is bound, binding of l-Thr or an inhibitor such as pyruvate to the active site would favor the closed form. The closed form could be initiated by binding these compounds and could therefore be regarded as a Michaelis complex structure within SDR-like l-ThrDH. This idea is consistent with the results of Yoneda et al. (17), who observed that NAD+- and l-Thr-bound TvThrDH (Y137F) also adopted a closed form and predicted that the reaction progresses via this form. A movie depicting the structural changes of CnThrDH (holo, with NAD+) from the closed to open form is provided as supplemental Movie S1.
Enzyme Kinetics Studies of CnThrDH Variants, L80G, G184A, and T186N
From the results of the crystal structures and the MD simulations, residues 80–87 and 180–186 are key positions for switching between the open and closed forms of CnThrDH. Therefore, we predicted that site-directed mutations in the regions should affect switching and thereby influence enzymatic activity toward the substrate, l-Thr. We could obtain three variants in soluble forms: L80G, G184A and T186N. The locations of the mutation sites are shown in Fig. 5A. In SDR-like l-ThrDH, the hydroxyl group of Thr186 forms a hydrogen bond with the amino group of l-Thr (Fig. 1C and Ref. 17) but is distant from the site of the reaction, i.e. the β-carbon and β-hydroxyl hydrogen atoms of l-Thr. Therefore, it would be difficult for the T186N variant to form a Michaelis complex (closed form) because of the steric repulsion resulting from the mutation, but it should retain activity because the T186N mutation does not affect the dehydrogenation reaction directly. Overall, this should lead to a decrease of kcat/Km, l-Thr value: the Km value toward l-Thr (Km, l-Thr) should be increased, but kcat should not be zero.
FIGURE 5.

Changes in enzymatic activity resulting from mutations in Leu80, Gly184, and Thr186 of CnThrDH. A, locations of the mutated sites (orange). Thr186 forms a hydrogen bond with l-Thr, the substrate, but there is no hydrogen bonding between l-Thr and Leu80 or Gly184. B and C, catalytic activity of wild type (B) and three variants (L80G, G184A, and T186N) (C) toward l-Thr. The data are shown as means ± S.D.
The enzyme kinetics of WT and variant enzymes toward l-Thr are plotted in Fig. 5 (B and C), and the kinetics parameters are shown in Table 2. The initial velocities of the variants (L80G, G184A, and T186N) did not reach maximum value (Fig. 5C) because of their significantly larger Km, l-Thr values and the solubility limit of the substrate, l-Thr. Consequently, the kcat and Km, l-Thr values for the variants could not be determined accurately. Therefore, these parameters are annotated as “not determined” (ND in Table 2), and the kcat/Km, l-Thr value is instead estimated (Table 2).
TABLE 2.
Enzymatic properties of CnThrDH WT and variants
| Variantsa | kcatb | Km, l-Thrc | Km, NAD+c | kcat/Km, l-Thr |
|---|---|---|---|---|
| s−1 | mm | mm | s−1 mm−1 | |
| WT | 50.0 ± 3.7 | 15.2 ± 2.6 | 0.120 ± 0.020 | 3.3 |
| L80G | NDd | ND | 0.115 ± 0.003 | 0.001 |
| G184A | ND | ND | 0.156 ± 0.012 | 0.01 |
| T186N | ND | ND | 0.097 ± 0.004 | 0.01 |
a Locations of mutated sites in the three-dimensional structure are shown in Fig. 5A. Recombinant enzymes were purified as described under “Experimental Procedures.”
b kcat values of the variants and WT were derived from the data shown in Fig. 5 (B and C).
c Km, l-Thr and Km, NAD+ values represent the Michaelis constant value toward l-Thr and NAD+, respectively.
d ND, not determined.
As predicted in the previous paragraph, the T186N variant had retained low activity: the kcat/Km, l-Thr value of the T186N variant was ∼330-fold lower than that of WT (Fig. 5C and Table 2). As for the G184A variant, the parameters were similar to that of T186N, and the kcat/Km, l-Thr values of both variants were nearly the same (Table 2). Judging from analyses of kinetic parameters of other NAD+-dependent enzymes, it seemed to be difficult for the G184A and T186N variants to adopt the closed forms. Consistent with this finding, ADH variants that have difficulty adopting the closed form exhibit significantly lower kcat/Km values toward the substrate than the wild-type ADH (18).
Like G184A and T186N, L80G also seemed to have difficulty in adopting the closed form: its kcat/Km, l-Thr value was 3300-fold lower than that of WT (Fig. 5C and Table 2). In addition, the kcat/Km, l-Thr value of L80G was ∼10-fold lower than those of the G184A and T186N variants (Table 2). From the kinetics plot (Fig. 5C), this reduction seemed to result not only from the larger Km, l-Thr value but also from the lower kcat value relative to the other variants. Because the side chain of Leu80 is one the residues forming the active site (Fig. 5A), the mutation of Leu80 to Gly may create a space and affect the motion of l-Thr, NAD+, and active site residues such as Tyr144 during the reaction. Such motion changes may affect the distances between the C4 atom of the pyridine ring of NAD+ and the β-carbon hydrogen atom of l-Thr and between the oxygen atom of Tyr144 and the β-hydroxyl hydrogen atom of l-Thr. This would shift the pKa values of the molecules and possibly alter the efficiency of hydride transfer, ultimately decreasing kcat. In ADH, shifting the pKa of alcoxide and NAD+ by mutating hydrophobic residues near NAD+ decreases the kcat value (35).
The mutated residues in the variants are distant from the NAD+ binding site (Fig. 5A). Therefore, we predicted that the mutations do not affect NAD+ binding. To confirm this, we measured the Km values toward NAD+ (Km, NAD+); they are shown in Table 2. As expected, the values for WT and the variants were almost the same, less than 0.2 mm (Table 2). This observation implied that NAD+ binds the variants as well as the WT protein and that the reduction in kcat/Km, l-Thr is brought about by less efficient binding of the substrate, l-Thr.
Coenzyme (NAD+) and Substrate (l-Thr) Recognition Mechanism of CnThrDH
Based on our crystal structures, MD simulations, and enzyme kinetics analyses, we propose a schematic model for the changes in flexibility and structure induced in SDR-like l-ThrDH by binding of NAD+ and l-Thr (Fig. 6). Previous work showed that NAD+ binds to the active site prior to l-Thr and that the product, 2-amino-3-keto butyrate, is released before NADH (11). Data summarizing these changes are shown in Table 3. In the CnThrDH (apo) structure, which does not contain either NAD+ or l-Thr, all four regions adopt flexible forms (Fig. 6A; corresponding to the CnThrDH (apo) in Table 3). Upon binding of NAD+, residues 38–59 and 77–79 adopt rigid forms (Fig. 6B). On the other hand, the other three regions adopt flexible forms, resulting in a mostly open overall structure (Fig. 6B; corresponding to CnThrDH (holo, with NAD+) in Table 3). Once NAD+ is already bound, binding of l-Thr provides a driving force to change from the open form to the closed form, in which all four regions have rigid structures (Fig. 6C; corresponding to the CnThrDH (holo) in Table 3). After completion of the reaction, CnThrDH adopts the open form to release the product, 2-amino-3-keto butyrate (2-AKB; Fig. 6D). These interactions, confirmed in the l-Thr- and NAD+-bound structure (Fig. 1C and Ref. 17), would be altered by production of 2-AKB and NADH because of the changes in the steric and electronic interactions among the components of the ternary complex (protein molecule, 2-AKB, and NADH). Furthermore, these changes would promote adoption of the open form by SDR-like l-ThrDH. At least two factors are predicted to provide this driving force, as follows: (i) the hybrid orbital of l-Thr at the Cβ atom changes from sp3 (l-Thr) to sp2 (2-AKB) as a result of the reaction, and this should induce a molecular geometry change, e.g. Cα-Cβ bond rotation and a change in the Cβ–Cα–C bond angle; this may alter the interactions confirmed in Fig. 1C and Ref. 17; and (ii) electronic interactions among the components of the ternary complex may be altered by conversion of the positively charged cofactor NAD+ into the uncharged cofactor NADH. More experimental data are required to reveal the cause of the driving force. Subsequently, NADH is emitted into the solvent, and the reaction cycle returns to the initial state (Fig. 6A). The existence of an NADH-bound state (Fig. 6D) was predicted from the binding order of SDR-like l-ThrDH and the flexibility of CnThrDH (holo, with NAD+) (Table 3).
FIGURE 6.

Schematic model of structural changes induced by binding of NAD+ and l-Thr. In the A, B, and D states, CnThrDH adopts an open form, whereas in the C state, CnThrDH adopts the closed form. The flexibility changes that occur in each domain upon binding of NAD+ and l-Thr are summarized in Table 3.
TABLE 3.
Dynamical structural changes upon binding of NAD+ and l-Thr
| No binding of NAD+ and l-Thr | Binding of NAD+ alone | Binding of NAD+ and l-Thr | |
|---|---|---|---|
| Corresponding to | CnThrDH (apo) | CnThrDH (holo, with NAD+) | CnThrDH (holo) |
| Crystal structure | MD simulation | Crystal structure | |
| Residues 38–59 | Flexible | Rigid | Rigid |
| Residues 77–87 | Flexible | Rigid (residues 77–79) | Rigid |
| Flexible (residues 80–87) | |||
| Residues 180–186 | Flexible | Flexible | Rigid |
| Catalytic domain | Flexible | Flexible | Rigid |
We are aware of no previous study that analyzed the structural changes of SDR-like l-ThrDH. Additional experimental studies could provide data that might support our model of these structural changes (Fig. 6). Several spectrometric methods, such as hydrogen-deuterium exchange and fluorescence methods (36), are helpful in understanding structural changes at various time scales (from μs to s) that are difficult to analyze by MD simulation. As for other members of the extended SDR superfamily, both crystallography and computer simulation studies (37–39) have suggested that GalE undergoes structural changes during its reaction. Specifically, x-ray crystal structures indicated that the catalytic domain of GalE is flexible, just as that of SDR-like l-ThrDH is, when substrate is not bound to the active site (39). Furthermore, using MD simulation, Friedman et al. (38) showed that induced fit and structural changes of the catalytic domain are likely to play a role in the binding of substrates to the active site of GalE. On the other hand, switching between open and closed forms was not mentioned in these studies. In addition to SDR-like l-ThrDH, other members of the extended SDR family may perform enzymatic reactions by switching between forms.
Based on the results of this study, we propose that the high specificity and reactivity of SDR-like l-ThrDH are controlled by the flexibility and structural changes induced by binding of both NAD+ and l-Thr (Fig. 6). Thus, catalysis by SDR-like l-ThrDH is accompanied by dynamical structural change. We believe that such changes are general to other SDR-like l-ThrDH enzymes. If these changes could be controlled by site-directed mutations, it is possible that mutated SDR-like l-ThrDH could acquire reactivity toward novel substrates.
Acknowledgments
The x-ray data were collected at the synchrotron facilities of the Photon Factory using Beamline BL-17A (proposal 2012U005). We are grateful to the beamline staff and to Dr. Yusuke Yamada for assistance with the experiments at Photon Factory. We also thank to Dr. Suyoung Re of RIKEN for helpful discussion about the manuscript.
This work was supported by a grant from the ERATO Asano Active Enzyme Molecule Project from the Japan Science and Technology Agency.

This article contains supplemental Movie S1.
The atomic coordinates and structure factors (codes 3WMW and 3WMX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- l-ThrDH
- l-threonine 3-dehydrogenase
- CnThrDH
- l-ThrDH from C. necator
- GalE
- UDP-galactose 4-epimerase
- ADH
- alcohol dehydrogenase
- MD
- molecular dynamics
- SDR
- short chain dehydrogenase/reductase
- MDR
- medium chain dehydrogenase/reductase
- RMSD
- root mean square distance
- RMSF
- root mean square fluctuation
- 2-AKB
- 2-amino-3-keto butyrate.
REFERENCES
- 1. Epperly B. R., Dekker E. E. (1991) l-Threonine dehydrogenase from Escherichia coli. Identification of an active site cysteine residue and metal ion studies. J. Biol. Chem. 266, 6086–6092 [PubMed] [Google Scholar]
- 2. Misono H., Shinagawa Y., Nagata S., Nagasaki S. (1987) Occurrence of d-threonine dehydrogenase in Pseudomonas cruciviae. Agric. Biol. Chem. 51, 1467–1469 [Google Scholar]
- 3. Bowyer A., Mikolajek H., Stuart J. W., Wood S. P., Jamil F., Rashid N., Akhtar M., Cooper J. B. (2009) Structure and function of the l-threonine dehydrogenase (TkTDH) from the hyperthermophilic archaeon Thermococcus kodakaraensis. J. Struct. Biol. 168, 294–304 [DOI] [PubMed] [Google Scholar]
- 4. Kavanagh K. L., Jörnvall H., Persson B., Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Persson B., Hedlund J., Jörnvall H. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the MDR superfamily. Cell. Mol. Life Sci. 65, 3879–3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nordling E., Jörnvall H., Persson B. (2002) Medium-chain dehydrogenases/reductases (MDR). Family characterizations including genome comparisons and active site modeling. Eur. J. Biochem. 269, 4267–4276 [DOI] [PubMed] [Google Scholar]
- 7. Higashi N., Fukada H., Ishikawa K. (2005) Kinetic study of thermostable l-threonine dehydrogenase from an archaeon Pyrococcus horikoshii. J. Biosci. Bioeng. 99, 175–180 [DOI] [PubMed] [Google Scholar]
- 8. Shimizu Y., Sakuraba H., Kawakami R., Goda S., Kawarabayasi Y., Ohshima T. (2005) l-Threonine dehydrogenase from the hyperthermophilic archaeon Pyrococcus horikoshii OT3: gene cloning and enzymatic characterization. Extremophiles 9, 317–324 [DOI] [PubMed] [Google Scholar]
- 9. Ishikawa K., Higashi N., Nakamura T., Matsuura T., Nakagawa A. (2007) The first crystal structure of l-threonine dehydrogenase. J. Mol. Biol. 366, 857–867 [DOI] [PubMed] [Google Scholar]
- 10. Bashir Q., Rashid N., Jamil F., Imanaka T., Akhtar M. (2009) Highly thermostable l-threonine dehydrogenase from the hyperthermophilic archaeon Thermococcus kodakaraensis. J. Biochem. 146, 95–102 [DOI] [PubMed] [Google Scholar]
- 11. Kazuoka T., Takigawa S., Arakawa N., Hizukuri Y., Muraoka I., Oikawa T., Soda K. (2003) Novel psychrophilic and thermolabile l-threonine dehydrogenase from psychrophilic Cytophaga sp. strain KUC-1. J. Bacteriol. 185, 4483–4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Millerioux Y., Ebikeme C., Biran M., Morand P., Bouyssou G., Vincent I. M., Mazet M., Riviere L., Franconi J. M., Burchmore R. J., Moreau P., Barrett M. P., Bringaud F. (2013) The threonine degradation pathway of the Trypanosoma brucei procyclic form: the main carbon source for lipid biosynthesis is under metabolic control. Mol. Microbiol. 90, 114–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ueatrongchit T., Asano Y. (2011) Highly selective l-threonine 3-dehydrogenase from Cupriavidus necator and its use in determination of l-threonine. Anal. Biochem. 410, 44–56 [DOI] [PubMed] [Google Scholar]
- 14. Ebara S., Toyoshima S., Matsumura T., Adachi S., Takenaka S., Yamaji R., Watanabe F., Miyatake K., Inui H., Nakano Y. (2001) Cobalamin deficiency results in severe metabolic disorder of serine and threonine in rats. Biochim. Biophys. Acta 1568, 111–117 [DOI] [PubMed] [Google Scholar]
- 15. Kamiya Y., Horikoshi T., Takagi H., Okada S., Hashimoto K., Kido C., Takehara K., Nagamine T., Sekiguchi T., Mori M. (1995) Type II citrullinemia associated with neutropenia. Intern. Med. 34, 679–682 [DOI] [PubMed] [Google Scholar]
- 16. Yoneda K., Sakuraba H., Muraoka I., Oikawa T., Ohshima T. (2010) Crystal structure of UDP-galactose 4-epimerase-like l-threonine dehydrogenase belonging to the intermediate short-chain dehydrogenase-reductase superfamily. FEBS J. 277, 5124–5132 [DOI] [PubMed] [Google Scholar]
- 17. Yoneda K., Sakuraba H., Araki T., Ohshima T. (2012) Crystal structure of binary and ternary complexes of archaeal UDP-galactose 4-epimerase-like l-threonine dehydrogenase from Thermoplasma volcanium. J. Biol. Chem. 287, 12966–12974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plapp B. V. (2010) Conformational changes and catalysis by alcohol dehydrogenase. Arch. Biochem. Biophys. 493, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramaswamy S., Park D. H., Plapp B. V. (1999) Substitutions in a flexible loop of horse liver alcohol dehydrogenase hinder the conformational change and unmask hydrogen transfer. Biochemistry 38, 13951–13959 [DOI] [PubMed] [Google Scholar]
- 20. Hayward S., Kitao A. (2006) Molecular dynamics simulations of NAD+-induced domain closure in horse liver alcohol dehydrogenase. Biophys. J. 91, 1823–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 23. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of COOT. Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 25. Lamzin V. S., Wilson K. S. (1993) Automated refinement of protein models. Acta Crystallogr. D Biol. Crystallogr. 49, 129–147 [DOI] [PubMed] [Google Scholar]
- 26. DeLano W. L. ( 2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 27.Molecular Operating Environment (2012) Chemical Computing Group, Inc., Montreal, QC, Canada [Google Scholar]
- 28. Li H., Robertson A. D., Jensen J. H. (2005) Very fast empirical prediction and rationalization of protein pKa values. Proteins 61, 704–721 [DOI] [PubMed] [Google Scholar]
- 29. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 31. Andersen H. C. (1983) Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34 [Google Scholar]
- 32. Seeber M., Cecchini M., Rao F., Settanni G., Caflisch A. (2007) Wordom: a program for efficient analysis of molecular dynamics simulations. Bioinformatics 23, 2625–2627 [DOI] [PubMed] [Google Scholar]
- 33. Tsutsui Y., Ramakrishnan B., Qasba P. K. (2013) Crystal structures of β-1,4-galactosyltransferase 7 enzyme reveal conformational changes and substrate binding. J. Biol. Chem. 288, 31963–31970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gil-Ortiz F., Ramón-Maiques S., Fernández-Murga M. L., Fita I., Rubio V. (2010) Two crystal structures of Escherichia coli N-acetyl-l-glutamate kinase demonstrate the cycling between open and closed conformations. J. Mol. Biol. 399, 476–490 [DOI] [PubMed] [Google Scholar]
- 35. Nagel Z. D., Meadows C. W., Dong M., Bahnson B. J., Klinman J. P. (2012) Active site hydrophobic residues impact hydrogen tunneling differently in a thermophilic alcohol dehydrogenase at optimal versus nonoptimal temperatures. Biochemistry 51, 4147–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nashine V. C., Hammes-Schiffer S., Benkovic S. J. (2010) Coupled motions in enzyme catalysis. Curr. Opin. Chem. Biol. 14, 644–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Timson D. J., Lindert S. (2013) Comparison of dynamics of wildtype and V94M human UDP-galactose 4-epimerase-A computational perspective on severe epimerase-deficiency galactosemia. Gene 526, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Friedman A. J., Durrant J. D., Pierce L. C., McCorvie T. J., Timson D. J., McCammon J. A. (2012) The molecular dynamics of Trypanosoma brucei UDP-galactose 4′-epimerase: a drug target for African sleeping sickness. Chem. Biol. Drug Design 80, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sakuraba H., Kawai T., Yoneda K., Ohshima T. (2011) Crystal structure of UDP-galactose 4-epimerase from the hyperthermophilic archaeon Pyrobaculum calidifontis. Arch. Biochem. Biophys. 512, 126–134 [DOI] [PubMed] [Google Scholar]