

Background: Genome stability is maintained in part by chromatin structure and checkpoint factors.
Results: Yeast cells harboring mutations in the Ddc1-Mec3-Rad17 checkpoint complex exhibit synthetic phenotypes with histone chaperone mutants.
Conclusion: The Ddc1-Mec3-Rad17 complex regulates nucleosome assembly.
Significance: Checkpoint factors and histone chaperones coordinate to maintain genome integrity.
Keywords: Checkpoint Control, Chromatin, Chromatin Regulation, DNA Damage Response, DNA Replication, Histone Chaperone, Histones, CAF-1, Rtt106, Nucleosome Assembly
Abstract
The maintenance of genome integrity is regulated in part by chromatin structure and factors involved in the DNA damage response pathway. Nucleosome assembly is a highly regulated process that restores chromatin structure after DNA replication, DNA repair, and gene transcription. During S phase the histone chaperones Asf1, CAF-1, and Rtt106 coordinate to deposit newly synthesized histones H3-H4 onto replicated DNA in budding yeast. Here we describe synthetic genetic interactions between RTT106 and the DDC1-MEC3-RAD17 (9-1-1) complex, a sliding clamp functioning in the S phase DNA damage and replication checkpoint response, upon treatment with DNA damaging agents. The DNA damage sensitivity of rad17Δ rtt106Δ cells depends on the function of Rtt106 in nucleosome assembly. Epistasis analysis reveals that 9-1-1 complex components interact with multiple DNA replication-coupled nucleosome assembly factors, including Rtt106, CAF-1, and lysine residues of H3-H4. Furthermore, rad17Δ cells exhibit defects in the deposition of newly synthesized H3-H4 onto replicated DNA. Finally, deletion of RAD17 results in increased association of Asf1 with checkpoint kinase Rad53, which may lead to the observed reduction in Asf1-H3 interaction in rad17Δ mutant cells. In addition, we observed that the interaction between histone H3-H4 with histone chaperone CAF-1 or Rtt106 increases in cells lacking Rad17. These results support the idea that the 9-1-1 checkpoint protein regulates DNA replication-coupled nucleosome assembly in part through regulating histone-histone chaperone interactions.
Introduction
Chromatin structure governs a number of cellular processes. The basic repeating unit of chromatin is the nucleosome, consisting of 147 base pairs of DNA wrapped around a protein octamer of one histone (H3-H4)2 tetramer and two H2A-H2B dimers. During DNA replication, DNA repair, and transcription, nucleosomes must be disassembled/remodeled in order for the machinery critical for the aforementioned processes to gain access to the DNA. After passage of the DNA replication, gene transcription, and DNA repair machineries, histones are deposited, nucleosomes assembled, and higher order chromatin structure restored. Nucleosome assembly is critical for epigenetic inheritance and the maintenance of chromatin structure (1, 2).
Histone chaperones are protein factors that regulate nucleosome assembly. In budding yeast the histone chaperones Asf1, CAF-1, and Rtt106 coordinate to deposit histones H3-H4 after DNA replication and DNA repair (3, 4). Asf1 binds newly synthesized H3-H4 dimers and facilitates acetylation of H3-H4 at histone H3 lysine 56 (H3K56ac) (5). H3K56ac promotes H3 ubiquitylation at lysine residues located close to the H3 interface mediating the Asf1-H3 interaction. H3 ubiquitylation disrupts the Asf1-H3 interaction and facilitates the transfer of H3-H4 dimers to the histone chaperones CAF-1 and Rtt106 for (H3-H4)2 tetramer formation and deposition (6). The ability of CAF-1 to deposit H3-H4 onto replicating DNA depends on its physical interaction with proliferating cell nuclear antigen (PCNA),2 a DNA polymerase clamp protein complex residing at and recruiting proteins to the DNA replication fork (7–10). Factors involved in DNA replication-coupled nucleosome assembly are important for the maintenance of genome stability as revealed by genetic analyses and growth in response to DNA damaging agents (11–14). For instance, cells lacking both Cac1, the large subunit of the CAF-1 complex, and Rtt106 show a dramatic increase in DNA damage sensitivity than either single mutant alone (15), most likely as a result of nucleosome assembly defects (3, 15).
DNA damage during S phase is recognized and repaired through activation of two different checkpoint pathways, the DNA damage checkpoint and the replication checkpoint. Checkpoint activation is initiated by replication protein A (RPA)-coated single-stranded DNA, which recruits the sensor checkpoint kinase Mec1 (16) and the 9-1-1 (Rad9-Hus1-Rad1 in humans, Ddc1-Mec3-Rad17 in budding yeast) clamp, a protein complex structurally resembling PCNA. The 9-1-1 complex is recruited to DNA in a reaction dependent on the clamp loader, Rad24 (16–18). The 9-1-1 complex stimulates Mec1 activity, which in turn phosphorylates and activates the downstream kinase Rad53 (19). The DNA replication checkpoint utilizes these same factors, except a different adaptor is used for Rad53 activation. Rad9 serves as the adaptor for the DNA damage checkpoint and Mrc1 for the DNA replication checkpoint. Together these two branches of the S phase checkpoint maintain genome integrity during S phase (20, 21).
Factors involved in nucleosome assembly and the DNA damage response exhibit physical and genetic interactions, suggesting that checkpoint proteins and nucleosome assembly are linked together. For instance, Rad53 interacts physically with Asf1 in budding yeast, and the Asf1-Rad53 interaction negatively regulates the interaction between Asf1 and free histones H3-H4 (22, 23). It is proposed that the Rad53-Asf1 interaction is regulated by Mec1, which in turn regulates the function of Asf1 and Hir1, a histone chaperone involved in DNA replication-independent nucleosome assembly, in the maintenance of chromatin integrity (24). In addition, deletion of RAD9 or RAD24 in cac1Δ mutant cells dramatically increases the gross chromosomal rearrangement accumulation rate (25), suggesting that cac1Δ mutant cells require the DNA damage checkpoint to maintain genome stability. Despite many documented interactions, it remains unclear how proteins involved in nucleosome assembly coordinate with DNA damage response proteins to maintain genome and chromatin integrity during S phase.
In an attempt to identify novel regulators of nucleosome assembly and the maintenance of genome integrity during DNA damage stress, we performed a synthetic genetic array analysis for factors that function in parallel to RTT106 in the response to the DNA damaging agent camptothecin (CPT), a topoisomerase I inhibitor. Here we describe a web of genetic interactions between subunits of the 9-1-1 complex and factors involved in nucleosome assembly, including RTT106, CAC1 (a subunit of CAF-1), and marks of newly synthesized histones. The DNA damage sensitivity phenotype of cells harboring mutations in RTT106 and the 9-1-1 complex depends upon the histone chaperone function of Rtt106. Importantly, cells lacking a functional 9-1-1 complex exhibit defects in the deposition of H3K56ac onto replicating DNA and altered interactions between histones and histone chaperone proteins. Together, our results suggest a novel role for the 9-1-1 complex in DNA replication-coupled nucleosome assembly.
EXPERIMENTAL PROCEDURES
Yeast Strains and Materials
Yeast strains for the synthetic genetic array analysis were of a BY4741 background. All other strains were of W303 background. Yeast strains and plasmids were constructed using standard methods.
Synthetic Genetic Array
The synthetic genetic array screen was performed according to published reports (26–28). After selection of double mutants, cells were dotted onto medium containing low concentrations of CPT. Double mutants were scored to identify those mutants with no growth defects on a control plate (0 μg/ml CPT) but a pronounced growth defect in medium containing CPT. First round hits were verified by a second round of screening comparing single and double mutants in a 384-well plate setting. Candidates from the second round of large-scale screening were verified by tetrad dissection and a standard DNA damage sensitivity dot assay. Finally, candidates were verified by analysis in the independent W303 background strain.
DNA Damage Sensitivity Spot Assay
Freshly grown yeast cells were diluted to A600 0.6. A 10-fold serial dilution was performed, and cells were spotted to regular growth medium or medium for plasmid selection with or without the indicated concentration of DNA damaging agent. Images were recorded at various time points, and select images are shown.
Whole Cell Extraction
Yeast cells were grown to A600 0.8–1.0 and harvested. Cell pellets were washed once, and the pellets were boiled for 3 min. Cells were resuspended in buffer (25 mm Tris, pH 8, 100 mm NaCl, 1 mm EDTA, 10 mm MgCl2, 0.01% Nonidet P-40, 1 mm DTT, 1 mm PMSF, 1 mm benzamidine) and subjected to beads beating. The collected lysate was prepared for SDS-PAGE analysis by the addition of 1× volume of 2× SDS sample buffer. Proteins were resolved by SDS-PAGE, and proteins of interest were detected by Western blot using the indicated antibodies. Alternatively, to detect checkpoint activation after DNA damage treatment, extracts were prepared as described (27). Briefly, cell pellets were boiled briefly followed by resuspension with buffer (1.85 m NaOH, 7.4% 2-mercaptoethanol) and protein precipitation with 20% trichloroacetic acid. The resultant pellets were washed with acetone, dried, and resuspended in 0.1 m NaOH and 2× SDS sample buffer. Proteins were resolved by SDS-PAGE and detected by Western blot.
Cell Cycle Analysis
Cell were arrested at G1 phase with α-factor followed by release into fresh medium or medium containing the indicated concentration of DNA damaging agent. Samples were taken at various time points, prepared for propidium iodide staining, and analyzed by flow cytometry.
Rad52-YFP Foci Formation
Cells expressing Rad52 tagged with YFP were grown to an A600 of 0.6–0.8 at 25 °C and treated for 30 min with the indicated concentration of CPT. Cells were prepared for live cell imaging by washing, once with water and three times with synthetic complete medium lacking tryptophan. Cells were imaged using a Zeiss fluorescence microscope, 100× oil lens, with Z-stack images (bright field and GFP) taken at 0.4-μm intervals. Only the percentage of S/G2/M phase cells containing foci was reported.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed as previously described (12). Briefly, cells were arrested at G1 using α-factor and then released into medium containing 0.2 m hydroxyurea (HU). Cells collected at the indicated time points after release from G1 phase were fixed with 1% paraformaldehyde and quenched with glycine. Cells were harvested, washed twice, and homogenized by bead beating. Chromatin DNA was sheared using a bioruptor (Diagenode) to a size of 0.5–1 kb. Chromatin immunoprecipitation was performed using antibodies against H3 and H3K56ac as indicated, and ChIP DNA was analyzed using real time quantitative PCR as previously described (12).
Tandem Affinity Purification (TAP)
TAP purification was performed as previously described (15). Proteins from soluble cell extracts and immunoprecipitated samples were resolved by SDS-PAGE and detected by Western blot using antibodies against the indicated proteins.
RESULTS
The Histone Chaperone RTT106 Exhibits a Synthetic Genetic Interaction with 9-1-1 Complex Components in Response to Camptothecin
Upon DNA damage and checkpoint activation, the cell undergoes a number of changes, including changes in gene expression, protein localization, and protein modifications (29–33). Novel protein functions may be revealed through analysis of genetic and physical interactions that occur under certain conditions, including DNA damage (22, 28, 34–36). Nucleosome assembly and disassembly, mediated by histone chaperone proteins, is important for regulating chromatin structure during DNA replication, transcription, and DNA repair as well as maintaining genome integrity (2, 37). Rtt106 and CAF-1 are histone H3-H4 chaperones that deposit newly synthesized histones onto DNA during S phase (15, 38, 39). Single rtt106Δ mutants exhibit only minor defects in response to CPT, a topoisomerase I poison, but cells with defects in both CAF-1 and Rtt106 are highly sensitive to DNA damage agents, including CPT. This suggests that CAF-1 and Rtt106 have non-overlapping functions in response to CPT treatment and that CAF-1 and Rtt106 function in parallel to regulate cellular growth under CPT-induced insult (15). To further investigate Rtt106 function in the maintenance of genome stability, we performed a yeast synthetic genetic array analysis combining the rtt106Δ mutation with mutations at each of ∼4700 yeast nonessential genes and testing the double mutant sensitivity to CPT (27, 40). Although genes exhibiting a synthetic growth phenotype with rtt106Δ were uncovered, these genetic interactions were not validated and, therefore, are not described in this report. Instead, our studies focused on viable double mutant cells that were sensitive to CPT (Fig. 1A). In particular, we were interested in those double mutant cells that lacked obvious growth defects on normal growth medium compared with single mutants and exhibited a synthetic slow growth phenotype in medium containing CPT during subsequent tests. Two genes, RAD17 and MEC3, were identified as having a synthetic genetic interaction with RTT106 in response to CPT treatment and no growth defects in normal growth medium. Rad17 and Mec3 are two components of the Ddc1-Mec3-Rad17 (9-1-1) sliding clamp involved in DNA damage response (20, 41). Further analysis confirmed that RTT106 exhibits a similar genetic interaction with all three components of the 9-1-1 complex, DDC1, MEC3, and RAD17, in response to CPT treatment in the W303 genetic background (Fig. 1B and Table 1). These results suggest that Rtt106 and the 9-1-1 complex have non-overlapping functions in maintaining cell survival and growth under CPT-induced genomic stress.
FIGURE 1.

RTT106 genetically interacts with each component of the RAD17-MEC3-DDC1 complex in response to the topoisomerase I inhibitor, CPT. A, identification of RTT106 genetic interactors in response to CPT treatment using the synthetic genetic array method. Double mutants containing rtt106Δ and each of ∼4700 nonessential deletion mutants were generated, and viable double mutants were assessed for growth defects in medium with or without low concentrations of CPT. Candidate genes were selected in which double mutants exhibited no apparent growth defects in normal growth medium but a severe growth defect in CPT-containing medium. B, RTT106 and the 9-1-1 complex exhibited a synthetic interaction in response to CPT. Fresh cells of the indicated genotype (WT = wild type) were spotted onto regular growth medium or medium containing low concentrations of CPT. Growth was assessed over several days with the representative images shown. C, the rtt106Δ mutant exhibited genetic interactions with mutations at genes involved in the S phase DNA damage checkpoint but not the DNA replication checkpoint. A schematic summary of the genetic analyses performed for rtt106Δ in combination with mutants of each of the factors indicated in response to CPT. Genes marked in red genetically interacted with RTT106, whereas genes marked in black did not. Genetic interactions were determined using a DNA damage sensitivity spot assay and are described in Table 1.
TABLE 1.
A summary of genetic interactions among rtt106Δ and different checkpoint mutants
Cells containing mutations at the genes indicated in the left column, alone or in combination with rtt106Δ, were plated in a 10-fold serial dilution onto regular growth medium (YPD) or medium containing low concentrations of the indicated DNA damage agent (CPT, Zeocin HU, and MMS). Cell viability and growth were assessed over several days of incubation. A 0 represents no phenotypic growth effect over either single mutant (no synergistic effect). A + represents a synergistic defect, with additional +'s indicating a more severe growth phenotype. NA = not assessed. Sup = growth defects of single mutant were suppressed in double mutants.
| Mutant | YPD | CPT | Zeocin | HU | MMS |
|---|---|---|---|---|---|
| rad17Δ | 0 | ++ | ++ | + | 0 |
| ddc1Δ | 0 | ++ | ++ | + | 0 |
| mec3Δ | 0 | ++ | ++ | + | 0 |
| mrc1Δ | 0 | 0 | 0 | 0 | 0 |
| Mrc1AQ | 0 | 0 | 0 | + | 0 |
| rad9Δ | 0 | + | +++ | 0 | Sup |
| mec1–1 | 0 | +++ | NA | NA | NA |
| rad53–1 | 0 | + | NA | NA | NA |
| rad24Δ | 0 | +++ | NA | NA | NA |
| mrc1Δ rad17Δ | 0 | 0 | 0 | 0 | 0 |
| Mrc1AQ rad17Δ | 0 | 0 | 0 | 0 | 0 |
An in-depth analysis of the genetic interactions for RTT106 and genes encoding factors involved in the S phase DNA damage and replication checkpoint response, including RAD24, RAD9, MEC1, RAD9, MRC1, and RAD53, revealed a synthetic interaction between RTT106 and RAD24, the 9-1-1 complex clamp loader, and checkpoint kinases MEC1 and RAD53 in response to CPT treatment. Rad9 and Mrc1 are adaptor proteins mediating Rad53 activation in the S phase DNA damage and replication checkpoints, respectively. No synthetic interaction in response to CPT treatment was observed between RTT106 and MRC1, the DNA replication checkpoint adaptor; however, RTT106 and RAD9 did exhibit a synthetic interaction (Fig. 1C, Table 1). Based on these genetic interactions, we suggest that Rtt106 functions in parallel to the S phase DNA damage checkpoint to maintain genome stability under CPT-induced insult.
Cells respond differently to DNA damage induced by distinct DNA damaging agents (30, 36, 42). Therefore, we tested whether the observed genetic interaction between RAD17 and RTT106 in response to CPT held across other DNA damaging agents, including Zeocin, HU, and methyl methanesulfonate (MMS). MMS, an alkylating agent, and HU, an agent that results in a reduction in dNTP levels, cause different forms of replication stress, whereas Zeocin is a DNA intercalating agent that leads to the generation of double-strand breaks (43–45). Mutant rad17Δ rtt106Δ cells exhibited pronounced Zeocin sensitivity over single mutants. Furthermore, compared with single mutants, rad17Δ rtt106Δ cells grew slower in response to HU at higher concentrations, and no growth defects were observed in response to low concentrations of MMS (Table 1, Fig. 2A). Similar growth effects were observed for the rtt106Δ mutation combined with mutations in DDC1 or MEC3, the other 9-1-1 complex components, as well as RAD24, the 9-1-1 complex loader. These results support the idea that synthetic genetic interactions between RTT106 and each component of the 9-1-1 complex are limited to certain types of genomic stress.
FIGURE 2.

RAD17 and RAD24 exhibit a web of genetic interactions with nucleosome assembly factors upon DNA damage stress. A, CAC1, RTT106, and 9-1-1 genetic interactions in response to CPT, Zeocin, HU, and MMS. B and C, RAD17 (B) and RAD24 (C) exhibit synthetic interactions with histone mutants harboring mutations at lysine residues that are acetylated on newly synthesized histones. H35KR: H3K9, -14, -18, -23, -27R. Genetic interactions in response to CPT, Zeocin, HU, and MMS were assessed as described in Fig. 1B. The day after plating for which the recorded image was taken is reported below the image.
To further differentiate between the S phase DNA damage and replication checkpoint response, we tested how rtt106Δ rad9Δ and rtt106Δmrc1Δ cells responded to different DNA damage agents. Compared with either single mutant, rtt106Δ rad9Δ cells were more sensitive to Zeocin, whereas deletion of RTT106 suppressed the MMS sensitivity of rad9Δ cells (Table 1). In general, RTT106 and MRC1 did not exhibit any synthetic interaction irrespective of the type of DNA damage. Similarly, rtt106Δ rad17Δ mrc1Δ triple cells did not have heightened sensitivity over double mutant rtt106Δ rad17Δ or rad17Δ mrc1Δ cells in response to any agent tested (Table 1). Based on this extensive genetic analysis, we suggest that RTT106 functions in a non-overlapping pathway with factors of the S phase DNA damage checkpoint to maintain genome stability when challenged with CPT and Zeocin.
Epistasis Analysis Reveals a Link between the 9-1-1 Complex and Nucleosome Assembly
Cells lacking both RTT106 and CAC1, the large subunit of CAF-1, exhibit growth defects and enhanced CPT sensitivity over either single mutant (15). Thus, we tested the genetic interactions among CAC1, the large subunit of CAF-1, and components of the 9-1-1 complex. Similar to the interactions observed with the rtt106Δ mutation, cac1Δ rad17Δ cells and cac1Δ ddc1Δ cells exhibited more dramatic growth defects in the presence of CPT compared with the corresponding single mutants (Fig. 2A, Table 2). Furthermore, triple mutant cells, rtt106Δ cac1Δ rad17Δ and rtt106Δ cac1Δ ddc1Δ, exhibited severe CPT sensitivity over single and double mutants. This suggests that the 9-1-1 complex functions in parallel with both CAF-1 and Rtt106 in response to CPT-induced genomic stress.
TABLE 2.
Summary of the genetic interactions observed among CAC1, RTT106, and RAD17
The experiments were performed as described in Fig. 1, and representative images are shown in Fig. 2. Growth on regular growth medium (YPD) or medium containing a low concentration of the indicated DNA damage agent was assessed after 2–5 days of incubation. A 0 indicates no phenotypic difference between the mutant and wild type cells; a + represents a growth defect on the indicated media, with additional +'s indicating a more severe growth phenotype. Note that similar interactions were observed among CAC1 or RTT106 with DDC1 or MEC3, two other components of the 9-1-1 complex (Fig. 2 and data not shown).
| Mutant | YPD | CPT | Zeocin | HU | MMS |
|---|---|---|---|---|---|
| rtt106Δ | 0 | + | + | 0 | 0 |
| cac1Δ | 0 | 0 | + | + | + |
| cac1Δ rtt106Δ | + | ++ | ++ | + | + |
| rad17Δ | 0 | + | 0 | ++ | + |
| rtt106Δ rad17Δ | 0 | ++ | ++ | +++ | + |
| cac1Δ rad17Δ | 0 | ++ | ++ | ++++ | +++ |
| cac1Δ rtt106Δ rad17Δ | + | +++ | ++++ | +++++ | +++ |
We also tested the genetic interactions among RTT106, CAC1, and the 9-1-1 complex in response to Zeocin, HU, and MMS. Compared with single mutants, enhanced growth defects were observed for cac1Δ rad17Δ cells under Zeocin and HU treatment (Table 2 and Fig. 2A). Although no genetic interaction was observed for RTT106 and RAD17 in response to MMS, cac1Δ rad17Δ cells had a more severe growth defect in response to MMS than cac1Δ or rad17Δ cells. Deletion of RTT106 in cac1Δ rad17Δ cells resulted in dramatic growth defects in response to Zeocin and HU but not MMS. Together, these results indicate that the 9-1-1 complex functions in parallel to both CAF-1 and Rtt106 in response to HU and Zeocin but not MMS-induced stress.
Newly synthesized histones H3 and H4 are acetylated at specific lysine residues, and these acetylation marks serve an important regulatory function during replication-coupled nucleosome assembly, including histone protein processing and folding, histone nuclear import, and the regulation of histone-histone chaperone interactions. In budding yeast, acetylation of histone H4 lysines 5, 8, and 12, histone H3 lysine 56, and some lysine residues at the H3 the N-terminal tail (H3 K9, 14, 18, 23, 27) regulates nucleosome assembly (1, 3). To determine whether the 9-1-1 complex exhibits genetic interactions with these histone marks regulating nucleosome assembly, mutant cells containing rad17Δ and a histone mutant preventing acetylation at important regulatory lysine residues (H3K56R, H3 5KR (H3 K9R, K14R, K18R, K23R, K27R), H4K5,12R or H3K5,8,12R) were analyzed in response to CPT, Zeocin, HU, and MMS. The rad17Δ H3K56R, rad17Δ H4K5,12R, and rad17Δ H4K5,8,12R cells were extremely sensitive to CPT, Zeocin, and HU compared with the corresponding single mutant (Fig. 2B). We also observed a similar synthetic effect in cells containing the rad24Δ mutation, where rad24Δ cells harboring the H3K56R or H4K5,12R mutations exhibited heightened sensitivity to DNA damaging agents compared with single mutants (Fig. 2C). In contrast, deletion of RAD24 or RAD17 did not increase the DNA damage sensitivity of H3 5KR mutant cells toward HU, MMS, or CPT but did increase sensitivity toward Zeocin. Therefore, in addition to two histone chaperones, CAF-1 and RTT106, components of the 9-1-1 complex and RAD24 exhibit synthetic genetic interactions with histone marks in response to DNA damaging agents, suggesting that the 9-1-1 complex or the Rad17 component tested here has a role in nucleosome assembly.
The 9-1-1 Complex Genetically Interacts with Rtt106 Mutants That Cannot Bind H3-H4
Rtt106 recognizes and binds (H3-H4)2 tetramers via two domains: an N-terminal dimerization domain recognizing unacetylated H3-H4 and a tandem PH domain that specifically binds histone H3K56ac (39). The ability of Rtt106 to bind new (H3-H4)2 is required for its role in nucleosome assembly and maintaining genome integrity under DNA damage insult (39, 46). To provide additional evidence that the 9-1-1 complex has a role in nucleosome assembly, we tested whether the genetic interactions between rtt106Δ and rad17Δ or rtt106Δ and rad24Δ depend on the ability of Rtt106 to bind (H3-H4)2. Wild-type Rtt106 or mutant forms of Rtt106 harboring mutations at sites important for (H3-H4)2 binding, including the dimerization domain (I30E) or sites within the PH domain (Y291A or Y261A) (39, 46), were expressed in rad17Δ rtt106Δ and rad24Δ rtt106Δ cells, and the cells were assessed for CPT sensitivity. Expression of wild-type Rtt106 rescued the CPT sensitivity of rad17Δ rtt106Δ cells to that of single mutant rad17Δ cells, whereas expression of the Rtt106 dimerization (I30E) or PH domain (Y291A) mutant was unable to rescue the rad17Δ rtt106Δ CPT sensitivity phenotype (Fig. 3A). Expression of various forms of Rtt106 was confirmed by Western blot analysis using antibodies against endogenous and exogenously expressed Rtt106 (Fig. 3B). A similar analysis was performed in rad24Δ rtt106Δ mutants. Consistent with the rad17Δ rtt106Δ experiment, the CPT sensitivity of rad24Δ rtt106Δ cells was rescued with expression of wild-type Rtt106 but not the Rtt106 mutants with defects in histone binding (Fig. 3, C and D). Therefore, Rtt106 ability to bind (H3-H4)2 and function in nucleosome assembly is important for the growth and survival of rad17Δ and rad24Δ cells in the presence of CPT. Together, these data provide additional evidence linking the 9-1-1 complex to replication-coupled nucleosome assembly.
FIGURE 3.
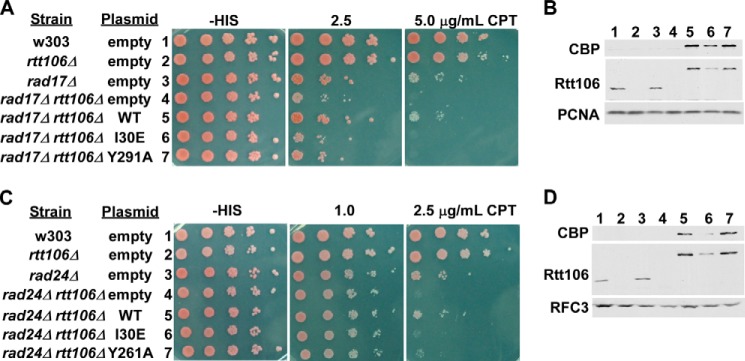
Rtt106 histone chaperone function is critical for the growth of rad17Δ and rad24Δ mutant cells in the presence of CPT. A and B, expression of Rtt106 oligomerization and histone binding point mutants did not rescue the CPT sensitivity phenotype of rad17Δ rtt106Δ cells. A, double mutant rad17Δ rtt106Δ cells were transformed with either empty vector or plasmid expressing WT or mutant forms of Rtt106 (oligomerization mutant, I30E, or PH domain mutant, Y291A) with disrupted histone chaperone function. CPT sensitivity was assessed by spot assay as described in Fig. 1B except using selection medium. B, WT and mutant forms of Rtt106 were expressed in rad17Δ rtt106Δ cells. The expression level of WT and mutant forms of Rtt106 was detected by Western blot using antibodies against calmodulin binding peptide (CBP, part of the tandem affinity purification tag on the exogenously expressed Rtt106) or endogenous Rtt106. PCNA was used as a loading control. C and D, expression of Rtt106 histone chaperone mutants did not rescue the CPT sensitivity phenotype of rad24Δ rtt106Δ cells. Experiments were performed as described in A and B using rad24Δ mutants and the Rtt106 Y261A PH domain mutant. RFC3 was used as a loading control for Western blots.
Cell Cycle Progression, DNA Damage Response, and Repair Protein Recruitment Are Not Altered in rad17Δ rtt106Δ Cells
Slow growth or cell death in the presence of low amounts of DNA damaging agents may result from defects in cell cycle progression, altered DNA damage response, or defects in the recruitment of repair proteins. Thus, we tested whether rad17Δ rtt106Δ cells had defects in cell cycle progression, repair protein recruitment, or checkpoint activation. First, cell cycle progression was analyzed in single and double mutant cells under normal and challenged growth conditions. Cells were arrested at G1 phase and then released into the cell cycle in fresh media. Cells lacking RTT106 had a slight S phase delay compared with wild-type, rad17Δ, and rad24Δ mutant cells (Fig. 4, A and B). Furthermore, rtt106Δ cells exhibited a 15-min delay in entry into the next cell cycle at the transition from G2/M to G1/S phase. Double mutant rad17Δ rtt106Δ and rad24Δ rtt106Δ cells had a cell cycle profile similar to rtt106Δ single mutant cells under normal conditions (Fig. 4B and data not shown). Because rad17Δ rtt106Δ synthetic growth defects were observed only when challenged with DNA damaging agents, we next tested S phase progression under DNA damage insult. After arrest at G1, wild-type, rad17Δ, rtt106Δ, and rad17Δ rtt106Δ cells were released into S phase and media containing a low concentration of either HU, CPT, or MMS. No significant S phase progression defects over single mutant cells were observed in double mutant cells released from G1 into S phase in media containing HU (not shown) or CPT (Fig. 4C). When released into media containing MMS, a slight S phase delay was observed in rad17Δ rtt106Δ cells compared with wild-type and single mutant cells (Fig. 4D). Because rad17Δ rtt106Δ double mutant cells were not sensitive to MMS compared with CPT and HU, these results suggest that the CPT sensitivity observed for rad17Δ rtt106Δ cells is unlikely due to altered cell cycle progression.
FIGURE 4.

Cell cycle progression is not dramatically altered in rad17Δ rtt106Δ cells under normal conditions or DNA damage stress. A, cells with defects in the 9-1-1 complex have normal cell cycle progression. B, similar defects in cell cycle progression are observed for rtt106Δ and rad17Δ rtt106Δ cells under normal growth conditions. C, the S phase progression of rad17Δ rtt106Δ cells was not disrupted with treatment of CPT. D, mutant rad17Δ rtt106Δ cells exhibit defects in S phase progression in the presence of MMS. Cells of the indicated genotype were arrested at G1 phase using α-factor and then released into S phase and the cell cycle in normal medium (A and B) or medium containing the indicated amount of CPT (C) or MMS (D).
After a chromosome break, the repair protein Rad52 is recruited to the double-strand break site to facilitate homologous recombination during S phase as well as DNA repair (47). Cells with defects in nucleosome assembly factors have increased levels of spontaneous chromosome breaks as detected by formation of Rad52 foci (12, 13, 48). We therefore investigated whether rad17Δ rtt106Δ cells had increased spontaneous damage and/or defects in repair protein recruitment compared with wild-type and single mutant cells by detecting Rad52 foci under normal growth conditions and DNA damage insult. Compared with wild-type cells, rtt106Δ cells did not have a significant change in the percentage of S/G2/M cells with Rad52 foci (Fig. 5A). Consistent with published reports, rad17Δ cells had roughly 20% of S/G2/M cells containing foci (48). Surprisingly, we observed that the deletion of RTT106 in rad17Δ cells resulted in partial rescue of the spontaneous DNA damage, with 10% of rad17Δ rtt106Δ double cells containing Rad52 foci. When mutant cells were challenged with CPT for 30 min, rtt1106Δ rad17Δ double-mutant cells had a similar percentage of cells with Rad52 foci compared with wild-type cells. Together, these results suggest that the increased CPT sensitivity observed in rtt106Δ rad17Δ double mutant cells is not likely due to initial recruitment of proteins to the DNA damage site in response to chromosome breaks.
FIGURE 5.

The DNA damage response is not altered in rad17Δ rtt106Δ cells. A, Rad52 recruitment during DNA damage was normal in rad17Δ rtt106Δ cells. Cells of the indicated genotype were left untreated or treated with CPT for 30 min. Rad52 foci, as determined from expression of Rad52 tagged with yellow fluorescent protein, Rad52-YFP, from S/G2/M cells were counted. B, checkpoint activation is normal in mec3Δ rtt106Δ cells after Zeocin treatment. Cells of the indicated genotype were treated with Zeocin for the indicated amounts of time, and protein extraction was performed. Western blot was used to detect phosphorylation of H2AS129 (γ-H2AX). Sir2 and PCNA were used as loading controls.
Phosphorylation of histone H2A is an early response to DNA damage in yeast and mammals and is critical for downstream repair factor recruitment (49, 50). We analyzed phosphorylation of histone H2A serine 129 (H2AS129ph, yeast homolog of mammalian phosphorylation of variant histone H2A, γ-H2AX) in double mutant cells lacking a component of the 9-1-1 complex and Rtt106. Epistasis analysis of γ-H2AX and S phase DNA damage checkpoint genes suggest that γ-H2AX is an important cellular response to S phase damage that is independent of the intra S phase checkpoint (51). Double mutant mec3Δ rtt106Δ cells were treated with Zeocin, a double-strand break agent for which a strong genetic interaction was observed for RTT106 and each member of the 9-1-1 complex, for various times, and whole cell extracts were prepared for analysis of H2AS129ph (γ-H2AX) by Western blot. Compared with wild-type and single mutant cells, the appearance of γ-H2AX kinetics in double mutant cells was similar to wild-type, mec3Δ, or rtt106Δ single mutant cells (Fig. 5B). Altogether, we present evidence that rad17Δ rtt106Δ cells do not have obvious defects in cell cycle progression, Rad52 recruitment, or checkpoint activation compared with single mutant cells. Together, these results indicate that the DNA damage sensitivity of double mutants containing the rtt106Δ or a component of the 9-1-1 complex is due to defects other than cell cycle progression and initial signaling of the DNA damage checkpoint, supporting the idea that the CPT damage sensitivity observed in cells lacking Rtt106 and components of 9-1-1 complex is most likely due to compromised nucleosome assembly in double mutant cells.
Rad17 Regulates the Deposition of Newly Synthesized Histones at Replicated DNA
Our genetic analyses along with published studies suggest that the function of the 9-1-1 complex and S phase damage checkpoint may be tightly linked with nucleosome assembly (23–25, 52). To test whether the 9-1-1 complex has a role in DNA replication-coupled nucleosome assembly, the deposition of newly synthesized histones, marked by H3K56ac, was monitored at early replication origins in wild-type and 9-1-1 mutant cells by ChIP assays. Cells were arrested at G1 phase and then released into medium containing HU, an agent that slows down replication fork progression. Using an antibody against H3K56ac, we detected deposition of newly synthesized histones in wild-type cells at 30 and 45 min after release of cells from G1 into S phase, with a peak deposition at 30 min after release. Histone deposition was detected at replicated DNA surrounding early replication origins ARS607, but not at the distal sites, ARS607+ 14 kb (15) (Fig. 6A). A significant defect in H3K56ac deposition at ARS607 was observed in rad17Δ cells compared with wild-type cells at 30 min after release into S phase. Furthermore, a more dramatic defect in H3K56ac deposition was observed in rad17Δ rtt106Δ cells compared with rad17Δ cells. Because cells lacking Rtt106 exhibit no apparent defects in H3K56ac deposition (15), the reduction observed in rad17Δ rtt106Δ mutant cells is likely due to inactivation of both Rad17 and Rtt106. Notably, H3K56ac deposition at 45 min was similar to wild-type levels, suggesting that H3K56ac deposition was delayed but not defective in rad17Δ and rad17Δ rtt106Δ mutants. No H3K56ac deposition was detected at the ARS607 + 14 kb locus in any strain or at any time point (Fig. 6B). Similar to rad17Δ rtt106Δ cells, defects in H3K56ac deposition were detected in rad24Δ rtt106Δ cells compared with wild-type and single mutants (Fig. 6, C and D). Together, these studies support the idea that Rad17 and Rad24 function with histone chaperone Rtt106 for efficient assembly of newly synthesized H3-H4 onto replicating DNA.
FIGURE 6.

The 9-1-1 complex regulates replication-coupled nucleosome assembly. A and B, the deposition of newly synthesized histone H3 is delayed in rad17Δ and rad17Δ rtt106Δ cells. Cells were arrested at G1 using α-factor and then released into fresh media containing 0.2 m HU. ChIP was performed using an antibody against H3K56ac, a mark of newly synthesized histone H3. ChIP DNA was analyzed by quantitative real-time PCR, amplifying the early replication origin, ARS607 (A), and a region downstream, ARS607 + 14 kb (B). The percentage of H3K56ac ChIP over input DNA was shown, with error bars representing the S.D. of three independent experiments. An asterisk represents a p value less than 0.05 compared with the WT signal as calculated using Student's t test. A carot (^) represents a p value less than 0.05 compared with rad17Δ single mutant. C and D, the deposition of newly synthesized histones is compromised in rad24Δ and rad24Δ rtt106Δ cells. ChIP was performed as described in A for both H3K56ac and unmodified H3. Data are shown as the ratio of H3K56ac signal to H3 signal for each time point. One of three independent experiments, for which the same trend was observed, is shown, with error bars representing the S.D. of triplicate quantitative real-time PCR samples. E, Rad17 regulates the interaction between histones and histone chaperone proteins regulating replication-coupled nucleosome assembly. Cells (WT or rad17Δ) expressing Asf1-TAP, Rtt106-TAP, or Cac2-TAP were used for tandem affinity purification of the indicated histone chaperone. Total (soluble cell extracts (SCE)) and immunoprecipitated (IP) proteins were detected by Western blot using antibodies as indicated. PCNA was used as a loading control for soluble cell extracts.
Rad17 Regulates the Interactions Asf1 and Rad53 as Well as Interactions between Histones and Histone Chaperones
Defects in H3K56ac deposition onto replicated DNA may reflect alterations in the interaction between histone and histone chaperone proteins (6, 12, 15). Histone chaperones Asf1, CAF-1, and Rtt106 function coordinately in nucleosome assembly (3). In addition, Asf1 physically interacts with the checkpoint kinase Rad53, and this interaction negatively regulates Asf1-histone H3-H4 interaction (22). Therefore, we tested how the rad17Δ mutation affected the interaction between H3-H4 with each histone chaperone, Asf1, Rtt106, and CAF-1. TAP was performed using wild-type and rad17Δ mutant strains expressing TAP-tagged Asf1, Rtt106, or Cac2 (subunit of CAF-1). After immunoprecipitation, co-precipitated proteins were determined by Western blot. Compared with wild-type cells, deletion of RAD17 resulted in an increase in the amount of Rad53 co-purifying with Asf1 with a concomitant reduction in the Asf1-H3 interaction (Fig. 6E, left panel). These results suggest that Rad17 regulates the Asf1-Rad53 interaction, in turn impacting Asf1-histone H3 interactions.
In contrast to the reduced association of Asf1 with H3-H4 in rad17Δ mutant cells, more histone H3 co-purified with histone chaperones Rtt106 and Cac2 in rad17Δ mutant cells than wild-type cells (Fig. 6E, middle and right panels). We suggest that the observed increase in interactions between histone H3 and histone chaperone CAF-1 and Rtt106 is due to free histones released from Asf1-H3-H4 complex in rad17Δ mutant cells. Therefore, the interactions among histones and histone chaperones are altered in rad17Δ mutant cells, suggesting that Rad17 plays a role in replication-coupled nucleosome assembly by regulating the balance of interactions between histone chaperone and histones.
DISCUSSION
Using a genetic screen, we found that RAD17 and MEC3 exhibit a genetic interaction with RTT106 in response to the DNA damage agent CPT. We presented the following lines of evidence to support the idea that the 9-1-1 complex has a role in DNA replication-coupled nucleosome assembly. First, we described a web of genetic interactions among CAF-1 and Rtt106, two histone chaperones involved in DNA replication-coupled nucleosome assembly, and components of the 9-1-1 complex. For instance, cells harboring mutations at components of the 9-1-1 complex exhibit synthetic interactions with mutations at RTT106 and CAC1 in response to CPT and Zeocin. Second, synthetic genetic interactions are also observed when mutations at RAD17 or the 9-1-1 complex loader, RAD24, were combined with mutations at histone lysine residues implicated in DNA replication-coupled nucleosome assembly, with most of these genetic interactions being revealed when cells are challenged with a DNA damaging agent. Third, expression of wild-type Rtt106 rescues the CPT sensitivity phenotype of rad17Δ rtt106Δ and rad24Δ rtt106Δ cells, whereas expression of mutant forms of Rtt106 that disrupt histone binding have no effect, suggesting that the synthetic phenotype observed in rad17Δ rtt106Δ and rad24Δ rtt106Δ cells is caused in part by impaired nucleosome assembly. Fourth, we show that cells with mutations in the 9-1-1 complex exhibit defects in the deposition of newly synthesized H3K56ac onto replicating DNA, and this defect is further exacerbated with deletion of RTT106. Finally, we show that the interaction between Asf1 and Rad53 as well as interactions between histones H3 and H4 with Asf1, Rtt106 and CAF-1 are altered in rad17Δ cells compared with wild-type cells.
It is well documented that factors involved in nucleosome assembly are linked physically and genetically to S phase DNA damage and replication checkpoint factors. For instance, yeast histone chaperone Asf1 physically interacts with the checkpoint kinase Rad53, and this interaction is regulated by the Mec1 checkpoint kinase (23, 24). Second, DNA damage caused by Asf1 deletion leads to activation of both the DNA damage and DNA replication checkpoint, whereas accumulation of DNA damage in cells lacking CAF-1 activates only the DNA damage checkpoint (25). Third, Asf1 and H3K56ac regulate nucleosome assembly, and completion of nucleosome assembly is needed for checkpoint recovery after DNA damage repair (53). Here, we provide additional evidence supporting the link between checkpoint activation and nucleosome assembly by describing a web of genetic interactions between RTT106 and factors involved in the DNA damage checkpoint, including RAD17, RAD24, MEC3, DDC1, RAD9, MEC1, and RAD53, upon exposure to DNA damage agents. This web of genetic interactions was further extended to include interactions between RAD17 and CAC1 and RAD17 and histone H3 lysine residues documented to regulate nucleosome assembly.
There are several non-exclusive interpretations for why cells lacking Rtt106 and a component of the 9-1-1 complex are more sensitive to DNA damage agents. First, it is possible that rtt106Δ mutant cells exhibit a low level of DNA damage, and this effect is exaggerated when the DNA damage checkpoint is mutated. Arguing against this interpretation, we did not observe a synergistic increase in spontaneous chromosome breaks as detected by the Rad52 foci assay. Second, like Asf1 and the histone acetyltransferase Rtt109 (53), Rtt106-mediated nucleosome assembly may be required for switching off checkpoint signaling after DNA damage. According to this model, both Rtt106 and the 9-1-1 complex have a role in DNA checkpoint recovery. Third, it is possible that the 9-1-1 complex regulates nucleosome assembly during DNA damage stress. In addition to the web of genetic interactions outlined above supporting this idea, we have shown that deposition of new H3 is compromised in cells lacking RAD17 or RAD24. Furthermore, we show that histone-histone chaperone interactions are altered in rad17Δ mutant cells. These results strongly support the idea that the DNA damage sensitivity observed in cells lacking a component of the 9-1-1 complex and Rtt106 is likely due to defects in nucleosome assembly.
How does Rad17 regulate nucleosome assembly? We suggest that the Rad17-containing clamp regulates the Asf1-Rad53 interaction, which in turn impacts histone-histone chaperone interactions. Consistent with this interpretation, Asf1-Rad53 interactions are increased in rad17Δ mutant cells. Rad53 is known to prevent Asf1 from interacting with H3-H4 (22). Therefore, in rad17Δ mutant cells, less Asf1 will function in delivery of H3-H4 to CAF-1 and Rtt106 at replication forks, which will lead to defects in deposition of H3-H4 at replication forks. Consistent with idea, we observed that less H3-H4 bound to Asf1 in rad17Δ mutant cells. We suggest that this will result in more free histones H3-H4 binding to CAF-1 and Rtt106 in the nuclear plasma in rad17Δ cells. This model could explain why the apparent increase in CAF-1 and Rtt106 interactions with H3-H4 observed in rad17Δ mutant cells did not lead to increased histone deposition in these mutant cells. In human cells, the histone chaperones NASP and Asf1a/b regulate the pool of free histone H3-H4 seen by CAF-1 and other histone chaperones and function to meet cellular demands for histones under various conditions (54). For instance, in cells under replication stress, histones accumulate in NASP containing complexes to stabilize histones that are unable to be incorporated (54, 55).
In addition to regulating histone-histone chaperone interactions, Rad17 may impact nucleosome assembly through other mechanisms. First, the 9-1-1 complex may regulate the recruitment of histone chaperones to DNA for nucleosome assembly. The ability of CAF-1 to deposit new H3-H4 onto replicating DNA depends on its interaction with PCNA (7, 8), but how Rtt106 is recruited to DNA replication fork for histone deposition is not clear. Second, yeast cells lacking CAF-1 and Rtt106 are still viable; thus, other histone chaperones must exist to promote nucleosome assembly of new H3 in the absence of CAF-1 and Rtt106. Our genetic studies suggest that the 9-1-1 complex functions in parallel with both CAF-1 and Rtt106 in response to CPT. It is possible that the 9-1-1 complex functions to recruit a histone chaperone to damaged DNA or the replication fork to promote nucleosome assembly in the absence of CAF-1 and Rtt106. Third, the 9-1-1 complex may function to recruit other factors that facilitate nucleosome assembly and regulation of chromatin structure under normal and stress conditions.
It is known that checkpoint proteins directly interact with the DNA replication machinery, including PCNA and ATM in mammalian cells as well as Mrc1 and MCM helicase (56–58). These interactions may be important for proper recruitment of repair factors to damaged or stalled replication forks. Recently, Mec1 was shown to regulate chromatin accessibility at the replication fork during replication stress (59). Mec1 is activated through recruitment of the 9-1-1 complex. Although the exact mechanism by which Mec1 regulates chromatin accessibility is not clear, it is suggested that Mec1 may phosphorylate chromatin remodeling factors or histone modifying enzymes (59). In mammalian cells, recruitment of the histone chaperone HIRA to sites of DNA damage for deposition of histone H3 variant H3.3 deposition and chromatin priming of transcriptional restart depends on the activity of early DNA damage response factors like Cullin 4a and DNA damage binding proteins 1 and 2 (60). Therefore, the 9-1-1 complex may regulate nucleosome assembly via multiple ways, including the regulation of interactions between histone and histone chaperones.
It is not unprecedented for the 9-1-1 complex to have alternative roles in the maintenance of genome integrity beyond its classic role in checkpoint activation. For instance, the 9-1-1 complex is known to directly interact with DNA polymerase Polzeta, involved in translesion synthesis, and may regulate its access to DNA (61). In addition, the 9-1-1 complex regulates DNA damage tolerance independent of the G1/S checkpoint, a function that is uncoupled from replication forks (62). Because the 9-1-1 complex is one of the first complexes recognizing DNA damage and replication stress, it is possible that 9-1-1 functions to link and coordinate checkpoint activation, DNA replication, and nucleosome assembly during S phase to maintain genome integrity.
Acknowledgment
We thank Dr. Michael Weinreich for yeast strains.
This work was supported, in whole or in part, by National Institutes of Health Grants GM72719 and GM81838.
- PCNA
- proliferating cell nuclear antigen
- CPT
- camptothecin
- HU
- hydroxyurea
- TAP
- tandem affinity purification
- MMS
- methyl methanesulfonate
- PH
- pleckstrin homology.
REFERENCES
- 1. Li Q., Burgess R., Zhang Z. (2012) All roads lead to chromatin. Multiple pathways for histone deposition. Biochim Biophys Acta 1819, 238–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Groth A., Rocha W., Verreault A., Almouzni G. (2007) Chromatin challenges during DNA replication and repair. Cell 128, 721–733 [DOI] [PubMed] [Google Scholar]
- 3. Burgess R. J., Zhang Z. (2013) Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 20, 14–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ransom M., Dennehey B. K., Tyler J. K. (2010) Chaperoning histones during DNA replication and repair. Cell 140, 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Han J., Zhou H., Li Z., Xu R. M., Zhang Z. (2007) Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J. Biol. Chem. 282, 28587–28596 [DOI] [PubMed] [Google Scholar]
- 6. Han J., Zhang H., Zhang H., Wang Z., Zhou H., Zhang Z. (2013) A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 155, 817–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shibahara K., Stillman B. (1999) Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96, 575–585 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Z., Shibahara K., Stillman B. (2000) PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 408, 221–225 [DOI] [PubMed] [Google Scholar]
- 9. Moggs J. G., Grandi P., Quivy J. P., Jónsson Z. O., Hübscher U., Becker P. B., Almouzni G. (2000) A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol. 20, 1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mailand N., Gibbs-Seymour I., Bekker-Jensen S. (2013) Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 14, 269–282 [DOI] [PubMed] [Google Scholar]
- 11. Masumoto H., Hawke D., Kobayashi R., Verreault A. (2005) A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436, 294–298 [DOI] [PubMed] [Google Scholar]
- 12. Burgess R. J., Zhou H., Han J., Zhang Z. (2010) A role for Gcn5 in replication-coupled nucleosome assembly. Mol. Cell 37, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han J., Zhou H., Horazdovsky B., Zhang K., Xu R. M., Zhang Z. (2007) Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315, 653–655 [DOI] [PubMed] [Google Scholar]
- 14. Tyler J. K., Adams C. R., Chen S. R., Kobayashi R., Kamakaka R. T., Kadonaga J. T. (1999) The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402, 555–560 [DOI] [PubMed] [Google Scholar]
- 15. Li Q., Zhou H., Wurtele H., Davies B., Horazdovsky B., Verreault A., Zhang Z. (2008) Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 134, 244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 17. Majka J., Binz S. K., Wold M. S., Burgers P. M. (2006) Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J. Biol. Chem. 281, 27855–27861 [DOI] [PubMed] [Google Scholar]
- 18. Ellison V., Stillman B. (2003) Biochemical characterization of DNA damage checkpoint complexes. Clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 1, E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Majka J., Niedziela-Majka A., Burgers P. M. (2006) The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol. Cell 24, 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harrison J. C., Haber J. E. (2006) Surviving the breakup. The DNA damage checkpoint. Annu. Rev. Genet. 40, 209–235 [DOI] [PubMed] [Google Scholar]
- 21. Nyberg K. A., Michelson R. J., Putnam C. W., Weinert T. A. (2002) Toward maintaining the genome. DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656 [DOI] [PubMed] [Google Scholar]
- 22. Emili A., Schieltz D. M., Yates J. R., 3rd, Hartwell L. H. (2001) Dynamic interaction of DNA damage checkpoint protein Rad53 with chromatin assembly factor Asf1. Mol. Cell 7, 13–20 [DOI] [PubMed] [Google Scholar]
- 23. Hu F., Alcasabas A. A., Elledge S. J. (2001) Asf1 links Rad53 to control of chromatin assembly. Genes Dev. 15, 1061–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharp J. A., Rizki G., Kaufman P. D. (2005) Regulation of histone deposition proteins Asf1/Hir1 by multiple DNA damage checkpoint kinases in Saccharomyces cerevisiae. Genetics 171, 885–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Myung K., Pennaneach V., Kats E. S., Kolodner R. D. (2003) Saccharomyces cerevisiae chromatin-assembly factors that act during DNA replication function in the maintenance of genome stability. Proc. Natl. Acad. Sci. U.S.A. 100, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tong A. H., Evangelista M., Parsons A. B., Xu H., Bader G. D., Pagé N., Robinson M., Raghibizadeh S., Hogue C. W., Bussey H., Andrews B., Tyers M., Boone C. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 [DOI] [PubMed] [Google Scholar]
- 27. Burgess R. J., Zhou H., Han J., Li Q., Zhang Z. (2012) The SCFDia2 ubiquitin E3 ligase ubiquitylates Sir4 and functions in transcriptional silencing. PLoS Genet 8, e1002846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burgess R. J., Zhang Z. (2010) Roles for Gcn5 in promoting nucleosome assembly and maintaining genome integrity. Cell Cycle 9, 2979–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tkach J. M., Yimit A., Lee A. Y., Riffle M., Costanzo M., Jaschob D., Hendry J. A., Ou J., Moffat J., Boone C., Davis T. N., Nislow C., Brown G. W. (2012) Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 14, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fry R. C., Begley T. J., Samson L. D. (2005) Genome-wide responses to DNA-damaging agents. Annu Rev Microbiol 59, 357–377 [DOI] [PubMed] [Google Scholar]
- 31. Povlsen L. K., Beli P., Wagner S. A., Poulsen S. L., Sylvestersen K. B., Poulsen J. W., Nielsen M. L., Bekker-Jensen S., Mailand N., Choudhary C. (2012) Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 14, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 32. Beli P., Lukashchuk N., Wagner S. A., Weinert B. T., Olsen J. V., Baskcomb L., Mann M., Jackson S. P., Choudhary C. (2012) Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 34. Bandyopadhyay S., Mehta M., Kuo D., Sung M. K., Chuang R., Jaehnig E. J., Bodenmiller B., Licon K., Copeland W., Shales M., Fiedler D., Dutkowski J., Guénolé A., van Attikum H., Shokat K. M., Kolodner R. D., Huh W. K., Aebersold R., Keogh M. C., Krogan N. J., Ideker T. (2010) Rewiring of genetic networks in response to DNA damage. Science 330, 1385–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu H., Galka M., Mori E., Liu X., Lin Y. F., Wei R., Pittock P., Voss C., Dhami G., Li X., Miyaji M., Lajoie G., Chen B., Li S. S. (2013) A method for systematic mapping of protein lysine methylation identifies functions for HP1β in DNA damage response. Mol. Cell 50, 723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guénolé A., Srivas R., Vreeken K., Wang Z. Z., Wang S., Krogan N. J., Ideker T., van Attikum H. (2013) Dissection of DNA damage responses using multiconditional genetic interaction maps. Mol. Cell 49, 346–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burgess R. J., Zhang Z. (2010) Histones, histone chaperones, and nucleosome assembly. Protein Cell 1, 607–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang S., Zhou H., Katzmann D., Hochstrasser M., Atanasova E., Zhang Z. (2005) Rtt106p is a histone chaperone involved in heterochromatin-mediated silencing. Proc. Natl. Acad. Sci. U.S.A. 102, 13410–13415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Su D., Hu Q., Li Q., Thompson J. R., Cui G., Fazly A., Davies B. A., Botuyan M. V., Zhang Z., Mer G. (2012) Structural basis for recognition of H3K56-acetylated histone H3-H4 by the chaperone Rtt106. Nature 483, 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tong A. H., Boone C. (2006) Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol. Biol. 313, 171–192 [DOI] [PubMed] [Google Scholar]
- 41. Majka J., Burgers P. M. (2003) Yeast Rad17/Mec3/Ddc1. A sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. U.S.A. 100, 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gasch A. P., Huang M., Metzner S., Botstein D., Elledge S. J., Brown P. O. (2001) Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell 12, 2987–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tercero J. A., Diffley J. F. (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412, 553–557 [DOI] [PubMed] [Google Scholar]
- 44. Lopes M., Cotta-Ramusino C., Pellicioli A., Liberi G., Plevani P., Muzi-Falconi M., Newlon C. S., Foiani M. (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412, 557–561 [DOI] [PubMed] [Google Scholar]
- 45. Liu L. F., Desai S. D., Li T. K., Mao Y., Sun M., Sim S. P. (2000) Mechanism of action of camptothecin. Ann. N.Y. Acad. Sci. 922, 1–10 [DOI] [PubMed] [Google Scholar]
- 46. Fazly A., Li Q., Hu Q., Mer G., Horazdovsky B., Zhang Z. (2012) Histone chaperone Rtt106 promotes nucleosome formation using (H3-H4)2 tetramers. J. Biol. Chem. 287, 10753–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lisby M., Rothstein R., Mortensen U. H. (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. U.S.A. 98, 8276–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alvaro D., Lisby M., Rothstein R. (2007) Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 3, e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Downs J. A., Lowndes N. F., Jackson S. P. (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408, 1001–1004 [DOI] [PubMed] [Google Scholar]
- 50. Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 51. Redon C., Pilch D. R., Rogakou E. P., Orr A. H., Lowndes N. F., Bonner W. M. (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep 4, 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramey C. J., Howar S., Adkins M., Linger J., Spicer J., Tyler J. K. (2004) Activation of the DNA damage checkpoint in yeast lacking the histone chaperone anti-silencing function 1. Mol. Cell. Biol. 24, 10313–10327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen C. C., Carson J. J., Feser J., Tamburini B., Zabaronick S., Linger J., Tyler J. K. (2008) Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cook A. J., Gurard-Levin Z. A., Vassias I., Almouzni G. (2011) A specific function for the histone chaperone NASP to fine-tune a reservoir of soluble H3-H4 in the histone supply chain. Mol. Cell 44, 918–927 [DOI] [PubMed] [Google Scholar]
- 55. Groth A., Ray-Gallet D., Quivy J. P., Lukas J., Bartek J., Almouzni G. (2005) Human Asf1 regulates the flow of S phase histones during replicational stress. Mol. Cell 17, 301–311 [DOI] [PubMed] [Google Scholar]
- 56. Gamper A. M., Choi S., Matsumoto Y., Banerjee D., Tomkinson A. E., Bakkenist C. J. (2012) ATM protein physically and functionally interacts with proliferating cell nuclear antigen to regulate DNA synthesis. J. Biol. Chem. 287, 12445–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Komata M., Bando M., Araki H., Shirahige K. (2009) The direct binding of Mrc1, a checkpoint mediator, to Mcm6, a replication helicase, is essential for the replication checkpoint against methyl methanesulfonate-induced stress. Mol. Cell. Biol. 29, 5008–5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Green C. M., Erdjument-Bromage H., Tempst P., Lowndes N. F. (2000) A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 10, 39–42 [DOI] [PubMed] [Google Scholar]
- 59. Rodriguez J., Tsukiyama T. (2013) ATR-like kinase Mec1 facilitates both chromatin accessibility at DNA replication forks and replication fork progression during replication stress. Genes Dev. 27, 74–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Adam S., Polo S. E., Almouzni G. (2013) Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 155, 94–106 [DOI] [PubMed] [Google Scholar]
- 61. Sabbioneda S., Minesinger B. K., Giannattasio M., Plevani P., Muzi-Falconi M., Jinks-Robertson S. (2005) The 9-1-1 checkpoint clamp physically interacts with polzeta and is partially required for spontaneous polzeta-dependent mutagenesis in Saccharomyces cerevisiae. J. Biol. Chem. 280, 38657–38665 [DOI] [PubMed] [Google Scholar]
- 62. Karras G. I., Fumasoni M., Sienski G., Vanoli F., Branzei D., Jentsch S. (2013) Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol. Cell 49, 536–546 [DOI] [PubMed] [Google Scholar]