

Background: Targeting disruption of STAT3 results in inhibition of tumor growth and survival in malignant glioma.
Results: Cucurbitacin I triggers protective autophagy through the AMPK/mTOR/p70S6K pathway and down-regulates HIF-1α.
Conclusion: Autophagy blockade sensitizes glioblastoma to cucurbitacin I treatment.
Significance: This study provides new insights into the biological and antiproliferative activities of cucurbitacin I against glioblastoma.
Keywords: Apoptosis, Autophagy, Glioblastoma, Hypoxia-inducible Factor (HIF), Signal Transduction, Chloroquine, Cucurbitacin I
Abstract
There is an urgent need for new therapeutic avenues to improve the outcome of patients with glioblastoma multiforme (GBM). Current studies have suggested that cucurbitacin I, a natural selective inhibitor of JAK2/STAT3, has a potent anticancer effect on a variety of cancer cell types. This study showed that autophagy and apoptosis were induced by cucurbitacin I. Exposure of GBM cells to cucurbitacin I resulted in pronounced apoptotic cell death through activating bcl-2 family proteins. Cells treatment with cucurbitacin I up-regulated Beclin 1 and triggered autophagosome formation and accumulation as well as conversion of LC3I to LC3II. Activation of the AMP-activated protein kinase/mammalian target of rapamycin/p70S6K pathway, but not the PI3K/AKT pathway, occurred in autophagy induced by cucurbitacin I, which was accompanied by decreased hypoxia-inducible factor 1α. Stable overexpression of hypoxia-inducible factor 1α induced by FG-4497 prevented cucurbitacin I-induced autophagy and down-regulation of bcl-2. Knockdown of beclin 1 or treatment with the autophagy inhibitor 3-methyladenine also inhibited autophagy induced by cucurbitacin I. A coimmunoprecipitation assay showed that the interaction of Bcl-2 and Beclin 1/hVps34 decreased markedly in cells treated with cucurbitacin I. Furthermore, knockdown of beclin 1 or treatment with the lysosome inhibitor chloroquine sensitized cancer cells to cucurbitacin I-induced apoptosis. Finally, a xenograft model provided additional evidence for the occurrence of cucurbitacin I-induced apoptosis and autophagy in vitro. Our findings provide new insights into the molecular mechanisms underlying cucurbitacin I-mediated GBM cell death and may provide an efficacious therapy for patients harboring GBM.
Introduction
A variety of cancers, including glioma, exhibits an aberrant activation of STAT3, which plays a pivotal role in malignant transformation and tumor cell survival, and blocking the aberrant activation of STAT3 results in the inhibition of tumor growth and survival and induction of apoptosis with few side effects to normal cells (1–7). Thus, abrogation of STAT3 activation is considered an effective cancer therapeutic approach (8). Cucurbitacin I, a selective inhibitor of JAK2/STAT3, is a natural plant product isolated from various plant families and has been used as folk medicine for centuries in China, India, Brazil, and Peru (9). Accumulating evidence shows that cucurbitacin I has a potent anticancer effect on a variety of cancer cell types, such as breast cancer, lung cancer, neuroblastoma, melanoma, and glioma (10–12).
Glioblastoma multiforme (GBM),2 classified as grade IV astrocytoma by the World Health Organization, is the most aggressive glioma and accounts for 54% of all gliomas (13). Even though a combination of surgery, chemotherapy, and radiotherapy is used, there has been only a minimal improvement in the median survival time of GBM patients (from ∼12 to 14 months) or in the 5-year survival rate (less than 5%) (14), which points to the critical need to identify and implement therapeutic strategies. Because JAK2/STAT3 has garnered significant interest as a key driver of tumor cell survival, proliferation, and invasion in GBM (5, 15–18), it may be exploitable for a novel therapy against GBM.
Macroautophagy (hereafter called autophagy), known as programmed cell death type II, has an important homeostatic role, mediating the removal of dysfunctional or damaged organelles that are digested and recycled for cellular metabolic needs (19). Consequently, autophagy might maintain cancer survival under metabolic stress conditions and mediate resistance to anticancer therapies such as radiation, chemotherapy, and some targeted therapies. Mounting evidence suggests that inhibition of autophagy promotes cancer cell death (20–23) and potentiates various anticancer therapies (24–26), implicating autophagy as a mechanism that enables tumor cells to survive antineoplastic therapy. Treatment of these cells with inhibitors of autophagy such as chloroquine or knockdown of essential autophagy genes (beclin-1, autophagy-associated gene) resulted in enhanced therapy-induced apoptosis (27, 28). These findings have led to the initiation of multiple clinical trials combining autophagy inhibitors and chemotherapeutic agents for diverse cancer types (29).
Here we unveil a hitherto rare described cellular response of cucurbitacin I-induced protective autophagy associated with HIF-1α and a dramatic cytotoxic effect following autophagy inhibition by CQ in GBM. Our studies could provide the groundwork for future investigation of the implication of cucurbitacin I-mediated anticancer activities against GBM
EXPERIMENTAL PROCEDURES
Reagents
Cucurbitacin I, 3-methyladenine, chloroquine (CQ), and Dimethyl sulfoxide (DMSO) were purchased from Sigma. FG-4497 was purchased from FibroGen. Antibodies used in the study were as follows: AMPK, phospho-AMPK (Thr-172), mTOR, phospho-mTOR (Ser-2448), AKT, phospho-AKT (Ser-473), Beclin 1, Bcl-2, Bcl-xL, Ki67, JAK2, STAT3, phospho-STAT3 (Ser-727), and LC3BI/II (Cell Signaling Technology); phospho-JAK2 (Tyr-1007/Tyr-1008) and Bax (Abcam); and cleaved caspase 3 (p17), HIF-1α, p70S6K, and phospho-p70 S6K (Santa Cruz Biotechnology); and hVps34 (Echelon Biosciences).
Cell Lines and Culture
The human GBM cell lines T98G and U251 were purchased from the ATCC and incubated in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 100 units/ml penicillin, and 100 μg/ml streptomycin in humidified air with 5% CO2 at 37 °C. Human astrocytes were purchased from ScienceCell and incubated in astrocyte medium consisting of 20% fetal bovine serum, 100 units/ml penicillin, and 1% astrocyte growth supplement (ScienceCell) in humidified air with 5% CO2 at 37 °C.
Cell Viability Assay
The cytotoxic effect of cucurbitacin I on GBM cell lines was determined using a CCK-8 assay (Dojindo, Japan). Tumor cells in medium containing 10% fetal bovine serum were seeded into 96-well, flat-bottomed plates at 5 × 103 cells/well and incubated at 37°C overnight. After the desired treatment, the cells were incubated for an additional 4 h with 100 μl of serum-free DMEM with 10 μl of CCK-8 at 37°C. The absorbance at 450 nm was measured using a microplate reader. The absorbance was measured at 450-nm wavelength.
Western Blot Analysis
After the desired treatment, cells were washed twice with cold PBS and harvested with a rubber scraper. Cell pellets were lysed and kept on ice for at least 30 min in a buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.5% Nonidet P-40, 50 mm NaF, 1 mm Na3 VO4, 1 mm phenylmethylsulfonyl fluoride, and 1 mm PMSF. The lysates were cleared by centrifugation, and the supernatants were collected. Cell lysates were then separated by SDS-PAGE and subjected to Western blot analysis with the primary antibodies and horseradish peroxidase-labeled secondary antibodies.
Coimmunoprecipitation Assay
Cells lysates were collected as described under “Western Blot Analysis.” Samples were immunoprecipitated with 1 μg of Bcl-2, Beclin 1, or hVps34 antibodies or irrelevant IgG at 4 °C overnight, and the immunoprecipitated protein was pulled down with protein A-agarose beads (Santa Cruz Biotechnology) at 4 °C for 6 h. The supernatant was removed, and proteins were boiled in 4× SDS loading buffer (Millipore) before SDS-PAGE electrophoresis.
Immunofluorescence Staining
GBM cells were plated on glass slides in 24-well culture plates at a concentration of 2 × 105 cells/well for 24 h and subsequently treated with drugs for an additional 48 h in serum-free DMEM. Thereafter, the cells were fixed with a 4% formaldehyde solution in PBS, permeabilized with 0.5% Triton X-100 in PBS, stained with the primary antibody overnight, and labeled with anti-mouse or anti-rabbit IgG conjugated with FITC (Santa Cruz Biotechnology). The cells were counterstained with DAPI and observed under an Olympus BX61 fluorescence microscope. Pictures were scanned with a DP71 CCD (charge-coupled device) digital camera.
GFP-LC3 Transient Transfection
Cells were transiently transfected with the pSELECT-GFP-LC3 plasmid (Invivogen) using Lipofectamine 2000 reagent (Sigma) according to the instructions of the manufacturer. To quantify autophagic cells after drug treatment, we counted the number of autophagic cells demonstrated by GFP-LC3 punctates (≥20 punctates as a positive cell) in 100 fields. Pictures were scanned with a DP71 CCD digital camera.
Transmission Electron Microscopy
Cells were fixed with 3% glutaraldehyde in PBS for 2 h, washed five times with 0.1 m cacodylate buffer, and postfixed with 1% OsO4 in 0.1 m cacodylate buffer containing 0.1% CaCl2 for 1.5 h at 4 °C. The samples were then stained with 1% Millipore-filtered uranyl acetate, dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in LX-112 medium (Ladd Research Industries, Inc.). After polymerization of the resin at 60 °C for 48 h, ultrathin sections were cut with a Leica Ultracut microtome (Leica). Sections were stained with 4% uranyl acetate and lead citrate, and images were obtained using a JEM-100cxII electron microscope (JEM).
Small Interfering RNA Transfection
Beclin 1 and negative control siRNA were synthesized by Genephama. The sequences of siRNA were as follows: human beclin 1, 5′-GGA GCC AUU UAU UGA AAC UTT-3′ (sense) and 5′-GU UUC AAU AAA UGG CUC CTT-3′ (antisense). The siRNAs were transfected with Lipofectamine 2000 for 48 h in U251 and T98G cells according to the protocol of the manufacturer.
TUNEL Assay
GBM cells were plated on glass slides in 24-well culture plates at 2 × 105 cells/well for 24 h and subsequently treated with drugs for an additional 48 h in serum-free DMEM. Glass slides with GBM cells and tumor paraffin-embedded sections were stained with the TUNEL technique using a TACS®2 TdT-Fluor in situ apoptosis detection kit (Trevigen, Inc.) according to the instructions of the manufacturer. TUNEL-positive cells were counted from at least 100 random fields under a fluorescence microscope. *TACS-NucleaseTM and buffer were used as positive controls to induce apoptosis.
Cell Death Detection ELISAPlus Assay
A cell death detection ELISAPlus assay (Roche) was performed to determine apoptosis by quantification of histone-complexed DNA fragments according to the instructions of the manufacturer, and absorbance was determined at 405-nm wavelength.
Immunohistochemistry
Solid tumors were removed from sacrificed mice and fixed with 4% formaldehyde. Paraffin-embedded tumor tissues were sectioned to 5-μm thickness and mounted on positively charged microscope slides, and 1 mm EDTA (pH 8.0) was used for antigen retrieval. Endogenous peroxidase activity was quenched by incubating the slides in methanol containing 3% hydrogen peroxide, followed by washing in PBS for 5 min, after which the sections were incubated for 2 h at room temperature with normal goat serum and subsequently incubated at 4 °C overnight with primary antibodies (1:100 Ki67, 1:200 LC3B, 1:100 bcl-2, 1:100 bcl-xL, and 1:100 p-caspase 3). Next, the sections were rinsed with PBS and incubated with horseradish peroxidase-linked goat anti-rabbit or anti-mouse antibodies, followed by reaction with diaminobenzidine and counterstaining with Mayer's hematoxylin.
Tumor xenograft Model
The experiments conformed to the Animal Management Rule of the Chinese Ministry of Health (documentation 55, 2001), and the experimental protocol was approved by the Animal Care and Use Committee of Shandong University. BALB/c nude (nu/nu) female mice were purchased from Vital River Laboratories. U251 cells (5 × 106 cells in 50 μl of serum-free DMEM) were inoculated subcutaneously into the right flank of 5-week-old female mice after acclimatization for a week. Tumor growth was measured daily with calipers. Tumor volume was calculated as (L × W2) / 2, where L is the length in millimeters and W is the width in millimeters. When the tumors reached a mean volume of 90–120 mm3, animals were randomized into groups. Two in vivo experiments were done: one to investigate the effect of cucurbitacin I and another one to assess the effects of CQ against cucurbitacin I treatment. In the first experiment, 16 mice were randomly assigned to cucurbitacin I (1 mg/kg/day in 20% DMSO in PBS) or drug vehicle control (20% DMSO in PBS) and dosed intraperitoneally with 100 μl of vehicle or drug once daily for 18 days, whereas, in the second, 20 mice were assigned to four groups. Control animals received 20% DMSO in PBS vehicle, whereas treated animals were injected with cucurbitacin I (1 mg/kg/day) in 20% DMSO in PBS, CQ (25 mg/kg/day) in 20% DMSO in PBS, and cucurbitacin I (1 mg/kg/day) plus CQ (25 mg/kg/day) in 20% DMSO in PBS and dosed intraperitoneally with 100 μl of vehicle or drug once daily for 15 days. Tumors were dissected and frozen in liquid nitrogen or fixed in formalin.
Statistical Analysis
The data were expressed as means ± S.D. Statistical analysis was performed with two-tailed Student's t test. Significance between groups was determined with the Kruskal-Wallis test and Mann-Whitney U test. The criterion for statistical significance was set at p < 0.05.
RESULTS
Cucurbitacin I Inhibited the Growth of GBM Cells in Vitro and in Vivo
To systematically address the inhibitory activity of cucurbitacin I on GBM cell growth, we first evaluated its cell viability by CCK-8 assay in vitro. The IC50 values of cucurbitacin I against U251 and T98MG cells were 170 and 245 nm, respectively. Treatment with cucurbitacin I resulted in growth inhibition of U251 and T98G cells in a dose-dependent manner, but their responses varied (Fig. 1A). Moreover, the slight effect of cucurbitacin I on human astrocytes was observed. To determine whether the in vivo effects of cucurbitacin I on GBM cells aligned with those in vitro, we conducted a series of therapeutic experiments using U251 cell xenograft mouse models. We found that intraperitoneal administration of cucurbitacin I (1 mg/kg/d, 18 days) markedly inhibited tumor volume and tumor weight compared with the counterparts treated with DMSO. The average tumor volume of solid tumors in cucurbitacin I-treated mice was 412 mm3 (± 82) compared with 1286 mm3 (± 251) for the control group. Moreover, the average tumor weights at study termination were 1340 mg (± 260) and 418 mg (± 80) in the control and cucurbitacin I groups, respectively (Fig. 1, B and C). Furthermore, Ki67 immunostaining confirmed a pronounced decrease in tumor cell proliferation (Fig. 1D). Together, our findings showed that cucurbitacin I significantly suppressed GBM growth in vitro and in vivo.
FIGURE 1.

Cucurbitacin I inhibited the growth of GBM cells in vitro and in vivo. A, a CCK-8 assay was performed to assess cell viability in GBM cells and human astrocytes treated with different concentrations of cucurbitacin I for 24 and 48 h. Cucurbitacin I markedly inhibited tumor growth in U251 cell xenografts as measured by tumor volume (B), tumor weight (C), and Ki67 immunohistochemical staining (D). All data are mean ± S.D. *, p < 0.05; ***, p < 0.001; compared with the control group. Scale bar = 50 μm.
Cucurbitacin I Induced Apoptosis in GBM Cells and a Xenograft Mouse Model Related to bcl-2 Family Proteins
To investigate the underlying mechanisms involved in cucurbitacin I-induced apoptosis against GBM, a cell death detection ELISAPlus assay, a TUNEL assay, and apoptosis-related protein analysis were performed. The percentage of TUNEL-positive cells increased remarkably in a dose-dependent manner in cucurbitacin I-treated U251 and T98G cells (Fig. 2A). Similar to this observation, the DNA fragmentation ratio of cucurbitacin I-treated groups was predominantly elevated, compared with the control group, in a dose-dependent manner, as examined by cell death detection ELISAPlus assay (Fig. 2B). For a further assessment of apoptosis induced by cucurbitacin I, we examined the expression of apoptosis-related molecules by Western blot analysis. GBM cells treated with cucurbitacin I for 48 h significantly up-regulated Bax and cleaved caspase 3 (p17) but decreased antiapoptotic proteins such as Bcl-2 and Bcl-xL in a dose-dependent manner (Fig. 2C). In agreement, intraperitoneal injections of cucurbitacin I resulted in massive apoptotic cell death, cleaved caspase 3 (p17) increase, and marked bcl-2 and bcl-xL decrease on xenograft sections (data not shown).
FIGURE 2.

Cucurbitacin I induced apoptosis in GBM cells and a xenograft mouse model related to bcl-2 family proteins. A, a TUNEL assay was performed to measure apoptosis in GBM cells treated with different concentrations of cucurbitacin I for 48 h. B, apoptotic ratios were assessed by a cell death detection ELISAPlus assay in GBM cells treated with different concentrations of cucurbitacin I for 48 h. C, Western blot analysis of Bcl-xL, Bcl2, Bax, and cleaved caspase 3 expression from lysates of GBM cells treated with different concentrations of cucurbitacin I for 48 h. β-actin served as the loading control. All data are mean ± S.D. **, p < 0.01 compared with the control group. Scale bar = 50 μm.
Cucurbitacin I Triggered Autophagy and Activated the Autophagy-related Gene Beclin 1 in GBM Cells
An increasing number of studies has shown that cancer cells, including GBM cells, undergo autophagy in response to various anticancer therapies (30, 31). We examined whether cucurbitacin I induced autophagy in GBM cells. To evaluate the activation of autophagy by cucurbitacin I, transmission electron microscopy, immunofluorescence staining of LC3B, and GFP-LC3 transient transfection were used to show expressing LC3 aggregation in U251 and T98G cells. Transmission electron microscopy revealed abundant characteristic autophagosomes in GBM cells treated with cucurbitacin I and scarce autophagosomes in control cells (Fig. 3A). To further confirm that cucurbitacin I induces autophagy in GBM cells, we determined the induction of autophagy by localizing an autophagosome-specific protein, LC3, by GFP-LC3 transient transfection. As shown in Fig. 3B, we observed abundant autophagosomes in a dose-dependent manner in 48-h cucurbitacin I-treated cells. We found similar results through immunofluorescence staining of LC3B (data not shown). Moreover, to examine whether cucurbitacin I treatment induced processing of LC3-I to LC3-II and activated the autophagy-related gene Beclin 1, a Western blot analysis was performed. As shown in Fig. 3C, LC3-II and Beclin 1 expression were more pronounced with an increased dose of cucurbitacin I. To test whether cucurbitacin I-induced up-regulation of LC3B is due to autophagy induction or the inhibition of autolysosomal function, bafilomycin A1 was used to inhibit autophagic flux. As shown in Fig. 3D, although increased LC3-II levels were detected in bafilomycin A1-treated cells because of inhibition of lysosomal degradation of LC3-II, LC3-II levels were even higher in the cucurbitacin I-treated cells. The reduced p62 level usually indicates the activation of autophagy in cells (32). We found that, in the absence of bafilomycin A1, the expression of p62 protein was decreased in cucurbitacin I-treated cells, suggesting that autophagy was activated and that the p62 protein was degraded via autophagy. The p62 level was obviously elevated in cells treated with bafilomycin A1 and cucurbitacin I, indicating that autophagy was blocked by bafilomycin A1 and that p62 was accumulated in GBM cells (Fig. 3D). Furthermore, a massive LC3B accumulation was noted on tumor sections in cucurbitacin I-treated xenografts (Fig. 3E). Taken together, these data indicated that cucurbitacin I induced autophagy and activated the autophagy-related gene beclin 1 in GBM in vitro and in vivo.
FIGURE 3.

Cucurbitacin I triggered autophagy and activated the autophagy-related gene beclin 1 in GBM cells. A, images from transmission electron microscopy showing characteristic autophagosomes (arrows) in GBM cells after treatment with DMSO (<0.1%) or 200 nm cucurbitacin I for 48 h. N, nucleus. B, pSELECT-GFP-LC3 transfection disclosed LC3 punctates in GBM cells treated with DMSO (<0.1%), rapamycin (1 μm), and various concentrations of cucurbitacin I for 48 h. Scale bar = 10 μm. C, Western blot analysis of LC3B and beclin1 expression from lysates of GBM cells treated with different concentrations of cucurbitacin I for 48 h. β-actin served as the loading control. D, LC3B and p62 levels were examined by Western blot analysis for GBM cells after treatment with 200 nm cucurbitacin I or DMSO (<0.1%) in the absence or presence of 100 nm bafilomycin A1 for 48 h. β-actin served as the loading control. E, immunohistochemical staining results of LC3B on tumor sections. Scale bar = 50 μm. All are mean ± S.D. *, p < 0.05; **, p < 0.01; compared with the control group.
Constitutive Activation of the AMPK/mTOR/p70S6K Pathway Was Involved in Cucurbitacin I-induced Autophagy in GBM Cells
PI3K/Akt/mTOR pathway is the main regulatory pathway by which autophagy is suppressed (33, 34). However, we observed that treatment with cucurbitacin I for 48h down-regulated p-mTOR expression in a dose-dependent manner but failed to note any alteration of p-AKT (Fig. 4A), which indicated that the PI3K/AKT pathway might not be the main signaling pathway when autophagy occurred after cucurbitacin I treatment. It has been shown recently that AMP-activated protein kinase activation leads to autophagy through the negative regulation of mTOR (35, 36). Therefore, we examined whether phosphorylation of AMPK was involved in cucurbitacin I-induced autophagy in GBM cells. As expected, cucurbitacin I-mediated phosphorylation of AMPK was more pronounced with the increased dose of cucurbitacin I. In addition, the decreased phosphorylation of p70S6K, a downstream target of mTOR, was observed dose-dependently in GBM cells. These findings indicated that AMPK/mTOR/p70S6K pathway was involved in cucurbitacin I-induced autophagy in GBM cells.
FIGURE 4.
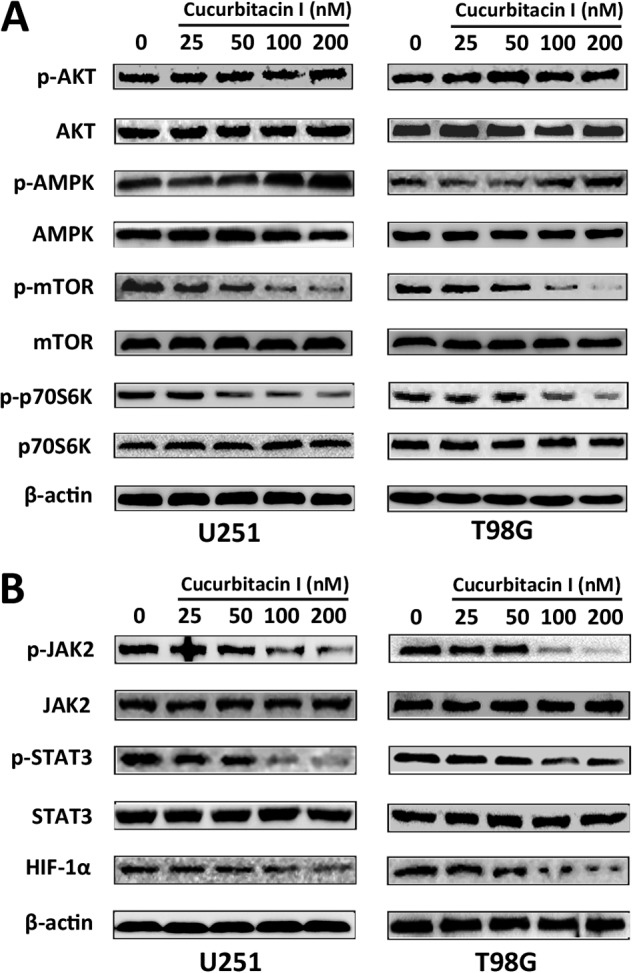
Constitutive activation of AMPK/mTOR/p70S6K and JAK2/STAT3/HIF-1α signaling was involved in cucurbitacin I-induced autophagy in GBM cells. Western blot analysis of p-AKT, AKT, p-AMPK, AMPK, p-mTOR, mTOR, p-p70S6K, and p70S6K (A) and p-JAK2, JAK2, p-STAT3, STAT3, and HIF-1α (B) expression from lysates of GBM cells treated with different concentrations of cucurbitacin I for 48 h. β-actin served as the loading control.
The JAK2/STAT3 pathway positively regulates HIF-1α in many cancer cell types, such as breast cancer, ovarian cancer, renal carcinoma, hepatocellular carcinoma, etc. (37–40). We wondered whether cucurbitacin I, a JAK2/STAT3 inhibitor, may inhibit this pathway. As shown in Fig. 4B, cucurbitacin I inhibited the JAK2/STAT3 cascade and resulted in HIF-1α down-regulation in a dose-dependent manner.
Down-regulation of HIF-1α Played Pivotal Roles in Cucurbitacin I-induced Autophagy in GBM Cells
We further investigated the functional role of HIF-1α down-regulation in cucurbitacin I-induced autophagy. FG-4497 was applied to prevent HIF-1α from quick degradation in normoxia (41, 42). Intriguingly, in the presence of FG-4497, U251 cells showed a significant decrease in the number of autophagosomes and the percentage of cells with GFP-LC3 dots after treatment with cucurbitacin I, which indicates that overexpression of HIF-1α might prevent autophagy occurrence after cucurbitacin I treatment (Fig. 5, A and B). To determine further interconnection between HIF-1α and autophagy in GBM cells treated with cucurbitacin I, a Western blot analysis was performed (Fig. 5C). Accordingly, in view of our above findings, U251 cells overexpressing HIF-1α induced by FG-4497 showed a significant decrease in the level of conversion of LC3B-II to LC3B-I. In addition, we found that cucurbitacin I decreased the level of bcl-2 protein that occurred, along with the induction of autophagy in U251 cells, and that overexpression of HIF-1α prevented cucurbitacin I-induced down-regulation of bcl-2 and autophagy. We then gained further insight into the mechanisms of cucurbitacin I-induced autophagy. Beclin 1 was up-regulated in cells treated by cucurbitacin I (Fig. 4C). Next, we examined the effects of knockdown of beclin 1 on cucurbitacin I-induced autophagy. As shown in Fig. 5C, compared with the results in siRNA controls, knockdown of beclin 1 prevented an increase in the level of LC3-II by cucurbitacin I. Similar results were observed after pretreating the cells with 3-methyladenine (Fig. 5D). It has been reported that bcl-2, a well known antiapoptosis protein that is transcriptionally regulated by HIF-1α (43), regulates autophagy by binding to beclin 1/hVps34, which leads to autophagy (44, 45). To determine the biological effect of cucurbitacin I on the Bcl-2·Beclin 1 (hVps34) complex, coimmunoprecipitation was performed to monitor the interaction of Bcl-2 with Beclin 1/hVps34. We found that, under basal conditions, Bcl-2 and Beclin1/hVps34 coimmunoprecipitated with each other in U251 cells, whereas the interaction markedly decreased in cucurbitacin I-treated cells (Fig. 5E).
FIGURE 5.

Down-regulation of HIF-1α played pivotal roles in cucurbitacin I-induced autophagy in GBM cells. A, autophagosomes (arrows) in U251 cells were observed using transmission electron microscopy. In the absence or presence of FG-4497 (50 μm), U251 cells were treated with cucurbitacin I (200 nm) or DMSO (<0.1%) for 48 h. N, nucleus. Data are mean ± S.D. **, p < 0.01. B, pSELECT-GFP-LC3 transfection showed LC3 punctates in U251 cells treated with DMSO (<0.1%), FG-4497 (50 μm), or cucurbitacin I (200 nm) or cotreated with cucurbitacin I and FG-4497 for 48 h. Data are mean ± S.D. **, p < 0.01. C, a Western blot analysis was utilized to analyze the expression of LC3B, HIF-1α, and Bcl-2 from lysates of U251 cells treated with cucurbitacin I (200 nm) or DMSO (<0.1%) in the absence or presence of FG-4497 (50 μm) for 48 h. D, a Western blot analysis was utilized to analyze the expression of LC3B and Beclin 1 in Beclin 1 knockdown or non-Beclin 1 knockdown U251 cells in the absence or presence of 200 nm cucurbitacin I for 48 h. E, Western blot analysis showing expression of LC3B and Beclin 1 in U251 cells treated with 200 nm cucurbitacin I or DMSO (<0.1%) in the absence or presence of 20 nm 3-methyladenine for 48 h. β-actin served as the loading control in the Western blot analysis. F, coimmunoprecipitation analysis of Bcl-2 and Beclin 1/hVps34 in U251 cells treated with 200 nm cucurbitacin I or DMSO (<0.1%) for 48 h. IP, immunoprecipitation.
Inhibition of Autophagy Enhanced Cucurbitacin I-induced Apoptosis in GBM Cells
Several studies have demonstrated that autophagy may serve as a protective mechanism in tumor cells and that therapy-induced cell death can be potentiated through autophagy inhibition (46, 47). To determine the biological significance of autophagy on cucurbitacin I-mediated apoptotic cell death, the autophagy inhibitor CQ was utilized to prevent autophagy at a later stage. As shown in Fig. 6A, CQ significantly enhanced cucurbitacin I-induced suppression of GBM cells. In agreement with this observation, cucurbitacin I-induced apoptotic cell death was augmented in the presence of CQ, which was demonstrated by cell death detection ELISAPlus assay (Fig. 6B) and TUNEL assay (Fig. 6 C).
FIGURE 6.

Inhibition of autophagy enhanced cucurbitacin I-induced apoptosis in GBM cells. A, a CCK-8 assay was performed to assess cell viability in GBM cells treated with different concentrations of cucurbitacin I in the presence or absence of CQ for 48 h. Data are mean ± S.D. *, p < 0.05 compared with the control group. B, apoptotic ratios were assessed by cell death detection ELISAPlus assay in GBM cells treated with cucurbitacin I in the presence or absence of CQ for 48 h. Data are mean ± S.D. **, p < 0.01. C, apoptotic ratios were assessed by TUNEL assay in GBM cells. Data are mean ± S.D. *, p < 0.05. GBM cells transiently transfected with control siRNA and Beclin 1 siRNA were treated with cucurbitacin I (100 nm) for 48 h. D, a CCK-8 assay was performed to assess cell viability. E, apoptosis was assessed by cell death detection ELISAPlus assay. Data are mean ± S.D. *, p < 0.05.
Considering the effects of CQ on lysosomes that are independent of autophagy, a genetic approach was applied to block the formation of autophagosomes by knocking down the expression of beclin 1 through RNA interference. A CCK-8 assay was utilized to determine cell viability, and a cell death detection ELISAPlus assay was performed to examine the apoptosis level in GBM cells. Our data demonstrated that silencing the expression of beclin 1 markedly enhanced the cucurbitacin I-induced inhibition of GBM cells growth (Fig. 6D) and promoted apoptotic cell death (Fig. 6E). These findings indicate that cucurbitacin I-induced autophagy plays a protective role against apoptotic cell death and that inhibition of cucurbitacin I-induced autophagy enhanced the potential for apoptosis after cucurbitacin I treatment in GBM cells.
CQ Enhanced Cucurbitacin I-induced Tumor Growth Inhibition in a Xenograft Tumor Model
To further determine whether autophagy blockade can enhance the effects of cucurbitacin I in vivo, the impact of the cucurbitacin I/CQ combination on the growth of U251 xenografts was determined. No major side effects were noted throughout the study. As shown in Fig. 7A, average tumor volumes at the end of the study were as follows: control, 616 mm3 (± 130); CQ, 580 mm3 (± 107); cucurbitacin I, 346 mm3 (± 79); and combination, 220 mm3 (± 62). Although no statistically significant difference was found between the CQ and control arms (p = 0.25), the differences in tumor volume between the cucurbitacin I and control, combination and control, and combination and cucurbitacin I arms were significant (p < 0.05). Furthermore, combination-treated tumors exhibited a significantly (p < 0.01) lower average tumor weight at study termination than the control (Fig. 7B). Moreover, there was no effect on the body weights of mice (Fig. 7C). Finally, a pronounced decrease in tumor cell proliferation (Ki67) and increase in apoptosis (TUNEL) were noted in combination-treated xenografts (Fig. 7D). These data recapitulated the observations made in vitro and showed that autophagy blockade sensitized the cucurbitacin I killing effect on GBM.
FIGURE 7.

CQ enhanced cucurbitacin I-induced tumor growth inhibition in a U251 cell xenograft tumor model. A–C, mice were sacrificed 15 days after the indicated treatments, and tumor volume, tumor weight, and mouse body weight were measured. D, TUNEL assay and immunohistochemical staining result of Ki67 on tumor sections. All data are mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar = 50 μm.
DISCUSSION
Novel therapeutic strategies that efficaciously target GBM are needed desperately to improve the currently unfavorable outcome of GBM patients. Multiple studies have provided compelling evidence of a critical role for aberrant JAK2/STAT3 pathway signaling in these aggressive malignancies (15–18). In this study, we first showed apoptotic cell death in GBM after cucurbitacin I treatment. Second, we offered that protective autophagy was induced in GBM treated with cucurbitacin I. Third, we explored the mechanisms by which autophagy was induced by cucurbitacin I. Finally, we studied the biological role of autophagy in the response of GBM cells to cucurbitacin I.
There is a lot of evidence showing that cucurbitacin I, a selective inhibitor of JAK2/STAT3, inhibits tumor cell survival, growth, and invasion in a large group of GBM (48–50). As found previously, our findings showed that cucurbitacin I significantly suppressed GBM growth and induced apoptosis through regulating bcl-2 family proteins in vitro and in vivo. Moreover, a slight effect was observed on human astrocytes after cucurbitacin I treatment.
Autophagy maintains cancer survival under metabolic stress conditions and mediates resistance to anticancer therapies such as radiation and chemotherapy (19, 51). Numerous anticancer agents have been reported to trigger cellular autophagy. In this study, the appearance of characteristic autophagosomes, pronounced conversion of LC3-I to LC3-II, massive accumulation of LC3B and GFP-LC3 punctuates, as well as LC3B expression increase on tumor sections provided strong evidence that autophagy was induced after cucurbitacin I treatment.
Notwithstanding mTOR acting as a major checkpoint in signaling pathways regulating autophagy-integrated signaling through the PI3K/AKT pathway (52), our studies relevant to the mechanisms of autophagy indicated that the PI3K/AKT pathway was not involved in cucurbitacin I-induced autophagy. Phosphorylation of AMPK activates downstream signaling that leads to mTOR inhibition and triggers autophagy (36). Our data showed that treatment with cucurbitacin I in GBM cells had a stronger effect on the activation of AMPK/mTOR/p70S6K signaling, indicating that the AMPK/mTOR/p70S6K pathway is involved in cucurbitacin I-induced autophagy.
Further investigation documented that cucurbitacin I treatment inhibited the JAK2/STAT3 pathway, accompanied by decreased levels of HIF-1α, which echoed the findings that the JAK2/STAT3 pathway positive regulates HIF-1α in a variety of cancer cell types (37–40). HIF-1α plays an important role in hypoxia-induced autophagy, requiring specific activation of an autophagy-inducing molecule, BNIP3, which is activated by HIF-1 only in hypoxic cells (53). However, under normoxic conditions, we found that overexpression of HIF-1α induced by FG-4497 strongly prevented cucurbitacin I-induced autophagy and down-regulation of bcl-2. Further data indicated that the interaction of Bcl-2 with Beclin 1/hVps34 in GBM cells was dissociated under cucurbitacin I treatment. Because bcl-2, transcriptionally regulated by HIF-1α, inhibits autophagy by binding to beclin 1/hVps34 (43–45), our study suggests that a decrease in bcl-2 resulting from transcription inhibition because of a cucurbitacin I-induced inhibition of HIF-1α protein synthesis diminished a bcl-2 and beclin 1/hVps34 association, thereby triggering autophagy.
Autophagy is known to have dual functions. On one hand, it can serve as a cytoprotective mechanism, allowing tumor cells to survive under conditions of metabolic stress and hypoxia and to escape anticancer treatment-induced cell death. On the other hand, autophagy induced by therapeutic interventions can cause the death of cancer cells that are resistant to apoptosis (54, 55). Certain anticancer agents, i.e. sirolimus, phenethyl isothiocyanate, and nilotinib, can induce autophagic cell death (56–58), whereas others, i.e. arginine deiminase, timosaponin A-III, and AZD8055, exert protective autophagy that antagonizes apoptotic cell death (59–61). In this study, we demonstrated that cucurbitacin I triggered autophagy in GBM cells both in vitro and in vivo. We found that inhibition of autophagy by CQ or beclin 1 knockdown markedly increased cucurbitacin I-induced apoptotic cell death, suggesting that cucurbitacin I-induced autophagy exerted a mechanism that enabled tumor cells to survive under anticancer therapy.
CQ, an autophagy inhibitor, prolongs median survival and decreases the rate of death for patients with GBM (62). Further experiments were performed to extend our in vivo results to evaluate the effect of cucurbitacin I and CQ treatment in mouse xenograft models. As a result, we recapitulated the observations made in vitro and showed that autophagy blockade enhances the anti-GBM treatment effects of cucurbitacin I.
In conclusion, this study demonstrated that cucurbitacin I induced autophagy that protected GBM cells from apoptotic death involving AMPK/mTOR/p70S6K signaling activation and decreased HIF-1α. The down-regulation of HIF-1α played pivotal roles in cucurbitacin I-induced autophagy. We also demonstrated that autophagy inhibition by knockdown of beclin 1 or treatment with CQ sensitized GBM cells to cucurbitacin I-induced apoptosis. These findings provide advantageous insights for the development of efficacious therapies for GBM by combining cucurbitacin I and CQ, which might represent a promising avenue with higher efficacy for GBM patients.
Acknowledgment
We thank Professor Xun Qu for comments and advice.
This work was supported by Natural Science Foundation of China Grant 81172403, by Independent Innovation Foundation of Shandong University Grant IIFSDU2009TS067, and by Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province Grant BS2010YY022.
- GBM
- glioblastoma multiforme
- HIF
- hypoxia-inducible factor
- CQ
- chloroquine
- DMSO
- dimethyl sulfoxide
- AMPK
- AMP-activated protein kinase
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1. Kim D. J., Chan K. S., Sano S., Digiovanni J. (2007) Signal transducer and activator of transcription 3 (STAT3) in epithelial carcinogenesis. Mol. Carcinog. 46, 725–731 [DOI] [PubMed] [Google Scholar]
- 2. Chan K. S., Sano S., Kataoka K., Abel E., Carbajal S., Beltran L., Clifford J., Peavey M., Shen J., Digiovanni J. (2008) Forced expression of a constitutively active form of STAT3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene 27, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 3. Ling X., Arlinghaus R. B. (2005) Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 65, 2532–2536 [DOI] [PubMed] [Google Scholar]
- 4. Li Y., Du H., Qin Y., Roberts J., Cummings O. W., Yan C. (2007) Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 67, 8494–8503 [DOI] [PubMed] [Google Scholar]
- 5. Garner J. M., Fan M., Yang C. H., Du Z., Sims M., Davidoff A. M., Pfeffer L. M. (2013) Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor κB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J. Biol. Chem. 288, 26167–26176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao W., Jaganathan S., Turkson J. (2010) A cell-permeable STAT3 SH2 domain mimetic inhibits STAT3 activation and induces antitumor cell effects in vitro. J. Biol. Chem. 285, 35855–35865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turkson J., Zhang S., Mora L. B., Burns A., Sebti S., Jove R. (2005) A novel platinum compound inhibits constitutive STAT3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 280, 32979–32988 [DOI] [PubMed] [Google Scholar]
- 8. Redell M. S., Ruiz M. J., Alonzo T. A., Gerbing R. B., Tweardy D. J. (2011) STAT3 signaling in acute myeloid leukemia. Ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule STAT3 inhibitor. Blood 117, 5701–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blaskovich M. A., Sun J., Cantor A., Turkson J., Jove R., Sebti S. M. (2003) Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 63, 1270–1279 [PubMed] [Google Scholar]
- 10. Jing N., Tweardy D. J. (2005) Targeting STAT3 in cancer therapy. Anticancer Drugs 16, 601–607 [DOI] [PubMed] [Google Scholar]
- 11. Molavi O., Ma Z., Hamdy S., Lai R., Lavasanifar A., Samuel J. (2008) Synergistic antitumor effects of CpG oligodeoxynucleotide and STAT3 inhibitory agent JSI-124 in a mouse melanoma tumor model. Immunol. Cell Biol. 86, 506–514 [DOI] [PubMed] [Google Scholar]
- 12. Chen J. C., Chiu M. H., Nie R. L., Cordell G. A., Qiu S. X. (2005) Cucurbitacins and cucurbitane glycosides. Structures and biological activities. Nat. Prod. Rep. 22, 386–399 [DOI] [PubMed] [Google Scholar]
- 13. Louis D. N., Ohgaki H., Wiestler O. D., Cavenee W. K., Burger P. C., Jouvet A., Scheithauer B. W., Kleihues P. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Meir E. G., Hadjipanayis C. G., Norden A. D., Shu H. K., Wen P. Y., Olson J. J. (2010) Exciting new advances in neuro-oncology. The avenue to a cure for malignant glioma. CA-Cancer J. Clin. 60, 166–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peñuelas S., Anido J., Prieto-Sánchez R. M., Folch G., Barba I., Cuartas I., García-Dorado D., Poca M. A., Sahuquillo J., Baselga J., Seoane J. (2009) TGF-β increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 15, 315–327 [DOI] [PubMed] [Google Scholar]
- 16. Wang H., Lathia J. D., Wu Q., Wang J., Li Z., Heddleston J. M., Eyler C. E., Elderbroom J., Gallagher J., Schuschu J., MacSwords J., Cao Y., McLendon R. E., Wang X. F., Hjelmeland A. B., Rich J. N. (2009) Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 27, 2393–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao Y., Lathia J. D., Eyler C. E., Wu Q., Li Z., Wang H., McLendon R. E., Hjelmeland A. B., Rich J. N. (2010) Erythropoietin receptor signaling through STAT3 is required for glioma stem cell maintenance. Genes Cancer 1, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carro M. S., Lim W. K., Alvarez M. J., Bollo R. J., Zhao X., Snyder E. Y., Sulman E. P., Anne S. L., Doetsch F., Colman H., Lasorella A., Aldape K., Califano A., Iavarone A. (2010) The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Z., Klionsky D. J. (2010) Eaten alive. A history of macroautophagy. Nat. Cell Biol. 12, 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rubinsztein D. C., Gestwicki J. E., Murphy L. O., Klionsky D. J. (2007) Potential therapeutic applications of autophagy. Nat. Rev. Drug Dis. 6, 304–312 [DOI] [PubMed] [Google Scholar]
- 21. Lum J. J., Bauer D. E., Kong M., Harris M. H., Li C., Lindsten T., Thompson C. B. (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 [DOI] [PubMed] [Google Scholar]
- 22. Degenhardt K., Mathew R., Beaudoin B., Bray K., Anderson D., Chen G., Mukherjee C., Shi Y., Gélinas C., Fan Y., Nelson D. A., Jin S., White E. (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim K. W., Mutter R. W., Cao C., Albert J. M., Freeman M., Hallahan D. E., Lu B. (2006) Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J. Biol. Chem. 281, 36883–36890 [DOI] [PubMed] [Google Scholar]
- 24. Apel A., Herr I., Schwarz H., Rodemann H. P., Mayer A. (2008) Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 68, 1485–1494 [DOI] [PubMed] [Google Scholar]
- 25. Qadir M. A., Kwok B., Dragowska W. H., To K. H., Le D., Bally M. B., Gorski S. M. (2008) Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res. Treat. 112, 389–403 [DOI] [PubMed] [Google Scholar]
- 26. Li J., Hou N., Faried A., Tsutsumi S., Takeuchi T., Kuwano H. (2009) Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 16, 761–771 [DOI] [PubMed] [Google Scholar]
- 27. Livesey K. M., Tang D., Zeh H. J., Lotze M. T. (2009) Autophagy inhibition in combination cancer treatment. Curr. Opin. Investig. Drugs 10, 1269–1279 [PubMed] [Google Scholar]
- 28. Maycotte P., Thorburn A. (2011) Autophagy and cancer therapy. Cancer Biol. Ther. 11, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen N., Karantza V. (2011) Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 11, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ogier-Denis E., Codogno P. (2003) Autophagy. A barrier or an adaptive response to cancer. Biochim. Biophys. Acta 1603, 113–128 [DOI] [PubMed] [Google Scholar]
- 31. Gozuacik D., Kimchi A. (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23, 2891–2906 [DOI] [PubMed] [Google Scholar]
- 32. Mizushima N., Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy 3, 542–545 [DOI] [PubMed] [Google Scholar]
- 33. Yang Z., Klionsky D. J. (2009) An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335, 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roca H., Varsos Z., Pienta K. J. (2008) CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J. Biol. Chem. 283, 25057–25073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He C., Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meley D., Bauvy C., Houben-Weerts J. H., Dubbelhuis P. F., Helmond M. T., Codogno P., Meijer A. J. (2006) AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 281, 34870–34879 [DOI] [PubMed] [Google Scholar]
- 37. Park J. H., Darvin P., Lim E. J., Joung Y. H., Hong D. Y., Park E. U., Park S. H., Choi S. K., Moon E. S., Cho B. W., Park K. D., Lee H. K., Kim M. J., Park D. S., Chung I. M., Yang Y. M. (2012) Hwanggeumchal sorghum induces cell cycle arrest, and suppresses tumor growth and metastasis through Jak2/STAT pathways in breast cancer xenografts. PloS ONE 7, e40531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kandala P. K., Srivastava S. K. (2012) Diindolylmethane suppresses ovarian cancer growth and potentiates the effect of cisplatin in tumor mouse model by targeting signal transducer and activator of transcription 3 (STAT3). BMC Med. 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anglesio M. S., George J., Kulbe H., Friedlander M., Rischin D., Lemech C., Power J., Coward J., Cowin P. A., House C. M., Chakravarty P., Gorringe K. L., Campbell I. G., Australian Ovarian Cancer Study Group, Okamoto A., Birrer M. J., Huntsman D. G., de Fazio A., Kalloger S. E., Balkwill F., Gilks C. B., Bowtell D. D. (2011) IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin. Cancer Res. 17, 2538–2548 [DOI] [PubMed] [Google Scholar]
- 40. Horiguchi A., Asano T., Kuroda K., Sato A., Asakuma J., Ito K., Hayakawa M., Sumitomo M. (2010) STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 102, 1592–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reischl S., Li L., Walkinshaw G., Flippin L. A., Marti H. H., Kunze R. (2014) Inhibition of HIF prolyl-4-hydroxylases by FG-4497 reduces brain tissue injury and edema formation during ischemic stroke. PloS ONE 9, e84767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robinson A., Keely S., Karhausen J., Gerich M. E., Furuta G. T., Colgan S. P. (2008) Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 134, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carmeliet P., Dor Y., Herbert J. M., Fukumura D., Brusselmans K., Dewerchin M., Neeman M., Bono F., Abramovitch R., Maxwell P., Koch C. J., Ratcliffe P., Moons L., Jain R. K., Collen D., Keshert E. (1998) Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394, 485–490 [DOI] [PubMed] [Google Scholar]
- 44. Wei Y., Pattingre S., Sinha S., Bassik M., Levine B. (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 [DOI] [PubMed] [Google Scholar]
- 46. Meijer A. J., Codogno P. (2009) Autophagy. Regulation and role in disease. Crit. Rev. Clin. Lab. Sci. 46, 210–240 [DOI] [PubMed] [Google Scholar]
- 47. Maiuri M. C., Zalckvar E., Kimchi A., Kroemer G. (2007) Self-eating and self-killing. Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752 [DOI] [PubMed] [Google Scholar]
- 48. Stechishin O. D., Luchman H. A., Ruan Y., Blough M. D., Nguyen S. A., Kelly J. J., Cairncross J. G., Weiss S. (2013) On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro-oncology 15, 198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Banerjee S., Byrd J. N., Gianino S. M., Harpstrite S. E., Rodriguez F. J., Tuskan R. G., Reilly K. M., Piwnica-Worms D. R., Gutmann D. H. (2010) The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res. 70, 1356–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Su Y., Li G., Zhang X., Gu J., Zhang C., Tian Z., Zhang J. (2008) JSI-124 inhibits glioblastoma multiforme cell proliferation through G2/M cell cycle arrest and apoptosis augment. Cancer Biol. Ther. 7, 1243–1249 [DOI] [PubMed] [Google Scholar]
- 51. Marino M. L., Pellegrini P., Di Lernia G., Djavaheri-Mergny M., Brnjic S., Zhang X., Hägg M., Linder S., Fais S., Codogno P., De Milito A. (2012) Autophagy is a protective mechanism for human melanoma cells under acidic stress. J. Biol. Chem. 287, 30664–30676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 53. Zhang H., Bosch-Marce M., Shimoda L. A., Tan Y. S., Baek J. H., Wesley J. B., Gonzalez F. J., Semenza G. L. (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54. Shintani T., Klionsky D. J. (2004) Autophagy in health and disease. A double-edged sword. Science 306, 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Høyer-Hansen M., Jäättelä M. (2008) Autophagy. An emerging target for cancer therapy. Autophagy 4, 574–580 [DOI] [PubMed] [Google Scholar]
- 56. Takeuchi H., Kondo Y., Fujiwara K., Kanzawa T., Aoki H., Mills G. B., Kondo S. (2005) Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 65, 3336–3346 [DOI] [PubMed] [Google Scholar]
- 57. Bommareddy A., Hahm E. R., Xiao D., Powolny A. A., Fisher A. L., Jiang Y., Singh S. V. (2009) Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 69, 3704–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu H. C., Lin C. S., Tai W. T., Liu C. Y., Shiau C. W., Chen K. F. (2013) Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation. J. Biol. Chem. 288, 18249–18259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim R. H., Coates J. M., Bowles T. L., McNerney G. P., Sutcliffe J., Jung J. U., Gandour-Edwards R., Chuang F. Y., Bold R. J., Kung H. J. (2009) Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 69, 700–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sy L. K., Yan S. C., Lok C. N., Man R. Y., Che C. M. (2008) Timosaponin A-III induces autophagy preceding mitochondria-mediated apoptosis in HeLa cancer cells. Cancer Res. 68, 10229–10237 [DOI] [PubMed] [Google Scholar]
- 61. Huang S., Yang Z. J., Yu C., Sinicrope F. A. (2011) Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J. Biol. Chem. 286, 40002–40012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sotelo J., Briceño E., López-González M. A. (2006) Adding chloroquine to conventional treatment for glioblastoma multiforme. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343 [DOI] [PubMed] [Google Scholar]