

Background: Triokinase, which phosphorylates dihydroxyacetone and fructose-derived glyceraldehyde, remains molecularly unidentified.
Results: Human DAK gene encodes homodimeric triokinase/FMN cyclase formed by two-domain subunits. Although kinase activity requires intact homodimers, cyclase requires only a truncated, single domain subunit.
Conclusion: Triokinase/FMN cyclase identity and bifunctionality are established.
Significance: This study molecularly dissects a bifunctional enzyme of unusual specificity and finishes the molecular identification of fructose pathway enzymes.
Keywords: FAD; Fructose Metabolism; Molecular Docking; Molecular Dynamics; Mutagenesis, Site-specific; Bifunctional Kinase/Cyclase; Cyclic FMN; DAK Gene; Dihydroxyacetone Phosphorylation; Glyceraldehyde Phosphorylation
Abstract
Mammalian triokinase, which phosphorylates exogenous dihydroxyacetone and fructose-derived glyceraldehyde, is neither molecularly identified nor firmly associated to an encoding gene. Human FMN cyclase, which splits FAD and other ribonucleoside diphosphate-X compounds to ribonucleoside monophosphate and cyclic X-phosphodiester, is identical to a DAK-encoded dihydroxyacetone kinase. This bifunctional protein was identified as triokinase. It was modeled as a homodimer of two-domain (K and L) subunits. Active centers lie between K1 and L2 or K2 and L1: dihydroxyacetone binds K and ATP binds L in different subunits too distant (≈14 Å) for phosphoryl transfer. FAD docked to the ATP site with ribityl 4′-OH in a possible near-attack conformation for cyclase activity. Reciprocal inhibition between kinase and cyclase reactants confirmed substrate site locations. The differential roles of protein domains were supported by their individual expression: K was inactive, and L displayed cyclase but not kinase activity. The importance of domain mobility for the kinase activity of dimeric triokinase was highlighted by molecular dynamics simulations: ATP approached dihydroxyacetone at distances below 5 Å in near-attack conformation. Based upon structure, docking, and molecular dynamics simulations, relevant residues were mutated to alanine, and kcat and Km were assayed whenever kinase and/or cyclase activity was conserved. The results supported the roles of Thr112 (hydrogen bonding of ATP adenine to K in the closed active center), His221 (covalent anchoring of dihydroxyacetone to K), Asp401 and Asp403 (metal coordination to L), and Asp556 (hydrogen bonding of ATP or FAD ribose to L domain). Interestingly, the His221 point mutant acted specifically as a cyclase without kinase activity.
Introduction
Triokinase (EC 2.7.1.28) catalyzes the third and final step of the classical Hers pathway for fructose metabolism: the 1-OH group of the sugar is phosphorylated by fructokinase, the product fructose 1-phosphate is converted to dihydroxyacetone phosphate and d-glyceraldehyde by aldolase B, and finally d-glyceraldehyde (GA)4 is phosphorylated to d-glyceraldehyde 3-phosphate by triokinase (1). In the so-called GA crossroads, triokinase phosphorylates GA, overriding competing enzymes (2). Despite the relevance of the Hers pathway in liver (1–5) and the much heralded concerns on the effects of the high consumption of fructose in human health (5–12), mammalian triokinase, contrary to fructokinase and aldolase B (13, 14), is still without an established molecular identity, and it is not yet firmly associated to a specific gene. However, there are a few reports suggesting that mammalian triokinase could be a product of DAK (15, 16), a gene named by homology to yeast and bacterial genes coding for ATP-dependent dihydroxyacetone (DHA) kinases (17, 18), some of which are known to be active as GA kinases too (18, 19).
FAD-AMP lyase (cyclic FMN-forming) or FMN cyclase (EC 4.6.1.15) was discovered in rat liver (20). It is a Mn2+-dependent phosphorus-oxygen lyase that catalyzes intramolecular reactions of some ribonucleoside diphosphate-X (NDP-X) compounds, yielding a ribonucleoside monophosphate (NMP) and a five-atom cyclic phosphodiester of X as products. Its best substrates are FAD and ADP-glucose (21). As the latter does not occur in mammals, the enzyme is named after its activity on FAD, which forms AMP and the cyclic phosphodiester riboflavin cyclic 4′,5′-phosphate (cyclic FMN (cFMN)). The biological role of this unusual flavin is unknown, but it is known to be present in rat liver (22) and in the posterior flagellum of swarmers of the brown alga Scytosiphon lomentaria (23). The peptide mass fingerprint of rat liver FMN cyclase identifies it as the ortholog of a protein product of the human DAK gene, which has been cloned as cDNA and expressed in bacteria. Both this human recombinant protein and the native protein purified from rat liver show activity as Mg2+-dependent DHA kinases and Mn2+-dependent FMN cyclases (24). In fact, this may be a general feature of DHA kinases because that from Citrobacter sp. also acts as FMN cyclase albeit with lesser catalytic efficiency than the mammalian enzymes. In relation to this, it has been argued that the FMN cyclase activity of DHA kinases represents an instance of metal-dependent catalytic promiscuity (25).
Besides the unexpected duality of DHA kinase/FMN cyclase, the biochemistry and the biological role of these proteins are intriguing. The crystal structure of Citrobacter sp. DHA kinase has been determined in complex with DHA and the ATP analog ANP (26). It is a homodimeric protein of two-domain (K and L) subunits (1 and 2) with two active sites per dimer, one located between K1 and L2 domains and the other located between K2 and L1. DHA binds covalently to the His210 side chain in the K domains, and the ATP analog binds noncovalently to the L domains. The ATP binding site and the L domain define a unique kinase fold (15, 27). However, according to the crystal structure, ATP and DHA would occupy positions too distant (≈14 Å) for the phosphoryl transfer to take place, and it has been suggested that domain mobility may be required for kinase activity (15). This is different from Escherichia coli and some other bacterial DHA kinases that are not dependent on ATP but on a phosphoprotein of the phosphoenolpyruvate:sugar phosphotransferase system (PTS), use ADP as a permanently bound cofactor and intermediate donor for DHA phosphorylation, and are structured as heterotetramers composed of two DhaK and two DhaL subunits (28–30). These separate subunits are homologous, both in sequence and function, to the K and L domains, respectively, of the subunits of the DHA kinase of Citrobacter sp. However, in the E. coli heterotetramer, the donor intermediate ADP and DHA are well positioned for the phosphoryl transfer to take place directly, contrary to the homodimeric DHA kinase of Citrobacter sp. (30).
In prokaryotes and lower eukaryotes, DHA kinase plays a role in the metabolism of DHA formed from glycerol (31–33) and in detoxification of high DHA doses. For instance, DHA is toxic for Saccharomyces cerevisiae devoid of DHA kinases by gene deletion (17) and for the parasites Trypanosoma brucei (34, 35) and Plasmodium falciparum (36), which are naturally devoid of any ATP-dependent DHA kinase ortholog in their genomes. In humans and in general in mammals where an endogenous source of DHA has not been reported, this compound is also potentially toxic (37), but when administered exogenously (38–42), it can be efficiently disposed of through DHA kinase (43).
Finally, another intriguing feature of DHA kinase/FMN cyclase is the known interaction of the human protein with the RNA helicase melanoma differentiation-associated gene 5 (MDA5), which blocks the signaling role of the helicase in the innate antiviral response mediated by β-interferon promoter activation (44, 45). In a recent study of liver protein profiling of chronic hepatitis C patients, two protein spots identified as DHA kinase/FMN cyclase isoforms were part of a small group of proteins that showed differential expression at the protein level when patients who were nonresponsive to interferon therapy were compared with responsive patients (46).
Driven by the diversity of interesting aspects of these dual activity enzymes, we performed an extensive study of the human protein, including biochemical and in silico experiments. Biochemical evidence is presented for the identity of the DAK gene-encoded DHA kinase/FMN cyclase with human triokinase, which phosphorylates both DHA and GA. Thus, in the following, the protein is named human triokinase/FMN cyclase (hTKFC). Enzyme characterization of hTKFC mutants provided insight into the differential roles of K and L domains and of specific amino acid residues in the kinase and cyclase activities. After modeling homodimeric hTKFC by homology to the crystal structure of Citrobacter DHA kinase, molecular dynamics simulations led to the active site closure with DHA reaching a near-attack conformation over ATP as needed to explain the kinase activity.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise stated, commercial products were purchased from suppliers mentioned elsewhere (21, 24). ITP, GTP, CTP, and UTP were from Sigma. GA was from Fluka, glycerol was from Merck, and glycolaldehyde was from Sigma. Phosphoenolpyruvate and CAPS were from Sigma, and arsenate was from Panreac. MgCl2, MnCl2, acetic acid, HCl, and NaOH were from Merck. Tris was from Roche Applied Science. Molecular weight markers were from Roche Applied Science. Auxiliary enzymes glycerol-3-phosphate dehydrogenase, lactate dehydrogenase, and alkaline phosphatase were from Roche Applied Science, and triose-phosphate isomerase, glyceraldehyde-3-phosphate dehydrogenase, and pyruvate kinase were from Sigma.
Recombinant hTKFC and Mutant Proteins
hTKFC was obtained by expression under isopropyl thiogalactoside induction in E. coli BL21 cells transformed with plasmid pGEX-6P-3-hF2, which encodes a GST fusion protein from which hTKFC was obtained separated from the GST tag by specific proteolysis with PreScission, all carried out essentially as described earlier (24). Mutant versions of hTKFC, either point mutants or truncations, were obtained similarly from pGEX-6P-3 constructs prepared as described below.
Mutagenesis
The coding sequence of the L domain (amino acids 1–339) was amplified from plasmid pGEX-6P-3-hF2 with a forward primer that added an EcoRI site before the initiation codon (CATCTCGAATTCCATGACCTCCAAGAAGCTG) and a reverse primer that added a stop codon and a SalI site after codon 339 (AGTAGCGTCGACTCACACGTTAGGCCAGGCTGCTGC). In a similar way, the coding sequence of the K domain (amino acids 359–575) was amplified with primers that added a BamHI site before amino acid codon 359 (CCGCATGGATCCGAGGCCCCTGATTCCACTGCTGC) and an EcoRI site after the hTKFC stop codon (TTACAGGAATTCCTAGCTCTGCAAGACCTCCAAG). The two amplicons were cut and separately inserted between the corresponding restriction sites of plasmid pGEX-6P-3 (Amersham Biosciences) to obtain plasmids pGEX-6P-3-hTKFC-L and pGEX-6P-3-hTKFC-K, respectively. Point mutants were constructed following the QuikChange protocol (Stratagene) using mutagenic primer pairs and pGEX-6P-3-hF2 as template. The correctness of the truncation constructs and all point mutants was confirmed by sequencing the two chains of the complete coding sequence (Servicio de Genómica, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid, Madrid, Spain).
Kinase Kinetic Assays
The kinase assays of wild-type and mutant hTKFC were performed continuously in a Shimadzu MPS-2000 ultraviolet-visible spectrophotometer, recording at 340 nm the conversion of NAD+ to NADH (or vice versa) with ϵ = 6220 m−1 cm−1 (or vice versa with ϵ = −6220 m−1 cm−1) in enzyme-coupled reactions at 37 °C. In every case, phosphorylation of 1 mol of substrate yielded 1 mol of converted coenzyme. Measurements were taken under conditions of linearity with respect to incubation time and to enzyme amount. Among other things, this ensured that the reaction rate was not limited by auxiliary enzymes in coupled assays.
In the DHA kinase assay, triose phosphorylation (DHA + ATP → DHAP + ADP) was coupled to glycerol-3-phosphate dehydrogenase as indicator enzyme (DHAP + NADH → glycerol 3-phosphate + NAD+). Unless otherwise stated, the standard reaction mixtures contained 100 mm Tris-HCl, pH 7.5 (adjusted at 37 °C), 0.18 mm NADH, 10 mm MgCl2, 0.5 mm DHA, 5 mm ATP, 3 units ml−1 glycerol-3-phosphate dehydrogenase, and 0.1 mg ml−1 bovine serum albumin. After equilibrating the mixtures for a few minutes at 37 °C while recording A340, the reaction was initiated by addition of DHA from a concentrated stock. The negative linear slope was recorded for several minutes to determine the initial rate of the kinase reaction. In this assay, GA phosphorylation does not give a measurable signal, so the assay was applied to evaluate DHA kinase inhibition by GA.
In most of the GA kinase assays, triose phosphorylation (GA + ATP → GAP + ADP) was coupled to triose-phosphate isomerase as auxiliary enzyme (GAP → DHAP) and glycerol-3-phosphate dehydrogenase as indicator enzyme (see above). Therefore, unless otherwise stated, the standard reaction mixtures and the procedure were like those for the DHA kinase assay except that GA substituted for DHA, and 15 units ml−1 triose-phosphate isomerase was included. Because DHA phosphorylation also gives a measurable signal in this assay, it was not applied to evaluate GA kinase inhibition by DHA.
In an alternative GA kinase assay, which is specific for GA phosphorylation, the reaction was coupled to d-glyceraldehyde-3-phosphate dehydrogenase in the presence of arsenate instead of phosphate (GAP + NAD+ + arsenate → 1-arseno-3-phosphoglycerate + NADH). The irreversibility of the reaction was ensured by the nonenzymatic hydrolysis of arsenophosphoglycerate (1-arseno-3-phosphoglycerate + H2O → 3-phosphoglycerate + arsenate) (47). Unless otherwise stated, the standard reaction mixtures contained 100 mm Tris-HCl, pH 7.5 (adjusted at 37 °C), 0.5 mm NAD+, 10 mm MgCl2, 0.5 mm GA, 5 mm ATP, 5 mm sodium arsenate, 5 units ml−1 d-glyceraldehyde-3-phosphate dehydrogenase, and 0.1 mg ml−1 bovine serum albumin. After equilibrating the mixtures for a few minutes at 37 °C while recording A340, the reaction was initiated by addition of ATP from a concentrated stock. The positive linear slope was recorded for several minutes to determine the initial rate of the kinase reaction. In this assay, DHA phosphorylation does not give a measurable signal, so the assay was applied to evaluate GA kinase inhibition by DHA.
To test the (lack of) phosphorylation activities on glycerol or glycolaldehyde, an acceptor-independent assay was used in which the formation of ADP was coupled to pyruvate kinase (ADP + phosphoenolpyruvate → ATP + pyruvate) and lactate dehydrogenase (pyruvate + NADH → lactate + NAD+). The reaction mixtures contained 100 mm Tris-HCl, pH 7.5 (adjusted at 37 °C), 0.18 mm NADH, 10 mm MgCl2, 1 mm phosphoenolpyruvate, up to 2.5 mm glycerol or glycolaldehyde, 5 mm ATP, 4 units ml−1 pyruvate kinase, 5 units ml−1 lactate dehydrogenase, and 0.1 mg ml−1 bovine serum albumin. After equilibrating the mixtures for a few minutes at 37 °C while recording A340, the reaction was initiated by addition of glycerol or glycolaldehyde from a concentrated stock. The negative linear slope was recorded for several minutes to determine the initial rate of the kinase reaction. Because phosphorylating activity was not detected, the same procedure was used to assay DHA kinase as a positive control.
Controls without hTKFC were processed in parallel, and any hTKFC-independent NAD+ reduction or NADH oxidation was used to correct data collected in the presence of hTKFC. One activity unit is defined as the amount of enzyme catalyzing the phosphorylation of 1 μmol of substrate/min.
Cyclizing Lyase Kinetic Assays
Biochemical details of all the cyclizing lyase reactions of hTKFC were discussed earlier (21). The enzyme assays of wild-type and mutant hTKFC were performed discontinuously, following by HPLC the conversion of the substrate to product at 37 °C, under conditions of linearity with respect to incubation time and to enzyme amount.
In the assay of FMN cyclase (FAD → cFMN + AMP), the product evaluated was cFMN. Unless otherwise indicated, the standard reaction mixtures contained 50 mm Tris-HCl, pH 7.5 (adjusted at 37 °C), 500 μm FAD, 6 mm MnCl2, and 1 mg ml−1 bovine serum albumin. In cyclizing lyase assays with other substrates (NDP-X → cyclic phosphodiester of X + NMP), the product evaluated was the NMP moiety formed, i.e. the leaving group of the reaction after its alkaline phosphatase-dependent conversion to the corresponding nucleoside (NMP + H2O → N + P where N represents nucleoside and P represents phosphate), which separates better from the NDP-X substrate in the HPLC assay. In the standard reaction mixtures, the NDP-X substrate substituted for FAD, and 7 units ml−1 alkaline phosphatase was included. All the reaction mixtures were preincubated for 10 min at 37 °C before initiating the reaction by addition of substrate from a concentrated stock. At predetermined intervals, aliquots of 20–100 μl were taken from the reaction mixtures, immediately mixed with 4–20 μl of 0.2 m EDTA, put on a boiling bath for 2 min, cooled down on ice, and centrifuged for 2 min at 11,000 rpm. The supernatants were frozen at −20 °C until ready for HPLC analysis (see below).
For every cyclizing lyase assay, initial rates of reaction were derived from the linear slopes of substrate-to-product conversions recorded at three to four incubation time intervals. Strict controls without enzyme were processed in parallel, and any nonenzymatic conversion of substrate to product was subtracted. One activity unit is defined as the amount of enzyme catalyzing the conversion of 1 μmol of substrate into product/min.
HPLC
As stated above, HPLC was used to assay the conversion of nucleotidic substrates to products by the cyclizing lyase activity of hTKFC. A Hewlett-Packard HP-1100 chromatograph was used with a manual injector equipped with a 20-μl injection loop and a diode array detector. Chromatogram analysis was performed with HP ChemStation software.
To assay the conversion of FAD to cFMN, a 150 × 4-mm Kromasil 100 column (Teknokroma, Spain) preceded by a 10 × 4-mm guard column of the same material was used. The isocratic mobile phase was run at a flow of 0.4 ml min−1 and consisted of 10 mm sodium phosphate, pH 7 with 40% (by volume) methanol. The detection was performed at 430 nm at which FAD and cFMN absorbed similarly, which allowed calculation of the concentration of cFMN by applying directly the percent area of the cFMN peak with respect to the sum of FAD and cFMN to the FAD concentration in the initial reaction mixture.
To assay the conversion of NDP-X to the corresponding N nucleoside (formed by alkaline phosphatase dephosphorylation of the NMP product), a 150 × 4-mm Hypersyl ODS column (Teknokroma) was used. The mobile phase was composed of two solutions: A, 5 mm sodium phosphate, pH 7, 20% (by volume) methanol, and 5 mm tetrabutylammonium bromide; B, same as A with 100 mm sodium phosphate. Depending on the NDP-X compound present in the assay, different chromatographic conditions were applied. The detection was performed at 260 nm at which the NDP-X compounds split and the respective N nucleoside products formed absorbed similarly, which allowed calculation of the concentration of N by applying directly the percent area of the N peak with respect to the sum of NDP-X and N to the NDP-X concentration in the initial reaction mixture.
Completion of DHA Kinase Templates for Modeling
The construction of a structural model of hTKFC (see below) was based on two crystal structures of DHA kinase from Citrobacter sp. available in the RCSB Protein Data Bank under accession codes 1UN8 and 1UN9 (26). Both structures are rather similar homodimers of the same 552-amino acid subunit that contains two domains, K or N terminus and L or C terminus, linked together by a long spacer. The K domain consists of two six-stranded α/β-folds, and the L domain is an eight-helix α-barrel. The two subunits are intertwined and adopt an almost linear arrangement of domains, L2-K1-K2-L1.
Protein Data Bank code 1UN8 corresponds to the apo form of the Citrobacter sp. protein crystallized in the absence of substrates and contains ordered water and two 2-myristoyl-3-palmitoyl-phosphatidic acid molecules, each within the α-barrel of one of the L domains. In this file, residues 518–525, 551–552, and some atoms of Met1 and Arg538 are missing. These omissions were added de novo by generating 500 conformations with the loop-modeling routines of Modeler 9.5 (48, 49). The best conformation was selected using the DOPE function for evaluation (50). The coordinates of the completed template are in supplemental File 1 (file 1UN8-c.pdb).
Protein Data Bank code 1UN9 corresponds to the crystal structure of protein bound to the ATP analog ANP, Mg2+ ions, and DHA but contains neither phospholipids nor ordered water. Two ANPs (one in anti and the other in syn conformation) are bound, each to the top of one of the L domains coordinated with two Mg2+ ions, and two DHA molecules are covalently bound in hemiaminal linkage, each to the Nϵ2 of one His220 residue. In this file, residues 516–528 and 551–552 are missing, whereas residues 1 and 538 are both identified as Ala instead of Met and Arg, respectively. These omissions were added or corrected with the loop-modeling routines of Modeler 9.5 except for the two loops 516–528 that, because they are too large for de novo modeling, were modeled later by homology to the completed 1UN8-c structure. In each modeling step, 500 structures were generated, and the best structure was selected using the DOPE function. Finally, the ANP ligand was changed to ATP by substituting oxygen for the imido nitrogen atom. The coordinates of the completed template are in supplemental File 2 (1UN9-c.pdb).
Homology Modeling
The construction of the structural model of dimeric hTKFC was performed with Modeler by homology to the completed templates 1UN8-c and 1UN9-c (see supplemental Files 1 and 2), which contain the structures of Citrobacter sp. apo-DHA kinase and of the same protein in complex with ATP-Mg and DHA, respectively. We wanted to obtain a model containing kinase substrates bound, so the use of 1UN9-c as template was needed. However, the quality of 1UN8-c, as estimated with WHAT_CHECK (51) or PROCHECK (52) programs, was higher. Therefore, the use of both structures as templates was preferred. Concerning other aspects of the procedure, different alternatives were tested: imposing symmetry or not on the Cα and Cβ carbons of the two chains of the dimer and including phospholipids or not within the two L domains. In addition, rather than considering the ligands as rigid bodies, it was preferable to allow some flexibility in their modeling for which their descriptions (topologies and parameters) were needed. ATP is already included in the library of residues of Modeler, but DHA bound to histidine (in fact, the trihydroxyprop-2-yl radical) is not, so it was prepared and added to the library. After constructing several sets of 200 dimeric models of hTKFC followed by their WHAT_CHECK and PROCHECK evaluation, the model chosen was the best evaluated within the set prepared by imposing Cα and Cβ symmetry and omitting phospholipids. This decision was also supported by the lack of evidence for the presence of phospholipids in hTKFC and by the fact that phospholipid removal scarcely affects the catalytic activity of Citrobacter sp. DHA kinase (26). The program Reduce (53) was used to add hydrogens and to establish the most likely protonation state of histidines in the model chosen. The file describing the final model is available as supplemental File 3 (hTKFC_2DHA_2ATP.pdb). The two ATPs are in different conformations, syn and anti, because these are the conformation of the ANPs in the original crystal structure, 1UN9 (26). The two ATPs in the completed 1UN9-c structure (supplemental File 2 (1UN9-c.pdb)) (see above) used as template are also in these conformations.
Docking
For this procedure, supplemental File 3 (hTKFC_2DHA_2ATP.pdb) obtained as described above was further modified as follows. ATP and the trihydroxyprop-2-yl radical were manually removed, and Kollman partial charges were added and nonpolar hydrogens were stripped off with AutoDockTools (or ADT) (54). The resulting file was saved in the pdbqt format, and charges (2+) were assigned to Mg2+ ions. Two sets of grid maps of 35.2 × 35.2 × 33.8 Å, centered on the L2-K1 and K2-L1 active sites, with elements spaced 0.375 Å were made with AutoGrid. The tridimensional model of FAD was generated with Marvin (available from ChemAxon) from the two-dimensional structure of the ligand. Gasteiger charges and atom types were assigned, nonpolar hydrogens were stripped off, and active torsions were defined. The resulting file was saved in the pdbqt format.
FAD docking to the active sites of hTKFC was carried out with AutoDock 4.0 (55). To obtain one FAD pose within each binding site, the Lamarckian genetic algorithm was applied to an initial randomly generated population of 300 conformations, and variation and selection were continued up to a maximum of 105 generations or 108 energy evaluations. The best adapted conformation (low energy) was conserved as a possible FAD pose. In total, 600 poses were obtained for each active site and analyzed as described under “Results and Discussion.” All docking procedures were implemented on an Apple Mac Pro desktop with two Quad-Core Intel Xeon processors and 8 GB of RAM. The computing time needed to obtain 600 FAD poses was about 150 h.
Molecular Dynamics Simulation
This procedure was undertaken to investigate whether, in the hTKFC dimeric model prepared with Modeler by homology, the large ATP-to-DHA distance could be favorably modified in the presence of explicit solvent as a consequence of domain mobility. The structure described in supplemental File 3 (hTKFC_2DHA_2ATP.pdb) was placed in a periodic cell shaped as a rhombic dodecahedron (to diminish the amount of solvent needed with respect to a cubic cell), keeping a minimum 10-Å distance from the protein atoms to the cell walls. Water, modeled as TIP3P molecules (56), and 50 mm NaCl were included in the system. Molecular dynamics with periodic boundary conditions was performed with Gromacs 4.0.4 (57) using the Amber03 force field (58). The topologies and parameters of Nτ-(1,2,3-trihydroxyprop-2-yl)-l-histidine and ATP are available under access codes F-86 and F-91 in the R.E.DD.B. database (59). The full system was first submitted to 2000 energy-minimizing iterations and then equilibrated in three steps of dynamics: 20 ps at constant volume with position restraints on the protein, metals, and ligand atoms; 200 ps at constant pressure (P) and T with the same position restraints; and a final 200 ps of restriction-free dynamics at constant P and T. During the trajectory production phase, P and T were also maintained constant at 1 atm and 300 K, respectively, with the Berendsen algorithms (60). An integration step of 2 fs was applied while keeping constant the length of the covalent bonds of hydrogen in the protein and ligands with the LINCS algorithm (61) and in the water with SETTLE (62). The potential energies of van der Waals interactions were evaluated in every integration step only for atomic pairs closer than 9 Å with smoothing between 8 and 9 Å. The list of the relevant atomic pairs was updated every 10 integration steps. Potential energies for electrostatic interactions were measured in every integration step by the particle mesh Ewald method (63). System coordinates were saved once each 500 steps, thus generating one snapshot per picosecond. The molecular dynamics simulation was run on the same computer used for docking, and the time needed to generate 1 ns of trajectory was about 39 h. A 75-ns trajectory was recorded and is presented as a supplemental movie (hTKFC_2DHA_2ATP.mpg). The analyses of the trajectory and the figures were done with VMD software (64).
RESULTS AND DISCUSSION
Catalytic Specificity of hTKFC as Triokinase and Cyclizing Lyase
The GST-hTKFC fusion protein, adsorbed by tag affinity to GSH-Sepharose, was treated by in-column specific proteolysis to separate hTKFC from the adsorbed GST tag (24). The purified hTKFC protein coeluted with ATP-dependent phosphorylating activities on GA and DHA and with cyclizing lyase activity on FAD, indicating that triokinase and FMN cyclase are manifestations of the same protein (Fig. 1A). Further exploration of hTKFC specificity is shown in Fig. 1B.
FIGURE 1.

Triokinase and cyclizing lyase activities of hTKFC. A, coexpression of GA kinase with DHA kinase and FMN cyclase activities in BL21 cells transformed with plasmid pGEX-6P-3-hF2 that encodes a GST-human DHA kinase/FMN cyclase fusion protein (24). The lysate supernatant was chromatographed on a GSH-Sepharose column to which the GST-labeled protein adsorbed. The recombinant protein was eluted from the column after an overnight incubation with PreScission protease. In the fractions collected, the three enzyme activities shown were assayed, and the presence of the recombinant 60-kDa protein band composed of the 575 amino acids of human DHA kinase/FMN cyclase plus an N-terminal extension of 11 amino acids left by the cut with PreScission was detected by SDS-PAGE. B, substrate specificity of the kinase and cyclizing lyase activities. Activity with S.D. (error bars) is the mean of three experiments, each performed with two different amounts of enzyme evaluated either by continuous spectrophotometric recording (kinase) or by HPLC at three time points (cyclizing lyase) under conditions of linear response with respect to incubation time and hTKFC amount. Kinase activities were assayed with 0.5 mm phosphoryl acceptor, 5 mm (deoxy)nucleoside triphosphate donor, and 10 mm MgCl2. Cyclizing lyase activities were assayed with 0.5 mm substrate and 6 mm MnCl2. Ap2A, diadenosine pyrophosphate.
The Mg2+/ATP-dependent kinase activity of hTKFC was assayed on GA, DHA, glycerol, and glycolaldehyde. Only GA and DHA were detectably phosphorylated. DHA phosphorylation was assayed with alternative donors of which the best was ATP followed by CTP and ITP, and lesser activities were seen with GTP, dATP, and UTP. This agrees with known characteristics of triokinase (65–68) except that pig kidney triokinase reportedly has a weak activity on glycolaldehyde (65) and rat liver triokinase has a stricter preference for ATP as donor (68). To our knowledge, phosphorylation with dATP as a donor has not been reported for triokinase. The absence of activity on glycerol distinguished hTKFC from glycerol kinase, which phosphorylates trioses too (69, 70).
The Mn2+-dependent cyclizing lyase activity was assayed at a fixed substrate concentration on a collection of 16 NDP-X compounds. hTKFC split only those NDP-X compounds able to undergo an inner nucleophilic attack of an X-OH hydroxyl over the proximal phosphorus atom to render as products NMP and a five-atom phosphodiester either as a monocyclic compound or a cis-bicyclic phosphodiester-pyranose fusion. For example, hTKFC split NDP-glucose but not NDP-mannose substrates, which would give trans-bicyclic fusions as products. The structural aspects of this remarkable cyclizing lyase specificity have been discussed earlier in relation with rat FMN cyclase (21).
The saturation kinetics of the kinase and cyclizing lyase activities were studied with a selection of hTKFC substrates (Table 1). Concerning the Mg2+/ATP-dependent kinase activity, DHA and GA were phosphorylated with similar kcat values, but DHA was processed 10-fold more efficiently due to its lower Km (1.6 versus 18 μm). Concerning the Mn2+-dependent lyase activity, ADP-glucose and FAD were the best substrates with similar efficiencies (kcat/Km), whereas UDP-glucose and UDP-galactose displayed considerably lower efficiencies due to their higher Km values. Interestingly, the catalytic efficiency of hTKFC with the best cyclizing lyase substrates was comparable with the GA kinase reaction (Table 1). This corresponded to reactions at pH 7.5, but because the kinase and cyclase activities displayed different pH optima (Fig. 2), at higher pH values the ratio of activities became favorable to the cyclizing lyase at least with FAD as the substrate.
TABLE 1.
Kinetic parameters of hTKFC and mutants
The kcat, Km, and inhibition values with S.D. are averages of triplicate measurements. Several forms of hTKFC analyzed but not included in the table (individual K domain and D401A, D403A, and D556A point mutants) showed molecular activities of kinases (≤1% of wild-type kinase assayed with 10 mm MgCl2, 5 mm ATP, and 500 μm DHA or GA) and FMN cyclase (<0.1% of wild-type cyclase assayed with 6 mm MnCl2 and100 μm FAD) that were not detectable (ND). NA, not assayed.
| Assay performed (substrate) | hTKFC forms |
||||||
|---|---|---|---|---|---|---|---|
| Wild type | L domain | H221A | C404A | S446A | T112A | K204A | |
| DHA kinase (DHA) activity | |||||||
| kcat (s−1) | 5.00 ± 1.55 | ND | ND | 2.26 ± 0.23 | 0.13 ± 0.01 | 0.51 ± 0.04 | 2.79 ± 0.38 |
| 106 Km (m) | 1.55 ± 0.17 | 1.91 ± 0.58 | 0.99 ± 0.32 | 6.63 ± 0.29 | 2.52 ± 0.50 | ||
| 10−4 kcat/Km (m−1 s−1) | 321 | 118 | 13.1 | 7.6 | 115 | ||
| DHA kinase (ATP) activity | |||||||
| kcat (s−1) | 4.59 ± 1.21 | ND | ND | 2.17 ± 0.13 | 0.15 ± 0.01 | 0.50 ± 0.05 | 2.34 ± 0.09 |
| 106 Km (m) | 43.2 ± 7.9 | 99.6 ± 11.9 | 5.74 ± 0.80 | 80.6 ± 21.7 | 63.7 ± 24.9 | ||
| 10−4 kcat/Km (m−1 s−1) | 10.5 | 2.18 | 2.6 | 0.64 | 4.2 | ||
| GA kinase (GA) activity | |||||||
| kcat (s−1) | 4.83 ± 1.25 | ND | ND | 2.27 ± 0.22 | 0.74 ± 0.03 | 0.16 ± 0.01 | 2.09 ± 0.17 |
| 106 Km (m) | 18.1 ± 4.7 | 16.2 ± 0.67 | 7.1 ± 0.6 | 13.7 ± 0.2 | 15.0 ± 3.3 | ||
| 10−4 kcat/Km (m−1 s−1) | 27.5 | 14.0 | 10.4 | 1.2 | 14.2 | ||
| GA kinase (ATP) activity | |||||||
| kcat (s−1) | 3.94 ± 1.25 | ND | ND | 2.10 ± 0.15 | 0.66 ± 0.07 | 0.16 ± 0.01 | 1.70 ± 0.05 |
| 106 Km (m) | 62.4 ± 8.6 | 183 ± 17 | 35.2 ± 5.4 | 85.3 ± 7.7 | 69.6 ± 0.2 | ||
| 10−4 kcat/Km (m−1 s−1) | 6.3 | 1.15 | 1.9 | 0.19 | 2.4 | ||
| Cyclizing lyase (FAD) activity | |||||||
| kcat (s−1) | 0.82 ± 0.21 | 0.39 ± 0.07 | 0.83 ± 0.02 | 0.39 ± 0.02 | 0.30 ± 0.01 | 0.67 ± 0.07 | 0.85 ± 0.04 |
| 106 Km (m) | 7.0 ± 2.5 | 79.3 ± 31.2 | 27.2 ± 5.1 | 52.2 ± 0.4 | 6.8 ± 2.6 | 10.8 ± 3.4 | 28 ± 5 |
| 10−4 kcat/Km (m−1 s−1) | 11.7 | 0.49 | 3.1 | 0.75 | 4.4 | 6.2 | 3.0 |
| Cyclizing lyase (ADP-glucose) activity | |||||||
| kcat (s−1) | 1.87 ± 0.49 | NA | 0.75 ± 0.13 | NA | NA | NA | NA |
| 106 Km (m) | 12.0 ± 1.0 | 20.0 ± 5.6 | |||||
| 10−4 kcat/Km (m−1 s−1) | 15.6 | 3.8 | |||||
| Cyclizing lyase (UDP-glucose) activity | |||||||
| kcat (s−1) | 1.19 ± 0.10 | NA | NA | NA | NA | NA | NA |
| 106 Km (m) | 317 ± 117 | ||||||
| 10−4 kcat/Km (m−1 s−1) | 0.38 | ||||||
| Cyclizing lyase (UDP-galactose) activity | |||||||
| kcat (s−1) | 2.51 ± 0.08 | NA | NA | NA | NA | NA | NA |
| 106 Km (m) | 263 ± 163 | ||||||
| 10−4 kcat/Km (m−1 s−1) | 0.95 | ||||||
| Cyclizing lyase (FAD) inhibition by kinase substrates | |||||||
| Inhibition by 10 μm ATP (%) | 92.5 ± 5.2 | 59.2 ± 4.1 | 88.2 ± 5.7 | 83.0 ± 3.3 | 87.6 ± 2.6 | 77.7 ± 2.7 | NA |
| Inhibition by 500 μm DHA (%) | 39.1 ± 3.7 | 1.7 ± 5.7 | 0.5 ± 2.4 | 38.3 ± 4.9 | 58.9 ± 4.7 | 16.0 ± 3.4 | NA |
| Inhibition by 500 μm GA (%) | 35.2 ± 4.4 | 8.8 ± 4.8 | 0.0 ± 6.2 | 44.9 ± 6.2 | 41.9 ± 9.8 | 24.5 ± 3.6 | NA |
FIGURE 2.
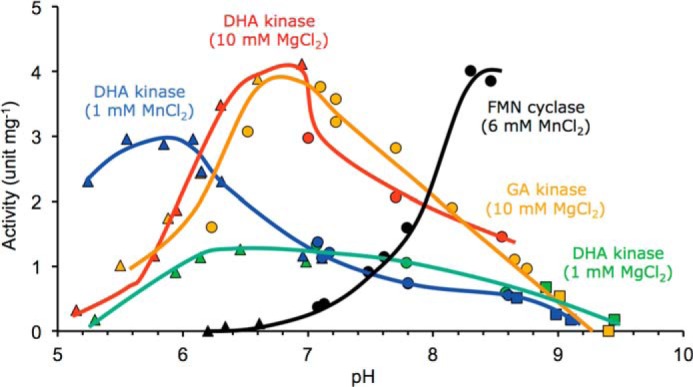
Activity-versus-pH profiles of the triokinase and FMN cyclase activities of hTKFC. The activities were measured by the standard assays at the MgCl2 or MnCl2 concentrations and the pH values indicated. The buffers used were either Tris acetate (pH ≤ 7), Tris-HCl (pH 7–8.8), or CAPS-NaOH (pH > 8.8). The pH values were directly measured with a glass electrode in mixtures like those used for activity assays.
Molecular Evidence for the Identity of the Swine Ortholog of hTKFC with Classical Triokinase
The specificity of the kinase activity of hTKFC confirmed it as the first molecularly identified triokinase of animal (human) origin. To our knowledge, there is a single previous report of highly purified triokinase from animal tissue: the pig kidney enzyme studied by Miwa et al .(65) in the pregenomics era. Some molecular data available for that protein confirmed that it coincides with the swine ortholog of hTKFC, a hypothetical protein (NCBI Reference Sequence XP_003122689) 92% identical to hTKFC (GenBankTM accession number ABA10576). The native molecular weight (122,000) and the dimeric character of pig kidney triokinase (65) agree with the subunit molecular weight predicted from the sequence of swine TKFC (59,100), assuming that it will form dimers like hTKFC does (Fig. 3). However, the most direct evidence for this identification came from the sequence of three lysyl endopeptidase peptides (Fig. 4A) and the full amino acid composition (Fig. 4B) of triokinase purified from pig kidney (65) that was found almost exactly reproduced in the hypothetical TKFC predicted from the Sus scrofa genome. In fact, an unrestricted search in the Swiss-Prot and TrEMBL databases run with AACompIdent (71) (ExPASy Bioinformatics Resource Portal) using the experimental amino acid composition of pig kidney triokinase (65) as the query gave the swine ortholog of hTKFC as the best hit.
FIGURE 3.

Dimeric molecular weight of native hTKFC. A 0.75-ml hTKFC sample was applied to an 87.5 × 1.2-cm Sephadex G-150 column equilibrated with 20 mm Tris-HCl, 0.5 mm EDTA, and 0.1 m KCl adjusted to pH 8.2 at 4 °C. The chromatography was run at 0.1 ml min−1 in the same buffer. Fractions were collected and weighed for precise volume determination, and DHA kinase and FMN cyclase activities were assayed. The column was calibrated with the following molecular weight standards: 1, catalase (240,000); 2, aldolase (158,000; 3, bovine serum albumin (66,000); 4, egg albumin (45,000); and 5, cytochrome c (12,400). This gel filtration experiment indicated an apparent molecular weight of 140,000 for native hTKFC with additional evidence for the formation of higher order aggregates. When compared with the monomeric mass of 60 kDa, the apparent molecular weight of the native protein is in agreement with the expected behavior of an elongated dimer like hTKFC forms (Fig. 5).
FIGURE 4.

Comparison of pig kidney triokinase and the hypothetical protein identified as the swine ortholog of hTKFC. A, ClustalW alignment of three lysyl endopeptidase peptides sequenced from triokinase purified from pig kidney (65) with the hypothetical S. scrofa protein XP_003122689. The asterisks mark identical amino acids. The numbering corresponds to the S. scrofa protein. B, comparison of the amino acid composition of pig kidney triokinase (65) with that of the hypothetical S. scrofa protein. Amino acids are identified by the single letter code (B, Asn or Asp; Z, Glu or Gln). The linear regression equation and the regression coefficient are shown within the graph.
Structural Model of Dimeric hTKFC with Kinase Substrates: ATP Bound to the L Domain and DHA Bound to the K Domain of Each Subunit
Besides the ability to phosphorylate DHA (26) and to split FAD yielding cFMN (25), Citrobacter sp. DHA kinase (Protein Data Bank code 1UN8) shares with hTKFC (GenBank accession number ABA10576) the dimeric character (26) and a 40% amino acid identity. Based on these common features, a model of dimeric hTKFC with bound ATP and DHA was constructed with Modeler using two crystal structures of the bacterial protein as templates after the addition of several missing features. The major characteristics of Citrobacter sp. DHA kinase (26) were conserved in the hTKFC model (Fig. 5): it corresponded to a homodimer with each subunit containing two domains, K or N terminus and L or C terminus, linked together by a 20-amino acid spacer. The K domain consisted of two α/β-folds, and the L domain was an eight-helix α-barrel. The two subunits (1 and 2) appeared intertwined, and the homodimer adopted an elongated form with the domains in the linear order L2-K1-K2-L1 with the K1 and K2 domains in close contact and the L1 and L2 domains totally apart from each other. Two complete active sites were formed between K and L domains of different subunits (L2-K1 and K2-L1; Fig. 5). DHA was covalently bound to each K domain in hemiaminal linkage to the Nϵ2 of His221. ATP was bound to the L domains on top of the barrels. According to the templates used for homology modeling (see “Experimental Procedures”), the adenosine moiety of ATP was either in anti (L2-K1 site) or syn (K2-L1 site) conformation, although during the molecular dynamics simulation (see below), the syn conformation was rapidly converted to anti and remained that way. Adenine was packed between the lateral chains of Leu450 on one side and Thr494 and Met495 on the other. The 2′-hydroxyl groups of the ribose were coordinated by Asp556, and in the K2-L1 site, the 5′-oxygen atom formed a hydrogen bond with Cys404. The phosphates were coordinated by two Mg2+ ions bound to the γ-carboxyl groups of Asp396, Asp401, and Asp403. Remarkably, the geometry of each active site left unclear how the phosphoryl transfer to DHA would take place because the donor sites in the L domains and their respective acceptor sites in the K domains lie 14 Å apart, which is too far for an in-line phosphoryl transfer.
FIGURE 5.

Theoretical model of dimeric hTKFC with kinase substrates bound. The homodimeric model of hTKFC with bound DHA, ATP, and two Mg2+ ions per active site was constructed with Modeler using two Citrobacter sp. ATP-dependent DHA kinase structures as templates. The file describing the full model is available as supplemental File 3 (hTKFC_2DHA_2ATP.pdb). In the figure, the point of view and the orientation of the model are different in each panel. A, schematic representation of the complete model. B, detail of ATP bound in the L2-K1 (left) and K2-L1 (right) active sites. C, detail of amino acids near the Mg2+ ions and ATP.
Structural Model of the Cyclizing Lyase Active Center of hTKFC with FAD Bound to the ATP Site in the L Domain with Little Contact to the K Domain
The binding of FAD to hTKFC was investigated by independent simulation of docking to both active sites of the dimeric protein after removing the kinase substrates DHA and ATP from the homology model of the protein. Six hundred poses were obtained with AutoDock for each site. Given the similarity between ATP and the ADP moiety of FAD and the nature of the FMN cyclase reaction, it was expected that any FAD pose candidate to reflect a productive ES complex would include a position of the ADP moiety of FAD like ATP and an FAD conformation with the 4′-OH of the ribitol chain approaching the distance and angle needed for an in-line internal attack over the β-phosphate of the ADP moiety, which would yield cFMN and AMP as products. These conditions were fulfilled by results obtained in the K2-L1 but not in the L2-K1 site. Upon ranking the 600 K2-L1 poses in order of increasing energies according to the AutoDock evaluation, those in the fifth and sixth rank positions showed the expected spatial arrangement. After reevaluation with the alternative functions X-Score (72) and DrugScorePDB (73) and the implementation of a consensus function (74, 75) comprising AutoDock, X-Score, and DrugScorePDB data, the top of the consensus rank was occupied by the pose shown in Fig. 6, which corresponds to the fifth AutoDock pose (the file describing this model is available as supplemental File 4 (hTKFC_FAD.pdb)). In it, most of the interactions of FAD occurred with the L1 subunit. The adenine base, the ribose ring, and the two phosphate groups interacted with Mg2+-hTKFC as the ADP moiety of ATP (see above), although in the ATP molecule bound to the K2-L1 site, adenosine was in the syn conformation, whereas in the FAD molecule bound to the same site, adenosine was in the anti conformation. Concerning the ribityl chain, the 4′-OH group appeared to be acceptor and donor in possible hydrogen bonds with the peptidic NH of Ser446 and the β-phosphate group, respectively. In this layout, the 4′-oxygen atom was 3.28 Å from the phosphorus atom and formed an angle of 164° with the P–O scissile bond of the lyase reaction. This may correspond to a near-attack conformation for the FMN cyclase reaction. Interestingly, a simulation of FAD docking to the DHA kinase from Citrobacter sp. does not show a near-attack conformation of the 4′-OH group, perhaps in agreement with the much lower catalytic efficiency of the FMN cyclase activity of the bacterial enzyme (25). Isoalloxazine was the only part of the FAD molecule that interacted with the K2 domain of hTKFC (and of Citrobacter DHA kinase), including a hydrogen bond of the C2 carbonyl oxygen with the side chain of Asn117 and possible contacts of the rings with Thr82, Ser83, and Gly113.
FIGURE 6.
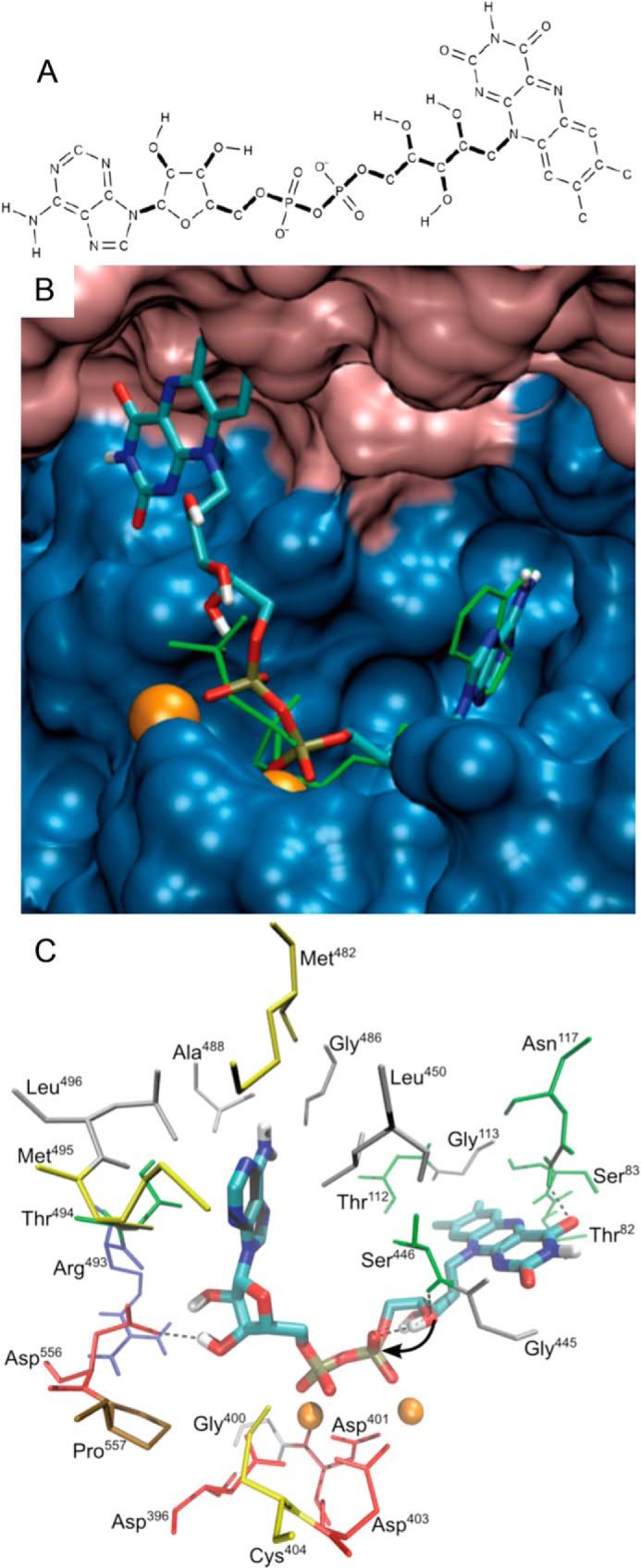
FAD docked to the K2-L1 active site of hTKFC. Docking to the L2-K1 and K2-L1 sites was modeled with AutoDock with good results only in the second case (see further details in the main text). The file describing the full model is available as supplemental File 4 (hTKFC_FAD.pdb). A, FAD structure used for docking. Thick bonds correspond to active torsions; i.e. they were allowed to rotate during docking. Only polar hydrogens are shown. B, FAD docked to the K2-L1 site shown in overlap with (green rods) the ATP bound to the same site as it appears in the right-hand side of Fig. 5B. C, detail of amino acids near the bound 2Mg2+-FAD. The curved arrow indicates the internal attack by the ribityl 4′-oxygen atom over the proximal phosphorus atom in the FMN cyclase reaction.
Inhibition of Cyclizing Lyase by Triokinase Substrates and Analogs
Several earlier kinetic experiments agree with the above structural models: ATP is a potent and complete inhibitor of the FMN cyclase activity, DHA is a much weaker and only partial inhibitor of the same activity, and FAD is a weak but complete inhibitor of DHA kinase activity (24, 25). Those observations were extended in several ways. Inhibition of FMN cyclase by adenosine phosphates required at least a diphosphate and the 2′-OH group because ADP was as potent an inhibitor as ATP, whereas inhibition by AMP was very weak (the same pattern was produced by uridine phosphates), and deoxyadenosine phosphates were much weaker inhibitors (Fig. 7A). Besides the FMN cyclase activity, DHA also partially inhibited the cyclizing lyase activity on ADP-glucose, and GA was also a partial inhibitor of FMN cyclase with somewhat lesser potency than DHA (Fig. 7B). At the same concentrations, glycerol, which was not phosphorylated by hTKFC, did not inhibit FMN cyclase (not shown). Each triose, DHA and GA, inhibited completely the kinase activity on the other one (Fig. 7B). Finally, the kinetic analyses of the inhibitions of FMN cyclase (Fig. 8) indicated that ATP was a complete competitive inhibitor with a 20 nm Ki, whereas DHA inhibition data were better explained by the equations for partial mixed inhibition. However, because in this case the two Ki values characteristic of a mixed inhibitor were very similar (i.e. α ≈ 1 in equations included in Fig. 8), DHA can be easily seen as a partial pure noncompetitive inhibitor with a 4.5 μm Ki. All the inhibition data agreed with the active sites of the kinase and lyase activities of hTKFC being partly coincident: FAD, the substrate of the lyase activity, binds in the ATP site such that binding of one of them prevents binding of the other, but the presence of DHA or GA does not prevent FAD binding but just modifies the kinetic parameters of the lyase activity. All of this is in full agreement with the structural models of the ES complexes of hTKFC with ATP and DHA (Fig. 5) or with FAD (Fig. 6) that include DHA linkage to the K domains and ATP or FAD binding to the L domains of hTKFC.
FIGURE 7.

Inhibitors of the activities of hTKFC. A, inhibition of the FMN cyclase activity by adenosine phosphates and analogs. FMN cyclase was assayed with 50 μm substrate and the indicated concentrations of inhibitors under otherwise standard conditions. The inset shows percent activities determined in the presence of a fixed 20 μm concentration of the compound indicated. Error bars represent S.D. B, inhibition by trioses. Cyclizing lyase activities were assayed as in A, and the kinase activities were assayed with 500 μm GA (GA kinase) or 25 μm DHA (DHA kinase) and the indicated concentrations of inhibitors under otherwise standard conditions. The lines are best fits of the model equations for the partial pure noncompetitive (cyclizing lyase) or the complete competitive inhibition (kinase activities) (see Fig. 8).
FIGURE 8.

Kinetic analyses of FMN cyclase inhibition by ATP and DHA. FAD saturation curves of the cyclizing lyase activity of hTKFC were obtained in the absence of inhibitors and in the presence of either ATP (complete FMN cyclase inhibitor; Fig. 7A) or DHA (partial FMN cyclase inhibitor; Fig. 7B) as indicated. The kinetic analyses were based on the general mechanism for reversible inhibition (top scheme) and on the model equations describing the competitive, general noncompetitive or mixed, and uncompetitive inhibitions either complete or partial (84, 85). In these equations, under the assumptions of rapid equilibrium, Km approximates KS. The equations were adjusted to the two data sets by simultaneous adjustment to the three curves of each data set using the Solver function of Microsoft Excel (global fit). The parameters left to fluctuate during the adjustments were Vmax, Km, Ki, α, and β whenever relevant. The goodness of fit was estimated by the χ2 parameter (85), which supported that inhibition by ATP was of the complete competitive type and inhibition by DHA was a partial mixed inhibition; however, because for the latter the best adjustment returned an α value close to the unit (α = 0.92), DHA inhibition can be assumed to be a partial inhibition of the classical pure noncompetitive type (i.e. an inhibition in which the presence of the inhibitor affects the apparent Vmax but not the apparent Km value).
Differential Roles of hTKFC Domains K and L Expressed Individually
The domain organization of dimeric hTKFC, the binding of the reactants of the bisubstrate kinase reaction (DHA and ATP) to different domains (K and L) of different subunits, the docking of the reactant of the monosubstrate cyclase reaction (FAD) to the L domain with few interactions with the K domain, and the partial noncompetitive or mixed type inhibition of FMN cyclase activity by the triose substrate of the kinase reaction pointed to different hTKFC domain requirements for the kinase and cyclase reactions. Therefore, plasmid constructs encoding the K domain (amino acids 1–339) or the L domain (amino acids 359–575) were prepared, and the domains were separately expressed and assayed for kinase and cyclase activities. The K domain did not detectably catalyze either reaction type, but the L domain, although being equally inactive as a triokinase, retained considerable FMN cyclase activity. The kcat value of domain L cyclase activity was only slightly reduced, just about 50%, with respect to the full hTKFC protein. The Km for FAD was more significantly affected by the absence of the K domain, being about 10-fold higher than that of full hTKFC (Table 1), indicating that the few interactions with the K domain contribute significantly to the affinity for FAD without being essential for cyclizing lyase activity. The FMN cyclase activity of the L domain was desensitized to the partial inhibition by DHA or GA concentrations that were inhibitory for the full dimeric hTKFC (Table 1).
The importance of the long spacer between the K and L domains of the same subunit was highlighted by the failure to recover kinase activity when individually expressed domains were mixed in solution. This is different from the behavior of the E. coli PTS-dependent DHA kinase (30) structured as a heterotetramer composed of two DhaK and two DhaL subunits (homologous to the K and L domains of the subunits of homodimeric hTKFC and Citrobacter sp. DHA kinase, respectively) and devoid of the 20-amino acid spacers present in the ATP-dependent DHA kinases.
Conformational Flexibility of hTKFC Leading to a Marked ATP-to-DHA Approximation and New Hydrogen Bonds Detected in Closed Active Center Conformations Simulated by Molecular Dynamics
Domain mobility in solution has been invoked to explain the ability of Citrobacter sp. DHA kinase to catalyze the phosphoryl transfer from ATP to DHA when in the crystal structure the phosphoryl donor and acceptor are about 14 Å apart (15, 26). This situation was reproduced in the hTKFC model of the dimeric ternary complex we constructed (Fig. 5). To explore the possible effect of hTKFC conformational flexibility on the distance between ATP and DHA, a molecular dynamics simulation was run for 75 ns in explicit solvent starting with the hTKFC complex, including substrates bound to both active sites (supplemental movie (hTKFC_2DHA_2ATP_MD.mpg)). Fig. 9 shows the distances measured between the γ-phosphorus atoms of the ATP bound to the L1(L2) domain and the hydroxyl O1(3) of DHA covalently bound to His221 in the K2(K1) domain. Initially, the ATP-to-DHA distance was practically the same in both active sites: 14.21 Å in K2-L1 and 14.30 Å in L2-K1. In the first 5 ns of the trajectory, that distance increased rapidly in K2-L1 but then showed a more or less steady decrease up to about 12 Å at 50 ns, remaining practically unchanged until the end of the simulation. In L2-K1, during 30 ns, the ATP-to-DHA distance changed little with respect to the initial state, but from this time onward, the ATP-to-DHA distance fluctuated down and up twice such that at 34–37 and 47–55 ns distances were about 5 Å. This represents very significant approximations to the requirements for a nucleophilic attack of the hydroxyl O1 over the γ-phosphorus as needed for the phosphoryl transfer to occur. As an illustration, Fig. 10 presents the conformation of the L2-K1 active site in the initial state (Fig. 10A) and in two trajectory frames showing relative minima of the ATP-to-DHA distances for the 34–37- and 47–55-ns periods: 4.83 Å at 34.860 ns (Fig. 10B) and 4.16 Å at 54.622 ns (Fig. 10C). These could be considered as near-attack conformations in closed or near-closed active sites. Interestingly, in these frames and/or in others next to them, two hydrogen bonds specific to the closed conformations were detected. One was formed between the hydroxyl group of Thr112 in the K1 domain and the N6 atom of ATP bound to the L2 domain and could contribute to keep the phosphoryl donor near the triose (Fig. 10B). The formation of a hydrogen bond with the N6 of adenine acting as acceptor implies the nonplanarity of the adenine amino group, a feature that is included in the topology file of ATP used for molecular dynamics simulation (access code F-91 in the R.E.DD.B. database) and supported by evidence (76–78). Another hydrogen bond of interest was formed between Lys204 Nϵ in the K1 domain and one of the carbonyl groups of the Arg397-Ala398 backbone in the L2 domain, i.e. between opposing sides of the outer ridge of the L2-K1 groove (Fig. 10, B and C). This bond could make some contribution to the closed conformation of the active site.
FIGURE 9.
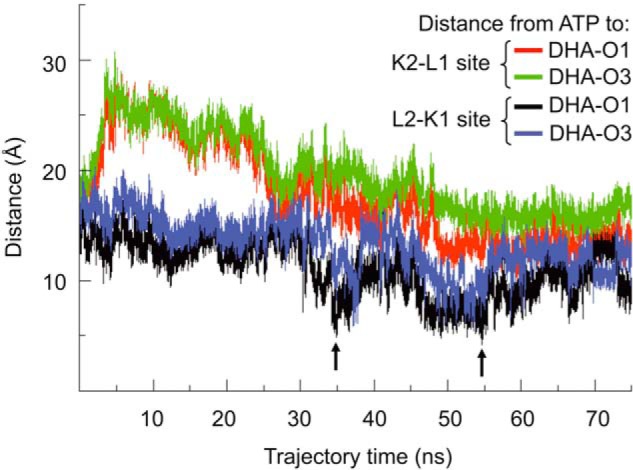
ATP-to-DHA distances in the two hTKFC active sites along the simulation of a molecular dynamics trajectory. A 75-ns molecular dynamics simulation was run in Gromacs with the Amber03 force field starting with the hTKFC-2DHA-2ATP complex constructed with Modeler (Fig. 5). The simulation covered a trajectory composed of 75,000 frames collected at intervals of 1 ps (see the supplemental movie (hTKFC_2DHA_2ATP_MD.mpg)). In the frames, the distances to each DHA O1 and O3 atoms were measured from the γ-phosphorus atom of ATP bound to the same active site. The arrows mark the relative minima of ATP-to-DHA distance, 4.83 and 4.16 Å, both recorded at the L2-K1 site in the frames corresponding to 34.860 and 54.622 ns, respectively (shown in Fig. 10, B and C).
FIGURE 10.
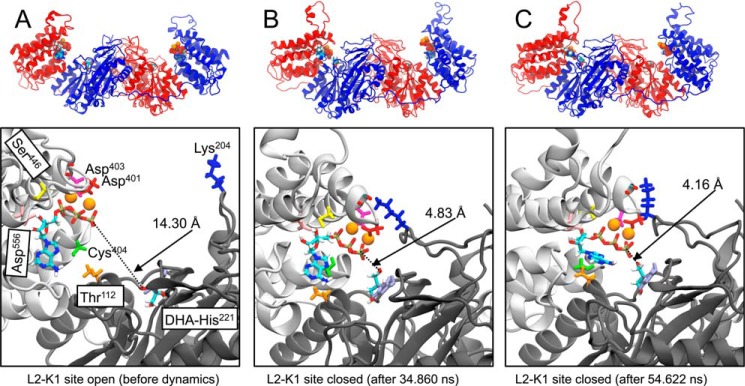
Conformations of hTKFC before dynamics and upon reaching two relative minima of ATP-to-DHA distance in the L2-K1 active site during the molecular dynamics simulation. Upper panels, schematic models of the conformations of hTKFC-2DHA-2ATP model before dynamics (see Fig. 5) (A) and the frames corresponding to the 34.860 (B) and 54.622 ns (C) of the molecular dynamics trajectory (marked by arrows in Fig. 9). Lower panels, detail of the L2-K1 site in the same models. The discontinuous lines mark the distances between the DHA O1 and the γ-phosphorus atom of ATP. In the left lower panel, the positions of the amino acids submitted to site-directed mutagenesis are also shown.
Comparisons of the Simulated Closed Conformations of hTKFC with the Closed Active Center of the E. coli DhaK-DhaL Complex
The closed conformations of the L2-K1 site of hTKFC formed during the molecular dynamics simulation were compared with the closed active center of the crystal structure of the E. coli DHA kinase complex composed of DhaK and DhaL subunits (30).
The structural comparison of DhaK with K1 and independently of DhaL with L2 in the open conformation of hTKFC before dynamics gave results similar to those of the comparisons of DhaK and DhaL with the K and L domains of the homodimeric DHA kinase from Citrobacter (30). Similar results were obtained when comparisons were performed with the K1 and L2 domains of the closed L2-K1 site (Fig. 10). However, the L2 loop formed by amino acids 535–554 showed large structural differences compared with the equivalent region of DhaL. It must be stressed that in Citrobacter DHA kinase crystals this loop is disordered (26), so it was missing in the templates used to model hTKFC by homology, and it was constructed de novo. During the dynamic simulation, this loop showed the largest internal mobility in L2 (supplemental movie (hTKFC_2DHA_2ATP_MD.mpg)). In contrast, the corresponding loop in the structure of E. coli DhaL (named α7/α8) is well ordered, both in the free (79) and the complexed state with DhaK (30), occupying a position that blocks the exit of the ADP prosthetic group and interacting extensively with the DhaK subunit (30). The mobility of the modeled loop of hTKFC is in agreement with the disordered state of its Citrobacter DHA kinase equivalent. The difference between the conformations of the α7/α8 loop of DhaL and its equivalent in the ATP-dependent Citrobacter DHA kinase or in hTKFC may be related to the different roles of the nucleotide as prosthetic group or cosubstrate.
Conversely, the comparison of the full DhaK-DhaL dimer with the K1-L2 moiety of hTKFC revealed a major difference in their closed conformations. When the DhaK subunit and the K1 domain were structurally aligned to each other, the orientation of DhaL relative to DhaK was very different from the orientation of L2 relative to the K1 domain (Fig. 11). Therefore, the closed conformations of the hTKFC L2-K1 site found by molecular dynamics simulation are quite different from that of the crystal structure of the E. coli DhaK-DhaL complex. Again, this could perhaps be related to the difference between an active site where ATP is a cosubstrate and one where ADP is a prosthetic group.
FIGURE 11.
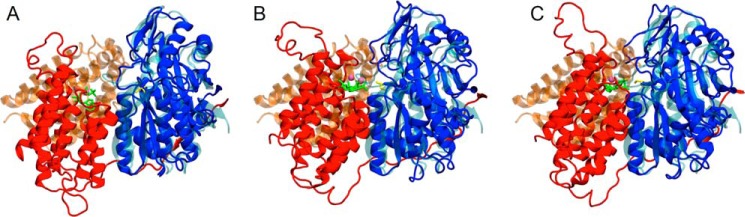
Comparison of the closed conformations of the L2-K1 site of hTKFC with the DhaK-DhaL complex of E. coli. Domains K1 (blue) and L2, including the L2-K2 linker (red), of the three hTKFC conformations of Fig. 10 are here shown in overlap with the DhaK (cyan) and DhaL (orange) subunits of the E. coli DhaK-DhaL complex. To emphasize the different relative orientations of L2 toward K1 and DhaL toward DhaK either in the open (A) or closed (B and C) hTKFC conformations, the K1 domain was structurally aligned with DhaK in each case.
As discussed by others (30), the binding interface of the DhaK and DhaL subunits in the E. coli DHA kinase is different from that of the K and L domains of the Citrobacter enzyme. In the first case, the contact areas have a predominantly polar nature, whereas in the second case, the interface is mainly hydrophobic. The same comment can be applied to the K and L domains of hTKFC in the open conformation (Fig. 10A). However, in the closed conformations of the L2-K1 site, as described above, new hydrogen bonds were observed between Thr112 in K1 and the N6 atom of ATP bound to L2 (Fig. 10B) and between Lys204 (K1) and the carbonyl groups of Arg397 or Ala398 (L2) (Fig. 10, B and C). Interestingly, the interaction between adenine and Thr112 also occurs in the active site of the E. coli DhaK-DhaL complex, although it is not a direct hydrogen bond but is mediated by a molecule of water (30).
Probing the Roles of Specific Amino Acids by Directed Mutagenesis
Three groups of hTKFC point mutants were constructed and assayed for kinase and cyclase activities and for inhibition of FMN cyclase by ATP, DHA, or GA (Table 1). The locations of the amino acids affected by the mutations are shown in Fig. 10.
The first mutant group includes only H221A-hTKFC, which lacks the side chain of His221 where DHA anchors in the K domains of native hTKFC. This amino acid is conserved in Citrobacter and E. coli DHA kinases, playing the same role (26, 30, 80). Mutagenesis experiments performed with the E. coli enzyme have shown that it is essential for kinase activity (30, 80). The same was true for hTKFC. However, the catalytic behavior of the mutant H221A-hTKFC resembled the individually expressed L domain in that both were inactive as kinases but retained FMN cyclase activity. In fact, the kcat for FAD splitting of the point mutant was indistinguishable from that of the wild-type dimeric hTKFC, whereas the mutant had a 4-fold higher Km than the wild type but still a 3-fold lower Km than the L domain. In agreement with the absence of the DHA anchor site, the FMN cyclase activity of H221A-hTKFC, like the L domain, did not show the partial inhibition by DHA or GA seen with the wild-type enzyme (Table 1).
The second group of mutants includes D401A-, D403A-, C404A-, S446A-, and D556A-hTKFC, which affect diverse L domain amino acids that interact with ATP or FAD either directly (Cys404, Ser446, and Asp556) or through coordination by the Mg2+ ions to which the phosphates bind (Asp401 and Asp403). The three aspartates and their roles are also conserved in the bacterial DHA kinases (26, 79). Cys404 is not conserved, but it is replaced by Thr and His, respectively, in the Citrobacter and E. coli DHA kinases; the latter amino acid is considered a differential feature of PTS-dependent DHA kinases critical for strong binding of the ADP cofactor (26, 28). Ser446 is conserved in the Citrobacter but not in the E. coli enzyme where it is replaced by Ala.
Mutation of any one of the two Mg2+-coordinated aspartates (D401A and D403A) led to a full inactivation of the kinase and cyclase activities of hTKFC. This agreed with the loss of kinase activity in the equivalent mutants of Citrobacter and, less drastically so, the E. coli DHA kinase (26). The mutation of Asp556 (D556A) was also fully inactivating, demonstrating the importance of the hydrogen bonds that this side-chain carboxylate formed with the ribose hydroxyls of ATP and FAD in the hTKFC models (Figs. 5C and 6C). In the Citrobacter DHA kinase, the interaction of the equivalent aspartate with ATP has also been noted but not tested by mutagenesis (26).
The C404A-hTKFC mutant retained considerable cyclase and kinase activities (Table 1). The removal of the thiol group due to the C404A mutation caused a small 2-fold decrease of kcat for all activities, a 2–3-fold increase of ATP Km values, and a stronger 7-fold increase of FAD Km. In contrast, this mutant showed little or no change of DHA and GA Km values with respect to the wild type. The pattern of effects on ATP and FAD Km values could not be accounted for in terms of hydrogen bonds of these substrates with Cys404 in the hTKFC models because, although the thiol group was not far from the 5′-oxygen atom of the ribose moiety in any case, a hydrogen bond was observed only with the ATP bound to the K2-L1 site (Fig. 5C). The small effect of mutating Cys404 on the kinase activity of hTKFC compares well with that of mutating the similarly placed and not essential Thr388 in Citrobacter DHA kinase, but it is in strong contrast with the inactivating effect of mutating His38 in the DhaL subunit of E. coli DHA kinase (26). On the other hand, from the kinetic parameters of the C404A-hTKFC mutant, it may be inferred that Cys404 plays a more significant role in FAD binding.
The removal of the hydroxyl group due to the S446A mutation caused a strong, ≈30-fold decrease of DHA kinase kcat but also a 7-fold decrease of ATP Km. Hence, in the absence of the Ser446 hydroxyl, the affinity for ATP binding is higher, but the resulting complex is less prone to undergo the transfer reaction; i.e. catalysis is slower. A similar but quantitatively less marked pattern of change related to the S446A mutation was observed on the GA kinase activity. The comparison with bacterial DHA kinases is not possible because neither the conserved residue of the Citrobacter nor the substitutive Ala of the E. coli enzyme has been mutated. Concerning the FMN cyclase activity, the S446A mutation caused only a small 2.5-fold decrease of kcat without effect on the Km of FAD (Table 1). Nonetheless, Ser446 may play a significant role in this reaction through its peptidic NH group, which is not affected by the mutation and forms a hydrogen bond with the nucleophilic 4′-oxygen of the ribityl chain in the near-attack conformation (Fig. 6). Altogether, the rationale for the effects of the S446A mutation in the kinetic parameters of hTKFC is unclear.
Finally, the third mutant group includes T112A- and K204A-hTKFC, which affect K domain amino acids (Thr112 and Lys204) that form hydrogen bonds upon closure of the active site to possible near-attack conformations. Thr112 is conserved in both bacterial enzymes, and Lys204 is conserved in the E. coli but replaced by Asp in the Citrobacter kinase.
The removal of the hydroxyl group of Thr112 due to the T112A mutation caused a 10–30-fold decrease of kinase kcat values with modest or no increases of Km values, whereas the removal of the Lys204 side chain due to the K204A mutation just reduced kinase kcat values 2-fold. The effects of both mutations on the FMN cyclase activity were inconspicuous. T112A and K204A mutations eliminate the possibility of hydrogen bonds that, being clearly absent in the hTKFC structure modeled by homology (Figs. 5 and 10A), formed at least in one of the conformations of minimal ATP-to-DHA distance reached during the molecular dynamics simulation (Fig. 10, B and C). These results, besides the dependence of hTKFC kinase activity on domain mobility, indicate that Thr112 bonding to the adenine group of ATP is significantly important to reach and/or stabilize the active conformation of the ES complex, whereas the hydrogen bonds of Lys204 with the Arg397-Ala398 backbone are at most minor players. In fact, Thr112 is conserved in ATP-dependent DHA kinases, whereas Lys204 is not. Interestingly, in the closed DhaK-DhaL complex of the PTS-dependent E. coli kinase, the Thr107 residue of DhaK forms a water-mediated hydrogen bond with the purine N6 of the DhaL-bound ADP (30).
Possible Catalytic Mechanisms of hTKFC
A mechanism for ATP-dependent DHA kinases has not been proposed in detail due to the lack of information about a closed active site conformation. However, a mechanism has recently been proposed for the PTS-dependent DHA kinase of E. coli based on the closed conformation of the active site formed between its DhaK and DhaL subunits in the crystallized complex (30). As discussed above, the closed conformations of hTKFC found by molecular dynamics simulation differ considerably from that of the E. coli DhaK-DhaL complex in the relative orientation of the ATP-binding domain/subunit with respect to the DHA-binding domain/subunit (Fig. 11). Despite such differences, the mechanism of the kinase reaction of hTKFC may be similar to the proposed three-step catalytic mechanism of E. coli DHA kinase: (i) formation of a covalent hemiaminal linkage between histidine and DHA in the DhaK subunit with intervention of a second histidine as a general acid, (ii) phosphoryl transfer from ATP bound to the DhaL subunit to DHA covalently bound to the DhaK subunit with a DhaK aspartate acting as a general base and a DhaL arginine stabilizing the γ-phosphoryl group during transfer, and (iii) breaking of the hemiaminal linkage to release DHAP with the second histidine mentioned in step i acting now as a general base all as described elsewhere (30). The two histidines and the aspartate are sequentially and structurally conserved in hTKFC (His221, His59, and Asp114 located in the K domain), whereas the arginine is sequentially but not structurally conserved (Arg543 included in the highly mobile loop of the L domain). During the simulation of molecular dynamics, the mobile loop did not reach a position in which Arg543 could interact with the γ-phosphoryl group of ATP. This could be due to insufficient length of the simulation. Altogether, the relevance of His59, Asp114, and Arg543 to the kinase activity of hTKFC remains to be tested by mutagenesis.
The catalytic mechanism of the FMN cyclase reaction seems quite different because the absence of the K domain reduced the kcat value just 2-fold (Table 1), indicating that important catalytic residues are probably not found in the K domain. Also, it should be considered that the Mn2+-dependent FMN cyclase reaction occurs in the absence of enzyme although at a much lower rate that the enzymatic reaction (21). The internal nucleophilic attack of the ribityl 4′-OH over the proximal phosphorus should benefit from groups helping the attacking hydroxyl to get rid of its proton and to orientate toward the reacting phosphorus atom and from groups stabilizing the transition state and the leaving group. In fact, the docking of FAD to hTKFC (Fig. 6) revealed no amino acid side chain with catalytic potential. Concerning the presence of a general base to facilitate the dissociation of the ribityl 4′-OH, no obvious candidate was apparent other than the oxygen atom of the cyclizing phosphoryl group that was hydrogen-bonded as acceptor to the 4′-OH. This possible substrate-assisted catalysis could be facilitated by the orientation effect of another hydrogen bond formed as donor by the peptidic NH of Ser446 with the 4′-oxygen atom (Fig. 6). With regard to the stabilization of the transition state or the leaving group of the FMN cyclase reaction, only the two metal ions complexed with aspartate residues occupy relevant positions by their interaction with the pyrophosphate group. However, in the docked structure of FAD, it is unclear whether, as in adenylyl cyclases (81–83), one of the metal ions facilitates the liberation of the proton from the nucleophilic OH by interacting with the oxygen atom because the distance was longer than 4 Å (Fig. 6). It remains to be tested whether the dynamics of the FAD complex could shorten that distance and/or bring other possible catalytic groups near the substrate. In this regard, it is tempting to consider a possible role for Arg543, which as discussed above is located in a highly mobile loop of the hTKFC L domain (disordered in Citrobacter DHA kinase crystals) and is required for DHA kinase activity in the E. coli enzyme but has not yet been tested by mutagenesis in the ATP-dependent DHA kinases. Nonetheless, two metal ion- and substrate-assisted catalysis, perhaps in combination, are good candidates to explain the acceleration of the hTKFC-catalyzed FMN cyclase reaction versus the nonenzymatic Mn2+-dependent splitting of FAD.
Concluding Remarks
The hTKFC protein previously shown to be a bifunctional DHA kinase/FMN cyclase encoded by the human DAK gene is here firmly identified as triokinase (hTKFC) based on the following. (i) In addition to ATP-dependent DHA kinase activity, it displays GA kinase but not glycerol kinase activity, altogether the hallmark of the classical triokinase participant in the Hers pathway for fructose metabolism. (ii) Amino acid composition and selected peptide sequences of pig kidney triokinase, the only enzyme of its kind for which significant molecular data have been obtained by biochemical means in the pregenomics era, show agreement with a hypothetical protein product encoded by the swine ortholog of DAK. These results finish the molecular identification of the Hers pathway enzymes.
With the aim to understand the kinase/cyclase bifunctionality of hTKFC, a homodimeric model was derived by homology to a bacterial ATP-dependent DHA kinase of known crystallographic structure. In the hTKFC model, like in the template, the kinase substrates (ATP and DHA) each bind to a different domain (L and K, respectively) of a different subunit that is too distant for the requirements of phosphoryl transfer. The mobility of the L domain toward the K domain provides a plausible solution to this enigma: during molecular dynamics simulation the active site of hTKFC undergoes a closing movement that brings ATP close to DHA, reaching an apparent near-attack conformation for the DHA kinase reaction. The simulated closed conformations of the hTKFC active site differ from that of the PTS-dependent E. coli DHA kinase, but the reaction mechanism of the ATP-to-DHA phosphoryl transfer could be similar for both enzymes. Conversely, the FMN cyclase substrate FAD binds to the same two metal ion-containing site as ATP in the L domain, although isoalloxazine makes a few additional contacts with the K domain. In the structure obtained by docking, FAD adopts a near-attack conformation. The ribityl 4′-OH is close to the proximal phosphorus atom almost in line with the scissile P–O bond. The same hydroxyl forms a hydrogen bond as donor with one phosphoryl oxygen atom suggestive of substrate-assisted catalysis and another hydrogen bond as acceptor with the NH of the Ser446 backbone, which can orientate the nucleophile.
The separate expression of hTKFC domains L and K confirms that the L domain suffices for the FMN cyclase activity because the absence of the K domain has little effect on its kcat value, although it causes an increase of Km for FAD attributable to the loss of favorable contacts of isoalloxazine with the K domain in the intact protein. In contrast, the kinase activity of hTKFC requires both L and K domains in dynamic interplay.
Molecular modeling combined with site-directed mutagenesis and enzymatic characterization of the hTKFC mutants clearly supports the specific intervention of Thr112 and His221 in the kinase activities. The T112A mutant, which cannot form the hydrogen bond with the amino group of the ATP adenine upon closure of the kinase active site, loses most of its kinase activity but maintains an intact FMN cyclase activity both in terms of catalysis and FAD affinity. More overwhelmingly, the H221A mutant, which lacks the anchor amino acid for trioses, acts specifically as a cyclase because it is devoid of kinase activity.
Acknowledgments
We give special thanks to María Antonia Günther Sillero and Antonio Sillero (Departamento de Bioquímica, Facultad de Medicina e Instituto de Investigaciones Biomédicas Alberto Sols, Universidad Autónoma de Madrid/Consejo Superior de Investigaciones Científicas, Madrid, Spain) for being an invaluable source of inspiration and gifts. We are also grateful to Y. Dupradeau (Université de Picardie-Jules Verne, Amiens, France) for making available the topological description of ATP before its inclusion in the R.E.DD.B. database and to the Instituto Politécnico de Leiria for permission granted to J. R. R. to be on study-and-research leave at our laboratory in Badajoz.
The work was supported in part by Ministerio de Educación y Ciencia or Ministerio de Ciencia e Innovación del Gobierno de España Grants BFU2006-510 and BFU2009-07296) and Junta de Extremadura or Gobierno de Extremadura Grants GRU08043, GRU09135, and GR10133 cofinanced by Fondo Europeo de Desarrollo Regional and Fondo Social Europeo.

- GA
- d-glyceraldehyde
- ANP
- adenylylimidodiphosphonate
- cFMN
- cyclic FMN or riboflavin cyclic 4′,5′-phosphate
- DHA
- dihydroxyacetone
- DHAP
- dihydroxyacetone phosphate
- GAP
- d-glyceraldehyde 3-phosphate
- hTKFC
- human triokinase/FMN cyclase
- NDP-X
- ribonucleoside diphosphate-X
- NMP
- ribonucleoside monophosphate
- PTS
- phosphoenolpyruvate:sugar phosphotransferase system
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- N
- nucleoside.
REFERENCES
- 1. Hers H. G., Kusaka T. (1953) The metabolism of fructose-1-phosphate in the liver. Biochim. Biophys. Acta 11, 427–437 [DOI] [PubMed] [Google Scholar]
- 2. Sillero M. A., Sillero A., Sols A. (1969) Enzymes involved in fructose metabolism in liver and the glyceraldehyde metabolic crossroads. Eur. J. Biochem. 10, 345–350 [DOI] [PubMed] [Google Scholar]
- 3. Mayes P. A. (1993) Intermediary metabolism of fructose. Am. J. Clin. Nutr. 58, 754S–765S [DOI] [PubMed] [Google Scholar]
- 4. Burns S. P., Murphy H. C., Iles R. A., Bailey R. A., Cohen R. D. (2000) Hepatic intralobular mapping of fructose metabolism in the rat liver. Biochem. J. 349, 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steinmann B., Gitzelmann R., Van den Berghe G. (2001) in The Metabolic and Molecular Basis of Inherited Disease (Scriver C., Beaudet A., Sly W., Valle D., Childs B., Vogelstein B., eds) 8th Ed., pp. 1489–1520, McGraw-Hill, New York [Google Scholar]
- 6. Sievenpiper J. L., Carleton A. J., Chatha S., Jiang H. Y., de Souza R. J., Beyene J., Kendall C. W., Jenkins D. J. (2009) Heterogeneous effects of fructose on blood lipids in individuals with type 2 diabetes: systematic review and meta-analysis of experimental trials in humans. Diabetes Care 32, 1930–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tappy L., Lê K. A. (2010) Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 90, 23–46 [DOI] [PubMed] [Google Scholar]
- 8. Lim J. S., Mietus-Snyder M., Valente A., Schwarz J. M., Lustig R. H. (2010) The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 7, 251–264 [DOI] [PubMed] [Google Scholar]
- 9. Johnson R. J., Sanchez-Lozada L. G., Nakagawa T. (2010) The effect of fructose on renal biology and disease. J. Am. Soc. Nephrol. 21, 2036–2039 [DOI] [PubMed] [Google Scholar]
- 10. Dekker M. J., Su Q., Baker C., Rutledge A. C., Adeli K. (2010) Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 299, E685–E694 [DOI] [PubMed] [Google Scholar]
- 11. Samuel V. T. (2011) Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol. Metab. 22, 60–65 [DOI] [PubMed] [Google Scholar]
- 12. Madero M., Perez-Pozo S. E., Jalal D., Johnson R. J., Sánchez-Lozada L. G. (2011) Dietary fructose and hypertension. Curr. Hypertens. Rep. 13, 29–35 [DOI] [PubMed] [Google Scholar]
- 13. Bonthron D. T., Brady N., Donaldson I. A., Steinmann B. (1994) Molecular basis of essential fructosuria: molecular cloning and mutational analysis of human ketohexokinase (fructokinase). Hum. Mol. Genet. 3, 1627–1631 [DOI] [PubMed] [Google Scholar]
- 14. Cross N. C., Cox T. M., de Franchis R., Sebastio G., Dazzo C., Tolan D. R., Gregori C., Odievre M., Vidailhet M., Romano V., Mascali G., Romano C., Musumeci S., Steinmann B., Gitzelmann R. (1990) Molecular analysis of aldolase B genes in hereditary fructose intolerance. Lancet 335, 306–309 [DOI] [PubMed] [Google Scholar]
- 15. Erni B., Siebold C., Christen S., Srinivas A., Oberholzer A., Baumann U. (2006) Small substrate, big surprise: fold, function and phylogeny of dihydroxyacetone kinases. Cell. Mol. Life. Sci. 63, 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Funari V. A., Crandall J. E., Tolan D. R. (2007) Fructose metabolism in the cerebellum. Cerebellum 6, 130–140 [DOI] [PubMed] [Google Scholar]
- 17. Molin M., Norbeck J., Blomberg A. (2003) Dihydroxyacetone kinases in Saccharomyces cerevisiae are involved in detoxification of dihydroxyacetone. J. Biol. Chem. 278, 1415–1423 [DOI] [PubMed] [Google Scholar]
- 18. Garcia-Alles L. F., Siebold C., Nyffeler T. L., Flükiger-Brühwiler K., Schneider P., Bürgi H. B., Baumann U., Erni B. (2004) Phosphoenolpyruvate- and ATP-dependent dihydroxyacetone kinases: covalent substrate-binding and kinetic mechanism. Biochemistry 43, 13037–13045 [DOI] [PubMed] [Google Scholar]
- 19. Hofmann K. H., Babel W. (1981) Dihydroxyacetone kinase of methanol-assimilating yeasts. II. Partial purification and some properties of dihydroxyacetone kinase from Candida methylica. Z. Allg. Mikrobiol. 21, 219–224 [DOI] [PubMed] [Google Scholar]
- 20. Fraiz F. J., Pinto R. M., Costas M. J., Aavalos M., Canales J., Cabezas A., Cameselle J. C. (1998) Enzymic formation of riboflavin 4′,5′-cyclic phosphate from FAD: evidence for a specific low-Km FMN cyclase in rat liver. Biochem. J. 330, 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cabezas A., Pinto R. M., Fraiz F., Canales J., González-Santiago S., Cameselle J. C. (2001) Purification, characterization, and substrate and inhibitor structure-activity studies of rat liver FAD-AMP lyase (cyclizing): preference for FAD and specificity for splitting ribonucleoside diphosphate-X into ribonucleotide and a five-atom cyclic phosphodiester of X, either a monocyclic compound or a cis-bicyclic phosphodiester-pyranose fusion. Biochemistry 40, 13710–13722 [DOI] [PubMed] [Google Scholar]
- 22. Canales J., Cabezas A., Pinto R. M., Cameselle J. C. (2005) Fluorimetric HPLC detection of endogenous riboflavin 4′,5′-cyclic phosphate in rat liver at nanomolar concentrations. Anal. Biochem. 341, 214–219 [DOI] [PubMed] [Google Scholar]
- 23. Yamano K., Saito H., Ogasawara Y., Fujii S., Yamada H., Shirahama H., Kawai H. (1996) The autofluorescent substance in the posterior flagellum of swarmers of the brown alga Scytosiphon lomentaria. Z. Naturforsch. C 51, 155–159 [Google Scholar]
- 24. Cabezas A., Costas M. J., Pinto R. M., Couto A., Cameselle J. C. (2005) Identification of human and rat FAD-AMP lyase (cyclic FMN forming) as ATP-dependent dihydroxyacetone kinases. Biochem. Biophys. Res. Commun. 338, 1682–1689 [DOI] [PubMed] [Google Scholar]
- 25. Sánchez-Moreno I., Iturrate L., Martín-Hoyos R., Jimeno M. L., Mena M., Bastida A., García-Junceda E. (2009) From kinase to cyclase: an unusual example of catalytic promiscuity modulated by metal switching. Chembiochem 10, 225–229 [DOI] [PubMed] [Google Scholar]
- 26. Siebold C., Arnold I., Garcia-Alles L. F., Baumann U., Erni B. (2003) Crystal structure of the Citrobacter freundii dihydroxyacetone kinase reveals an eight-stranded α-helical barrel ATP-binding domain. J. Biol. Chem. 278, 48236–48244 [DOI] [PubMed] [Google Scholar]
- 27. Cheek S., Ginalski K., Zhang H., Grishin N. V. (2005) A comprehensive update of the sequence and structure classification of kinases. BMC Struct. Biol. 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bächler C., Flükiger-Brühwiler K., Schneider P., Bähler P., Erni B. (2005) From ATP as substrate to ADP as coenzyme: functional evolution of the nucleotide binding subunit of dihydroxyacetone kinases. J. Biol. Chem. 280, 18321–18325 [DOI] [PubMed] [Google Scholar]
- 29. Zurbriggen A., Jeckelmann J. M., Christen S., Bieniossek C., Baumann U., Erni B. (2008) X-ray structures of the three L. lactis dihydroxyacetone kinase subunits and of a transient intersubunit complex. J. Biol. Chem. 283, 35789–35796 [DOI] [PubMed] [Google Scholar]
- 30. Shi R., McDonald L., Cui Q., Matte A., Cygler M., Ekiel I. (2011) Structural and mechanistic insight into covalent substrate binding by Escherichia coli dihydroxyacetone kinase. Proc. Natl. Acad. Sci. U.S.A. 108, 1302–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Norbeck J., Blomberg A. (1997) Metabolic and regulatory changes associated with growth of Saccharomyces cerevisiae in 1.4 m NaCl. Evidence for osmotic induction of glycerol dissimilation via the dihydroxyacetone pathway. J. Biol. Chem. 272, 5544–5554 [DOI] [PubMed] [Google Scholar]
- 32. Gancedo C., Llobell A., Ribas J. C., Luchi F. (1986) Isolation and characterization of mutants from Schyzosaccharomyces pombe defective in glycerol catabolism. Eur. J. Biochem. 159, 171–174 [DOI] [PubMed] [Google Scholar]
- 33. Daniel R., Stuertz K., Gottschalk G. (1995) Biochemical and molecular characterization of the oxidative branch of glycerol utilization by Citrobacter freundii. J. Bacteriol. 177, 4392–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uzcátegui N. L., Denninger V., Merkel P., Schoenfeld C., Figarella K., Duszenko M. (2007) Dihydroxyacetone induced autophagy in African trypanosomes. Autophagy 3, 626–629 [DOI] [PubMed] [Google Scholar]
- 35. Uzcátegui N. L., Carmona-Gutiérrez D., Denninger V., Schoenfeld C., Lang F., Figarella K., Duszenko M. (2007) Antiproliferative effect of dihydroxyacetone on Trypanosoma brucei bloodstream forms: cell cycle progression, subcellular alterations, and cell death. Antimicrob. Agents Chemother. 51, 3960–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pavlovic-Djuranovic S., Kun J. F., Schultz J. E., Beitz E. (2006) Dihydroxyacetone and methylglyoxal as permeants of the Plasmodium aquaglyceroporin inhibit parasite proliferation. Biochim. Biophys. Acta 1758, 1012–1017 [DOI] [PubMed] [Google Scholar]
- 37. Petersen A. B., Wulf H. C., Gniadecki R., Gajkowska B. (2004) Dihydroxyacetone, the active browning ingredient in sunless tanning lotions, induces DNA damage, cell-cycle block and apoptosis in cultured HaCaT keratinocytes. Mutat. Res. 560, 173–186 [DOI] [PubMed] [Google Scholar]
- 38. Pilkis S. J., Riou J. P., Claus T. H. (1976) Hormonal control of [14C]glucose synthesis from [U-14C]dihydroxyacetone and glycerol in isolated rat hepatocytes. J. Biol. Chem. 251, 7841–7852 [PubMed] [Google Scholar]
- 39. Warnette-Hammond M. E., Lardy H. A. (1985) Catecholamine and vasopressin stimulation of gluconeogenesis from dihydroxyacetone in the presence of atractyloside. J. Biol. Chem. 260, 12647–12652 [PubMed] [Google Scholar]
- 40. Argaud D., Roth H., Wiernsperger N., Leverve X. M. (1993) Metformin decreases gluconeogenesis by enhancing the pyruvate kinase flux in isolated rat hepatocytes. Eur. J. Biochem. 213, 1341–1348 [DOI] [PubMed] [Google Scholar]
- 41. Ivy J. L. (1998) Effect of pyruvate and dihydroxyacetone on metabolism and aerobic endurance capacity. Med. Sci. Sports Exerc. 30, 837–843 [DOI] [PubMed] [Google Scholar]
- 42. Taguchi T., Murase S., Miwa I. (2002) Glyceraldehyde metabolism in human erythrocytes in comparison with that of glucose and dihydroxyacetone. Cell Biochem. Funct. 20, 223–226 [DOI] [PubMed] [Google Scholar]
- 43. Hers H. G., Hue L. (1983) Gluconeogenesis and related aspects of glycolysis. Annu. Rev. Biochem. 52, 617–653 [DOI] [PubMed] [Google Scholar]
- 44. Diao F., Li S., Tian Y., Zhang M., Xu L. G., Zhang Y., Wang R. P., Chen D., Zhai Z., Zhong B., Tien P., Shu H. B. (2007) Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc. Natl. Acad. Sci. U.S.A. 104, 11706–11711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Komuro A., Bamming D., Horvath C. M. (2008) Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine 43, 350–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perdomo A. B., Ciccosanti F., Iacono O. L., Angeletti C., Corazzari M., Daniele N., Testa A., Pisa R., Ippolito G., Antonucci G., Fimia G. M., Piacentini M. (2012) Liver protein profiling in chronic hepatitis C: identification of potential predictive markers for interferon therapy outcome. J. Proteome Res. 11, 717–727 [DOI] [PubMed] [Google Scholar]
- 47. Harris J. I., Waters M. (1976) in The Enzymes (Boyer P. D., ed) 3rd Ed., Vol. 13, pp. 1–49, Academic Press, New York [Google Scholar]
- 48. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 49. Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen M. Y., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hooft R. W., Vriend G., Sander C., Abola E. E. (1996) Errors in protein structures. Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 52. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 53. Word J. M., Lovell S. C., Richardson J. S., Richardson D. C. (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285, 1735–1747 [DOI] [PubMed] [Google Scholar]
- 54. Sanner M. F. (2005) A component-based software environment for visualizing large macromolecular assemblies. Structure 13, 447–462 [DOI] [PubMed] [Google Scholar]
- 55. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 [Google Scholar]
- 56. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 57. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 58. Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 [DOI] [PubMed] [Google Scholar]
- 59. Dupradeau F. Y., Cézard C., Lelong R., Stanislawiak E., Pêcher J., Delepine J. C., Cieplak P. (2008) R.E.DD.B.: a database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 36, D360–D367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berendsen H. J. C., Postma J. P. M., Van Gunsteren W. F., DiNola A., Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 61. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 62. Miyamoto S., Kollman P. A. (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 63. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 64. Humphrey W., Dalke A., Schulten K. (1996) VMD—visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 65. Miwa I., Kito Y., Okuda J. (1994) Purification and characterization of triokinase from porcine kidney. Prep. Biochem. 24, 203–223 [DOI] [PubMed] [Google Scholar]
- 66. MacDonald M. J. (1989) Does glyceraldehyde enter pancreatic islet metabolism via both the triokinase and the glyceraldehyde phosphate dehydrogenase reactions? A study of these enzymes in islets. Arch. Biochem. Biophys. 270, 15–22 [DOI] [PubMed] [Google Scholar]
- 67. Beutler E., Guinto E. (1973) Dihydroxyacetone metabolism by human erythrocytes: demonstration of triokinase activity and its characterization. Blood 41, 559–568 [PubMed] [Google Scholar]
- 68. Frandsen E. K., Grunnet N. (1971) Kinetic properties of triokinase from rat liver. Eur. J. Biochem. 23, 588–592 [DOI] [PubMed] [Google Scholar]
- 69. Hayashi S. I., Lin E. C. (1967) Purification and properties of glycerol kinase from Escherichia coli. J. Biol. Chem. 242, 1030–1035 [PubMed] [Google Scholar]
- 70. Schomburg D., Schomburg I., Chang A. (eds) (2007) in Springer Handbook of Enzymes, Class 2, Transferases, 2nd Ed., Vol. 35, pp. 351–365, Springer, Berlin [Google Scholar]
- 71. Golaz O., Wilkins M. R., Sanchez J. C., Appel R. D., Hochstrasser D. F., Williams K. L. (1996) Identification of proteins by their amino acid composition: an evaluation of the method. Electrophoresis 17, 573–579 [DOI] [PubMed] [Google Scholar]
- 72. Wang R., Lai L., Wang S. (2002) Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 16, 11–26 [DOI] [PubMed] [Google Scholar]
- 73. Gohlke H., Hendlich M., Klebe G. (2000) Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 295, 337–356 [DOI] [PubMed] [Google Scholar]
- 74. Clark R. D., Strizhev A., Leonard J. M., Blake J. F., Matthew J. B. (2002) Consensus scoring for ligand/protein interactions. J. Mol. Graph. Model. 20, 281–295 [DOI] [PubMed] [Google Scholar]
- 75. Charifson P. S., Corkery J. J., Murcko M. A., Walters W. P. (1999) Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 42, 5100–5109 [DOI] [PubMed] [Google Scholar]
- 76. Luisi B., Orozco M., Sponer J., Luque F. J., Shakked Z. (1998) On the potential role of the amino nitrogen atom as a hydrogen bond acceptor in macromolecules. J. Mol. Biol. 279, 1123–1136 [DOI] [PubMed] [Google Scholar]
- 77. Dong F., Miller R. E. (2002) Vibrational transition moment angles in isolated biomolecules: a structural tool. Science 298, 1227–1230 [DOI] [PubMed] [Google Scholar]
- 78. Zierkiewicz W., Komorowski L., Michalska D., Cerny J., Hobza P. (2008) The amino group in adenine: MP2 and CCSD(T) complete basis set limit calculations of the planarization barrier and DFT/B3LYP study of the anharmonic frequencies of adenine. J. Phys. Chem. B 112, 16734–16740 [DOI] [PubMed] [Google Scholar]
- 79. Oberholzer A. E., Schneider P., Baumann U., Erni B. (2006) Crystal structure of the nucleotide-binding subunit DhaL of the Escherichia coli dihydroxyacetone kinase. J. Mol. Biol. 359, 539–545 [DOI] [PubMed] [Google Scholar]
- 80. Siebold C., García-Alles L. F., Erni B., Baumann U. (2003) A mechanism of covalent substrate binding in the x-ray structure of subunit K of the Escherichia coli dihydroxyacetone kinase. Proc. Natl. Acad. Sci. U.S.A. 100, 8188–8192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tesmer J. J., Sunahara R. K., Johnson R. A., Gosselin G., Gilman A. G., Sprang S. R. (1999) Two-metal-ion catalysis in adenylyl cyclase. Science 285, 756–760 [DOI] [PubMed] [Google Scholar]
- 82. Hurley J. H. (1999) Structure, mechanism, and regulation of mammalian adenylyl cyclase. J. Biol. Chem. 274, 7599–7602 [DOI] [PubMed] [Google Scholar]
- 83. Linder J. U. (2006) Class III adenylyl cyclases: molecular mechanisms of catalysis and regulation. Cell. Mol. Life Sci. 63, 1736–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fontes R., Ribeiro J. M., Sillero A. (2000) Inhibition and activation of enzymes. The effect of a modifier on the reaction rate and on kinetic parameters. Acta Biochim. Pol. 47, 233–257 [PubMed] [Google Scholar]
- 85. Copeland R. A. (2000) Enzymes: a Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., pp. 268–282, John Wiley & Sons, New York [Google Scholar]