

Background: Ligand-gated ionotropic glutamate receptors conduct current in response to binding of synaptically released neurotransmitter.
Results: A point mutation (R628E) of GluA2 alters ligand binding and the processes of channel deactivation and desensitization.
Conclusion: Residues within the extracellular vestibule may serve as an intermolecular latch, stabilizing the closed state of the pore.
Significance: The extracellular vestibule is a viable target for new positive allosteric modulators.
Keywords: Computer Modeling; Electrophysiology; Gating; Glutamate Receptors, Ionotropic (AMPA, NMDA); Glutamate Receptors, Metabotropic; Structural Biology
Abstract
AMPA receptors are gated through binding of glutamate to a solvent-accessible ligand-binding domain. Upon glutamate binding, these receptors undergo a series of conformational rearrangements regulating channel function. Allosteric modulators can bind within a pocket adjacent to the ligand-binding domain to stabilize specific conformations and prevent desensitization. Yelshansky et al. (Yelshansky, M. V., Sobolevsky, A. I., Jatzke, C., and Wollmuth, L. P. (2004) J. Neurosci. 24, 4728–4736) described a model of an electrostatic interaction between the ligand-binding domain and linker region to the pore that regulated channel desensitization. To test this hypothesis, we have conducted a series of experiments focusing on the R628E mutation. Using ultrafast perfusion with voltage clamp, we applied glutamate to outside-out patches pulled from transiently transfected HEK 293 cells expressing wild type or R628E mutant GluA2. In response to a brief pulse of glutamate (1 ms), mutant receptors deactivated with significantly slower kinetics than wild type receptors. In addition, R628E receptors showed significantly more steady-state current in response to a prolonged (500-ms) glutamate application. These changes in receptor kinetics occur through a pathway that is independent of that of allosteric modulators, which show an additive effect on R628E receptors. In addition, ligand binding assays revealed the R628E mutation to have increased affinity for agonist. Finally, we reconciled experimental data with computer simulations that explicitly model mutant and modulator interactions. Our data suggest that R628E stabilizes the receptor closed cleft conformation by reducing agonist dissociation and the transition to the desensitized state. These results suggest that the AMPA receptor external vestibule is a viable target for new positive allosteric modulators.
Introduction
AMPA receptors mediate synaptic transmission and can be modulated to facilitate synaptic plasticity that can be the basis for learning and memory. These channels are composed of four subunits (GluA1–4),2 combinations of which impart different biophysical characteristics to the intact channel (1). In addition, RNA editing and alternative splicing in each of these subunits can further diversify channel function. For example in GluA2, the “flip” isoform exhibits slower onset of desensitization than the alternatively spliced “flop” isoform, which differs by only 9 residues (2–4). This region defines a flexible hinge connecting the upper and lower sections of the ligand-binding domain (LBD) where agonist binds and initiates gating of the channel (1, 5).
Studies using targeted receptor mutations have played a major role in elucidating GluA2 activation and desensitization. Notably, the threonine 686 to alanine (T686A) mutation located within the ligand-binding cleft identified cross-cleft bonds that form during glutamate binding and contribute to receptor desensitization (6). Other studies have investigated intradimer interactions within the LBD using substituted cysteines, demonstrating that a decoupling of the LBD interface is necessary for the receptor to enter the desensitized state (7, 8). Although these mutant studies have described both structural and physiological changes that result from agonist binding, they have remained focused on the LBD and an isolated LBD crystal structure. Thus, it is not well understood how the energy of agonist binding is transferred to the channel gate to induce opening; nor are the allosteric transitions of the intact protein allowing termination of agonist-evoked responses fully understood. These can be measured by assessing the kinetics of desensitization (reduction of current during prolonged exposure to agonist) and deactivation (reduction of current due to channel closing and subsequent dissociation of agonist).
Yelshansky et al. (9) investigated a charge-inverting mutation of a residue thought to be in the linker between the LBD and the gate near the pore. This arginine to glutamate mutation (R628E in GluA2) slows the onset of desensitization through a mechanism involving electrostatic interactions. The more recent solution of the crystal structure for the intact AMPA receptor highlighted the potential significance of this region (10). We wished to determine whether this mutation solely affects the desensitized state by influencing a global rearrangement of extracellular domains or is the result of localized interactions between M3 helices. Receptor function measured with electrophysiology was supplemented with radioligand binding assays to probe receptor states and analyzed with mathematical modeling of the receptor to evaluate the impact of the R628E mutation on channel gating.
MATERIALS AND METHODS
DNA Constructs
Rat wild type GluA2 flip and flop cDNA-encoding plasmids were gifts from Dr. Peter Seeburg (University of Heidelberg, Heidelberg, Germany). In this study, wild type constructs were modified to include the R607Q mutation to enhance currents for patch clamp electrophysiology as described previously (11). Thus, the term wild type (“WT”) used in this study refers to GluA2 R607Q. The arginine 628 to glutamate (R628E) flip and flop mutants were made from the respective wild type plasmids using the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) using primers from Integrated DNA Technologies (Coralville, IA). Thus, the term “mutant” in this study refers to GluA2 R607Q,R628E. Because the edited form (Arg-607) is more highly expressed in the mammalian brain (12), experiments using Arg-607 may be more physiologically relevant; however, the low conductance of this isoform (2) precludes patch clamp electrophysiology experiments. However, given the proximity of the Arg-607 pore residue to the external vestibule residue studied here (Arg-628), it is possible that the activity of the mutant receptor could be different if expressed in the more physiologically relevant receptor background.
Transient Transfections for Electrophysiology
Passage 38–45 human embryonic kidney 293 (HEK 293) cells (CRL 1573, American Type Culture Collection, Manassas, VA) were cultured on 35-mm polystyrene dishes (BD Biosciences). Cells were transiently transfected using PolyJet reagent (SignaGen Laboratories, Gaithersburg, MD) with yellow fluorescent protein (YFP) fused in-frame to the C terminus of wild type GluA2 flip, wild type GluA2 flop, GluA2 R628E flip, or GluA2 R628E flop receptor DNA (1 μg/dish). The kinetics of deactivation and desensitization for fluorophore-tagged receptors do not differ from untagged receptors. Medium was changed 4–5 h post-transfection, and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline-2,3-dione (NBQX) was added to a final concentration of 10–20 μm to prevent cell toxicity.
Patch Clamp Electrophysiology
Currents were recorded 24–60 h after transfection from cells expressing a moderate level of fluorescence from the YFP tag. Outside-out membrane patches were held under voltage clamp at −60 mV using an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Patchmaster software (version 2.43, HEKA Instruments Inc., Bellmore, NY) controlled data acquisition and movement of a two-barrel flow pipe perfusion system driven by a piezoelectric device (Burleigh Instruments, Fishers, NY). Responses were digitized at 20 kHz and stored on an iMac computer (Apple, Inc., Cupertino, CA) using an ITC-16 interface (HEKA Instruments Inc.) connected through a USB-16 adapter (HEKA Instruments Inc.). Glass electrodes (TW150F, World Precision Instruments, Sarasota, FL) contained the following intracellular solution: 135 mm CsCl, 10 mm CsF, 10 mm HEPES, 5 mm Cs4BAPTA, 1 mm MgCl2, and 0.5 mm CaCl2, pH 7.2. Outside-out patches were perfused with solutions at 0.2 ml/min through a flow pipe made from θ tubing (BT150-10, Sutter Instruments, Novato, CA). After attaining whole-cell voltage clamp, outside-out patches were pulled from cells, raised off the dish, and positioned near the interface of the two flow pipe solution streams. One stream contained control solution (145 mm NaCl, 5.4 mm KCl, 5 mm HEPES, 1 mm MgCl2, and 1.8 mm CaCl2 with 0.01 mg/ml phenol red, pH 7.3). The other stream contained l-glutamate (10 mm) dissolved in the control solution. For modulator studies, each stream additionally contained either 100 μm cyclothiazide (Tocris Bioscience, Bristol, UK) or 100 μm CX614 (Cortex Pharmaceuticals, Inc., Irvine, CA). Agonist applications were achieved by stepping into and out of the glutamate-containing solution for either 1 or 500 ms to test for deactivation and desensitization, respectively. Solution exchange was measured for each patch by measurement of junction potentials after obliterating the patch at the end of the experiment. Open tip solution exchange times were between 150 and 200 μs.
Confocal Imaging
For confocal microscopy, transfections were performed as described above using 35-mm poly-d-lysine-coated dishes with cutouts and glass coverslips (MatTek Corp., Ashland, MA). Prior to plating, the glass coverslips were treated with entactin-collagen IV-laminin cell (ECL) attachment matrix (Millipore, catalog number 08-110, lot number DAM1661086) for 1 h at 37 °C. 24–48 h after transfection, cells were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2–7.4 for 1–3 h and then washed with 0.1 m phosphate buffer prior to imaging. Confocal analyses were done using a Zeiss LSM 510 Meta microscope with a Plan-Achromat 63×/1.4 numerical aperture oil differential interference contrast objective. An argon laser was used to excite YFP (excitation maximum, 514 nm), and YFP emission was detected with an LP 530 filter. Images were collected using the LSM software and then exported as tiff files.
Ligand Binding Assays with the Agonist [3H]Fluorowillardiine ([3H]FW)
Cells were harvested 24–48 h after transfection by scraping them into HEPES-buffered saline (HBS; 150 mm NaCl and 20 mm HEPES, pH 7.4) and homogenized using a Powergen tissue homogenizer. The resulting membranes were extensively washed by six centrifugations and suspended in HBS. Binding was measured by incubating 10–20 μg of protein for 30 min at 0 or 25 °C in a volume of 20–40 μl with a selected concentration of [3H]FW with or without modulatory drug. [3H]FW was used because affinities for glutamate and AMPA are generally too low to obtain reliable measurements without the aid of agents like thiocyanate that by themselves alter receptor kinetics (13). Samples were then diluted with ice-cold HBS plus 50 mm potassium thiocyanate (wash buffer) and immediately filtered through GF/C glass fiber filters. The filters were rapidly washed with wash buffer. [3H]FW was obtained from American Radiolabeled Chemicals (St. Louis, MO; 30 Ci/mmol). For drug tests, cyclothiazide and CX614 were diluted into warmed HBS from stock solutions in DMSO to obtain concentrations of 400 and 800 μm, respectively; then serially diluted with HBS/DMSO; and further diluted 1:2 into the incubations. DMSO concentration was kept the same in all samples and did not exceed 0.4% (for further details, see Ref. 14).
Simulations of AMPA Receptor Activity
Simulations were conducted with IGOR Pro 6.22 software (Wavemetrics, Lake Oswego, OR) using a custom written plug-in file with code originally written by John Clements and modified extensively by Benveniste et al. (15) as described previously (16). Briefly, receptor state occupancies were calculated from predefined transition rates into and out of each receptor state (see Fig. 7A and Table 2). Voltage clamp simulations of AMPA receptor current activated by glutamate in the presence and absence of modulator differed only by the concentration of modulator predefined for that simulation. Because experiments mimicked by these simulations cannot explicitly resolve many of these rate constants, only one of either the forward or reverse rate constants was manipulated for a particular transition unless otherwise warranted by the results. For Fig. 8, binding data were fit with a model similar to that presented in Fig. 7A except that fluorowillardiine was used as an agonist and occupancy of all agonist-bound states was summed. The fit was accomplished by minimizing the χ2 error using a Levenberg-Marquardt algorithm resident in IGOR. Agonist binding in the presence of modulator was normalized to the result obtained in the absence of modulator. To limit the number of possible solutions, we used the following strategy for fitting. First, fluorowillardiine binding isotherms were fit for both wild type and mutant. The results of these fits for wild type were then put into a model to display simulated currents in voltage clamp elicited by fluorowillardiine and compared with data obtained by Patneau et al. (17). In subsequent fits with modulator, these parameters remained unmodified for transitions without modulator bound. In addition, association rates for agonist were held constant, and in some cases, modulator affinity was held constant at values approximating their apparent affinity by electrophysiological measurements. For desensitization transitions, parameters determined for current responses to glutamate were used and held constant in the fit unless poor fits were obtained in which case these parameters were also allowed to vary. Multiple starting values and fitting strategies were tried until a consensus for parameter values was reached with regard to the values under each condition. Gibbs free energy of activation, ΔG‡, was calculated according to the following equation: ΔG‡ = RT ln(kBT/hk) where R is the universal gas constant, T is the temperature (293 K), kB is the Boltzmann constant, h is the Planck constant, and k is the rate constant of interest (11, 18).
FIGURE 7.

R628E physiological data can be replicated using a kinetic rate model. A, model schematic showing receptor states and the transitions between them (black). A simplified agonist binding site model was used in which either one (R-Glu) or two molecules (R-Glu2) of glutamate can bind to the AMPA receptor (R) allowing transition to an open state (R*-Glu2). Alternatively, receptors can enter desensitized states (Rd-Glu and Rd-Glu2). Identical states have been constructed for receptor in the presence of allosteric modulators (light gray; RM, RM-Glu, RM-Glu2, R*M-Glu2, RdM-Glu, and RdM-Glu2) with receptors states transitioning between modulator-bound and -unbound (gray arrows). The accompanying table describes relationships between receptor states and transitions and the nomenclature of rates used for modeling (actual rates are provided in Table 2): kon, agonist binding; koff, agonist dissociation; β, channel opening; α, channel closing; δ, entrance into desensitization; γ, recovery from desensitization. Rates denoted as prime (e.g. k′on) represent modulator-bound transitions; k+ denotes modulator on-rate constant; and k−, k′−, and k″− denote dissociation rate constants of modulator. All modulator on-rate constants were set equal to simplify the model, and modulator dissociation rate constants do not vary independently but are dependent on other parameter changes to maintain microscopic reversibility. Thus, due to redundancy between the transitions, the rates that could be manipulated were kon, koff, α, β, δ, and γ for control wild type. B and C, simulations of flip (upper) and flop (lower) deactivation (B) and desensitization (C) using wild type (gray) and R628E (black) transition rates (bar above trace, 10 mm glutamate application; scale bars, 5 ms for deactivation and 100 ms for desensitization). D, simulated paired pulse recovery of R628E flop using an initial 200-ms desensitizing pulse of 10 mm glutamate followed by a second 10 mm pulse at increasing time intervals (scale bar, 100 ms). E, recovery from desensitization measured by the ratio of the second peak amplitude to the first peak amplitude with increasing interpulse interval (D). A single exponential function was fit to the resulting values, yielding a time constant for recovery from desensitization of τrecovery = 29.3 ms.
TABLE 2.
Parameter values for transition rates used in kinetic modeling of current responses under voltage clamp for wild type and R628E flop receptors
Rate constants for the 12-state model (Fig. 7A) used for simulations of electrophysiological current responses are depicted for WT and R628E mutant receptors. The column on the left shows the rate constants for transitions 1–6 (see Fig. 7) of the receptor when no modulator is bound. The other columns show the rate constants of the equivalent transitions 8–13 when CTZ (middle column) or CX614 (right column) is bound to the receptor (indicated with a prime symbol; e.g. k′on) plus the rate constants for binding (k+) and unbinding (k−) of the modulator. Naturally, rate constants for transitions 7–17 only play a role when the modulator concentration is >0.

FIGURE 8.

Fitting of fluorowillardiine binding indicates that R628E stabilizes the agonist-bound closed state of the AMPA receptor. A, fits to the 12-state binding model for equilibrium binding of [3H]FW to the flop isoform of either wild type (open circles and gray line) or R628E mutant (filled circles and black line) AMPA receptors. Experimental data for saturation binding curves for [3H]FW are the same as in Fig. 6A (right panel). Best fit rate constant parameters for FW binding in the absence of modulator were used as constants for transitions among states 1–6 for subsequent fits with different concentrations of modulator. B, fits (solid lines) of the 12-state binding model to the modulation of [3H]FW binding of flop isoform receptors by cyclothiazide. Experimental data are the same as in Fig. 6B (right panel). C, fits (solid lines) of the 12-state binding model to the modulation of [3H]FW binding of flop isoform receptors by CX614. Experimental data are the same as in Fig. 6C (right panel). For B and C, data and fits have been normalized to FW binding in the absence of modulator. Error bars are S.E. D, Gibbs free energy diagrams calculated using fitted rate constant parameters determined for wild type (gray lines) and R628E mutant (black lines) with the 12-state model solutions for flop isoform binding data. Troughs from left to right of the upper trace represent the unbound receptor, the agonist-bound closed channel receptor, and the agonist-bound open channel. The lower trace represents the transition between the agonist-bound, closed channel receptor and the agonist-bound, desensitized receptor. Peaks represent the transitions between the states and are marked by their rate constants as defined in Fig. 7A (with FW substituted for glutamate). Forward rate constants are positioned above the line; reverse rate constants are below the line. These diagrams show that the fitted solution to the binding data indicates that the mutant stabilizes the agonist-bound, closed channel conformation of the receptor.
Data Analysis and Statistics
Digitized current recordings were imported to IGOR Pro 6.22 software using custom macros written by M. Benveniste. Traces of receptor deactivation and desensitization were averaged over 4–10 sweeps, and the time constant (τ) was calculated from a single exponential fit. Peak current responses from paired pulse recordings were plotted as the ratio of Peak2/Peak1 on the y axis over the interpulse interval on the x axis, and τrecovery was determined from a single exponential fit. Statistical means and S.E. were calculated from raw data imported into Prism 6 software and analyzed for significant effects compared with wild type using two-way analysis of variance with Sidak's correction.
RESULTS
The Arg-628 Side Chain in One Dimer of the Closed State Receptor Forms an Intersubunit Molecular Interaction with the Adjacent Arg-628 Side Chain
The crystal structure of the intact GluA2 receptor (Fig. 1A) revealed that the receptor has regions of 2- and 4-fold symmetry (10). Using Protein Data Bank code 3KG2 and PyMOL for structural analysis of the antagonist-bound closed state, we observed that residue Arg-628 resides within the 2- to 4-fold symmetry transition region (Fig. 1B). Requisite for this transition is an asymmetry of the pore-forming, membrane-spanning M3 helices with the A (green) and C (blue) subunits of the tetramer having a longer helical coil than the B (yellow) and D (red) subunits (Fig. 1B). This asymmetry results in a less ordered structure of the B and D subunits, consequently orienting the A/B and C/D Arg-628 residues with their guanidino groups opposite each other (Fig. 1C). This structure can be further visualized by looking down through the pore-occluding methionine residues (Met-629) from an extracellular viewpoint (Fig. 1D). Here, the 4-fold symmetry of the transmembrane region is seen as a central square with proline residues (Pro-632) having a 2-fold, parallelogram arrangement. Looking through the upper B/D Arg-628 subunits (translucent), we can see how the arginine guanidino groups interact with backbone oxygen atoms to form a “latch” between Arg-628 residues on adjacent subunits. We hypothesized that this arginine latch in the closed state would be disrupted by a charge-inverting mutation to glutamate (R628E) and that these proximal interactions may play an essential role in stabilizing certain conformational states of the receptor on a global scale. As such, we sought to re-evaluate the role this residue could play in receptor gating with a specific focus on how R628E affects the rate of information transfer from the ligand-binding core to the channel pore.
FIGURE 1.
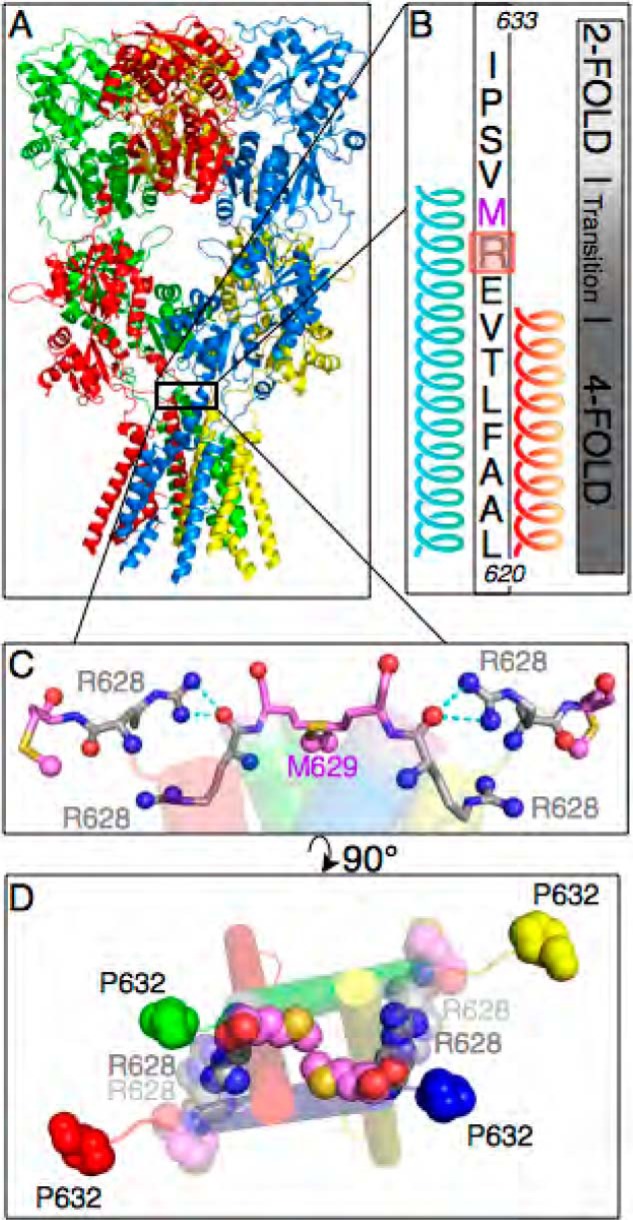
Arg-628 lies at a transition in receptor symmetry and forms the latch in the closed pore receptor state. A, GluA2 crystal structure (15) showing receptor subunits by color (A, green; B, yellow; C, blue; D, red). B, GluA2 amino acid sequence from Leu-620 in M3 (bottom) to Ile-633 in the linker region (top). Subunits A and C feature an extended M3 helix, creating a 4- to 2-fold symmetry transition over a region inclusive of Arg-628 (highlighted in red) and Met-629 (magenta text). C, the symmetry transition orients the Arg-628 side chain guanidino groups of subunits B and D within hydrogen bonding distance to the backbone oxygens of subunits C and A, respectively. D, the Arg-628 residues of subunits B and D (translucent) form a latch with Arg-628 residues of subunits A and C (opaque). This latch is adjacent to the pore-constricting Met-629 residues of subunits A and C (opaque pink spheres; Ref. 10).
The R628E Mutation Alters Kinetics of GluA2
Our first approach was to understand the functional consequence of the mutation on receptor activation. To characterize the effect of the R628E mutant on gating, we used ultrafast perfusion to record wild type and mutant receptor responses to glutamate in outside-out patches pulled from transiently transfected HEK 293 cells. For WT GluA2, the time course of deactivation is faster than that of desensitization for both the flip and flop isoforms, although this difference is more pronounced in flip versus flop (Table 1). In response to a 1-ms pulse of glutamate to probe receptor deactivation, the R628E mutant receptor response decayed significantly more slowly than that of wild type in both flip (p = 0.001) and flop (p = 0.004) receptor isoforms (Fig. 2C and Table 1). Although wild type receptor traces were easily fit to a single exponential time constant, R628E mutant receptor decays exhibited fast and slow exponential components. The amplitude of the slow component was less than 25% (Table 1). However, in most cases, both fast and slow components of mutant receptor deactivation were slower than the single exponential decay observed for wild type receptors (Table 1).
TABLE 1.
Mean data for wild type and R628E GluA2 deactivation and desensitization
Homomeric WT and R628E GluA2 flip and flop isoform receptor currents were recorded from outside-out patches of transiently transfected HEK 293 cells (Fig. 2). Deactivation and desensitization, respectively, were simulated by 1- and 500-ms applications of 10 mm glutamate. Currents from R628E receptors were best fit by two exponential functions (τFast and τSlow), which were weighted (Weighted τ) by percent amplitude (% Fast). The mutant receptors also showed greater steady state/peak ratios (SS/peak). Peak amplitudes did not differ significantly between WT and R628E for either deactivation or desensitization.

FIGURE 2.
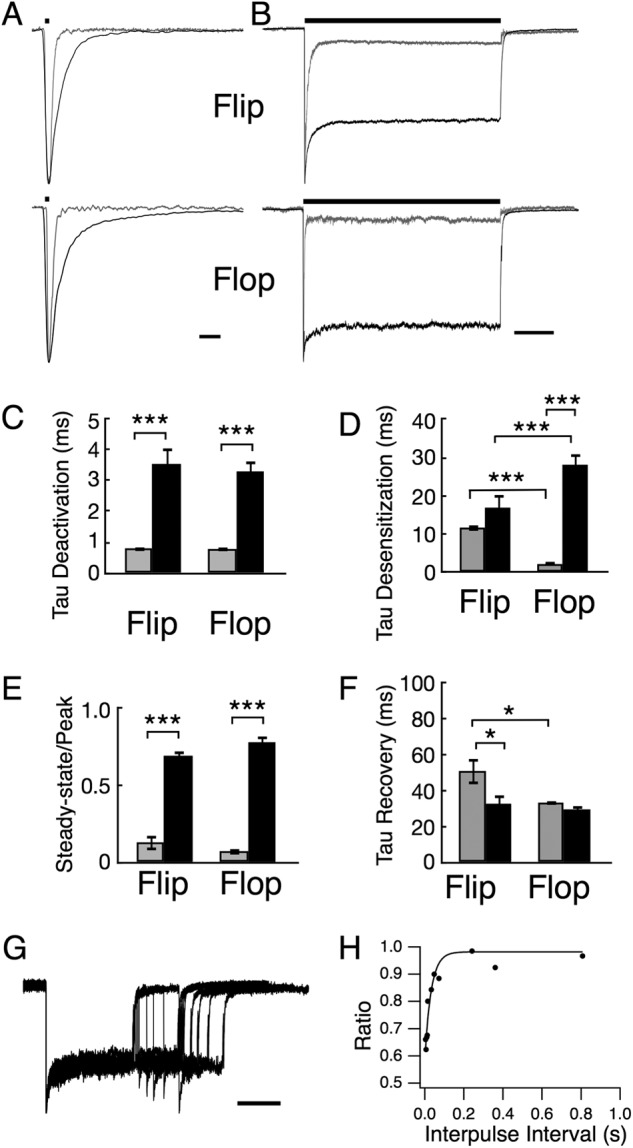
Electrophysiological recordings of R628E currents show altered receptor kinetics compared with wild type GluA2. A and B, representative current traces of outside-out patches pulled from HEK 293 cells expressing homomeric GluA2 flip (upper traces) or GluA2 flop (lower traces). R628E mutant (black) was normalized to wild type (gray) responses to a 10 mm pulse of glutamate (bar above trace) for 1 (A; deactivation) or 500 ms (B; desensitization). Scale bars, 5 ms for deactivation and 100 ms for desensitization. C–F, comparison of mean data between wild type (gray) and R628E (black) shows that the R628E mutant has significantly slower onset of deactivation (C) and increased steady-state current (E) for both flip and flop isoforms of GluA2. Only the flop isoform of R628E showed significantly slower desensitization (D), whereas only the flip isoform had significantly faster recovery (F). R628E deactivation data show the weighted mean of two exponential functions as shown in Table 1. G and H, representative paired pulse recovery sweep for R628E flop (G; scale bar, 100 ms) and the corresponding plot of the second peak to first peak ratio with increasing interpulse intervals (H). *, p < 0.05; ***, p < 0.001 using two-way analysis of variance with Sidak's correction. Error bars represent S.E. A paired pulse sweep entails a desensitizing pulse of glutamate with a second pulse following an incrementally increased recovery duration to measure percent recovery over time.
In addition to slowed deactivation, we observed kinetic differences between wild type- and R628E-mediated responses when currents were evoked with a more prolonged, 500-ms pulse of glutamate (Fig. 2B). A significantly larger steady-state current (equilibrium desensitization) was observed for mutant receptor responses for both flip (p = 0.001) and flop (p = 0.001) receptor isoforms (Fig. 2E and Table 1). In addition to increasing the steady-state current, the R628E mutant also slowed desensitization for the flop isoform of GluA2 (p = 0.001; Fig. 2D and Table 1). Interestingly, there was no difference in the desensitization time constant for the flip isoform (p = 0.51; Fig. 2D and Table 1). These results are qualitatively consistent with those of Yelshansky et al. (9), although we found a larger change in the time constant for onset of desensitization of the mutant on the flop isoform with little to no effect on the flip isoform.
The dramatic increase in the steady state/peak ratio (ss/peak) during prolonged glutamate exposure could arise from a slowed onset of macroscopic desensitization or a more rapid recovery from desensitization (19). To better understand this change, recovery from desensitization was measured using a paired pulse protocol where a 200-ms desensitizing pulse of 10 mm glutamate was followed by a second glutamate application at increasing interpulse intervals (Fig. 2G). The ratio of the second current peak to the first current response was plotted with respect to the interpulse interval and fit with a single exponential function (Fig. 2H). We discovered an isoform-dependent effect of the R628E mutant on recovery from desensitization (τrecovery: R628E flip, 32.63 ± 5.34 ms (n = 3; p = 0.036) compared with wild type flip, 50.83 ± 7.34 (n = 4); R628E flop, 29.48 ± 2.15 ms (n = 4) compared with wild type flop, 33.28 ± 0.72 ms (p = 0.17; n = 4); Fig. 2F). These data confirm that Arg-628, positioned at the outer vestibule of the pore, plays an important role in channel desensitization kinetics as reported previously (9) but also significantly slows the onset of receptor deactivation, a previously unappreciated finding.
The R628E Mutation Alters Receptor Trafficking
Many patches from fluorescent cells transfected with the R628E construct did not express current under voltage clamp (<5% success rate). The frequency of obtaining healthy patches was much higher for fluorescent cells expressing the wild type construct, suggesting that the membranes of cells expressing the mutant channel were more fragile. Because receptor expression at the cell surface is essential for recording currents, we investigated the possibility that the R628E mutation influences receptor trafficking. We transiently transfected HEK 293 cells with YFP-tagged wild type or R628E mutant cDNA and assessed their subcellular fluorescence distribution over time using confocal microscopy. Wild type receptors displayed distinct “rings” of fluorescence coincident with the plasma membrane as early as 24 h post-transfection, indicating successful trafficking of the receptor to the plasma membrane. This fluorescence remained stable for 48 h following transfection, consistent with previous reports (20–22). In contrast, cells transfected with cDNA encoding R628E receptors developed areas of dense fluorescence (“aggresomes”; Refs. 23 and 24). These aggresomes became more numerous as time progressed and were accompanied by a decreased accumulation at the plasma membrane surface (Fig. 3). The trafficking effect of the mutation was observed for expression of both flip and flop receptor cDNAs, with aggresomes observed at a seemingly faster rate with flop more than flip receptors (data not shown). These results indicate that the R628E mutant causes a perturbation in receptor trafficking and provide at least a partial explanation for the difficulty in recording functional data for the mutant receptor. The poor trafficking of this mutation likely reflects a reduced stability of the tertiary protein structure.
FIGURE 3.

The R628E mutant shows impaired trafficking and formation of aggresomes. Confocal images of HEK 293 cells transiently transfected with YFP-tagged flip wild type (left) or R628E (right) constructs are shown. Top row, images taken 24 h after cell transfection; bottom row, images taken 48 h post-transfection. The inset in upper right corner of each panel shows magnification of the area drawn by the dotted white box. Note the increased presence of aggresomes (white arrows) in HEK 293 cells transfected with the R628E construct indicative of disrupted channel trafficking.
Allosteric Modulation of R628E Is Preserved
The ability of the R628E mutation to perturb both receptor deactivation and desensitization is reminiscent of the actions of positive allosteric modulators that bind at the dimer interface of the ligand-binding core (11, 16, 25–28) and is also similar to the allosteric modulation of receptor gating conferred by transmembrane accessory receptor proteins such as stargazin (29, 30). Therefore, we hypothesized that assessing the efficacy of positive allosteric modulators on gating of the R628E mutation might provide insight into the molecular mechanism through which the mutation perturbs channel gating compared with wild type receptors.
For this approach, we utilized the modulators cyclothiazide (CTZ), a commonly used benzothiadiazide, and the pyrrolidinone CX614. These drugs were chosen because of their contrasting isoform and deactivation/desensitization efficacies. CTZ has been shown to be a flip-selective modulator of GluA2 desensitization with little effect on deactivation kinetics (27, 31–33), whereas CX614 is more selective for GluA2 flop, modulating both deactivation and desensitization of that isoform (16, 32).
Recalling that the R628E mutation resulted in the slowing of deactivation for the flip isoform of GluA2 (Fig. 2A), we note that addition of either modulator had no additional effect on deactivation kinetics (Fig. 4A). Deactivation time constants (τdeact) for wild type receptor responses were: CTZ, 0.94 ± 0.11 ms (n = 5; p = 0.98) and CX614, 0.79 ± 0.11 ms (n = 4; p = 0.99) compared with no modulator, 0.81 ± 0.09 ms (n = 6). Deactivation time constants for the mutant receptor responses were: CTZ, 4.10 ± 0.59 ms (n = 4; p = 0.72) and CX614, 4.26 ± 1.06 ms (n = 3; p = 0.65) compared with no modulator, 3.53 ± 0.55 ms (n = 12) (Fig. 4B). In contrast, desensitization of flip receptors in the presence of CTZ was non-existent for both wild type (ss/peak = 1.01 ± 0.02 (n = 5; p = 0.001) compared with no modulator, 0.13 ± 0.05 (n = 7)) and R628E mutant responses (ss/peak = 1.01 ± 0.04 (n = 4; p = 0.003) compared with no modulator, 0.69 ± 0.03 (n = 11)) (Fig. 4, A and C). Lastly, the effect of CX614 on R628E flip receptors showed nearly complete loss of desensitization (ss/peak = 0.96 ± 0.17 (n = 4; p = 0.02)) (Fig. 4, A and C).
FIGURE 4.
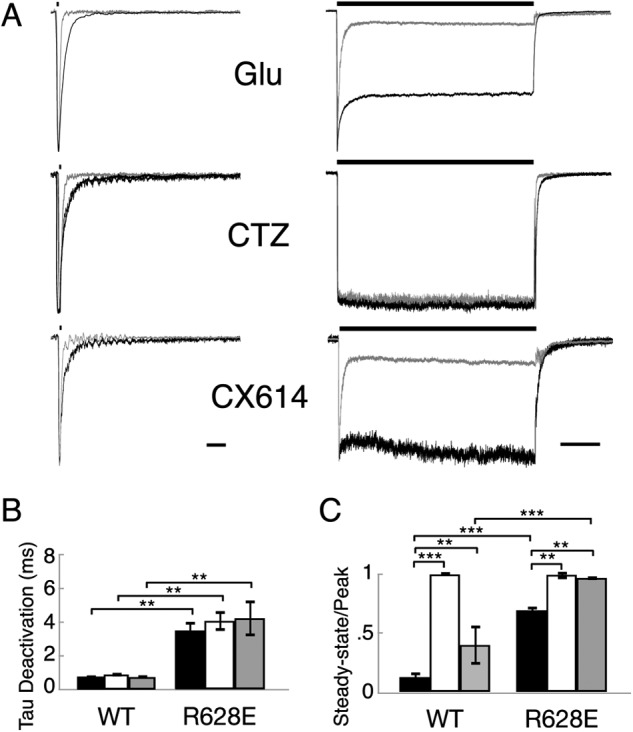
The R628E mutant does not occlude effects of modulator for the flip isoform of GluA2. A, representative current traces of outside-out patches pulled from HEK 293 cells transfected with homomeric GluA2 flip cDNA. R628E mutant (black) was normalized to wild type (gray) responses to a 10 mm pulse of glutamate (bar above trace) for 1 (left; deactivation) or 500 ms (right; desensitization). Currents were recorded with glutamate only (Glu) or in the continuous presence of 100 μm CTZ or 100 μm CX614. Scale bars, 10 ms for deactivation and 100 ms for desensitization. B and C, graphical comparisons of WT and R628E weighted deactivation (B) and ss/peak (C) in the absence of modulator (black) or presence of 100 μm CTZ (white) or 100 μm CX614 (gray). **, p < 0.01; ***, p < 0.001 using two-way analysis of variance with Sidak's correction. Error bars represent S.E.
We then tested the effects of the two modulators on the flop isoforms of the R628E mutant receptors (Fig. 5A). CTZ, which did not modulate wild type flop channel deactivation (τdeact: CTZ, 0.83 ± 0.09 ms (n = 6; p = 0.99) compared with no modulator, 0.79 ± 0.07 ms (n = 5)), also did not modulate deactivation of R628E flop receptors (τdeact: CTZ, 2.77 ± 0.76 ms (n = 4; p = 0.56) compared with no modulator, 3.29 ± 0.37 ms (n = 11)) (Fig. 5B). In contrast, CX614 was an efficacious modulator of flop deactivation for wild type receptors (τdeact: CX614, 1.98 ± 0.18 ms (n = 9; p = 0.048)). Likewise, deactivation for R628E flop receptors slowed significantly in the presence of CX614 (τdeact: CX614, 6.8 ± 0.40 (n = 4; p = 0.001)) (Fig. 5B, bold), demonstrating that the R628E mutation does not prevent CX614 modulation. As expected, desensitization of R628E flop channels was completely blocked by both CTZ and CX614 (ss/peak: CTZ, 0.98 ± 0.01 (n = 4; p = 0.004) and CX614, 0.96 ± 0.03 (n = 4; p = 0.009) compared with no modulator, 0.78 ± 0.04 (n = 12)). Thus, positive allosteric modulators, which bind to the dimer interface of the ligand-binding core, and the R628E mutation have additive rather than occluding effects on slowing of channel deactivation and desensitization. Radioligand binding assays provide an avenue to elaborate upon this finding.
FIGURE 5.
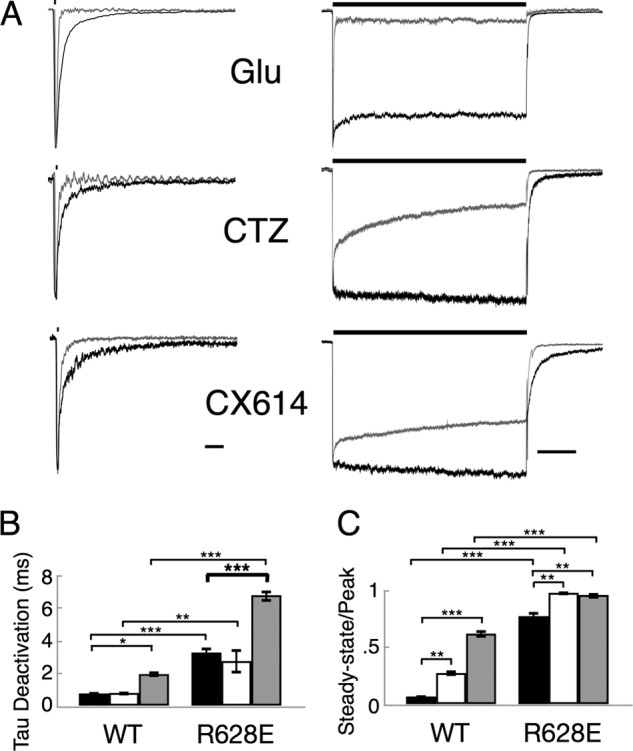
The R628E mutant does not occlude effects of allosteric modulators for the flop isoform of GluA2. A, representative current traces of outside-out patches pulled from HEK 293 cells transfected with homomeric GluA2 flοp cDNA. R628E mutant (black) was normalized to wild type (gray) responses to a 10 mm pulse of glutamate (bar above trace) for 1 (left; deactivation) or 500 ms (right; desensitization). Currents were recorded with glutamate only (Glu) or in the continuous presence of 100 μm CTZ or 100 μm CX614. Scale bars, 10 ms for deactivation and 100 ms for desensitization. B and C, graphical comparisons of WT and R628E weighted deactivation (B) and ss/peak (C) in the absence of modulator (black) or presence of 100 μm CTZ (white) or 100 μm CX614 (gray). *, p < 0.05; **, p < 0.01; ***, p < 0.001 using two-way analysis of variance with Sidak's correction. Error bars represent S.E.
Binding Assays Show Enhanced Agonist Affinity and Altered Efficacy of Allosteric Modulators
Radioligand binding assays were conducted on transiently transfected HEK 293 cells using the agonist [3H]FW to determine whether the R628E mutation influences agonist affinity and changes in affinity imposed on the receptor by CTZ or CX614. [3H]FW was used because it binds with higher affinity than tritiated AMPA and has been used as the model agonist binding reagent in many of the classical AMPA receptor binding studies (14). [3H]FW binding to wild type GluA2 was fit with a single binding site logistic equation for KD, Bmax, and the Hill coefficient (nH). The binding parameters for wild type flip receptors were: KD = 113.9 ± 20.9 nm, Bmax = 2.96 ± 0.20 pmol/mg of protein, and nH = 0.76 ± 0.03. For wild type flop receptors, the parameters were: KD = 60.0 ± 21.7 nm, Bmax = 2.04 ± 0.22 pmol/mg of protein, and nH = 0.83 ± 0.07 (Fig. 6A, open circles). For the GluA2 R628E mutant, the parameters were: flip receptors, KD = 24.7 ± 1.5 nm, Bmax = 2.72 ± 0.08 pmol/mg of protein, and nH = 1.03 ± 0.04; R628E flop receptors, KD = 23.9 ± 7.1 nm, Bmax = 2.02 ± 0.21 pmol/mg of protein, and nH = 0.97 ± 0.08 (Fig. 6A, dark circles). Thus, these data suggest that the R628E mutation enhances the apparent affinity for the agonist despite the fact that the mutation is not located near the ligand-binding core of the receptor.
FIGURE 6.

The R628E mutant shows altered agonist affinity with an isoform-specific effect in the presence of allosteric modulators. A, saturation binding curves for [3H]FW binding to wild type (open circles and gray line) and R628E receptors (closed circles and black line). Binding was measured at 1–400 nm radioligand at 0 °C. The data are shown as averages and S.E. of two (flip) or five (flop) experiments in which WT and mutant receptors were tested in tandem (absence of error bars indicates that the error was smaller than the symbol size). Data points were fitted with a logistic equation. KD, Bmax, and nH values are presented in the text. These data show that both flip (left) and flop (right) R628E receptors exhibit a shift toward higher agonist affinity. B and C, the effects of modulators CTZ (B) and CX614 (C) were measured using a fixed 5 nm [3H]FW concentration and varying concentrations of modulator (with maximum concentration limited by drug solubility). Binding at each modulator concentration was normalized to that without modulator and averaged across experiments. In the flop isoform (right panels), the CTZ-induced reduction in [3H]FW binding was greatly attenuated in R628E (−27% compared with −60% in WT). Conversely, the CX614-induced increase in [3H]FW binding was significantly enhanced in R628E receptors (+62% compared with +30% in WT). In the flip isoform (left panels), these changes in the effectiveness of CTZ and CX614 were much smaller. Error bars represent S.E.
Modulatory drugs were shown earlier to alter agonist binding in specific ways. For instance, CTZ and CX614 tend to cause opposite changes in agonist binding with CTZ reducing binding and CX614 enhancing binding. It has been argued that this reflects differences observed in electrophysiology where CTZ primarily acts by blocking desensitization whereas CX614 has prominent effects also on current deactivation and hence on ligand dissociation (14). Therefore, we tested the effects of CTZ and CX614 on [3H]FW binding in both wild type and R628E receptors (Fig. 6, B and C). These studies were achieved by using a constant [3H]FW concentration far below its KD and varying the modulator concentration. The data were then fitted with a three-parameter logistic equation to obtain an apparent affinity for the modulator (EC50). In addition, this fit also provides an asymptote that indicates the maximal obtainable change in agonist binding at saturating modulator concentration, which may also be described as drug “efficacy.”
As shown in Fig. 6, the efficacy of CTZ and CX614 was changed significantly in the flop isoform of the mutant receptor. At high concentrations of CTZ, binding was reduced to 40% of control binding in wild type receptors but to only 73% in R628E receptors. With CX614, the opposite effect was observed in that binding was increased from 131% of control in wild type receptors to 162% for mutant receptors. Thus, with both drugs, the asymptote was shifted upward for the R628E mutant receptors. Because the decrease of fluorowillardiine binding probably reflects shifting equilibria between the higher affinity desensitized states and lower affinity sensitized states where mutant receptors show greatly reduced desensitization (Fig. 2), the ability of CTZ to modulate agonist binding is reduced. In contrast, CX614 has multiple effects on the flop isoform by blocking desensitization but also slowing deactivation such that the effect on agonist binding is enhanced with increasing concentrations of modulator. Having both functional measurements and binding data, we proceeded to use computational modeling to recapitulate these data to gain insight into a molecular mechanism for the actions of the R628E mutation on receptor gating.
Replicating Electrophysiological Data Using a Kinetic Rate Model
Having a model that can reproduce both binding and electrophysiological data will limit the number of potential kinetic parameter solution sets. To simulate our electrophysiological data, we used our previously published kinetic model (Ref. 16 and Fig. 7A), which manipulates the proportion of modulator-bound and modulator-unbound populations. Thus, the model used here consists of 12 receptor states: six of these states (Fig. 7A) represent conformations of the receptor as it binds agonist leading to channel opening and desensitization in the absence of modulator, and six states (Fig. 7A) represent these receptor conformations with modulator bound. Forward and reverse transition rates between these receptor states can be found in Table 2.
In simulating AMPA receptor currents under voltage clamp for wild type flop channels, calculated kinetic parameters yielded a deactivation time constant of 0.7 ms and a steady state to peak ratio of 5% when using a 1- and 500-ms pulse of 10 mm glutamate, respectively. These values corresponded well with our experimental data (Table 1 and Fig. 7, B and C). Currents from wild type flop receptors in the presence of CTZ could be reproduced by slowing the rate for onset of desensitization (δ) by 100-fold (Table 2), and the resulting waveform accurately simulated the experimental findings (data not shown). Simulation of CX614 modulation required changes to δ (reduced 500-fold), recovery from desensitization (γ; reduced 15-fold), and agonist dissociation (koff; reduced 20-fold); the waveforms for CX614 also accurately simulated the experimental findings (data not shown).
Having identified kinetic parameters that reproduce wild type flop currents in the presence and absence of the two modulators, we simulated R628E flop receptor currents in the absence and presence of each modulator. Because the mutation changes receptor kinetics in the absence of modulator, transitions 1–6 were first targeted (Fig. 7A). Two different solutions could successfully reproduce our electrophysiological data in the absence of modulator (Fig. 7, B and C), each of which required changing multiple parameters. The first solution required slowing channel closure (α) 10-fold and δ ∼20-fold. The other solution required slowing koff 40-fold and δ 100-fold (as shown in the lower part of Table 2). Additionally, both of these solutions were able to appropriately recapitulate paired pulse recovery of R628E flop receptors (Fig. 7, D and E). As such, modeling electrophysiology alone was unable to discriminate the better exemplar. Given that one of the solutions suggests a change to agonist binding rates, we applied our model to the ligand binding data to discern which solution was preferable.
Fitting Radioligand Binding Data with the 12-State Model
In an attempt to further discern between possibilities for how the R628E mutation modifies transitions between receptor states, we fitted data from fluorowillardiine binding experiments on the flop isoform (Fig. 6) to the 12-state model. This required modification of the 12-state model used in Fig. 7 because the higher affinity ligand fluorowillardiine was used in binding experiments instead of glutamate. This was achieved by first fitting the fluorowillardiine binding isotherms depicted in Fig. 8A. The best fit to wild type flop binding data that also was consistent with electrophysiological data (see “Materials and Methods”) required modification of koff, γ, and δ and a slight change in kon from the original rate constants determined for glutamate (Tables 2 and 3). These constants (Table 3, WT Control) were then used as a starting point for fitting the binding data for R628E flop receptors. The best fit to these data required that koff, γ, and δ be allowed to vary during the fit (Fig. 8A). It should be noted that changing α, δ, or koff alone or koff and α together did not yield satisfactory fits. Comparison of the best fit parameters between the wild type and R628E binding data indicated a striking reduction (26.6-fold) in koff and both a decrease in δ (10-fold) and an increase in γ (5-fold) values for R628E. These results suggest that the solution set in which both agonist dissociation (koff) and desensitization (γ and δ) are altered more accurately describes the observed effects of the R628E mutation on agonist binding.
TABLE 3.
Parameter values for transition rates used in fitting of fluorowillardiine binding for wild type and R628E flop receptors to the 12-state model
Rate constants for the 12-state model (Fig. 7A) used for fitting of equilibrium binding data are depicted for WT and R628E mutant receptors either in the absence of modulator or in the presence of either CTZ or CX614. The column on the left shows the rate constants for transitions 1–6 (see Fig. 7) of the receptor when no modulator is bound. The other columns show the rate constants of the equivalent transitions 8–13 when CTZ (middle column) or CX614 (right column) is bound to the receptor (indicated with a prime symbol) plus the rate constants for binding (k+) and unbinding (k−) of the modulator. Rate constants for transitions 7–17 only play a role when the modulator concentration is >0.

The experimental data in Fig. 6, B and C, had further indicated that increasing concentrations of CTZ and CX614 also differentially change agonist binding. The mutant and modulator rate constants determined in the fluorowillardiine binding isotherms (Fig. 8A) were used as constants for receptor states not bound with modulator while allowing a minimum of rate constants associated with modulator binding/bound states to vary in fits for data in Fig. 8, B and C (see “Materials and Methods”). Fits to decreasing fluorowillardiine binding with increasing CTZ concentrations for WT receptors required a striking 33-fold decrease in δ′ in comparison with the parallel transition (δ) in the absence of modulator in addition to a slight slowing of k′off and γ′ (Table 3, WT CTZ). This is consistent with previous reports indicating that CTZ slows entry into the desensitized state and may slightly increase agonist affinity (27, 34). Fitting the R628E mutant data for fluorowillardiine binding in the presence of different concentrations of CTZ (Fig. 8B) also only required a 100-fold reduction in the rate constant for entering the desensitized state (Table 3, R628E CTZ, δ′).
A good fit of the 12-state model to wild type flop receptor binding data in which fluorowillardiine increased with increasing concentrations of CX614 (Fig. 8C) required a 24-fold reduction in k′off, a 100-fold reduction in δ′, and a 5-fold increase in γ′ in comparison with the parallel rate constants in the absence of modulator (Table 3, WT CX614). In the fit of the R628E mutant binding data in Fig. 8C, the major parameter affected when comparing CX614-modulated transitions with the parallel modulator-free transitions is a 10-fold slowing of δ′. There is also a small 2.4-fold reduction in k′off in comparison with the already slow koff for the parallel transition not bound with CX614. Interestingly, when comparing WT CX614 fit parameters with those of R628E fit parameters (Table 3), the only change to parameters is a slight reduction in k′off.
To better understand how an external vestibule mutant affects ligand binding, we used model parameters to construct Gibbs free energy diagrams depicting receptor conformation states and transitions between them (Fig. 8D). The first “peak” represents a combination of agonist binding and dissociation coupled with the transitions between the apo state and the closed cleft conformation (modeled as kon and koff). From the agonist-bound, closed cleft state, the receptor can either transition to the open state through β and α transitions or desensitize through δ and γ transitions. Changing koff alone stabilizes the closed cleft conformation along with all subsequent ligand-bound states. Interestingly, the additional slowing of δ and increasing of γ, which best recapitulated the data, yielded a desensitized state for the R628E mutant that had a ΔG value similar to that of wild type, leaving only the agonist-bound, closed cleft conformation and subsequent channel open states as more stable. Thus, our modeled solution for R628E, which accurately simulates experimentally observed changes to both electrophysiology and agonist binding, predicts that the predominant energetic change responsible for these effects is a stabilization of the agonist-bound, closed cleft state.
DISCUSSION
In this study, we revealed mechanistic insight into AMPA receptor gating by combining selective pharmacology with three complementary techniques: patch clamp electrophysiology with ultrafast perfusion, radioligand binding assays, and computational modeling of ion channel function. These methodologies, together with the full-length AMPA receptor crystal structure (10), have allowed us to reinterpret the role of outer vestibule residue R628E in AMPA receptor gating.
According to the crystal structure, the Arg-628 residue is located in the extracellular vestibule near the selectivity filter. Our electrophysiological results indicate that this residue is pivotal in the gating process because the R628E mutant slowed deactivation of GluA2 receptor channels in addition to blocking desensitization (Fig. 2). The concomitant slowing of deactivation and block of desensitization cannot be explained solely by modifying transitions into and out of the desensitized state (δ and γ) widely accepted to reflect rearrangements of the LBD dimer interface. Simulations utilizing the model depicted in Fig. 7 indicate that voltage clamp data acquired for the R628E mutant can only be recapitulated by the additional modification of either koff or the channel closing transition (α).
As macroscopic currents under voltage clamp yield information primarily about transitions near the open states, we further differentiated these possibilities by examining how their alternative mechanistic modifications predict changes in agonist binding in the absence and presence of known modulators of desensitization. [3H]Fluorowillardiine binding to mutant R628E receptors had ∼5-fold higher affinity in comparison with binding to wild type receptors. This finding is also supported by the fact that this mutation increased the apparent affinity for glutamate as measured using electrophysiology (9). Using two different approaches (Figs. 7 and 8), the R628E-induced increase in agonist affinity combined with the block of desensitization could be mimicked by decreasing koff and altering the γ/δ ratio. Conversely, decreasing both α and δ, which was similarly effective in mimicking electrophysiological data, produced an approximate 3.5-fold decrease in affinity (data not shown), which is inconsistent with experimental binding data (Fig. 6).
Further insight can be gained by modulation of AMPA receptor gating with CTZ and CX614. These two modulators bind to a common pocket between LBD dimers (33). However, although CTZ reduces desensitization putatively through modulation of δ and/or γ (27), CX614 additionally slows deactivation (14, 35). Increasing concentrations of these modulators also yield unique binding profiles for agonist that can further validate or discount mechanistic implications from computer simulations (14). Increasing the concentration of a modulator that is expected to solely block entry into the desensitized state should cause a shift in the proportion of agonist-bound receptors from high affinity desensitized receptors to lower affinity non-desensitized receptors, yielding a decrease in the amount of agonist bound in subsaturating conditions. This can be observed for wild type receptors with CTZ (Fig. 6) and was mimicked in simulations by lowering δ 100-fold (Fig. 8 and Table 2).
CX614 is known to slow deactivation as well as desensitization (Refs. 11 and 32 and Fig. 4). This produced an increase in agonist binding with increasing concentration of CX614 that was further enhanced for the R628E mutation (Ref. 14 and Fig. 6). Our simulations accurately reproduced this enhancement qualitatively and quantitatively (Fig. 8) by changing koff and γ/δ.
There are important technical limitations to our study that bear consideration. First, although we performed the study on a model system of homo-oligomeric GluA2 receptors, there is sound evidence that AMPA receptors exist as hetero-oligomeric assemblies in native tissue (36, 37). Furthermore, measurement of the function of the mutant receptors relies on patch clamp electrophysiology, which necessitates the use of the higher conducting (12) but potentially less physiologically relevant unedited isoform of GluA2 as discussed under “Materials and Methods.” Although this study focuses on the biophysics of channel activation and desensitization rather than physiology, this issue is important because the Gln/Arg editing site has been shown to be involved in a number of important physiological processes other than calcium permeability including receptor trafficking (38, 39) and association with accessory regulatory proteins (40). Thus, the function of the mutant receptor could be further influenced by the presence of an arginine within the pore, and our studies would not reveal that interaction. Finally, the mutant receptor we studied was a fusion protein with the enhanced GFP reporter fused to the C terminus of the AMPA receptor. This experimental strategy was used to permit the acquisition of the electrophysiological data despite technical challenges caused by the mutation itself. To address whether the addition of the fluorophore impacted our results, we compared the kinetics of the fusion protein with that of untagged recombinant receptors and concluded that the addition of the C-terminal fluorophore to GluA2 did not alter receptor kinetics. Specifically, the time constants for deactivation and desensitization of the WT receptor, 0.79 and 1.64 ms, respectively, are very consistent with time constants reported in other published reports (our own and others) using untagged receptors with deactivation kinetics in those reports ranging from 0.71 to 0.92 ms and desensitization kinetics ranging from 1.18 to 1.87 ms (3, 11, 41, 42). Thus, although there is a possibility that the unique functional properties of R628E may have been influenced by the C-terminal reporter protein, this is a less likely interpretation of our results as both the WT and mutant proteins contained the enhanced GFP sequences.
The unique orientation of Arg-628 residues within the tetramer of GluA2 subunits differs in several ways from that anticipated when the R628E mutant was first published (9). Although the majority of the receptor transmembrane region has 4-fold symmetry about the pore, this symmetry transitions to 2-fold symmetry in the region of the Arg-628 residue. One residue in each dimer of the dimer pair has a less ordered, extended conformation, permitting the Arg-628 residue of one subunit to interact with the less extended Arg-628 residue of the subunit in the other dimer of the dimer pair (Fig. 1).
Although electrostatic interactions along an extended conformation were originally predicted for this region, the subunit asymmetry and subsequent alignment of Arg-628 could not be deduced without the now available structural information. Theoretically, it is possible that electrostatic interference caused by R628E near the channel pore might slow channel closing (α). Indeed, the leftward shift in the glutamate concentration-response curve measured by patch clamp (9) is consistent with this conclusion because a longer sojourn in the open state causes an increase in apparent agonist affinity (43). However, our simulations indicate that slowing α does not reproduce the data.
Alternatively, the resulting charge inversion may stabilize the ligand-bound, closed cleft conformation represented in the present model by slowing koff. Although cleft opening and agonist dissociation are modeled as separate rates elsewhere (6), here we simulated cleft opening and agonist dissociation as a single rate. We simplified our model to have the ability to simulate changing proportions of modulator-bound and -unbound populations (Fig. 7). According to crystallographic studies, the R628E residue does not interact directly with agonist or associated water molecules in its binding site but rather resides in the extracellular vestibule (10). Thus, we can assume that agonist dissociation is not directly affected by this mutation and instead suggest that opening of the agonist-binding cleft is affected.
Support for this is also apparent when kinetic rate constants are transformed into Gibbs free energy values (Fig. 8). Simulations where koff and δ are slowed and γ is increased to mimic the comparison of wild type with R628E mutant receptors demonstrate a stabilization of the agonist-bound closed state (Fig. 7). It is interesting to note that this extracellular vestibule residue stabilizes the closed clamshell position even though it is far from the interface between the ligand-binding domains where modulators of desensitization bind. Therefore, we propose that the stability of the M3 bundle within the pore directly impacts the stability of the LBD intradimer interface (regulating desensitization) as well as the stability of the agonist-bound, closed cleft conformation (regulating deactivation). However, although we cannot rule out the possibility that the stability of the pore impacts only one of these interfaces, in such a case, the energetics of the two interfaces must be tightly coupled.
An alternative explanation proposed by Dong and Zhou (44) is that the Arg-628 mutation to negatively charged glutamate creates an unfavorable electrostatic interaction with negatively charged lipids in the cell membrane upon entering the desensitized state. Destabilization of the desensitized state would cause a decrease in desensitization. However, our recapitulation of electrophysiological and binding data by simulations and fitting requires a reduction in koff in addition to a slowing of δ (Fig. 8), suggesting that destabilization of the desensitized state is not sufficient to explain the results.
Mutation of the Arg-628 residue is reminiscent of the lurcher mutation found in GluD2 (A636T; Refs. 45 and 46), aspects of which are recapitulated in GluA1 and GluA2 (A622T). Unlike R628E, the lurcher mutation has a remarkable leak current (inward current in the absence of adding exogenous glutamate) that led to the speculation that the channel is constitutively open (47, 48). However, both residues are present in the M3 transmembrane region, and mutating either residue slows the time course of deactivation and decreases desensitization as well as increases agonist affinity (Refs. 9 and 49 and this study). It has been proposed that the lurcher mutation could cause a destabilization of “the tight helix crossing associated with the resting, closed state of the receptor” interpreted to be a local change reflecting pore gating (10). Klein and Howe (49) extended this supposition by showing that the lurcher mutation increased the probability of activation of GluA1 by increasing the apparent affinity for glutamate to the nanomolar range with a concomitant reduction in desensitization. This conclusion is more parsimonious with the findings in this study.
It is interesting to note that we consistently saw an additive effect of modulators on the R628E mutant, suggesting that the mutant and modulators alter GluA2 kinetics through different, non-interfering mechanisms. This result, in support of previous findings (50), identifies the AMPA receptor outer vestibule as a potential target for the design of future modulatory drugs. Furthermore, the results here suggest that compounds acting at the outer vestibule can be co-administered with currently established modulators to enhance observed effects. Additionally, it may also be possible to design non-competitive antagonists with a compensatory effect at this site, similar to the GYKI compounds (35, 51).
Acknowledgments
We thank Amanda Dudek for assistance with mutagenesis and John Gieser for contributions to the artwork. We thank Cortex Pharmaceutical, Inc. for generously providing CX614.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 MH064700 (to K. M. P.), S11NS055883 and U54NS083932 from the NINDS, and SC1AG046907 from the NIA (to M. B.).
- GluA
- AMPA receptor subunit homomer
- LBD
- ligand-binding domain
- CTZ
- cyclothiazide
- CX614
- 2H,3H,6aH-pyrrolidino(2,1–3′,2′)1,3-oxazino(6′,5′-5,4)benzo(e)1,4-dioxan-10-one
- FW
- fluorowillardiine
- HBS
- HEPES-buffered saline
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- ss/peak
- steady state/peak ratio.
REFERENCES
- 1. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Swanson G. T., Kamboj S. K., Cull-Candy S. G. (1997) Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J. Neurosci. 17, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koike M., Tsukada S., Tsuzuki K., Kijima H., Ozawa S. (2000) Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J. Neurosci. 20, 2166–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Partin K. M., Mayer M. L. (1996) Negative allosteric modulation of wild-type and mutants AMPA receptors by GYKI 53655. Mol. Pharmacol. 49, 142–148 [PubMed] [Google Scholar]
- 5. Armstrong N., Gouaux E. (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181 [DOI] [PubMed] [Google Scholar]
- 6. Robert A., Armstrong N., Gouaux J. E., Howe J. R. (2005) AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J. Neurosci. 25, 3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin R., Banke T. G., Mayer M. L., Traynelis S. F., Gouaux E. (2003) Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 6, 803–810 [DOI] [PubMed] [Google Scholar]
- 8. Plested A. J., Mayer M. L. (2009) AMPA receptor ligand binding domain mobility revealed by functional cross linking. J. Neurosci. 29, 11912–11923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yelshansky M. V., Sobolevsky A. I., Jatzke C., Wollmuth L. P. (2004) Block of AMPA receptor desensitization by a point mutation outside the ligand-binding domain. J. Neurosci. 24, 4728–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sobolevsky A. I., Rosconi M. P., Gouaux E. (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Timm D. E., Benveniste M., Weeks A. M., Nisenbaum E. S., Partin K. M. (2011) Structural and functional analysis of two new positive allosteric modulators of GluA2 desensitization and deactivation. Mol. Pharmacol. 80, 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puchalski R. B., Louis J. C., Brose N., Traynelis S. F., Egebjerg J., Kukekov V., Wenthold R. J., Rogers S. W., Lin F., Moran T., Morrison J. H., Heinemann S. F. (1994) Selective RNA editing and subunit assembly of native glutamate receptors. Neuron 13, 131–147 [DOI] [PubMed] [Google Scholar]
- 13. Arai A., Silberg J., Kessler M., Lynch G. (1995) Effect of thiocyanate on AMPA receptor mediated responses in excised patches and hippocampal slices. Neuroscience 66, 815–827 [DOI] [PubMed] [Google Scholar]
- 14. Kessler M., Arai A. C. (2006) Use of [3H]fluorowillardiine to study properties of AMPA receptor allosteric modulators. Brain Res. 1076, 25–41 [DOI] [PubMed] [Google Scholar]
- 15. Benveniste M., Clements J., Vyklický L., Jr., Mayer M. L. (1990) A kinetic analysis of the modulation of N-methyl-D-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J. Physiol. 428, 333–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harms J. E., Benveniste M., Maclean J. K., Partin K. M., Jamieson C. (2013) Functional analysis of a novel positive allosteric modulator of AMPA receptors derived from a structure-based drug design strategy. Neuropharmacology 64, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patneau D. K., Mayer M. L., Jane D. E., Watkins J. C. (1992) Activation and desensitization of AMPA/kainate receptors by novel derivatives of willardiine. J. Neurosci. 12, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eyring H. (1935) The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115 [Google Scholar]
- 19. Robert A., Howe J. R. (2003) How AMPA receptor desensitization depends on receptor occupancy. J. Neurosci. 23, 847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsubara A., Laake J. H., Davanger S., Usami S., Ottersen O. P. (1996) Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J. Neurosci. 16, 4457–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nusser Z. (2000) AMPA and NMDA receptors: similarities and differences in their synaptic distribution. Curr. Opin. Neurobiol. 10, 337–341 [DOI] [PubMed] [Google Scholar]
- 22. Meyer A. C., Frank T., Khimich D., Hoch G., Riedel D., Chapochnikov N. M., Yarin Y. M., Harke B., Hell S. W., Egner A., Moser T. (2009) Tuning of synapse number, structure and function in the cochlea. Nat. Neurosci. 12, 444–453 [DOI] [PubMed] [Google Scholar]
- 23. Corboy M. J., Thomas P. J., Wigley W. C. (2005) Aggresome formation. Methods Mol. Biol. 301, 305–327 [DOI] [PubMed] [Google Scholar]
- 24. Bedoukian M. A., Weeks A. M., Partin K. M. (2006) Different domains of the AMPA receptor direct stargazin-mediated trafficking and stargazin-mediated modulation of kinetics. J. Biol. Chem. 281, 23908–23921 [DOI] [PubMed] [Google Scholar]
- 25. Xia Y. F., Kessler M., Arai A. C. (2005) Positive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor modulators have different impact on synaptic transmission in the thalamus and hippocampus. J. Pharmacol. Exp. Ther. 313, 277–285 [DOI] [PubMed] [Google Scholar]
- 26. Sun Y., Olson R., Horning M., Armstrong N., Mayer M., Gouaux E. (2002) Mechanism of glutamate receptor desensitization. Nature 417, 245–253 [DOI] [PubMed] [Google Scholar]
- 27. Partin K. M., Fleck M.W., Mayer M. L. (1996) AMPA receptor flip/flop mutants affecting deactivation, desensitization and modulation by cyclothiazide, aniracetam and thiocyanate. J. Neurosci. 16, 6634–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hald H., Ahring P. K., Timmermann D. B., Liljefors T., Gajhede M., Kastrup J. S. (2009) Distinct structural features of cyclothiazide are responsible for effects on peak current amplitude and desensitization kinetics at iGluR2. J. Mol. Biol. 391, 906–917 [DOI] [PubMed] [Google Scholar]
- 29. Tomita S., Sekiguchi M., Wada K., Nicoll R. A., Bredt D. S. (2006) Stargazin controls the pharmacology of AMPA receptor potentiators. Proc. Natl. Acad. Sci. U.S.A. 103, 10064–10067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cho C. H., St-Gelais F., Zhang W., Tomita S., Howe J. R. (2007) Two families of TARP isoforms that have distinct effects on the kinetic properties of AMPA receptors and synaptic currents. Neuron 55, 890–904 [DOI] [PubMed] [Google Scholar]
- 31. Kessler M., Rogers G., Arai A. (2000) The norbornenyl moiety of cyclothiazide determines the preference for flip-flop variants of AMPA receptor subunits. Neurosci. Lett. 287, 161–165 [DOI] [PubMed] [Google Scholar]
- 32. Jin R., Clark S., Weeks A. M., Dudman J. T., Gouaux E., Partin K. M. (2005) Mechanism of positive allosteric modulators acting on AMPA receptors. J. Neurosci. 25, 9027–9036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ptak C. P., Ahmed A. H., Oswald R. E. (2009) Probing the allosteric modulator binding site of GluR2 with thiazide derivatives. Biochemistry 48, 8594–8602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitchell N. A., Fleck M. W. (2007) Targeting AMPA receptor gating processes with allosteric modulators and mutations. Biophys. J. 92, 2392–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arai A. C., Kessler M., Rogers G., Lynch G. (2000) Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: interactions with cyclothiazide and GYKI 52466. Mol. Pharmacol. 58, 802–813 [DOI] [PubMed] [Google Scholar]
- 36. Geiger J. R., Melcher T., Koh D. S., Sakmann B., Seeburg P. H., Jonas P., Monyer H. (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principle neurons and interneurons in rat CNS. Neuron 15, 193–204 [DOI] [PubMed] [Google Scholar]
- 37. Ripellino J. A., Neve R. L., Howe J. R. (1998) Expression and heteromeric interactions of non-N-methyl-D-aspartate glutamate receptor subunits in the developing and adult cerebellum. Neuroscience 82, 485–497 [DOI] [PubMed] [Google Scholar]
- 38. Greger I. H., Khatri L., Kong X., Ziff E. B. (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763–774 [DOI] [PubMed] [Google Scholar]
- 39. Greger I. H., Khatri L., Ziff E. B. (2002) RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34, 759–772 [DOI] [PubMed] [Google Scholar]
- 40. Körber C., Werner M., Kott S., Ma Z. L., Hollmann M. (2007) The transmembrane AMPA receptor regulatory protein γ4 is a more effective modulator of AMPA receptor function than stargazin (γ2). J. Neurosci. 27, 8442–8447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leever J. D., Clark S., Weeks A. M., Partin K. M. (2003) Identification of a site in GluR1 and GluR2 important for modulation of deactivation and desensitization. Mol. Pharmacol. 64, 5–10 [DOI] [PubMed] [Google Scholar]
- 42. Krampfl K., Schlesinger F., Wolfes H., Dengler R., Bufler J. (2001) Functional diversity of recombinant human AMPA type glutamate receptors: possible implications for selective vulnerability of motor neurons. J. Neurol. Sci. 191, 19–23 [DOI] [PubMed] [Google Scholar]
- 43. Cachelin A. B., Colquhoun D. (1989) Desensitization of the acetylcholine receptor of frog end-plates measured in a Vaseline-gap voltage clamp. J. Physiol. 415, 159–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dong H., Zhou H. X. (2011) Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat. Commun. 2, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kashiwabuchi N., Ikeda K., Araki K., Hirano T., Shibuki K., Takayama C., Inoue Y., Kutsuwada T., Yagi T., Kang Y., Aizawa S., Mishina M. (1995) Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluRδ2 mutant mice. Cell 81, 245–252 [DOI] [PubMed] [Google Scholar]
- 46. Zuo J., De Jager P. L., Takahashi K. A., Jiang W., Linden D. J., Heintz N. (1997) Neurodegeneration in Lurcher mice caused by mutation in δ2 glutamate receptor gene. Nature 388, 769–773 [DOI] [PubMed] [Google Scholar]
- 47. Taverna F., Xiong Z.-G., Brandes L., Roder J. C., Salter M. W., MacDonald J. F. (2000) The Lurcher mutation of an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor subunit enhances potency of glutamate and converts an antagonist to an agonist. J. Biol. Chem. 275, 8475–8479 [DOI] [PubMed] [Google Scholar]
- 48. Kohda K., Wang Y., Yuzaki M. (2000) Mutation of a glutamate receptor motif reveals its role in gating and δ2 receptor channel properties. Nat. Neurosci. 3, 315–322 [DOI] [PubMed] [Google Scholar]
- 49. Klein R. M., Howe J. R. (2004) Effects of the Lurcher mutation on GluR1 desensitization and activation kinetics. J. Neurosci. 24, 4941–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Balannik V., Menniti F. S., Paternain A. V., Lerma J., Stern-Bach Y. (2005) Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron 48, 279–288 [DOI] [PubMed] [Google Scholar]
- 51. Donevan S. D., Rogawski M. A. (1993) GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron 10, 51–59 [DOI] [PubMed] [Google Scholar]