

Background: In vitro studies suggested the chemical chaperone 4-PBA as treatment of uromodulin-associated kidney disease (UAKD).
Results: 4-PBA did not ameliorate uromodulin maturation defects in two UAKD mouse models. Renal abundance/phosphorylation of several NF-κB pathway components was increased.
Conclusion: 4-PBA failed to cure UAKD in vivo but deteriorated kidney function.
Significance: NF-κB pathway was revealed as a potential target for UAKD treatment.
Keywords: Endoplasmic Reticulum Stress, Genetic Diseases, Histone Deacetylase Inhibitors, Kidney, Molecular Chaperone, NF-κB (NF-KB), 4-PBA, RelB, UAKD, Uromodulin (UMOD)
Abstract
Uromodulin (UMOD)-associated kidney disease (UAKD) belongs to the hereditary progressive ER storage diseases caused by maturation defects of mutant UMOD protein. Current treatments of UAKD patients are symptomatic and cannot prevent disease progression. Two in vitro studies reported a positive effect of the chemical chaperone sodium 4-phenylbutyrate (4-PBA) on mutant UMOD maturation. Thus, 4-PBA was suggested as a potential treatment for UAKD. This study evaluated the effects of 4-PBA in two mouse models of UAKD. In contrast to previous in vitro studies, treatment with 4-PBA did not increase HSP70 expression or improve maturation and trafficking of mutant UMOD in vivo. Kidney function of UAKD mice was actually deteriorated by 4-PBA treatment. In transfected tubular epithelial cells, 4-PBA did not improve maturation but increased the expression level of both mutant and wild-type UMOD protein. Activation of NF-κB pathway in thick ascending limb of Henle's loop cells of UAKD mice was detected by increased abundance of RelB and phospho-IκB kinase α/β, an indirect activator of NF-κB. Furthermore, the abundance of NF-κB1 p105/p50, NF-κB2 p100/p52, and TRAF2 was increased in UAKD. NF-κB activation was identified as a novel disease mechanism of UAKD and might be a target for therapeutic intervention.
Introduction
Uromodulin-associated kidney disease (UAKD)2 is a dominant hereditary disorder caused by mutations of the uromodulin (UMOD) gene and comprises medullary cystic kidney disease type 2 (MCKD2; OMIM #603860), familial juvenile hyperuricemic nephropathy (FJHN; OMIM #162000), and glomerulocystic kidney disease (GCKD; OMIM #609886) (1–3). In about 80% of UAKD cases, earliest symptoms are hypouricosuric hyperuricemia, frequently associated with gout, commonly diagnosed in the late teenage years or in young adults (1, 4, 5). Furthermore, a mild defect in urinary concentration ability and a usually strong decrease of urinary uromodulin excretion are present. UAKD is progressive and can lead to end-stage renal disease at an age ranging between 25 and >70 years with the need of renal replacement therapy (4, 6, 7). Histomorphological kidney alterations usually comprise diffuse tubulointerstitial fibrosis with moderate inflammatory cell infiltration, tubular atrophy, and intracytoplasmic accumulation of uromodulin in tubular cells (4, 8). In about one-third of the patients, uni- or bilateral renal cysts are diagnosed (7).
Allelic UMOD variants causing UAKD are rare and have to be differentiated from common allelic variants of the UMOD gene. In various genome-wide association studies, common allelic UMOD promoter variants were identified to be associated with increased risk of chronic kidney disease and other complex traits, such as hypertension and kidney stones (for review, see Ref. 4). These risk variants of the UMOD promoter were recently identified to increase UMOD expression and secretion, which leads, by influencing salt reabsorption in the kidney, to increased risk of developing hypertension and chronic kidney disease (9).
Uromodulin is selectively expressed in cells of the thick ascending limb of Henle's loop (TALH) and of the early distal convoluted tubules (10). It is a highly glycosylated protein processed in the endoplasmic reticulum (ER) and Golgi apparatus, translocated to the luminal cell membrane, and released in the urine by proteolytic cleavage. So far, >70 different UAKD-causing UMOD mutations have been published, of which all but four (in-frame deletions) lead to amino acid substitutions (4, 7, 11). Mutant uromodulin isoforms exhibit a protein maturation defect and are retained in the ER (12, 13). ER retention of mutated UMOD is considered as a key step in the pathogenesis of UAKD (4). Analyses of kidney biopsies of UAKD patients revealed a hyperplastic ER in TALH cells with overexpression of BiP and PDI, indicative of ER stress (8, 14, 15). ER hyperplasia and activation of ER-resident molecular chaperones (e.g. BiP) and foldases represent cellular counteractions of unfolded protein response to increase folding capacity of the ER (16). Gain-of-toxic function of uromodulin mutations may lead to disturbance of ER homeostasis in tubular cells, to kidney dysfunction, and finally to UAKD (17).
Therapies of UAKD other than the symptomatic corrections of hyperuricemia and of water and electrolyte imbalance are not available (4). Treatment of hyperuricemia for prevention of gout can be efficiently done by xanthine oxidase inhibitors like allopurinol and febuxostat, by uricosuric drugs like probenecid, or by benzbromarone (5, 18). However, treatment of hyperuricemia does not prevent disease progression and deterioration of kidney function in UAKD (5). UMOD maturation retardation in the ER and ER overload represent the central pathomechanism in UAKD, a member of the ER storage diseases (4). Thus, novel therapeutic strategies for UAKD focus on improvement of ER capacity and function, and this approach might delay disease progression. Two independent in vitro studies postulated a positive effect of the chemical chaperone sodium 4-phenylbutyrate (4-PBA) on mutant UMOD protein maturation, suggesting that 4-PBA might reduce ER retention of UMOD and might ameliorate UAKD (19, 20). The present study evaluated the effects of 4-PBA in UmodA227T and UmodC93F mutant mice, two recently described models of UAKD (17, 21).
EXPERIMENTAL PROCEDURES
Animals
UmodA227T and UmodC93F mutant mice were generated by N-ethyl-N-nitrosourea (ENU) mutagenesis as described previously (17, 21). Both Umod mutant mouse lines exhibit the key features of UAKD like maturation defect and retention of uromodulin in the hyperplastic endoplasmic reticulum of TALH cells and impaired kidney function with mild defect in urinary concentration ability, reduced fractional excretion of uric acid, and reduced uromodulin excretion. Both mouse lines were maintained on the C3HeB/FeJ (C3H) genetic background and housed in a specific pathogen-free mouse facility as previously described (17, 21). All mice received food (V1124–3, Ssniff, Soest, Germany) and water ad libitum, and mouse husbandry was performed under standard environment conditions (22 ± 2 °C, 40–50% relative humidity, 12-h light/dark cycle). All animal experiments were approved and carried out in accordance with the German Animal Welfare Act with permission from the responsible veterinary authority.
Experimental Schedule of Administration of 4-PBA in Vivo
In vivo administration of the chemical chaperone 4-PBA with the drinking water in male homozygous UmodA227T mutants, male homozygous UmodC93F mutants, and their male wild-type littermates started at an age of 2 months. At this age the UAKD phenotype is already clinically present in Umod mutant mice of both lines, but is in an early disease stage where no progressed morphological kidney alterations like interstitial fibrosis, tubular atrophy, or infiltrates of inflammatory cells are present. Drug administration was performed for 60 ± 4 days. Pharmaceutical-grade 4-phenylbutyric acid sodium salt (Scandinavian Formulas, water solubility 0.62 g/ml) was dissolved in drinking water and administered at a daily dosage of 1 g/kg body weight. 4-PBA solutions were renewed every third day. Mice of the placebo groups received drinking water without the supplement. Body weights of mice were measured weekly, and water intake was measured per cage after each solution change. Blood sampling was performed at day 0 before the start of the experiment and at days 30 ± 4 and 60 ± 4 at the mid and the end of the experiment. On day 56 ± 4, the mice were individually transferred to metabolic cages (Tecniplast, no. 3600M021). After a conditioning period of 2 days to the metabolic cages, water consumption and urine excretion was assessed for 2 days. On day 60 ± 4, the animals were anesthetized for retro-orbital blood collection followed by necropsy.
Clinical Chemical Analyses
Analyses of blood and urine parameters were carried out as described previously (17, 22). Plasma creatinine values were determined enzymatically (Biomed), whereas urine creatinine concentration was determined by the Jaffé method.
Analyses of Urine and Kidney Function
Urine osmolality was determined by freezing point depression analysis. Daily excretion of solutes was measured. Calculations of creatinine clearance and of fractional excretion of a solute x (FEx) was performed as described previously (17, 23).
Administration of 4-PBA in Vitro
Immortalized murine proximal tubular epithelial cells (MTCs) (24) were donated by G. Wolf and cultured on collagen-coated plates in DMEM high glucose medium containing 10% fetal calf serum (FCS), 1% penicillin/streptomycin, and 1% insulin/transferrin/selenium. Transfection of MTCs (passage 6) was performed by Nucleofection™ (Lonza, program U12) with the constructs pcDNA3-Umodwt, pcDNA3-UmodA227T, and pcDNA3-UmodC93F, and stable clones were generated by G418 selection (2 mg/ml). Two stable clones per construct (with high and with medium UMOD expression) were selected for further analyses.
Isolation of primary kidney cells was performed by digest with 0.1% (w/v) collagenase II (Invitrogen) for 30 min at 37 °C after mincing kidneys of homozygous UmodA227T mutant, homozygous UmodC93F mutant, or wild-type mice. After gently pressing the digested renal tissue through a 100-μm cell strainer, a second digest with 0.1% (w/v) collagenase II and 7.5 units/ml DNase I for 30 min at 37 °C was performed. Finally, undigested tubular fragments and glomeruli were removed by filtration through a 40-μm cell strainer, and cell suspension was seeded on collagen-coated plates or 8-well slides (BD Biosciences) in DMEM/Ham's F-12 supplemented with 20% FCS, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% nonessential amino acids, and 1% insulin/transferrin/selenium.
Treatment of MTC clones with 4-PBA was performed at passages 12–19. The doses of drug supplement were tested in pilot studies. Seeding of cells on collagen-coated 12-well plates, volume of culture medium, and duration of treatment were standardized. Treatment of MTC clones was performed on two different stably transfected Umodwt-MTC clones and on each of three different MTC clones stable-transfected with UmodA227T or UmodC93F. Treatment of primary kidney cells was performed on eight-well slides.
Morphological Analysis
Histological analyses of kidneys were performed as described previously (21). Paraffin sections were stained with hematoxylin and eosin, Masson-Trichrom, and periodic acid-Schiff-silver. Immunohistochemistry was performed using the following primary antibodies: rabbit polyclonal antibody against human Tamm-Horsfall protein (H-135; Santa Cruz Biotechnology), rat monoclonal antibody against mouse uromodulin (clone 774056; R&D Systems), rabbit polyclonal antibody against HSP70 (Cell Signaling), rabbit monoclonal antibody against RelB (C1E4, no. 4922, Cell Signaling), and rabbit monoclonal antibody against phospho-IKKα/β (Ser-176/180) (no. 2697, Cell Signaling). Immunoreactivity was visualized using 3,3-diaminobenzidine tetrahydrochloride dihydrate (DAB) (brown color) or using Vector Red Alkaline Phosphatase Substrate Kit I (red color). Nuclear counterstaining was done with hemalum.
For immunofluorescence analyses of cells, the following primary antibodies were used: polyclonal goat antiserum against human uromucoid (MP Biomedicals) and polyclonal rabbit anti-GRP78/BiP IgG (Abcam). Fluorescence analyses were performed using a confocal laser scanning microscope (LSM 710, Zeiss).
Western Blot Analyses
Twenty-four-hour urine samples, standardized for equal creatinine levels, were analyzed for uromodulin content as described previously (21). MTCs were lysed in Laemmli extraction buffer, protein concentration was determined by a BCA assay, and equal protein amounts (10 μg) of denatured cell lysate per lane were separated in 8% SDS-polyacrylamide minigels. Equal amounts of supernatant of cells cultured under standardized conditions were used for analysis of their UMOD content.
TALH segment density in the kidney is highest in the outer medullar region. The outer medulla was obtained after cutting each fresh kidney of 4-month-old mice along the midsagittal plane into two halves and further into transverse sections and by removing the cortex and inner medulla. After homogenization of the outer medulla in Laemmli extraction buffer and determination of protein concentration by BCA assay, 15 μg of denatured tissue lysate per lane was separated in 10% or 12% SDS-polyacrylamide minigels and blotted on PVDF membranes. Equal loading was controlled by Ponceau staining.
The following primary antibodies were used: rabbit polyclonal antibody against human Tamm-Horsfall protein (H-135; Santa Cruz Biotechnology), rabbit polyclonal antibody against BiP (no. 3183), rabbit monoclonal antibody against GAPDH (no. 2118), rabbit polyclonal antibody against IKKα (no. 2682), rabbit monoclonal antibody against phospho-IKKα/β (Ser-176/177) (C84E11, no. 2078), rabbit polyclonal antibody against NF-κB1 p105/p50 (no. 12540), rabbit polyclonal antibody against NF-κB2 p100/p52 (no. 4882), rabbit monoclonal antibody against NF-κB p65 (no. 4764), rabbit monoclonal antibody against RelB (C1E4, no. 4922), rabbit polyclonal antibody against TRAF2 (no. 4712), and rabbit polyclonal antibody against TRAF3 (no. 4729) (all antibodies except for Tamm-Horsfall protein were bought from Cell Signaling). Bound antibodies were visualized using ECL reagent (GE Healthcare). Signal intensities were quantified using ImageQuant (GE Healthcare). Standardization of equal loading was referred to the signal intensities of GAPDH of the corresponding PVDF membrane.
Statistical Analysis
Data are shown as the means ± S.D. as indicated. Data were analyzed by using an unpaired Student's t test or two-way ANOVA as indicated using Sigma Plot 12.0. Holm-Bonferroni correction for multiple comparisons was performed where indicated.
RESULTS
Clinical and Clinical-Chemical Findings in Placebo and 4-PBA-treated UAKD Mouse Models and Wild-type Controls
Umod Mutant Versus Wild-type Mice after Placebo Treatment
Four-month-old homozygous mutants (UmodA227T and UmodC93F) receiving drinking water without supplement (placebo treatment group) had significantly increased plasma urea levels and plasma lipase activities compared with corresponding wild-type control animals, whereas plasma triglyceride concentrations were significantly decreased (Table 1, Fig. 1A). Additionally, plasma calcium levels were mildly increased in UmodC93F mutant mice compared with wild-type mice. Umod mutants of both lines exhibited distinctly reduced body weight, significantly reduced urine osmolality combined with slightly increased urine volume, significantly increased urinary calcium excretion and fractional excretion (FE) of calcium as well as reduced urinary excretion of phosphate and reduced FEP and FEurea when compared with wild-type mice (Table 2, Fig. 1B). Furthermore, UmodC93F mutants exhibited increased 24-h urinary potassium excretion versus wild-type mice. These findings are in line with earlier published effects of mutant UMOD expression on clinical-chemical parameters of plasma and urine in both UmodA227T and UmodC93F mutant mice representing models for UAKD (17, 21). UmodC93F mutants exhibited a more severe UAKD phenotype than mutants of the UmodA227T mouse line (21).
TABLE 1.
Plasma data at an age of 4 months of placebo mice or after 2 months of 4-PBA administration
Age of mice analyzed: 4 months. Data are the means ± S.D. n = 6–19 per genotype. Pi, inorganic phosphate; ALAT, alanine aminotransferase (EC 2.6.1.2); ASAT, aspartate aminotransferase (EC 2.6.1.1). 2-Way ANOVA: a, b, c, p < 0.05; aa, bb, cc, p < 0.01; aaa, bbb, ccc, p < 0.001; a, 4-PBA versus placebo of the same genotype; b, homozygous mutant versus wild type of the same treatment group; c, homozygous UmodC93F mutant versus homozygous UmodA227T mutant of the same treatment group.
| Plasma parameters | Wild-type |
UmodA227T homozygote |
UmodC93F homozygote |
|||
|---|---|---|---|---|---|---|
| Placebo | 4-PBA | Placebo | 4-PBA | Placebo | 4-PBA | |
| Na+ (mmol/liter) | 155.9 ± 3.1 | 154.3 ± 3.6 | 151.7 ± 6.6b | 156.4 ± 2.3aa.b | 155.5 ± 4.3c | 156.1 ± 1.9 |
| K+ (mmol/liter) | 4.07 ± 0.29 | 3.94 ± 0.19 | 4.15 ± 0.40 | 4.21 ± 0.32bb | 4.22 ± 0.59 | 3.98 ± 0.26 |
| Cl− (mmol/liter) | 114.6 ± 1.9 | 113.5 ± 2.8 | 111.8 ± 4.1b | 114.5 ± 1.5a | 113.9 ± 3.7 | 116.2 ± 1.1a,bb |
| Ca2+ (mmol/liter) | 2.30 ± 0.07 | 2.21 ± 0.08a | 2.32 ± 0.15 | 2.34 ± 0.09bbb | 2.37 ± 0.07bbb | 2.36 ± 0.11bbb |
| Pi (mmol/liter) | 2.39 ± 0.44 | 2.11 ± 0.31 | 2.64 ± 0.70 | 1.94 ± 0.36aaa | 2.30 ± 0.56 | 1.92 ± 0.26a |
| Total protein (g/liter) | 51.1 ± 1.8 | 51.3 ± 2.7 | 49.6 ± 3.8 | 52.3 ± 1.7a | 52.1 ± 2.4c | 52.6 ± 1.7 |
| Creatinine (μmol/liter) | 9.00 ± 1.92 | 8.00 ± 1.32 | 9.86 ± 0.96 | 9.93 ± 1.30bbb | 9.46 ± 1.83 | 9.45 ± 1.11b |
| Urea (mmol/liter) | 10.15 ± 1.16 | 11.02 ± 1.29 | 20.54 ± 2.36bbb | 20.49 ± 2.21bbb | 23.08 ± 2.53bbb,c | 25.99 ± 2.95aaa,bbb,ccc, |
| Cholesterol (mmol/liter) | 3.56 ± 0.42 | 3.20 ± 0.42a | 3.41 ± 0.16 | 3.31 ± 0.24 | 3.76 ± 0.25cc | 3.45 ± 0.25a |
| Triglycerides (mmol/liter) | 1.72 ± 0.87 | 1.67 ± 0.60 | 1.04 ± 0.27b | 1.25 ± 0.51b | 0.80 ± 0.24bbb | 0.93 ± 0.23bb,cc |
| ALAT (units/liter) | 32.1 ± 14.2 | 38.7 ± 24.0 | 29.3 ± 8.7 | 43.1 ± 44.8 | 28.1 ± 6.4 | 31.4 ± 5.2 |
| ASAT (units/liter) | 56.1 ± 14.5 | 68.7 ± 48.9 | 48.5 ± 5.4 | 72.3 ± 55.2 | 65.8 ± 29.5 | 58.3 ± 8.1 |
| Lipase (units/liter) | 45.6 ± 7.5 | 43.9 ± 7.8 | 54.2 ± 5.5bb | 49.8 ± 8.2b | 56.1 ± 6.0bbb | 55.9 ± 4.7bbb,c |
FIGURE 1.

Effect of placebo and 4-PBA treatment on plasma and urine parameters in wild-type and Umod mutant mice. A, clinical-chemical plasma parameters. B, urinary parameters of kidney function. Data are presented in Tukey's box plot (whiskers go 1.5 times the interquartile range or to the highest or lowest point, whichever is shorter), with outliers represented as dots. n = 6–19 per genotype and treatment group. 2-way ANOVA: a,b,c, p < 0.05; aa,bb,cc, p < 0.01; aaa,bbb,ccc, p < 0.001; a, 4-PBA versus placebo of the same genotype; b, homozygous mutant versus wild-type of the same treatment group; c, homozygous UmodC93F mutant versus homozygous UmodA227T mutant of the same treatment group.
TABLE 2.
Urine parameters at an age of 4 months of placebo mice or after 2 months of 4-PBA administration
Age of mice analyzed: 4 months. Data are the means ± S.D., standardized to 25 g body weight. n = 6–19 per genotype. 2-Way ANOVA: a, b, c, p < 0.05; aa, bb, cc, p < 0.01; aaa, bbb, ccc, p < 0.001; a, 4-PBA versus placebo of the same genotype; b, homozygous mutant versus wild type of the same treatment group; c, homozygous UmodC93F mutant versus homozygous UmodA227T mutant of the same treatment group.
| Urine parameters | Wild-type |
UmodA227T homozygote |
UmodC93F homozygote |
|||
|---|---|---|---|---|---|---|
| Placebo | 4-PBA | Placebo | 4-PBA | Placebo | 4-PBA | |
| Body weight (g) | 29.7 ± 2.0 | 30.7 ± 2.7 | 26.9 ± 1.2bb | 26.3 ± 1.5bbb | 24.2 ± 2.1bbb,ccc | 22.4 ± 0.7bbb,ccc |
| Urine volume (ml/day) | 1.2 ± 0.3 | 1.4 ± 0.3 | 1.5 ± 0.6 | 2.3 ± 0.5aa,bbb | 2.9 ± 0.7bbb,cc | 2.7 ± 1.0bb |
| Urine osmolality (mosmol/kg H2O) | 3667 ± 576 | 3175 ± 370aa | 2919 ± 494bb | 2350 ± 390aa,bbb | 1667 ± 327bbb,ccc | 2073 ± 437a,bbb |
| Na+ (μmol/day) | 286.3 ± 61.7 | 337.4 ± 84.1 | 289.4 ± 53.2 | 400.3 ± 87.8aa,b | 313.1 ± 53.5 | 424.6 ± 94.9aaa.bb, |
| K+ (μmol/day) | 495.3 ± 92.3 | 535.8 ± 121.8 | 508.0 ± 147.5 | 659.7 ± 141.6a,bb | 652.1 ± 84.4bb,c | 704.9 ± 161.1bbb |
| Cl− (μmol/day) | 508.8 ± 86.3 | 486.6 ± 114.5 | 453.4 ± 84.8 | 512.6 ± 114.3 | 485.0 ± 62.7 | 509.2 ± 98.9 |
| Ca2+ (μmol/day) | 2.6 ± 0.3 | 3.0 ± 1.1 | 7.8 ± 1.5bbb | 9.5 ± 3.4bbb | 7.6 ± 2.3bbb | 10.3 ± 3.4aa,bbb |
| Mg2+ (μmol/day) | 42.5 ± 7.8 | 39.7 ± 22.1 | 40.4 ± 14.6 | 38.1 ± 20.0 | 46.6 ± 15.4 | 48.7 ± 18.6 |
| Pi (μmol/day) | 160.3 ± 33.2 | 104.5 ± 55.5aa | 95.8 ± 64.1b | 58.4 ± 43.5bb | 108.4 ± 59.5b | 20.7 ± 31.2aaa,bbb |
| Creatinine (μmol/day) | 6.0 ± 1.2 | 5.9 ± 2.0 | 5.9 ± 0.8 | 6.8 ± 1.8 | 6.5 ± 0.8 | 7.3 ± 1.9b |
| Urea (mmol/day) | 2.7 ± 0.3 | 2.6 ± 0.6 | 2.6 ± 0.5 | 2.9 ± 0.6 | 2.9 ± 0.4 | 3.1 ± 0.5b |
| Glucose (μmol/day) | 3.1 ± 0.4 | 3.5 ± 1.1 | 2.3 ± 0.4 | 3.6 ± 1.9 | 3.3 ± 1.6 | 3.3 ± 2.8 |
| Creatinine clearance | 867.4 ± 457.1 | 932.8 ± 342.3 | 653.2 ± 113.5 | 725.5 ± 206.7 | 688.2 ± 166.8 | 696.7 ± 178.4 |
| FENa | 0.29 ± 0.12 | 0.31 ± 0.08 | 0.31 ± 0.03 | 0.40 ± 0.12b | 0.29 ± 0.06 | 0.36 ± 0.08a |
| FEK | 18.6 ± 6.1 | 19.3 ± 4.7 | 20.3 ± 5.1 | 24.3 ± 6.9b | 22.9 ± 5.8 | 23.5 ± 5.2b |
| FECl | 0.71 ± 0.26 | 0.61 ± 0.16 | 0.67 ± 0.08 | 0.68 ± 0.16 | 0.62 ± 0.14 | 0.58 ± 0.12 |
| FECa | 0.18 ± 0.06 | 0.19 ± 0.05 | 0.56 ± 0.10bbb | 0.63 ± 0.26bbb | 0.46 ± 0.14bbb | 0.59 ± 0.22a,bbb |
| FEPi | 10.86 ± 3.56 | 7.57 ± 5.23 | 6.07 ± 3.88b | 5.29 ± 4.75 | 7.12 ± 4.34b | 1.42 ± 1.99aa,bbb,c |
| FEurea | 40.9 ± 12.9 | 34.1 ± 8.3a | 21.7 ± 5.4bbb | 21.9 ± 4.6bbb | 18.5 ± 3.5bbb | 16.2 ± 4.3bbb,cc |
Umod Mutant Versus Wild-type Mice after 4-PBA Treatment
After 2 months of administration of 4-PBA at an age of 4 months, UmodA227T mutants and UmodC93F mutants displayed distinctly higher plasma levels of urea, creatinine, and calcium as well as higher plasma lipase activities and decreased plasma triglyceride levels compared with wild-type controls (Table 1, Fig. 1A). Furthermore, 4-PBA-treated UmodC93F mutant mice exhibited a significantly increased plasma chloride concentration compared with wild-type controls. A mild increase of plasma potassium concentration was detected in 4-PBA-treated UmodA227T mutants compared with corresponding wild-type controls.
Most of the urinary parameters of 4-PBA-treated Umod mutants were similarly altered as in the placebo treatment group (Table 2, Fig. 1B). 4-PBA-treated Umod mutants displayed reduced body weight, reduced urine osmolality combined with slightly increased 24-h urine volume, increased urinary excretion and FE of calcium, reduced urinary excretion of phosphate, and reduced FEurea in comparison to 4-PBA-treated wild-type mice. Furthermore, daily excretions of sodium and potassium as well as FEK were increased in Umod mutants of both mouse lines. Additionally, UmodC93F mutants, which exhibited a more severe UAKD phenotype than mutants of the UmodA227T mouse line, excreted more urea and creatinine in their 24-h urine and had a strongly decreased FEP when compared with 4-PBA-treated wild-type controls.
Umod Mutant Mice after 4-PBA Versus Placebo Treatment
Additional clinical-chemical plasma parameters showed trends to be altered by 4-PBA treatment of Umod mutants compared with their genotype-matched placebo-treated counterparts (Tables 1 and 2, Fig. 1). In Umod mutant mice of both lines, 4-PBA treatment led to a mild but significant increase of plasma chloride levels and to a distinct decrease of the plasma concentrations of inorganic phosphate. Furthermore, 4-PBA-treated UmodC93F mutant mice exhibited a significantly higher plasma urea concentration compared with placebo-treated mice of the same genotype, indicating worsening of renal function by 4-PBA in the more severely UAKD affected mutant mouse line UmodC93F.
In the setting of UAKD, long term administration of 4-PBA led to significantly increased 24-h urinary excretion of sodium in Umod mutants from both lines when compared with the placebo-treated Umod mutants of the same genotype. Urine osmolality was significantly reduced, and urine volume excretion was increased in 4-PBA-treated versus placebo-treated UmodA227T mutant mice. In contrast, urine osmolality of UmodC93F mutants receiving 4-PBA was significantly increased when compared with placebo-treated mice of the same genotype. However, as the UAKD phenotype is more pronounced in UmodC93F mutants compared with UmodA227T mutants, urine excretion volume was still distinctly higher and urine osmolality was distinctly lower in UmodC93F mutants when compared with UmodA227T mutants after placebo treatment, and this genotype difference was attenuated after 4-PBA treatment. Further striking alterations of kidney function of UmodC93F mutants due to 4-PBA treatment were a significant reduction of urinary excretion of phosphate and of FEP. Additionally, urinary excretion of potassium was increased in 4-PBA-treated UmodA227T mutants. In 4-PBA-treated UmodC93F mutants, 24-h urinary excretion of calcium, FECa, and FENa were increased when compared with the values of the genotype-matched placebo-treated mice.
Wild-type Mice after 4-PBA Versus Placebo Treatment
In 4-PBA-treated wild-type mice, plasma calcium and cholesterol levels tended to be lower than in wild-type mice after placebo treatment (Table 1, Fig. 1A). Furthermore, long term oral administration of 4-PBA in young adult wild-type mice resulted in a slight but significant decrease of urine osmolality and in a distinct decrease of 24-h urinary excretion of phosphate and of FEurea (Table 2, Fig. 1B).
4-PBA-treated UmodC93F Versus 4-PBA-treated UmodA227T Mutant Mice
UmodC93F mutants of the 4-PBA treatment group exhibited, besides the more prominent reduction of body weight also present in mice of the placebo group, significantly stronger reductions of FEP and FEurea when compared with 4-PBA-treated UmodA227T mutant mice (Table 2, Fig. 1B). These results indicate the more pronounced UAKD phenotype in UmodC93F mutants and a stronger effect of 4-PBA treatment in this line.
Progression of UAKD during 4-PBA and Placebo Treatment
At an age of 2 months, before testing the putative therapeutic effect of 4-PBA, renal dysfunction of homozygous mutants (UmodA227T and UmodC93F) was already clinically evident by distinctly increased plasma urea concentrations in mutants compared with age-matched wild-type controls (Table 3). With increasing age, mutant mice of both lines exhibited a continuous increase of plasma urea concentrations irrespective of treatment group (Fig. 2). However, the relative increase of plasma urea levels between 2 and 4 month of age was significantly more pronounced in 4-PBA-treated versus placebo-treated UmodC93F mutant mice. Of note, plasma urea concentrations of UmodC93F mutants were distinctly higher than those of UmodA227T mutants in both the 4-PBA and the placebo treatment groups. This reflects the more severe UAKD phenotype in the UmodC93F mutant mouse line, which is in line with previous results indicating that renal dysfunction is more pronounced in UmodC93F mutant mice than in age-matched UmodA227T mutants.
TABLE 3.
Plasma data of 2-month-old mice
Data are the means ± S.D. n = 19–33 animals per genotype. Pi, inorganic phosphate; ALAT, alanine aminotransferase (EC 2.6.1.2); ASAT, aspartate aminotransferase (EC 2.6.1.1). Student's t test with Holm-Bonferroni correction: b, c, p < 0.05; bb, cc, p < 0.01; bbb, ccc, p < 0.001; b, homozygous mutant versus wild type; c, homozygous UmodC93F mutant versus homozygous UmodA227T mutant.
| Plasma parameters | Wild-type | UmodA227T homozygote | UmodC93F homozygote |
|---|---|---|---|
| Na+ (mmol/liter) | 148.5 ± 3.8 | 148.5 ± 3.0 | 148.5 ± 4.5 |
| K+ (mmol/liter) | 3.93 ± 0.34 | 4.21 ± 0.29bb | 4.00 ± 0.33 |
| Ca2+ (mmol/liter) | 2.34 ± 0.07 | 2.34 ± 0.07 | 2.36 ± 0.11 |
| Cl− (mmol/liter) | 109.8 ± 2.2 | 110.2 ± 1.9 | 110.8 ± 3.7 |
| Pi (mmol/liter) | 2.58 ± 0.36 | 2.57 ± 0.36 | 2.33 ± 0.29b |
| Total protein (g/liter) | 49.8 ± 2.6 | 48.3 ± 1.9b | 49.1 ± 2.3 |
| Creatinine (μmol/liter) | 8.57 ± 1.59 | 9.46 ± 1.61 | 8.64 ± 1.53 |
| Urea (mmol/liter) | 9.67 ± 1.17 | 14.0 ± 1.9bbb | 16.1 ± 2.6bbb,cc |
| Cholesterol (mmol/liter) | 3.40 ± 0.28 | 3.25 ± 0.24 | 3.21 ± 0.20b |
| Triglycerides (mmol/liter) | 1.31 ± 0.53 | 1.24 ± 0.48 | 1.07 ± 0.33 |
| ALAT (units/liter) | 28.2 ± 10.0 | 30.4 ± 16.1 | 34.4 ± 10.0b |
| ASAT (units/liter) | 41.2 ± 4.9 | 41.8 ± 5.1 | 46.3 ± 6.0bb,c |
| Lipase (units/liter) | 52.8 ± 10.2 | 51.9 ± 11.0 | 54.3 ± 8.6 |
FIGURE 2.
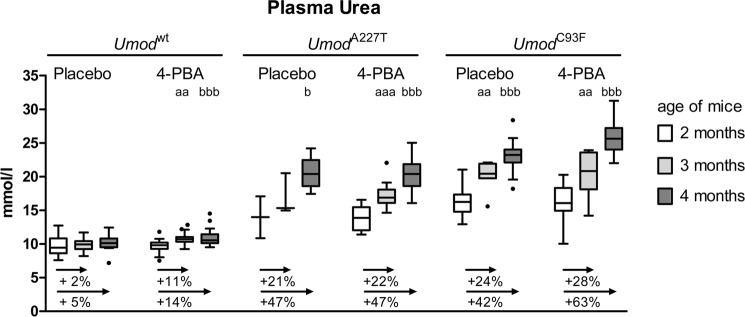
Development of plasma urea levels during 4-PBA treatment. Comparison of the plasma urea concentrations of mice at the beginning of the trial (age of mice: 2 months), after 1 month of treatment (age of mice: 3 months), and at the end of the 2-month duration of therapeutic testing of 4-PBA (age of mice: 4 months). Data are presented in Tukey's box plot (whiskers go 1.5 times the interquartile range or to the highest or lowest point, whichever is shorter), with outliers represented as dots. n = 11–19 per genotype, treatment group, and age group, except n = 8 of placebo group of homozygous UmodC93F mutants (age 2 and 3 months), n = 2–3 of placebo group of 2- and 3-month-old homozygous UmodA227T mutants, and n = 6 of placebo group of 4-month-old homozygous UmodA227T mutants. The relative increases of plasma urea concentrations with age compared with that of an age of 2 months are indicated. Student's t test with Holm-Bonferroni correction: a,b, p < 0.05; aa,bb, p < 0.01; aaa,bbb, p < 0.001; a, plasma urea concentration of 3-month-old versus 2-month-old mice of the same genotype and treatment group; b, plasma urea concentration of 4-month-old versus 2-month-old mice of the same genotype and treatment group.
Effect of 4-PBA Administration on Urinary Uromodulin Excretion and TALH Morphology
One characteristic of UAKD represents the reduced urinary uromodulin excretion due to the dominant negative effects of amino acid-changing UMOD mutations on protein maturation and secretion. As shown earlier, urinary UMOD excretion was strongly decreased in Umod mutant mice, with the extent of decrease being more pronounced in UmodC93F mutant mice than in UmodA227T mutant mice of the same allelic status (17, 21). After 4-PBA treatment for 2 months, homozygous mutant mice had a reduced UMOD excretion compared with wild-type mice, and the reduction of UMOD excretion was more pronounced in mutants of the UmodC93F mouse line than in the UmodA227T line (not shown). A putative beneficial therapeutic effect through a molecular chaperone is expected to attenuate the protein maturation defect and to increase the urinary UMOD excretion. However, Umod mutant mice of both lines treated with the molecular chaperone 4-PBA had similar urinary uromodulin contents as genotype-matched mutant mice of the placebo group (Fig. 3A).
FIGURE 3.
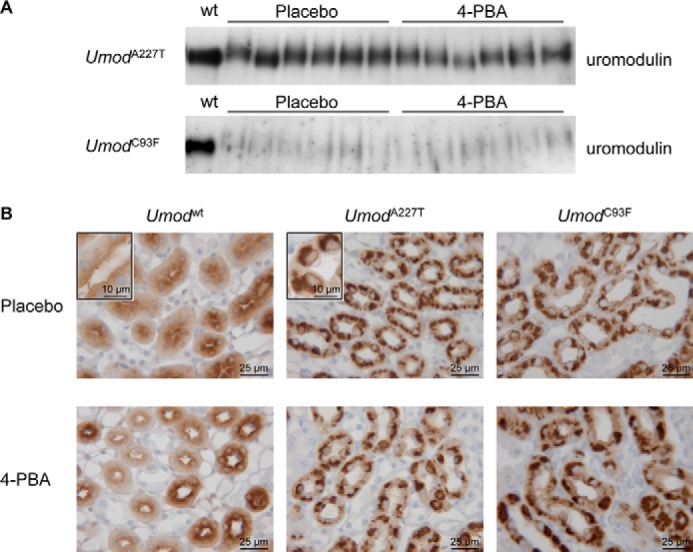
Evaluation of the in vivo effect of 4-PBA treatment on mutant uromodulin maturation and trafficking defect. A, on Western blot analysis, uromodulin abundance in urine, standardized on equal creatinine, of six mice per Umod mutant mouse line and treatment group were analyzed and compared with the UMOD abundance of a wild-type mouse of the placebo treatment group. Urinary uromodulin protein excretion was reduced in 4-PBA and placebo-treated Umod mutant mice compared with placebo-treated wild-type mice. Furthermore, the uromodulin excretion with urine was much more decreased in homozygous UmodC93F mutant mice than in homozygous mutant mice of the UmodA227T mutant mouse line. Compared with genotype-matched mice of the placebo control group, Umod mutant mice of both lines receiving 4-PBA for 2 months excreted similar amounts of UMOD protein with urine as placebo control mice. B, staining patterns of uromodulin in TALH cells were similar in placebo control mice and 4-PBA-treated mice of the same genotype group. TALH cells of wild-type mice exhibited a diffuse cytoplasmic uromodulin immunopositivity with enforcement of the luminal membrane. In contrast, TALH cells of Umod mutant mice of both lines displayed a strong paranuclear immunopositivity for UMOD irrespective of kind of treatment. The chromogen was DAB; nuclear staining was done with hemalum.
A morphological characteristic of UAKD is the intracytoplasmic accumulation of uromodulin in the hyperplastic endoplasmic reticulum in TALH cells. Uromodulin accumulation in the perinuclear compartment of TALH cells was demonstrated in placebo-treated Umod mutant mice of both lines but also after 4-PBA treatment (Fig. 3B). Furthermore, ultrastructural alterations of TALH cells such as perinuclear stacked lamellar structures were similar in 4-PBA and placebo-treated Umod mutant mice (not shown). In earlier analyses, these perinuclear lamellar structures were identified as hyperplastic endoplasmic reticulum where intracytoplasmic uromodulin retention and accumulation take place (21).
In Vitro Assays
Effect of 4-PBA on Uromodulin Secretion and Expression in Stably Transfected MTCs
In view of published in vitro effects of 4-PBA (19, 20), stably transfected clones of the immortalized murine kidney cell line MTC (24) expressing wild-type or mutant UMOD were generated. Mature uromodulin protein was detected in supernatant of MTCs stably transfected with wild-type, A227T mutant, or C93F mutant uromodulin expression vectors. There were differences of the uromodulin content in the supernatant between different stably transfected cell clones irrespective of the type of transfected Umod variant (not shown). 4-PBA administration (1 mm-5 mm) to UMOD expressing MTCs for 48 h led to an increase of uromodulin content in supernatant compared with control containing no 4-PBA in the culture medium (Fig. 4A). There was a correlation between concentration of 4-PBA and amount of uromodulin in the culture medium with higher concentrations of up to 5 mm 4-PBA, resulting in a higher uromodulin content in the supernatant. However, the increased uromodulin secretion was not due to improved protein maturation efficiency but was paralleled by an increase of uromodulin expression as demonstrated by increased uromodulin content in cell lysates. This effect was observed for stably transfected MTC clones irrespective of the type of transfected Umod variant. The intracellular content of both mature and immature uromodulin was increased, and this was particularly seen in Umod mutant-transfected MTCs.
FIGURE 4.
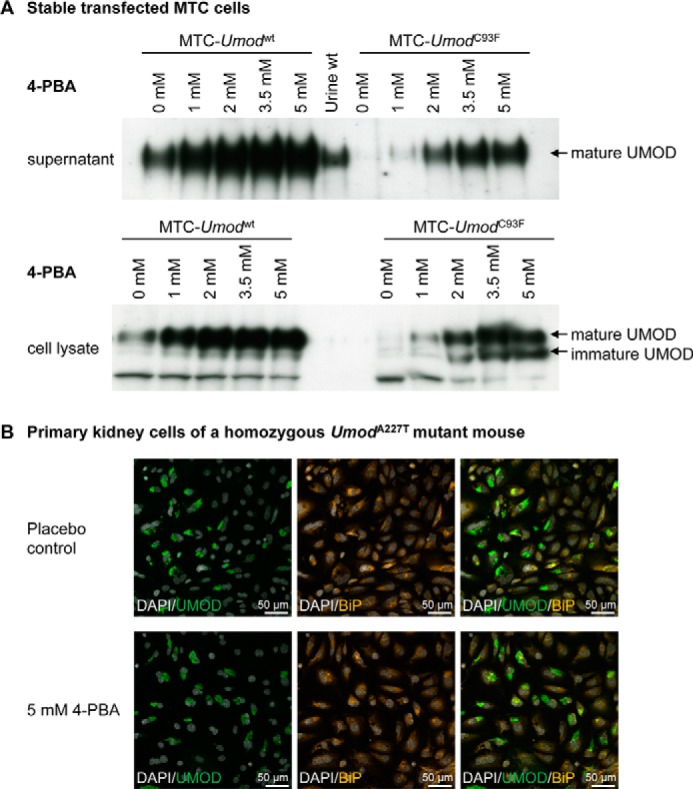
Evaluation of the in vitro effect of 4-PBA on mutant uromodulin-expressing cells. A, an increased concentration of administered 4-PBA to UMOD-expressing MTCs for 48 h led to an increased uromodulin content in supernatant compared with control containing no 4-PBA in the culture medium irrespective of the type of UMOD protein (native or mutant) expressed. In parallel to the increased UMOD content in supernatant by 4-PBA administration, the abundance of UMOD protein in cell lysate also increased irrespective of the type of UMOD protein (native or mutant) expressed. Furthermore, immature UMOD signals appeared to be stronger in the mutant protein expressing MTCs than in the wild-type UMOD protein-expressing cells and was increased after administration of higher 4-PBA concentrations. B, 4-PBA administration (5 mm) for 48 h on primary kidney cells of a homozygous UmodA227T mutant mouse did not lead to obvious changes of prominent perinuclear UMOD (green color) and BiP (orange color) immunofluorescent signals in TALH cells compared with placebo control cells. Thus, no obvious effect of 4-PBA on uromodulin retention and BiP signal intensity in TALH cells of Umod mutant mice was seen in vitro.
Effect of 4-PBA on Uromodulin Expression in Kidney Cells of Umod Mutant Mice in Vitro and on HSP70 Expression in TALH Cells in Vivo
To further elucidate the putative effect of 4-PBA to ameliorate mutant uromodulin maturation and to resolve ER hyperplasia, primary kidney cells of Umod mutant mice were cultured for 48 h in the presence of either 2 mm 4-PBA, 5 mm 4-PBA, or without drug supplementation. Double-immunofluorescence analyses were performed to identify UMOD-positive TALH cells, to localize UMOD, and to visualize the ER by the ER chaperone BiP (Fig. 4B). Of note, prominent BiP signals localizing to the enlarged ER were present in TALH cells of Umod mutant mice. No obvious effect of 4-PBA on uromodulin retention and BiP signal in TALH cells of Umod mutant mice was observed.
Ma et al. (20) reported an impact of the heat shock protein HSP70 in amelioration of protein maturation of mutant uromodulin in transfected MDCK cells by 4-PBA in vitro. Therefore, we analyzed the protein abundance of HSP70 in kidneys of UAKD-affected Umod mutant and wild-type mice. HSP70 was predominantly expressed in collecting duct cells of the renal papilla, but its abundance was only very weak or absent in TALH cells irrespective of genotype (Fig. 5). After administration of 4-PBA for 2 months, HSP70 immunostaining pattern and intensity were similar irrespective of genotype when compared with placebo control mice.
FIGURE 5.
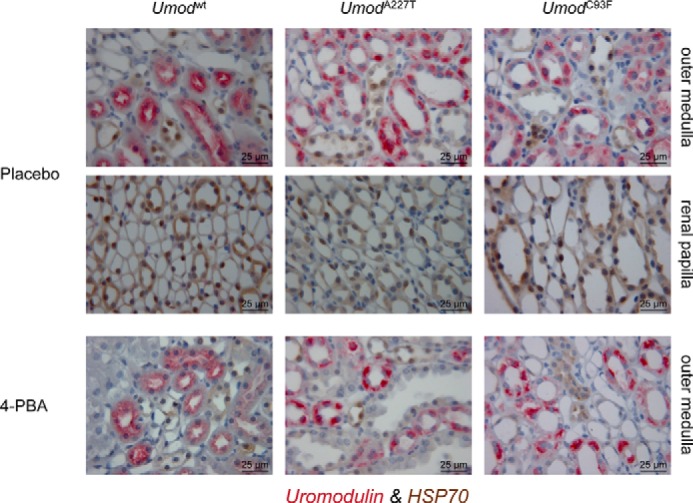
Effect of 4-PBA on HSP70 protein abundance in TALH cells of Umod mutant mice. HSP70 protein immunopositivity was predominantly detected in collecting duct cells of the renal papilla. In the outer medulla, the HSP70 protein was predominantly detected in collecting duct cells, but (nearly) no expression of HSP70 could be observed in uromodulin-expressing TALH cells. There were no obvious differences in HSP70 immunostaining pattern and intensity between different Umod genotypes or treatment groups (placebo control group versus 4-PBA group). The chromogen DAB for HSP70 and Vector RED for UMOD were used; nuclear staining was done with hemalum. The age of the mice analyzed was 4 month.
Analysis of Unfolded Protein Response Pathway in TALH Cells of Umod Mutant Mice
To elucidate the mechanism of unfolded protein response pathway in TALH cells in the setting of UAKD, protein abundances of factors of the NF-κB pathway were analyzed. Therefore, the renal segment with predominance of TALH cells, the outer medulla, was prepared (Fig. 6A). The lysates contained a high amount of TALH segments, which was confirmed by the >2-fold increase of the ER protein BiP in Umod mutant mice of both lines compared with wild-type controls. In Umod mutant mice, protein abundances of p105 and p50 of NF-κB1 were significantly increased in the outer medulla compared with that of wild-type mice (Fig. 6B). A 2-fold increased abundance of p100 and p52 of NF-κB2 and an at least 3-fold increased abundance of RelB was detected in UAKD-affected mice compared with wild-type controls, whereas minor alterations of protein abundance of NF-κB p65 were observed. Analyses of upstream factors of the NF-κB pathway revealed increased abundances of phospho-IKKα/β and TRAF2 in Umod mutant mice of both lines compared with wild-type mice. Protein abundances of IKKα and TRAF3 were similar in all genotype groups.
FIGURE 6.

Analysis of unfolded protein response pathway in the outer medulla of Umod mutant mice. A, the high density of TALH segments in the outer medulla was demonstrated by protein abundances of mature and immature UMOD and the increased abundance of BiP in Umod mutant mice compared with wild-type mice. B, protein abundances of NF-κB1 p105/p50, NF-κB2 p100/p52, NF-κB p65, RelB, phospho-IKKα/β, IKKα, TRAF2, and TRAF3. Signal intensities of the factors of NF-κB pathway were corrected for GAPDH signal intensities of the same PVDF-membrane, which was stripped several times to facilitate the detection of multiple proteins. The mean of protein abundance of wild-type mice was set on a value of 1 (mean (wild-type) = 1). Student's t test with Holm-Bonferroni correction: p versus wild-type, *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To have a more detailed view on the role of NF-κB pathway in UAKD, the abundance of phospho-IKKα/β (Ser-176/180), representing the active form of IKKα and IKKβ, was analyzed immunohistochemically (Fig. 7A). In wild-type mice, phospho-IKKα/β was predominantly and strongly detected in the basolateral cytoplasmic compartment of collecting duct cells, but its abundance in TALH cells was low. In Umod mutant mice, the abundance of phospho-IKKα/β in TALH cells appeared similar as in collecting duct cells. Furthermore, the staining pattern in TALH cells differed between Umod mutant mice and wild-type controls. A strong cytoplasmic basal and paranuclear staining pattern of phospho-IKKα/β was present in TALH cells of Umod mutant mice, and in wild-type mice the staining was weak and apical-accentuated. RelB was immunohistochemically localized with variable staining intensity in the cytoplasm and the nucleus of tubular cells (Fig. 7B). Whereas only few tubular cells were detected containing RelB in wild-type mice, the majority of tubular cells of the outer medulla of Umod mutant mice were immunopositive for RelB, and the staining intensity was stronger than in the corresponding region of wild-type mice. Identification of TALH segment was enabled by detection of UMOD.
FIGURE 7.
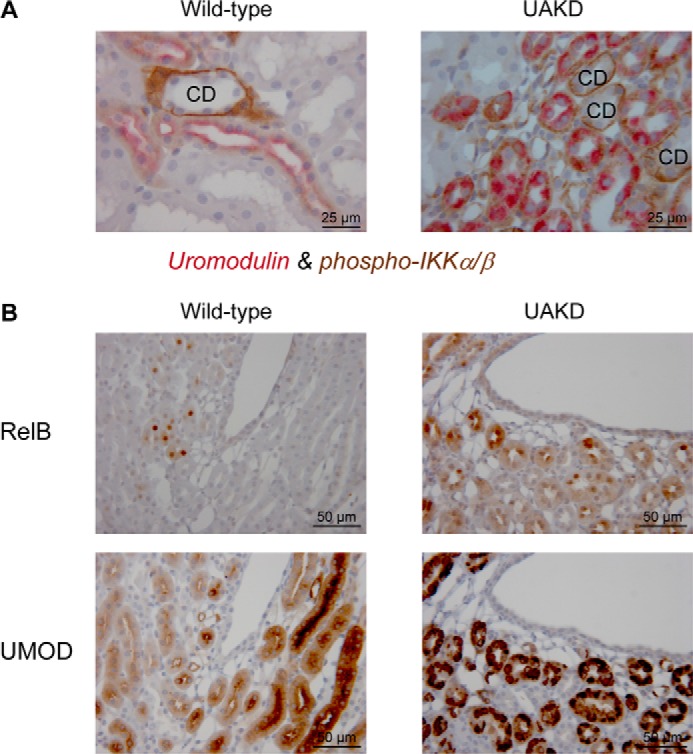
Immunohistochemical localization of factors of NF-κB pathway in TALH cells of Umod mutant mice. A, phospho-IKKα/β was predominantly detected in the basolateral cytoplasmic compartment of collecting duct cells. In TALH cells of wild-type mice, a weak diffuse cytoplasmic immunopositivity for phospho-IKKα/β was observed. In contrast, TALH cells of Umod mutant mice exhibited a strong signal of phospho-IKKα/β in the cytoplasmic basal and paranuclear compartment that signal intensity was in a similar range as that of collecting duct cells. The chromogens DAB was used for phospho-IKKα/β and Vector RED for UMOD; nuclear staining was done with hemalum. The age of mice analyzed was 4 month. CD, collecting duct profiles. Wild-type, Umodwt mouse; UAKD, homozygous UmodA227T mutant mouse. B, in wild-type mice RelB was detected in few tubular cells in the cytoplasm with a variable but predominantly weak staining intensity and in some nuclei. In contrast to wild-type mice, tubular cells of Umod mutant mice exhibited a stronger cytoplasmic and nuclear RelB staining intensity, and more tubular cells were stained. UMOD immunohistochemistry enabled identification of TALH segments. Serial kidney sections were used for RelB and UMOD immunohistochemistry, and corresponding kidney regions are shown. The chromogen was DAB, nuclear staining: hemalum. The age of the mice analyzed was 4 months. Wild-type, Umodwt mouse; UAKD, homozygous UmodC93F mutant mouse.
DISCUSSION
Specific curative therapies for UAKD are not available. Symptomatic treatment addresses correction of water and electrolyte imbalances and of hyperuricemia but does not prevent disease progression. The central disease mechanism of UAKD is the protein maturation defect caused by mutant uromodulin leading to UMOD retention in the ER and ER overload. Thus, amelioration of protein maturation capacity by molecular chaperons might represent a promising therapeutic approach to reduce UMOD retention in ER of TALH cells and consequently to reduce TALH dysfunction in UAKD. In vitro studies demonstrated a positive effect of the chemical chaperone 4-PBA on mutant uromodulin maturation (19, 20).
4-PBA is an orally bioavailable aromatic short-chain fatty acid and is used as United States Food and Drug Administration-approved drug (Buphenyl®, Ucyclyd Pharma Inc.; Ammonaps®, Orphan Europe of Immeuble “Le Guillaumet”) for clinical use as an ammonia scavenger in long term treatment of hyperammonemia in urea cycle disorders (25, 26). 4-PBA reduces ammonia levels in blood as it offers itself as an alternative to urea synthesis to excrete waste nitrogen from the body. It is converted to 2-phenylacetate and subsequently excreted with urine as nitrogen-containing phenylacetylglutamine in humans. Furthermore, a clinical effect of 4-PBA in treatment of homozygous mutants suffering from β-thalassemia was demonstrated (27) and also a putative curative effect of 4-PBA in spinal muscular atrophy is currently under critical discussion (28, 29). Various studies demonstrated the effect of the chemical chaperone 4-PBA in stabilizing protein conformation, improving ER folding capacity, facilitating the trafficking of mutant proteins, reducing ER stress, modulating the unfolded protein response, and thus improving protein homeostasis in ER storage diseases (25, 30, 31). For instance, 4-PBA has been shown to enhance in vitro and in vivo the adaptive capacity of the ER and to improve systemic insulin action in the setting of obesity-induced insulin resistance in type 2 diabetes (30). Furthermore, trafficking of the cystic fibrosis transmembrane regulator with the mutation ΔF508 (CFTRΔF508) was increased, and secretion of the mutant α1-ATZ protein in α1-antitrypsin deficiency was enhanced by 4-PBA (32, 33). Regarding the putative therapeutic effect of 4-PBA in the setting of UAKD, two independent in vitro studies demonstrated that mutant UMOD-transfected cells exhibited an increased secretion of uromodulin in the supernatant and had an enforced immunofluorescent uromodulin staining signal on their cell membrane (19, 20). In the present study, however, we were not able to demonstrate a positive effect of 4-PBA to improve mutant uromodulin maturation in vivo in our UAKD mouse models UmodA227T and UmodC93F. Intracellular accumulation of uromodulin in TALH cells was still present in Umod mutant mice after two months of treatment with water-soluble and orally bioavailable 4-PBA, and uromodulin excretion with urine remained low at a similar range as in genotype-matched placebo-treated mice. 4-PBA is rapidly metabolized to phenylacetate, and both components have short elimination half-lives of 0.76 and 1.29 h in humans, respectively. Therefore, missing bioavailability of 4-PBA to TALH cells has to be taken into account as a potential explanation for the lack of a therapeutic effect in our UAKD mouse models. However, this is unlikely as administration with the drinking water ensured continuous intake of 4-PBA throughout the day. Furthermore, effects of 4-PBA on some kidney parameters, like urine osmolality and urinary excretion of phosphate, were clearly detected. Basseri et al. (31) demonstrated an in vivo effect of 4-PBA on modulating unfolded protein response and attenuating ER stress in adipocytes using similar preparation, dosage, and oral administration of 4-PBA like we used in our study but changed drinking water only weekly in contrast to half-weekly fresh preparation made in our study. To further exclude the possibility that the missing effect of 4-PBA in our in vivo study was due to insufficient bioavailability of the drug at the site of action, the TALH cells, 4-PBA, was administered in vitro to culture medium of freshly isolated kidney cells of Umod mutant mice for 48 h. Again, 4-PBA was ineffective in attenuating uromodulin retention and ER hyperplasia in TALH cells isolated from kidneys of Umod mutant mice.
To evaluate the effect of 4-PBA on secretion of murine UMOD A227T and UMOD C93F mutant protein and to repeat published experiments indicating a positive effect of 4-PBA on secretion of human uromodulin in vitro, we performed assays on stably transfected MTCs. MTCs do not express endogenous uromodulin protein but provide the maturation capacity for GPI-linked secreted proteins thereby facilitating the interpretation of maturation ability of the mutant protein. We obtained various stable Umod-transfected MTC clones for each construct (Umodwt, UmodA227T, and UmodC93F) secreting variable amounts of uromodulin into the supernatant. There was no relationship between the type of Umod mutation and maturation efficiency of stably transfected cells. There were numerous in vitro studies published analyzing the maturation time of uromodulin until secretion and the amount of protein secreted into the supernatant by transfected cells. A common finding of these studies was that all UMOD mutations analyzed needed a longer time period for protein maturation and that the efficiency of protein maturation was different between different UMOD mutations (3, 12–14, 34). However, these in vitro models were described to be not suitable to analyze potential disease mechanisms downstream of mutant uromodulin retention as the cellular models reported so far did not reproduce key cellular hallmarks of UAKD, which are mutant uromodulin aggregation, ER membrane expansion, and ER dilation (4). In vitro 4-PBA administration to supernatant was reported to increase secretion of both wild-type and mutant uromodulin, which was also associated with stronger membrane labeling of uromodulin of stably transfected cells (19, 20). In our in vitro studies we also detected a strong increase of uromodulin secretion of both wild-type and mutant protein after 4-PBA stimulation. But in parallel the amount of intracellular uromodulin in MTCs was also increased, indicating an increase of uromodulin synthesis rather than changes in maturation efficiency of uromodulin. There are numerous reports of 4-PBA as a well known histone deacetylase inhibitor, which increases transcription of a number of genes (25, 35, 36). For instance, beneficial therapeutic effects of 4-PBA due to its histone deacetylase inhibitor function were demonstrated for motor neuron survival in mice with amyotrophic lateral sclerosis (35) or for treatment of different types of tumors (25). The therapeutic effect of 4-PBA to correct trafficking of mutant CFTRΔF508 in cystic fibrosis epithelial cells could be attributed to increased expression of some cytosolic molecular chaperones like heat shock protein 70 (HSP70) (37). In consequence, increased UMOD content in supernatant as well as in cell lysate of UMOD-expressing MTC clones in our study might be caused by the histone deacetylase inhibition due to 4-PBA administration increasing UMOD expression in a dose-dependent manner.
The in vitro results obtained by 4-PBA testing on UMOD-transfected cells might be highly dependent on the cell type used for in vitro analyses. Ma et al. (20) reported a similar steady-state expression of the molecular chaperone HSP70 in mock-transfected and wild-type UMOD-transfected MDCK cells, an immortalized collecting duct cell line, but a decrease of HSP70 expression in UMOD mutant-transfected MDCK cells and an increase of HSP70 expression in mutant UMOD-expressing cells up to the level of wild-type UMOD-expressing cells after incubation with 10 mm 4-PBA. The heat shock protein HSP70 is predominantly expressed in the kidney in papillary-collecting duct cells to protect these cells from hypertonic stress present in the hypertonic environment of the renal papilla (38). HSP70 protein abundance was low or absent in TALH cells of both wild-type mice and Umod mutant mice, indicating a minor relevance of this molecular chaperone in uromodulin protein maturation in TALH cells in vivo. Therefore, in vitro models of UMOD-transfected cells might be unsuitable for testing putative therapeutic effects of drugs, as Rampoldi et al. (4) already described that these cell models are not useful to analyze the molecular intracellular pathways in UAKD.
Long term administration of 4-PBA had, besides changes in phosphate levels (lower plasma phosphate levels and lower 24-h phosphate excretion with urine) and mild decrease of urine osmolality, only minor effects on renal function of healthy wild-type mice compared with untreated wild-type controls. However, in the setting of UAKD, long term treatment with 4-PBA changed additional parameters of renal function, like blood chloride and phosphate levels, urine osmolality, and renal handling of some electrolytes like sodium. The most prominent changes caused by 4-PBA treatment represented the strongly reduced fractional excretion of phosphate in homozygous UmodC93F mutant mice, exhibiting a more severe uromodulin maturation defect and UAKD phenotype as compared with UmodA227T homozygotes. Although a known function of 4-PBA is to act as nitrogen scavenger and thus to be able to decrease plasma ammonia and urea levels, plasma urea concentrations of 4-PBA-treated mice were similar to those of placebo-treated genotype-matched controls or were even increased as in homozygous UmodC93F mutant mice. Thus, 4-PBA treatment rather resulted in a further deterioration of renal function in both mouse models of UAKD than improving it.
To elucidate further therapeutic approaches to perform curative treatment of UAKD, one open question is the analysis of the predominant stress pathways that are activated by mutant uromodulin maturation retardation and ER retention. In our mouse models of UAKD, numerous factors of the NF-κB pathway were increased abundant and/or activated, and the activation of NF-κB pathway was localized in TALH cells of Umod mutant mice. RelB was identified as the factor with the highest increase of protein abundance in Umod mutant mice compared with wild-type controls. Together with the increased abundance of NF-κB2 p52 in Umod mutant mice, our data indicate that the non-canonical NF-κB pathway, which predominantly targets activation of the p52/RelB NF-κB complex (39, 40), might be of special importance as disease mechanism in UAKD.
In conclusion, our study demonstrated that, in contrast to the reported amelioration of the maturation and trafficking defect of mutant uromodulin in vitro, the chemical chaperone 4-PBA was ineffective to improve mutant uromodulin maturation and TALH function in two mouse models of UAKD in vivo. Activation of the NF-κB pathway in the thick ascending limb of Henle's loop cells of UAKD mice was demonstrated, identifying a novel disease mechanism of UAKD and a potential target for therapeutic intervention.
Acknowledgments
We thank Angela Siebert for excellent technical assistance on electron microscopic analyses, Simona Horn and Stephanie Schmidt for excellent support on immunohistological analyses, Elfi Holupirek for clinical-chemical analyses carried out at the German Mouse Clinic, and the mouse facility of the Moorversuchsgut Oberschleissheim for excellent animal care.
This work was supported by the German Research Foundation (DFG grant KE1673/1-1) and by Infrafrontier Grant 01KX1012.
- UAKD
- uromodulin-associated kidney disease
- UMOD
- uromodulin
- IKK
- IκB kinase
- TALH
- thick ascending limb of Henle's loop
- ER
- endoplasmic reticulum
- 4-PBA
- 4-phenylbutyrate acid
- DAB
- 3,3-diaminobenzidine tetrahydrochloride dihydrate
- FE
- fractional excretion
- HSP70
- heat shock protein 70
- MTC
- murine proximal tubular epithelial cell
- ANOVA
- analysis of variance.
REFERENCES
- 1. Hart T. C., Gorry M. C., Hart P. S., Woodard A. S., Shihabi Z., Sandhu J., Shirts B., Xu L., Zhu H., Barmada M. M., Bleyer A. J. (2002) Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J. Med. Genet. 39, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scolari F., Caridi G., Rampoldi L., Tardanico R., Izzi C., Pirulli D., Amoroso A., Casari G., Ghiggeri G. M. (2004) Uromodulin storage diseases. Clinical aspects and mechanisms. Am. J. Kidney Dis. 44, 987–999 [DOI] [PubMed] [Google Scholar]
- 3. Rampoldi L., Caridi G., Santon D., Boaretto F., Bernascone I., Lamorte G., Tardanico R., Dagnino M., Colussi G., Scolari F., Ghiggeri G. M., Amoroso A., Casari G. (2003) Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol. Genet. 12, 3369–3384 [DOI] [PubMed] [Google Scholar]
- 4. Rampoldi L., Scolari F., Amoroso A., Ghiggeri G., Devuyst O. (2011) The rediscovery of uromodulin (Tamm-Horsfall protein). From tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. 80, 338–347 [DOI] [PubMed] [Google Scholar]
- 5. Bleyer A. J., Zivná M., Kmoch S. (2011) Uromodulin-associated kidney disease. Nephron Clin. Pract. 118, c31–c36 [DOI] [PubMed] [Google Scholar]
- 6. Bleyer A. J., Woodard A. S., Shihabi Z., Sandhu J., Zhu H., Satko S. G., Weller N., Deterding E., McBride D., Gorry M. C., Xu L., Ganier D., Hart T. C. (2003) Clinical characterization of a family with a mutation in the uromodulin (Tamm-Horsfall glycoprotein) gene. Kidney Int. 64, 36–42 [DOI] [PubMed] [Google Scholar]
- 7. Bollée G., Dahan K., Flamant M., Morinière V., Pawtowski A., Heidet L., Lacombe D., Devuyst O., Pirson Y., Antignac C., Knebelmann B. (2011) Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin. J. Am. Soc. Nephrol. 6, 2429–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nasr S. H., Lucia J. P., Galgano S. J., Markowitz G. S., D'Agati V. D. (2008) Uromodulin storage disease. Kidney Int. 73, 971–976 [DOI] [PubMed] [Google Scholar]
- 9. Trudu M., Janas S., Lanzani C., Debaix H., Schaeffer C., Ikehata M., Citterio L., Demaretz S., Trevisani F., Ristagno G., Glaudemans B., Laghmani K., Dell'Antonio G., Swiss Kidney Project on Genes in Hypertension (SKIPOGH) team, Loffing J., Rastaldi M. P., Manunta P., Devuyst O., Rampoldi L. (2013) Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat. Med. 19, 1655–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Serafini-Cessi F., Malagolini N., Cavallone D. (2003) Tamm-Horsfall glycoprotein. Biology and clinical relevance. Am. J. Kidney Dis. 42, 658–676 [DOI] [PubMed] [Google Scholar]
- 11. Zaucke F., Boehnlein J. M., Steffens S., Polishchuk R. S., Rampoldi L., Fischer A., Pasch A., Boehm C. W., Baasner A., Attanasio M., Hoppe B., Hopfer H., Beck B. B., Sayer J. A., Hildebrandt F., Wolf M. T. (2010) Uromodulin is expressed in renal primary cilia and UMOD mutations result in decreased ciliary uromodulin expression. Hum Mol. Genet. 19, 1985–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernascone I., Vavassori S., Di Pentima A., Santambrogio S., Lamorte G., Amoroso A., Scolari F., Ghiggeri G. M., Casari G., Polishchuk R., Rampoldi L. (2006) Defective intracellular trafficking of uromodulin mutant isoforms. Traffic. 7, 1567–1579 [DOI] [PubMed] [Google Scholar]
- 13. Williams S. E., Reed A. A., Galvanovskis J., Antignac C., Goodship T., Karet F. E., Kotanko P., Lhotta K., Morinière V., Williams P., Wong W., Rorsman P., Thakker R. V. (2009) Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum. Hum Mol. Genet. 18, 2963–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vylet'al P., Kublová M., Kalbácová M., Hodanová K., Baresová V., Stiburková B., Sikora J., Hulková H., Zivný J., Majewski J., Simmonds A., Fryns J. P., Venkat-Raman G., Elleder M., Kmoch S. (2006) Alterations of uromodulin biology. A common denominator of the genetically heterogeneous FJHN/MCKD syndrome. Kidney Int. 70, 1155–1169 [DOI] [PubMed] [Google Scholar]
- 15. Adam J., Bollée G., Fougeray S., Noël L. H., Antignac C., Knebelman B., Pallet N. (2012) Endoplasmic reticulum stress in UMOD-related kidney disease. A human pathologic study. Am. J. Kidney Dis. 59, 117–121 [DOI] [PubMed] [Google Scholar]
- 16. Schröder M., Kaufman R. J. (2005) ER stress and the unfolded protein response. Mutat Res. 569, 29–63 [DOI] [PubMed] [Google Scholar]
- 17. Kemter E., Rathkolb B., Rozman J., Hans W., Schrewe A., Landbrecht C., Klaften M., Ivandic B., Fuchs H., Gailus-Durner V., Klingenspor M., de Angelis M. H., Wolf E., Wanke R., Aigner B. (2009) Novel missense mutation of uromodulin in mice causes renal dysfunction with alterations in urea handling, energy, and bone metabolism. Am. J. Physiol. Renal Physiol. 297, F1391–F1398 [DOI] [PubMed] [Google Scholar]
- 18. Khanna D., Fitzgerald J. D., Khanna P. P., Bae S., Singh M. K., Neogi T., Pillinger M. H., Merill J., Lee S., Prakash S., Kaldas M., Gogia M., Perez-Ruiz F., Taylor W., Liote F., Choi H., Singh J. A., Dalbeth N., Kaplan S., Niyyar V., Jones D., Yarows S. A., Roessler B., Kerr G., King C., Levy G., Furst D. E., Edwards N. L., Mandell B., Schumacher H. R., Robbins M., Wenger N., Terkeltaub R.; American College of Rheumatology (2012) 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. (Hoboken) 64, 1431–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choi S. W., Ryu O. H., Choi S. J., Song I. S., Bleyer A. J., Hart T. C. (2005) Mutant tamm-horsfall glycoprotein accumulation in endoplasmic reticulum induces apoptosis reversed by colchicine and sodium 4-phenylbutyrate. J. Am. Soc. Nephrol. 16, 3006–3014 [DOI] [PubMed] [Google Scholar]
- 20. Ma L., Liu Y., El-Achkar T. M., Wu X. R. (2012) Molecular and cellular effects of Tamm-Horsfall protein mutations and their rescue by chemical chaperones. J. Biol. Chem. 287, 1290–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kemter E., Prueckl P., Sklenak S., Rathkolb B., Habermann F. A., Hans W., Gailus-Durner V., Fuchs H., Hrabě de Angelis M., Wolf E., Aigner B., Wanke R. (2013) Type of uromodulin mutation and allelic status influence onset and severity of uromodulin-associated kidney disease in mice. Hum Mol. Genet. 22, 4148–4163 [DOI] [PubMed] [Google Scholar]
- 22. Rathkolb B., Hans W., Prehn C., Fuchs H., Gailus-Durner V., Aigner B., Adamski J., Wolf E., Hrabě de Angelis M. (2011) Clinical chemistry and other laboratory tests on mouse plasma or serum. Current Protocols in Mouse Biology, John Wiley & Sons, Inc., pp. 69–100 [DOI] [PubMed] [Google Scholar]
- 23. Kemter E., Rathkolb B., Bankir L., Schrewe A., Hans W., Landbrecht C., Klaften M., Ivandic B., Fuchs H., Gailus-Durner V., Hrabé de Angelis M., Wolf E., Wanke R., Aigner B. (2010) Mutation of the Na+-K+-2Cl− cotransporter NKCC2 in mice is associated with severe polyuria and a urea-selective concentrating defect without hyperreninemia. Am. J. Physiol. Renal Physiol. 298, F1405–F1415 [DOI] [PubMed] [Google Scholar]
- 24. Wolf G., Neilson E. G. (1994) Isoproterenol induces mitogenesis in MCT and LLC-PK1 tubular cells. J. Am. Soc. Nephrol. 4, 1995–2002 [DOI] [PubMed] [Google Scholar]
- 25. Iannitti T., Palmieri B. (2011) Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 11, 227–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brusilow S. W., Maestri N. E. (1996) Urea cycle disorders. Diagnosis, pathophysiology, and therapy. Adv. Pediatr. 43, 127–170 [PubMed] [Google Scholar]
- 27. Collins A. F., Pearson H. A., Giardina P., McDonagh K. T., Brusilow S. W., Dover G. J. (1995) Oral sodium phenylbutyrate therapy in homozygous β thalassemia. A clinical trial. Blood 85, 43–49 [PubMed] [Google Scholar]
- 28. Wadman R. I., Bosboom W. M., van der Pol W. L., van den Berg L. H., Wokke J. H., Iannaccone S. T., Vrancken A. F. (2012) Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 4, CD006282. [DOI] [PubMed] [Google Scholar]
- 29. Mercuri E., Bertini E., Messina S., Pelliccioni M., D'Amico A., Colitto F., Mirabella M., Tiziano F. D., Vitali T., Angelozzi C., Kinali M., Main M., Brahe C. (2004) Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul. Disord. 14, 130–135 [DOI] [PubMed] [Google Scholar]
- 30. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basseri S., Lhoták S., Sharma A. M., Austin R. C. (2009) The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid Res. 50, 2486–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burrows J. A., Willis L. K., Perlmutter D. H. (2000) Chemical chaperones mediate increased secretion of mutant α1-antitrypsin (α1-AT) Z. A potential pharmacological strategy for prevention of liver injury and emphysema in α1-AT deficiency. Proc. Natl. Acad. Sci. U.S.A. 97, 1796–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lim M., McKenzie K., Floyd A. D., Kwon E., Zeitlin P. L. (2004) Modulation of deltaF508 cystic fibrosis transmembrane regulator trafficking and function with 4-phenylbutyrate and flavonoids. Am. J. Respir. Cell Mol. Biol. 31, 351–357 [DOI] [PubMed] [Google Scholar]
- 34. Jennings P., Aydin S., Kotanko P., Lechner J., Lhotta K., Williams S., Thakker R. V., Pfaller W. (2007) Membrane targeting and secretion of mutant uromodulin in familial juvenile hyperuricemic nephropathy. J. Am. Soc. Nephrol. 18, 264–273 [DOI] [PubMed] [Google Scholar]
- 35. Ryu H., Smith K., Camelo S. I., Carreras I., Lee J., Iglesias A. H., Dangond F., Cormier K. A., Cudkowicz M. E., Brown R. H., Jr., Ferrante R. J. (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93, 1087–1098 [DOI] [PubMed] [Google Scholar]
- 36. Ricobaraza A., Cuadrado-Tejedor M., Pérez-Mediavilla A., Frechilla D., Del Río J., García-Osta A. (2009) Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology 34, 1721–1732 [DOI] [PubMed] [Google Scholar]
- 37. Suaud L., Miller K., Panichelli A. E., Randell R. L., Marando C. M., Rubenstein R. C. (2011) 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J. Biol. Chem. 286, 45083–45092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neuhofer W., Fraek M. L., Ouyang N., Beck F. X. (2005) Differential expression of heat shock protein 27 and 70 in renal papillary collecting duct and interstitial cells. Implications for urea resistance. J. Physiol. 564, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun S. C. (2011) Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smale S. T. (2011) Hierarchies of NF-κB target-gene regulation. Nat. Immunol. 12, 689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]