

Background: The molecular tweezer, CLR01, binds to Lys and prevents aggregation of α-synuclein.
Results: CLR01 binds directly to monomeric α-synuclein near the N terminus and changes the charge distribution in the sequence, swelling the chain, and increasing the protein reconfiguration rate.
Conclusion: Aggregation is inhibited by making the protein more diffusive.
Significance: The most effective aggregation inhibitors may change monomer dynamics rather than structure.
Keywords: α-Synuclein, Mass Spectrometry (MS), Parkinson's Disease, Protein Aggregation, Spectroscopy, Inhibitor, Intramolecular Diffusion
Abstract
Recent work on α-synuclein has shown that aggregation is controlled kinetically by the rate of reconfiguration of the unstructured chain, such that the faster the reconfiguration, the slower the aggregation. In this work we investigate this relationship by examining α-synuclein in the presence of a small molecular tweezer, CLR01, which binds selectively to Lys side chains. We find strong binding to multiple Lys within the chain as measured by fluorescence and mass-spectrometry and a linear increase in the reconfiguration rate with concentration of the inhibitor. Top-down mass-spectrometric analysis shows that the main binding of CLR01 to α-synuclein occurs at the N-terminal Lys-10/Lys-12. Photo-induced cross-linking of unmodified proteins (PICUP) analysis shows that under the conditions used for the fluorescence analysis, α-synuclein is predominantly monomeric. The results can be successfully modeled using a kinetic scheme in which two aggregation-prone monomers can form an encounter complex that leads to further oligomerization but can also dissociate back to monomers if the reconfiguration rate is sufficiently high. Taken together, the data provide important insights into the preferred binding site of CLR01 on α-synuclein and the mechanism by which the molecular tweezer prevents self-assembly into neurotoxic aggregates by α-synuclein and presumably other amyloidogenic proteins.
Introduction
The protein α-synuclein is highly aggregation-prone and is involved in a number of neurodegenerative diseases, the best known of which is Parkinson disease (PD).3 The 140-residue protein, whose function is still unknown, has no stable structure in vitro, but can adopt an α-helical structure in association with membranes and possibly exists as a helical tetramer in vivo (1, 2). However, all evidence indicates that the aggregation pathway passes through an at least partially unstructured state. Thus, much work has focused on which subset of the conformation ensemble is prone to nucleating aggregation (3–6).
Ahmad et al. recently have shown that aggregation of α-synuclein may be determined not by formation of certain structures but rather, by the reconfiguration of the entire unfolded ensemble (7). When reconfiguration is fast relative to intermolecular collision, as is the situation at low temperature, then during an encounter between two proteins, each chain reconfigures too rapidly to make stabilizing intermolecular interactions. In contrast, if reconfiguration occurs at about the same rate as bimolecular association, which is the case at physiological temperatures, then aggregation is more likely to occur. Thus, a potential therapeutic strategy to prevent PD is to use small molecules that bind to α-synuclein and increase its reconfiguration rate under physiological conditions, thereby preventing aggregation. This strategy was demonstrated with curcumin, a naturally occurring compound in the spice turmeric, which was found to bind strongly to α-synuclein, prevent fibrilization, and increase the rate of protein reconfiguration at physiological temperatures (8). However, it is still not known where on α-synuclein curcumin binds and whether there are multiple binding sites, though the observation that the reconfiguration rate increased at molar ratios greater than 1:1 suggests that multiple curcumin molecules bind each α-synuclein monomer.
In this work, we show that the molecular tweezer, CLR01, known to bind predominantly to Lys side chains, behaves similarly to curcumin in increasing protein reconfiguration and preventing fibrilization, but binds more strongly than curcumin. Similarly to curcumin, CLR01 binds at multiple sites, yet the predominant binding site is located at the N terminus as identified by mass spectrometry. Fluorescence changes show that increasing CLR01 concentration slows down the early oligomerization process. These results suggest that reversing the net charge on certain residues from positive to negative modulates intramolecular interactions in a manner that prevents collapse and speeds up reconfiguration. The data also suggest a possible roadmap for therapeutic development.
EXPERIMENTAL PROCEDURES
α-Synuclein
For mass spectrometry (MS) experiments, recombinant α-synuclein was purchased from rPeptide (Bogart, GA). The protein was dissolved in distilled water and desalted by using 10-kDa molecular weight cutoff centrifugal filter devices (Amicon Ultra, Millipore, Billerica, MA) with 20 mm ammonium acetate, pH 6.8. The final protein concentration was 10 μm.
For PICUP experiments, recombinant WT α-synuclein was produced using cDNA in which the original TAC codon for Tyr was mutated to TAT to avoid erroneous translation to Cys (9). Competent BL21(DE3) Escherichia coli bacteria were transformed with this plasmid, allowed to grow in 3 liters of Luria broth to A600 ≈ 0.6, induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside and collected after 3 additional hours of incubation. The bacteria were collected by centrifugation for 15 min at 4,690 × g and resuspended in 100 ml of lysis buffer containing 0.2 m Tris, pH 8.0, 1 mm EDTA, 5 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride. The bacteria then were lysed on ice using a tip sonicator set to 3 KJoule for 3 × 2 min cycles of 3 s power on and 3 s power off. The lysate was centrifuged at 31,920 × g for 30 min and the supernatant was collected and supplemented slowly with 10 mg/ml streptomycin. The solution then was stirred on ice for 45 min and centrifuged again at 31,920 × g for 30 min. The proteins were precipitated from the supernatant by addition of 0.23 g/ml ammonium sulfate. The solution was stirred on ice for 30 min and centrifuged at 31,920 × g for 30 min. The supernatant was discarded, and the protein pellet was dried by inverting the centrifuge tube on paper towels. The pellet was resuspended in 20 ml of 25 mm Tris, pH 8.0, and the resulting solution dialyzed overnight against 4 liters of the same buffer. The crude protein mixture was fractionated using ion-exchange Q columns (GE Healthcare, Piscataway, NJ) and a 100-ml gradient ranging from 0 to 0.5 m NaCl in 25 mm Tris, pH 8.0. The fraction collected between 0.17 and 0.39 m NaCl was purified further by size-exclusion chromatography using a 2.15 × 600 mm TSK-gel G3000SW column (Tosoh Biosciences, San Francisco, CA) with elution buffer comprising 100 mm sodium sulfate, 25 mm sodium phosphate, and 1 mm sodium azide, pH 6.5. Finally, the fractions containing purified α-synuclein were dialyzed against a buffer containing 20 mm sodium phosphate, pH 7.4, 0.1 m NaCl. The purity of the protein was assessed by SDS-PAGE and Coomassie Blue staining, and the final concentration of purified α-synuclein was 0.42 mm.
α-[Cys69, Trp94]synuclein for Trp-Cys quenching experiments and fluorescence binding studies was expressed similarly in E. coli BL21(DE3) cells transformed with a T7-7 vector containing the cognate gene for expression of α-synuclein with the appropriate mutations. After cell lysis, the cell suspension was boiled for 20 min and centrifuged at 4,724 × g. The supernatant was collected, and the protein was precipitated by addition of 0.36 g/ml ammonium sulfate. The pellet was resuspended in 25 mm Tris-HCl, pH 7.4, chromatographed on a Q-Sepharose Fast Flow column equilibrated with 25 mm Tris, pH 7.4, and eluted with a linear NaCl gradient (0–1,000 mm). α-Synuclein-containing fractions were pooled and further purified by gel filtration on a Hi-Prep Sephacryl S200 column. α-Synuclein eluted as a single peak and was found to be >95% pure as assessed by SDS-PAGE and Coomassie Blue staining. The protein concentration was determined from the absorbance at 280 nm using extinction coefficient of 11,460 m−1 cm−1. The stock solution of ∼0.3 mm was stored at −80 °C in 25 mm sodium phosphate, pH 7.4, in the presence of 1 mm tris(2-carboxyethyl)phosphine (TCEP) to prevent disulfide bond formation.
Binding and Oligomerization Experiments Using Fluorescence
Trp and CLR01 fluorescence measurements were carried out on a PTI QW4 spectrofluorimeter equipped with a temperature-controlled cuvette holder and stirrer kit. For binding, the Trp fluorescence spectra were all measured at room temperature in a 1-cm path length cuvette exposed to 1.25-mm slit widths. Trp-94 in the mutant α-synuclein was excited at 280 nm and binding of CLR01 was measured by monitoring the change in the intrinsic fluorescence between 300 and 450 nm. The α-synuclein concentration was kept fixed at 3 μm, and CLR01 was added to the protein with increasing concentrations, ranging from 0.02 to 25.6 μm. Increase of fluorescence as well as spectral blue shifts were observed with increasing CLR01 concentration, from which the binding constant was determined. It was not possible to use higher CLR01 concentrations because the compound has significant fluorescence in the 300–450 nm range. CLR01 fluorescence was excited at 285 nm, and fluorescence measured between 300–450 nm. The CLR01 concentration was kept constant at 15 μm, and wild-type α-synuclein was added in increasing concentrations, ranging from 1–100 μm.
To measure early oligomerization, 45-μm α-[Cys69, Trp94]synuclein were incubated at 37 °C with constant stirring in the cuvette holder of the fluorometer. The protein was excited at 280 nm, and emission was measured between 300–450 nm with 1-mm excitation slit width. Data were collected every ∼10 min with the excitation light blocked between measurements to minimize photobleaching. 15 mm TCEP were added to the cuvette to prevent disulfide formation during the experiment.
Native Electrospray Ionization Mass Spectrometry
Electrospray ionization (ESI) mass spectra were acquired using a solariX hybrid 15-Tesla Fourier transform ion-cyclotron resonance (FT-ICR) mass-spectrometer (Bruker Daltonics, Billerica, MA) in positive-ion mode. The estimated resolving power was 400,000 at m/z 400. A nanoESI source using borosilicate glass capillaries with Au/Pd coatings (Proxeon Biosystems/Thermo Scientific) was operated at low analyte flow conditions (100–200 nL/min) supported by a syringe pump. The ESI voltage used was 1,100–1,300 V. The capillary inlet temperature was set to 160 °C, and the skimmer voltage was set to 30–60 V. For top-down MS/MS experiments of the intact protein, precursor ions were first isolated in the quadrupole region and then transferred to the ICR cell where the protein ions interact with low energy electrons (1.1 eV, 1 ms pulse duration) to perform electron-capture dissociation (ECD). MS/MS product ions were identified against the protein sequence using tools provided by ProteinProspector.
Trp-Cys Contact Quenching
For these experiments, 300-μl aliquots of 300 μm protein were diluted 10-fold in 100-mm sodium phosphate, pH 7.4, 10-mm TCEP, and 0, 10, 20, or 30% sucrose (w/w) to change the solution's viscosity, along with the desired concentration of CLR01, making the final concentration of the protein 30 μm. The solution was stirred continuously during measurements. CLR01 concentrations used were 15, 30, or 60 μm. The solution without the protein was bubbled with N2O for 1 h, to eliminate oxygen and scavenge solvated electrons created in the UV laser pulse. Triplet lifetime decay kinetics of Trp as a result of Cys quenching was measured using an instrument described previously (10). Briefly, the Trp triplet was excited by a 10-ns laser pulse at 289 nm created from the fourth harmonic of an Nd:YAG laser (Continuum) and a 1-meter Raman cell filled with 450 p.s.i of D2 gas. The triplet population was probed at 441 nm by a HeCd laser (Kimmon). The probe and the reference beam were measured with silicon detectors and then combined in a differential amplifier (DA 1853A, LeCroy) with an additional stage of a 350-MHz preamplifier (SR445A, Stanford Research Systems). The total gain was 50-fold. The temperature was controlled as described previously (10). Measurement of each sample at six temperatures - 0, 10, 15, 20, 30, and 40 °C) took about 20 min, so aggregation during this time was negligible. The viscosity of each solvent at each temperature was measured independently using a cone-cup viscometer (Brookfield engineering).
PICUP
PICUP reactions were performed with minor modifications on previously described protocols (11, 12). Briefly, purified α-synuclein was aliquoted in 15 or 60 μm stock solutions, for the 3 and 30 μm reactions, respectively, in 100 mm sodium phosphate, pH 7.4, and stored at −80 °C. CLR01 was dissolved at 600 or 1,000 μm, respectively, in the same buffer. The appropriate α-synuclein and CLR01 solutions were mixed to provide the final concentrations in 18 μl in PCR tubes followed by addition of 1 μl Ru(Bpy) and 1 μl APS, irradiation for 1 s, and quenching immediately with 20 μl of Tricine sample buffer (Invitrogen) containing 5% β-mercaptoethanol. Before loading on gels, 30 μm solutions were diluted 10-fold with sample buffer. Samples were fractionated by SDS-PAGE using 1-mm-thick, 10–20% Tricine gradient gels (Invitrogen). Non-cross-linked proteins were used as a control for the cross-linked proteins in each condition for each experiment and ran as monomers in all cases (data not shown). Protein bands were visualized by silver staining (Invitrogen). Gels were scanned and the abundance of each band was calculated densitometrically relative to the entire lane using Image J.
RESULTS
Binding of CLR01 to Monomeric α-Synuclein
For the Trp-Cys quenching experiments in this work, the wild-type (WT) sequence of α-synuclein was modified to contain a Phe→Trp substitution at position 94 and an Ala→Cys substitution at position 69 so that Trp-Cys quenching could be measured (7). Previously, Ahmad et al. showed that these mutations did not change the aggregation rate, as measured by ThT fluorescence or the CD spectrum, significantly relative to the WT sequence (7). Using this mutant sequence, Ahmad et al. measured intramolecular contact rates as a function of temperature and solution viscosity and found that intramolecular diffusion slowed significantly with increasing temperature between T = 0–40 °C. The introduction of the Trp also allows using UV fluorescence to investigate the binding of the molecular tweezer by its effect on the quantum yield and maximum emission wavelength, which in turn reveals information about the protein conformation. It was shown previously that CLR01 bound to Lys with a dissociation constant of 4–30 × 10−6 m using fluorescence at relatively high concentrations (13).
Here we investigated the binding of CLR01 to α-synuclein, which contains 15 Lys (Fig. 1) in the sequence, starting at substantially lower concentrations taking advantage of the relatively high quantum yield of Trp fluorescence. Fig. 2, a and b show an initial increase in fluorescence of Trp-94 in α-synuclein upon addition of CLR01, which fits well to a hyperbolic curve that saturates before 1 μm, the concentration of the protein, indicating high-affinity binding. Binding also was measured by native mass-spectrometry, which allows observation of free and bound α-synuclein directly (total protein concentration: 10 μm), yielding a binding constant KD = 0.16 μm as shown in supplemental Fig. S2. However, increasing the concentration of CLR01 revealed additional binding events. Mass-spectrometry indicated a second binding event with KD = 3 μm. Analysis of the intrinsic fluorescence of CLR01 when bound to wild-type α-synuclein suggested a binding event with KD ∼1.8 μm (at a CLR01 concentration: 15 μm), in agreement with the mass-spectrometry results. At CLR01 concentrations > 10 μm, the fluorescence spectrum of Trp-94 becomes contaminated by the fluorescence from the CLR01 itself, which peaks at ∼340 nm, causing an increase in background fluorescence and making it difficult to fit dissociation constants. However, if the entire fluorescence spectrum is analyzed with singular value decomposition (svd), the shift in the Trp-94 spectrum due to CLR01 binding can be well separated from the overall changes in total fluorescence, as shown in Fig. 2, b and c and yields two transitions at KD = 0.79 μm and KD = 7 μm. CLR01 fluorescence also suggested a transition with KD > 100 μm that was not well resolved over the concentrations studied. Thus there appear to be binding events with KD values ranging from 10−7 to 10−4 m.
FIGURE 1.
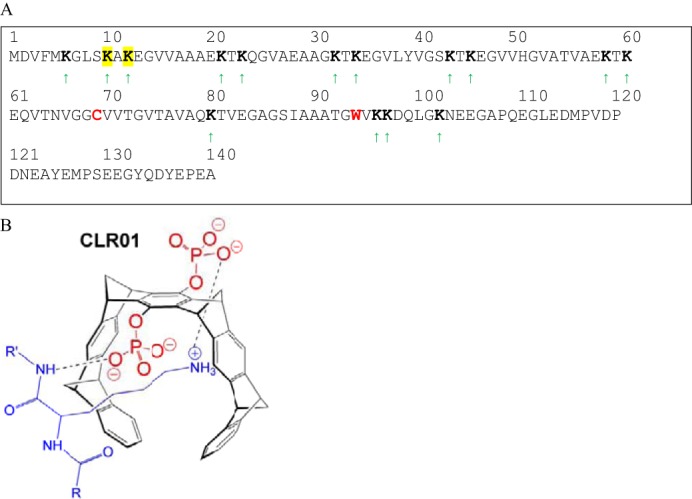
Schematic structures of α-synuclein and CLR01. A, in the α-synuclein sequence, the Ala-69→Cys and Tyr-94→Trp substitutions are shown in red. The Lys residues are marked by green arrows and boldface as possible binding sites for CLR01. The identified putative binding sites at Lys-10 and Lys-12 are highlighted in yellow. B, diagram of CLR01 bound to a lysine side-chain.
FIGURE 2.

Effect of CLR01 binding on Trp-94 fluorescence and CLR01 fluorescence. A, fluorescence spectra of 1 μm α-synuclein in the presence of increasing concentrations of CLR01. B, total fluorescence between 300–450 nm versus [CLR01]. The line is the fit of the first seven points to a hyperbola. C, first (black) and second (red) most significant spectra from single value decomposition of all spectra. D, corresponding amplitudes of each spectrum versus [CLR01]. The black lines are sigmoidal fits to sections of the data.
The spectral shift of Trp-94 upon binding of CLR01 to α-synuclein indicates that the Trp becomes slightly more shielded from the surrounding solvent, and the overall increase in intensity also indicates that the side chain is sequestered from solvent molecules and other side chains that might quench it. This suggests that Trp-94 is sequestered in a small hydrophobic pocket. Note that in a previous study, binding of α-synuclein to curcumin caused a decrease in Trp-94 fluorescence, so it is likely that the binding site of curcumin is different than that of CLR01 (8). Importantly, although the Trp is less solvent-exposed, this does not mean the entire chain is more compact, as shown below.
To elucidate the predominant binding site of CLR01 to α-synuclein, we used native electrospray-ionization mass-spectrometry (ESI-MS); a technique that can measure non-covalent ligand binding to large macromolecules (14). Previously, this approach was used to study relatively weak solution binding of small molecule ligands to α-synuclein (15). By using neutral pH solutions and relatively gentle conditions in the mass spectrometer, weak non-covalent interactions can be preserved for their measurement by ESI-MS. Under these conditions, the sites of ligand binding can be deciphered by tandem mass-spectrometry (MS/MS) or “top-down” MS of the protein-ligand complex. Using ECD, covalent backbone bonds of the polypeptide are cleaved while non-covalent forces holding the ligand bound to the macromolecule are maintained. ECD-MS/MS of protein:ligand complexes yields fragments or product ions of the protein chain, some of which are still bound to the ligand. The sequence of these ligand-bound fragments can be mapped onto the full-length protein sequence to determine the ligand binding region(s) (15–17). This strategy was used previously to measure the binding of CLR01 to amyloid β-protein (18) and therefore we applied ESI-MS/MS with ECD here to determine the sites of CLR01 binding to α-synuclein.
Using an equimolar, 10-μm solution of α-synuclein and CLR01, ESI-MS showed a stable 1:1 protein-ligand complex (Fig. 3A). Approximately 20% of α-synuclein was complexed with CLR01 in this solution. The 13+-charged ions for the 1:1 α-synuclein-CLR01 complex were subjected to ECD-MS/MS fragmentation. Protein fragments of the c- (retaining the N terminus) and z• - (retaining the C terminus) product-ion series were observed. Some product ions from dissociation of the polypeptide backbone corresponded to unbound protein (Fig. 3B, black lines), whereas other product ions retained binding to CLR01 (Fig. 3B, red lines). At some positions along the polypeptide chain, product ions were observed in both CLR01-bound and unbound states. Compared with our previous study with amyloid β-protein (Aβ) (18), ECD appears to induce more dissociation of CLR01 from α-synuclein. Nonetheless, the specific observation of a CLR01-bound c20-product ion (a 20-residue fragment from the N terminus) and a CLR01-bound z•131-product ion (a 131-residue fragment from the C terminus) suggests that the binding is located in the α-synuclein region spanning residues 10–20, most likely at Lys-10 and/or Lys-12.
FIGURE 3.

Binding sites of CLR01 on α-synuclein investigated by ECD-MS/MS. A, ESI mass spectrum of 10-μm α-synuclein and 10-μm CLR01 shows 1:1 binding stoichiometry (singly charged unbound CLR01 was observed at m/z 744). B, schematic of the ECD-MS/MS fragmentation profile for the 13+-charged 1:1 α-synuclein:CLR01 complex. Protein fragments of the c- (retaining the N terminus) and z*- (retaining the C terminus) product-ion series were observed. Some product ions from dissociation of the polypeptide backbone corresponded to unbound peptide (black line), whereas other product ions retained binding to CLR01 (red line). At some positions along the peptide chain, product ions were observed in both CLR01-bound and unbound states. From the fragmentation profile, residues 10–20 that include Lys-10 and Lys-12 are suggested to be the site(s) of CLR01 binding.
α-Synuclein Is Primarily Monomeric with and without CLR01
To test experimentally whether under the conditions used for the Trp-Cys quenching experiments α-synuclein was monomeric or consisted of higher-order oligomers, we applied photo-induced cross-linking of unmodified proteins (PICUP) (11, 19). This method uses visible light to produce Ru3+, a strong one-electron oxidizer that can then abstract an electron from a susceptible side chain in a protein, producing a radical, which in turn can covalently bond to another side chain on another protein. Fig. 4a shows that at room temperature, PICUP produces a range of oligomers (using the WT sequence of α-synuclein) with higher oligomer orders more abundant at 30 μm, than at 3 μm α-synuclein. Importantly, however, observation of cross-linked oligomers in PICUP experiments does not necessarily mean that these oligomers existed before the cross-linking. Distinguishing pre-existing oligomers from those generated by random collisions of monomers during the cross-linking reaction requires analysis of the entire population and comparison to theoretical models (20). Densitometric analysis of the observed distributions (Fig. 4, d and e) shows that they are consistent with those of monomeric peptides and proteins and suggests that at the concentrations used here, the observed cross-linked oligomers are a result of stochastic collisions among monomers, rather than pre-formed oligomer (20). Thus, the data strongly support the conclusion that under the conditions used here, α-synuclein was predominantly monomeric. The higher collision rate at 30 μm α-synuclein led to cross-linking of ∼60% of the protein, whereas at 3 μm only ∼25% of the protein formed intermolecular cross-links. Interestingly, oligomers higher than a dimer appeared to comprise multiple species, which in the case of the putative trimer and tetramer, were resolved into 3 distinct bands (Fig. 4a). In the case of the trimer, the ratio of these bands was ∼7:2:1 (Fig. 4b), whereas for the tetramer, the ratio was ∼3.5:2.5:4 (Fig. 4c). The addition of CLR01 did not significantly shift the observed oligomer distribution or the ratio of the bands comprising the trimer and tetramer.
FIGURE 4.

PICUP analysis of α-synuclein oligomerization. PICUP was performed on α-synuclein in the absence or presence of 1 or 10 molar equivalents of CLR01. a, following cross-linking, the products were fractionated by SDS-PAGE and visualized by silver staining. Positions of molecular weight markers are shown on the left. The gel is representative of each of three independent experiments done in triplicates. b, percent abundance of the three bands of the trimer and c, tetramer products for 30 μm protein. d and e, measured abundances of N-mers after PICUP for various ratios of CLR01:α-synuclein. The protein concentrations are (d) 3 μm and (e) 30 μm.
CLR01 Alters α-Synuclein Oligomerization
In addition to reporting on the interaction with inhibitors, the Trp-94 fluorescence also provides a unique probe for studying the early stages of α-synuclein oligomerization. Fig. 5a shows the fluorescence spectrum of Trp-94 shifting slowly over the first 6 h of incubation at 50 μm. The red shift suggests that Trp is becoming more solvent exposed in oligomers than in the monomer. After 6 h, the solution became turbid, indicating formation of large aggregates. When incubated under the same conditions in the presence of 1 equivalent CLR01 (Fig. 5b), a distinctly different behavior was observed. As expected from the binding studies, the spectrum showed a blue shift relative to α-synuclein alone and no red shift developed over time. The overall intensity increased with time, suggesting that CLR01 bound to the oligomers as well as to the monomer. The lack of spectral shift suggested that CLR01 changed the oligomer structure such that solvent exposure of Trp did not increase during oligomerization. However, the rate of increase in Trp fluorescence decreased at 2:1 CLR01:α-synuclein concentration ratio, suggesting that the inhibitor also slowed down the initial oligomerization step. The observed increase in α-synuclein fluorescence intensity over time when bound to CLR01 is consistent with earlier work on CLR01. Using native- and SDS-PAGE, Prabhudesai et al. showed evidence that to a small extent, CLR01 promotes the formation and/or stabilization of high molecular weight oligomers (21) following incubation for 1–3 days.
FIGURE 5.
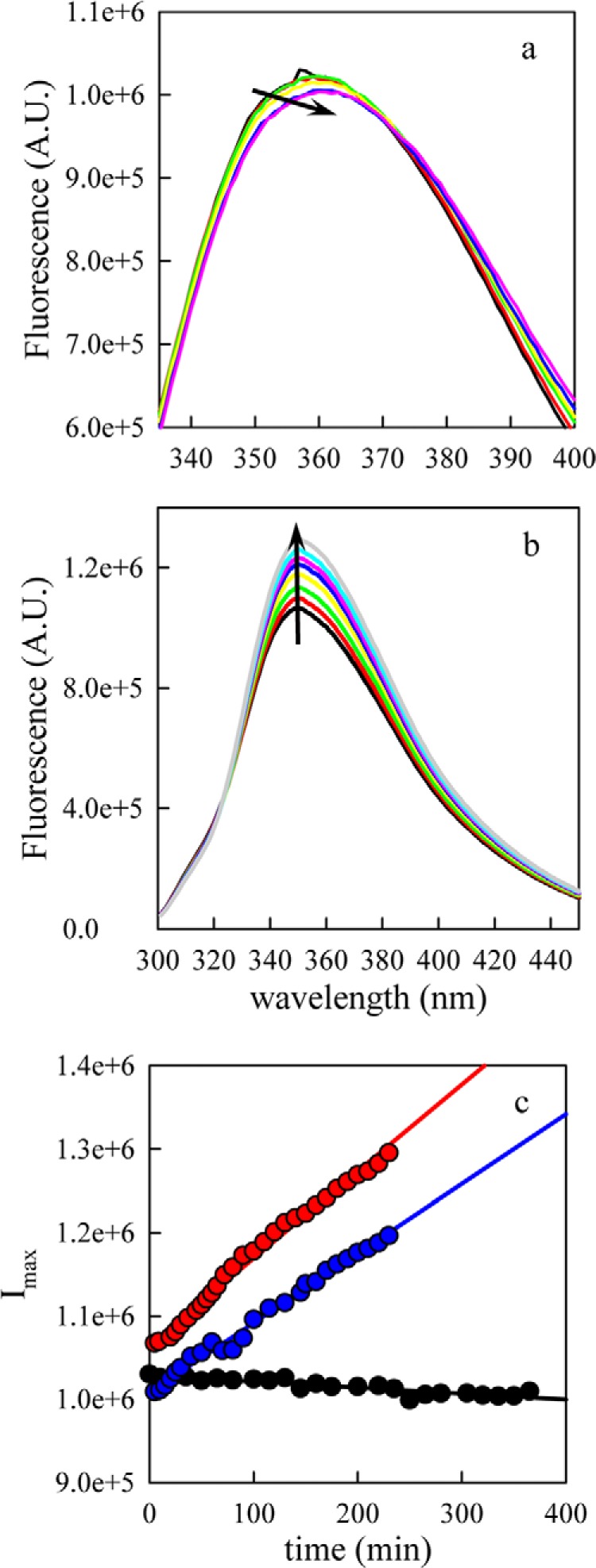
Measurement of α-synuclein oligomerization by Trp fluorescence. a, 45 μm [Cys69,Trp94] α-synuclein were incubated at 37 °C with stirring in a) the absence, or b) the presence of equimolar CLR01 and Trp fluorescence was measured at various times points. The arrows indicate increasing time. c, maximum intensity versus time for various molar ratios of CLR01 (0:1, black points; 1:1, red points; 2:1, blue points). The lines are linear fits to the data.
CLR01 Binding Increases α-Synuclein Intramolecular Diffusion
To measure intramolecular diffusion, we excited the Trp-94 to a long-lived triplet state, which is quenched upon close contact with Cys-69, 400-times more efficiently than with any other amino acid. The observed rate of triplet decay consists of two processes, intramolecular diffusion and irreversible quenching of the triplet by Cys on close contact. If stable oligomers are present in the measured sample, intramolecular diffusion within the oligomers would be slow and therefore, two decays would be observed; one for monomers and the other for oligomers, which likely would be close to the natural lifetime of the Trp triplet, 40 μs. Fig. 6a shows that only one decay was observed in the α-synuclein sample and that the lifetime is substantially shorter than 40 μs, suggesting that oligomers were not present to a significant extent in this sample. In equilibrium, intramolecular diffusion brings the Trp and Cys within the same polypeptide together with a diffusion-limited forward rate kD+, where it may be quenched with rate q or diffuse away with rate kD−. The observed rate is given by Equation 1 (22).
 |
If q ≫ kD− then kobs≈kD+ and the observed rate is diffusion-limited. However Cys is not a diffusion-limited quencher of free Trp in water so q≈kD−, and Equation 1 can be rewritten as Equation 2.
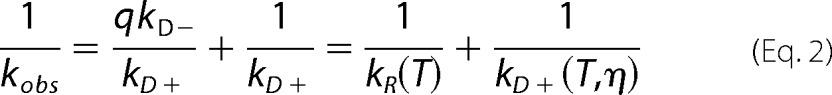 |
We assume that the reaction-limited rate kR depends only on the temperature (T), whereas kD+ depends on both the temperature and the viscosity of the solvent (η). Therefore, by making measurements at different viscosities for a constant temperature we can extract both kR and kD+ by fitting a plot of 1/kobs versus η at a given temperature to a line in which the intercept is 1/kR and the slope is 1/ηkD+. Fig. 6b shows a typical data set for one CLR01 concentration. As the temperature increases, the intercept decreases significantly but the slope increases, indicating that kR increases with temperature whereas kD+ decreases. Comparing the rates for 0:1 and 2:1 CLR01:protein we find that the intercepts are substantially higher, and the slopes are lower for each temperature, indicating the CLR01 lowers the reaction-limited rate and raises the diffusion-limited rate. These rates are plotted in Fig. 6, c and d for each temperature and CLR01 concentration.
FIGURE 6.
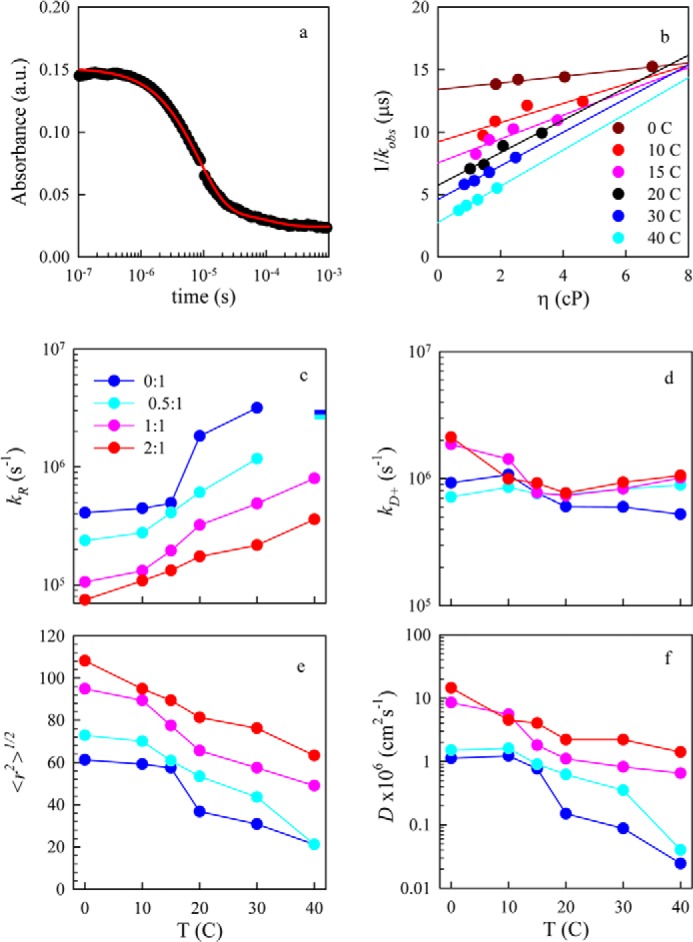
Measurement of Trp-94-triplet quenching. a, typical kinetic decay of the Trp triplet-state as measured by transient optical absorption. The data (black points) are fit to two exponential decays. The slower decay corresponds to various photophysical processes created by the UV pulse (e.g. solvated electrons), whereas the faster decay corresponds to intramolecular contact between Trp-94 and Cys-69. b, observed decay times versus solution viscosity for various temperatures. The lines are independent fits for each temperature. Reaction-limited (c) and diffusion-limited (d) rates of the Trp-Cys quenching measurement versus temperature for various molar ratios of CLR01:α-synuclein. The bars in c represent the lower limit of kR as determined by the inverse error of the intercept from Equation 2. e, average root mean square distance between Trp-94 and Cys-69 determined for each reaction-limited rate using Equations 3 and 5. f, intramolecular diffusion coefficients determined for each diffusion-limited rate using Equation 4.
To interpret these rates we use a theory by Szabo, Schulten and Schulten (SSS), which models intramolecular diffusion as diffusion on a one-dimensional potential of mean force determined by the probability of intrachain distances (P(r)) (23). The measured reaction-limited and diffusion-limited rates are given by Equations 3 and 4 (24),
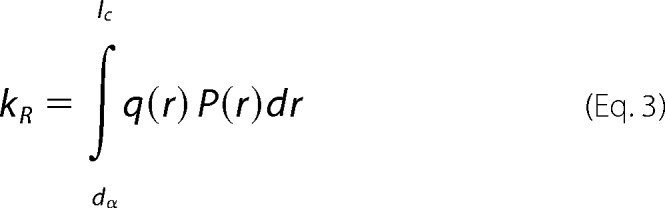 |
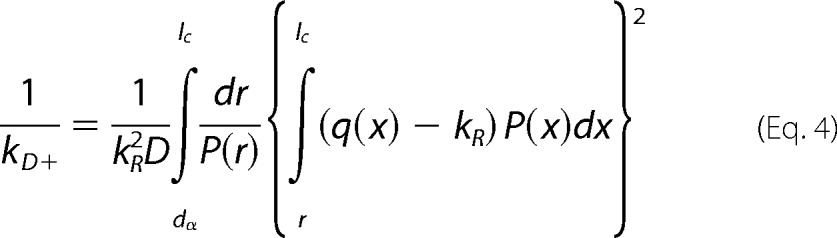 |
where r is the distance between the Trp and Cys, D is the effective intramolecular diffusion constant and q(r) is the distance-dependent quenching rate. The limits of integration are the distance of closest approach, dα = 4.0 Å and the contour length of the Trp-Cys loop, lc. The distance-dependent quenching rate for the Trp-Cys system drops off very rapidly beyond 4.0 Å, so the reaction-limited rate is mostly determined by the probability of the shortest distances (25). Very generally, kR and kD+ are both inversely proportional to the average volume of the chain and kD+ is directly proportional to D. To determine the diffusion constant we assume the probability distribution, P(r) is given by a Gaussian in Equation 5,
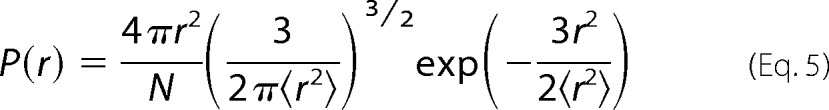 |
where <r2>, the average Trp-Cys distance, is an adjustable parameter, and N is a normalization constant such that ∫P(r)=1. For each measured kR, <r2> was found such that it matched the measured rate using Equation 3. The average r.m.s. distance is plotted in Fig. 5e. The Gaussian distribution likely is not a good estimate of real intrachain distances, but is a useful way of comparing changes in chain volume as a function of temperature and CLR01 concentration. Using the correct P(r) and the measured kD+, the effective intramolecular diffusion coefficient, D, is found from Equation 4 and is plotted in Fig. 6f.
Fig. 6, e and f, show that CLR01 makes the α-synuclein chain less compact and more diffusive at physiological temperatures. At 30 °C, the diffusion coefficient increases by 20-fold at 2:1 molar ratio (60 μm CLR01) relative to α-synuclein alone, and the average distance between the Trp and Cys increases by 2.5-fold. These changes are comparable to those observed upon addition of curcumin to α-synuclein (8).
Modeling How Increasing Reconfiguration Decreases Aggregation
To understand the conformational and dynamic changes in the context of aggregation, we employ a model that relates the rate of bimolecular association between two monomers to the rate of monomeric reconfiguration. We estimate the reconfiguration rate as the rate at which one part of the unstructured chain diffuses across the cross section of the chain volume, kr = 4D/(2RG)2. Because there are no estimates for the radius of gyration of α-synuclein in the presence of CLR01, we use the average mean squared Trp-Cys distance <r2> as an approximation, assuming that rates determined using these values are correct within ∼2×. The reconfiguration rates determined from kr = D/<r2> are plotted in Fig. 7a for 30 °C. This rate increases linearly with CLR01 concentration. Using these reconfiguration rates we solve the model shown in Reaction 1,
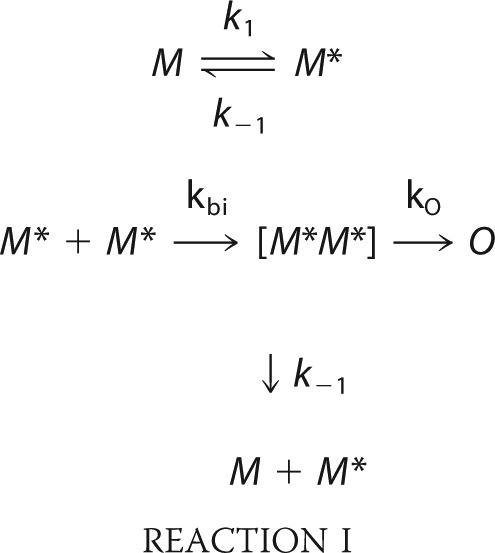 |
where M and M* are monomeric protein chains and M* is a form of the monomer capable of forming bimolecular contacts, [M*M*] is a transient bimolecular complex in which each chain is still highly diffusive, and O is a covalently cross-linked dimer that has extremely slow intramolecular diffusion. We assume k1 = kr = k-1 because the process has no significant free energy barrier, so the rate of adopting any particular conformation is just the rate of reconfiguration. kbi = 7 × 104 s−1 (the bimolecular diffusion rate for 30 μm protein) and ko = 100 s−1; an arbitrary rate that is slower than bimolecular association and fast enough to solve the model with reasonable computing time. Solving the model for the various reconfiguration rates, we determine that the second-order rate of oligomerization decreases hyperbolically with the CLR01:α-synuclein molar ratio (Fig. 7b). Clearly, the aggregation pathway is more complex than Reaction 1. For example, higher order oligomers could form from the aggregation of two or more O or they could form by the addition of diffusive monomers (M*). However, this is the simplest model that accounts for the correlation between reconfiguration and aggregation rates. Furthermore, real rates of reconfiguration and escape could be different, but these estimates serve to illustrate how aggregation can decrease dramatically with increasing reconfiguration rate.
FIGURE 7.
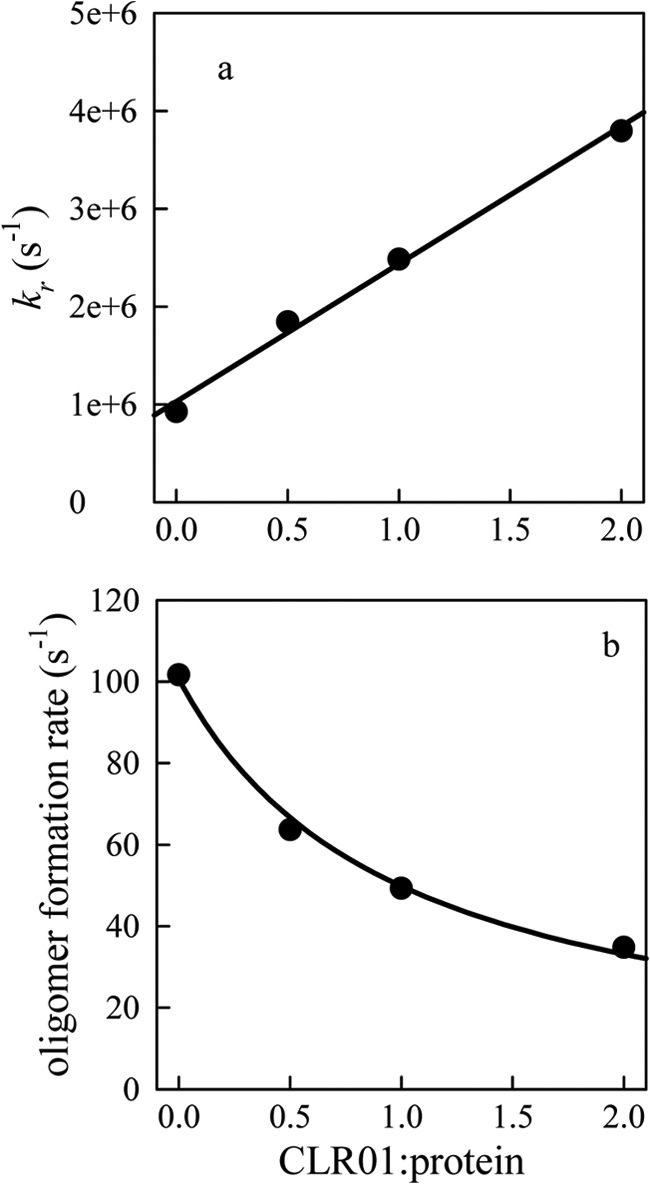
Effect of CLR01 on α-synuclein oligomerization. a, reconfiguration rates at 30 °C versus molar ratio of CLR01:α-synuclein calculated as kr = D/<r2>. The black line is a linear fit to the data. b, second-order formation rates of oligomer (O) calculated from the model in Reaction Scheme 1 using rates as noted in the text. The black line is a fit to a hyperbola.
DISCUSSION
Several neurodegenerative disorders, such as Parkinson's disease, multiple system atrophy, and dementia with Lewy bodies, are associated with the aggregation of α-synuclein. Aggregation is a multi-step process beginning with low order oligomers, proceeding through higher order oligomers and then transitioning to fibrillar structures. It is often postulated that the protein is misfolded before aggregation begins, but most biophysical studies find α-synuclein unstructured prior to aggregation and that the unstructured ensemble is highly diverse and freely diffusing among multiple conformations. Thus, with the exception of recent reports suggesting that α-synuclein may exist, at least partially, as a tetramer comprising extended α-helices (1, 2, 26), a view that has not yet reached consensus (27), the search for a well-defined structure has not been fruitful. We postulate that the physical basis for the first step of aggregation is not a set of one or more well-defined structures but the rate at which the monomer chain samples various conformations, some of which may be capable of forming a dimer, probably because of solvent exposure of hydrophobic residues on both chains. However if reconfiguration is rapid, these conformations are not maintained long enough to make sufficient stabilizing intermolecular interactions to form a dimer and the complex falls apart. Any alteration of the reconfiguration rate, e.g. by temperature or pH, can alter the aggregation rate. Thus, a small molecule that can bind to the chain and change the reconfiguration rate may be of therapeutic benefit in neurodegeneration.
Previous work using the molecular tweezer CLR01 showed that it selectively binds to Lys side chains (18, 28, 29), of which there are 15 in α-synuclein. Prabhudesai et al. used thioflavin T fluorescence and electron microscopy to show that in the presence of CLR01, α-synuclein did not form amyloid fibrils, and demonstrated that CLR01 inhibited α-synuclein-mediated toxicity in cell cultures and zebrafish embryos (21). In agreement with those findings, the data presented here suggest that to a large extent, CLR01 keeps α-synuclein monomeric by increasing its reconfiguration rate. However, the Trp fluorescence changes during early oligomerization also show that CLR01 interacts with small oligomers, altering the solvent exposure of Trp and possibly the oligomerization pathway. It is also possible that interaction of CLR01 with oligomers at a later assembly stage than studied here contributes to the inhibition of fibril formation and α-synuclein toxicity.
A still unanswered question is exactly how CLR01, when bound to one or two Lys in α-synuclein, increases the reconfiguration rate. Upon CLR01 binding, an increase in the chain volume is observed, suggesting that Lys-bound CLR01 interrupts intramolecular interactions that compact the α-synuclein chain. While bound, CLR01 changes the charge on Lys from +1 to −1 because each CLR01 molecule has two charged phosphate groups, so the addition of each CLR01 further decreases the net charge of the protein, which under physiologic conditions is -9.1. Pappu et al. have proposed that there is a phase transition from a disordered globule to a swollen coil as the fraction of positively or negatively charged residues increases while mean hydropathy remains constant (30) and that the pattern of charges in the sequence affects the size of the chain significantly (31). The charged Lys residues located throughout the first 100 residues in the sequence of α-synuclein likely preferentially associate with the multiple negatively charged residues, mostly located in the C terminus, thus creating many close associations between distant residues in the α-synuclein sequence. With CLR01 bound, Lys may prefer association with positively charged Lys or Arg residues, ostensibly creating close associations among residues located closer to each other in the sequence. Presumably, these local associations lead to conformations that are less compact and more diffusive as there are fewer long-range associations to break.
Considering intramolecular interactions in more detail, Chen et al. have proposed a model of unstructured proteins in which the preferred conformations are those in which non-contiguous residues of similar hydropathy are in close proximity (32). Because charged residues generally are hydrophilic, residues of both charges are preferentially associated under this model. Here, we expand the model of Chen et al. to also specifically include the effect of the sign of the charge in the sequence. The model of Chen et al. uses as a starting point a large number (10,000,000) of wormlike-chains created by a Monte Carlo method, setting the persistence length to 4 Å. These chains are created with an excluded diameter for each link on the chain of 4 Å. These parameters have been shown to model unstructured polypeptides well (10). From these chains, we can create a normalized probability distribution, P(r), for the distance, r, between Cys-69 and Trp-94.
To create compact ensembles, we re-weight the probability distribution to favor conformations in which residues of similar hydropathy are near each other. We assign hydropathy, h, for each residue in the sequence using the Miyazawa-Jernigan scale and assume that two residues with h values within 30% of each other contribute to hydrophobic or hydrophilic interactions, reducing the free energy of the system and therefore making conformations containing such interactions favorable.
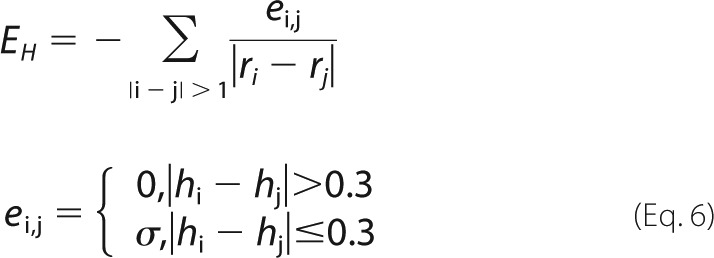 |
We apply a distance cut-off to EH such that EH = 0 for ri − rj > 6.5 Å. To account for charged residues, we find the charge, q, on each residue at pH 7.5 using the Henderson-Hasselbach equation and calculate the Coulombic energy in Equation 7.
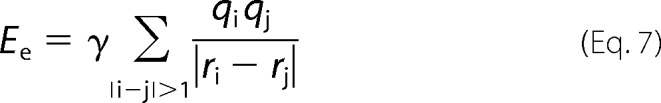 |
γ and σ are adjustable parameters for the Coulombic and hydrophobic interactions, respectively.
Using the total interaction energy ETOT = EH + Ee, we can re-weight the probability distribution P(r) to yield a new distribution Z(r).
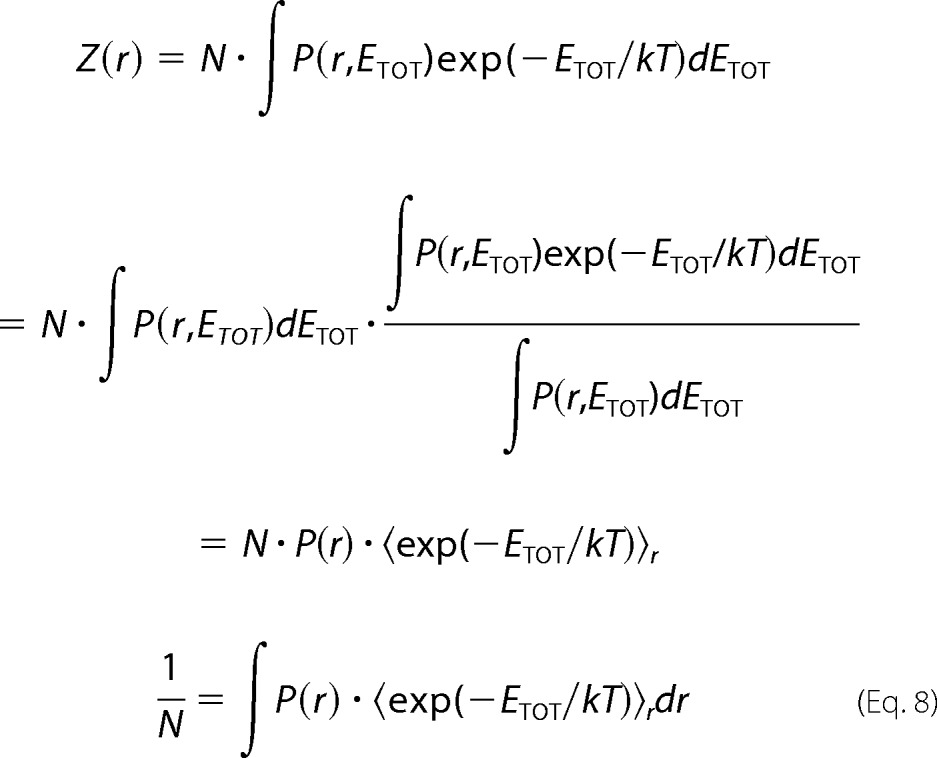 |
The reaction-limited rate then is calculated from the new distribution as Equation 9.
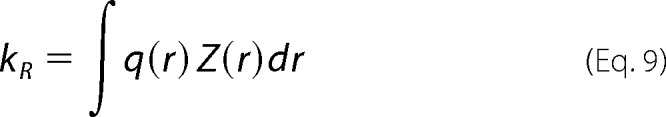 |
The tuning parameters, γ and σ, can be adjusted to make the calculated kR agree with the measured reaction-limited rate at any particular temperature.
To account for the binding of CLR01 to a Lys, we assume that the hydropathy is unchanged upon binding but that the charge reverses from ∼+1 to ∼−1. Based on the ECD-MS data (Fig. 3) the most likely residues to bind CLR01 are Lys-10 and Lys-12. Therefore, we reverse the charge on these residues and calculate Ee to compare with unbound α-synuclein. Fig. 8 shows the re-weighted probability distributions for α-synuclein alone or with CLR01 bound to Lys-10 and Lys-12. As each CLR01 molecule binds to one or both residues, the distribution shifts to larger r. Thus, it is clear that simply reversing the charge on lysine side chains near the N terminus is sufficient to expand the chain. The effect on the entire chain can be seen in contact maps presented in supplemental Fig. S3, which plot the average intramolecular distances under various conditions. The un-weighted chain contact map (supplemental Fig. S3a) appears to be completely featureless, with residues far apart in sequence farther apart in space than those close in sequence. Compared with the un-weighted chain, the weighted sequence (supplemental Fig. S3b) has significantly closer contacts between the N-terminal and C-terminal regions (residues 1–40 and 120–140). These close contacts are inhibited upon binding of the first CLR01 molecule (supplemental Fig. S3c) and decrease further upon binding of a second CLR01 (supplemental Fig. S3d).
FIGURE 8.
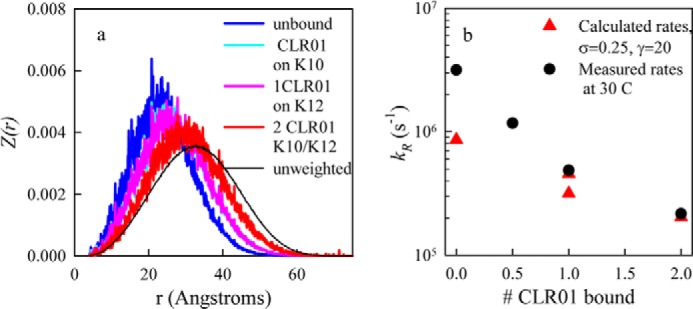
Calculated effect of CLR01 binding on α-synuclein conformation. a, re-weighted probability distributions, Z(r), for σ = 0, γ = 20 for α-synuclein alone (blue), α-synuclein with one CLR01 bound at Lys-10 (cyan), α-synuclein with one CLR01 bound at Lys-12 (magenta), and α-synuclein bound to two CLR01 molecules in positions 10 and 12 (red). In each case, the bound CLR01 are approximated by Lys→Glu substitutions. b, calculated (red triangles) reaction-limited rates using Equation 9 and the probability distributions calculated in a. The measured reaction-limited rates are plotted as black circles.
To achieve quantitative agreement between the model and experiment, the tuning parameters, σ and γ, were adjusted such that the reaction-limited rate determined by Equation 9 agreed with experimental values. First, γ was determined by adjusting the ratio of the calculated reaction-limited rates for 1 and 2 CLR01 bound to match the measured ratio. Then σ was adjusted to match the absolute rates for those sequences. The best fit for these parameters is γ = 20 and σ = 0.25, and the rates are plotted in Fig. 8b.
The model suggests that α-synuclein behaves as a random chain restrained by both repulsive and attractive interactions (excluded volume repulsion, hydrophobic attraction, and charge repulsion and attraction). Disruption of this balance of interactions, in this case by reversing the charge on certain residues, substantially changes the ensemble properties and therefore the dynamics. A more expanded chain is more diffusive and therefore more likely to avoid bimolecular association and subsequent oligomerization. The work presented here shows how CLR01 binds to α-synuclein and prevents its aggregation by perturbing key hydrophobic and electrostatic interactions, resulting in an increased reconfiguration rate of the protein. The data provide important insights into the mechanism of aggregation of α-synuclein and suggests that compounds that increase the reconfiguration rate may lead to efficient therapeutic drugs for Parkinson's disease and other synucleinopathies.
The work was supported in part by National Institutes of Health Grants R01 GM103479 (to J. A. L.); NIH High-End Shared Instrumentation Program S10 RR028893 (to J. A. L.); NIH R01 GM100908 (to L. J. L.); University of California-Los Angeles Jim Easton Consortium for Alzheimer's Drug Discovery and Biomarker Development (to G. B.); RJG Foundation Grant 20095024 (to G. B.); Team Parkinson/Parkinson Alliance Grant (to G. B.); NSF MCB-0825001 (to L. J. L.); and Development and Promotion of Science and Technology Talents Project (DPST), Royal Thai Government (to P. W.).

This article contains supplemental Figs. S1–S3.
- PD
- Parkinson disease
- TCEP
- tris(2-carboxyethyl)phosphine
- ESI
- electrospray ionization
- FT-ICR
- Fourier transform ion-cyclotron resonance
- ECD
- electron-capture dissociation
- ESI-MS
- electrospray-ionization mass-spectrometry.
REFERENCES
- 1. Bartels T., Choi J. G., Selkoe D. J. (2011) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dettmer U., Newman A. J., Luth E. S., Bartels T., Selkoe D. (2013) In Vivo Cross-linking Reveals Principally Oligomeric Forms of α-Synuclein and β-Synuclein in Neurons and Non-neural Cells. J. Biol. Chem. 288, 6371–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertoncini C. W., Jung Y. S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M., Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U. S. A. 102, 1430–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cho M.-K., Nodet G., Kim H.-Y., Jensen M. R., Bernado P., Fernandez C. O., Becker S., Blackledge M., Zweckstetter M. (2009) Structural characterization of α-synuclein in an aggregation prone state. Protein Sci. 18, 1840–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trexler A. J., Rhoades E. (2010) Single Molecule Characterization of α-Synuclein in Aggregation-Prone States. Biophys. J. 99, 3048–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uversky V. N., Li J., Fink A. L. (2001) Evidence for a Partially Folded Intermediate in α-Synuclein Fibril Formation. J. Biol. Chem. 276, 10737–10744 [DOI] [PubMed] [Google Scholar]
- 7. Ahmad B., Chen Y., Lapidus L. J. (2012) Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc. Natl. Acad. Sci. U. S. A. 109, 2336–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahmad B., Lapidus L. J. (2012) Curcumin Prevents Aggregation in α-Synuclein by Increasing Reconfiguration Rate. J. Biol. Chem. 287, 9193–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masuda M., Dohmae N., Nonaka T., Oikawa T., Hisanaga S., Goedert M., Hasegawa M. (2006) Cysteine misincorporation in bacterially expressed human α-synuclein. FEBS Lett. 580, 1775–1779 [DOI] [PubMed] [Google Scholar]
- 10. Singh V. R., Kopka M., Chen Y., Wedemeyer W. J., Lapidus L. J. (2007) Dynamic Similarity of the Unfolded States of Proteins L and G. Biochemistry 46, 10046–10054 [DOI] [PubMed] [Google Scholar]
- 11. Bitan G. (2006) Structural study of metastable amyloidogenic protein oligomers by photo-induced cross-linking of unmodified proteins. Methods Enzymol. 413, 217–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopes D. H., Sinha S., Rosensweig C., Bitan G. (2012) Application of photochemical cross-linking to the study of oligomerization of amyloidogenic proteins. Methods Mol. Biol. 849, 11–21 [DOI] [PubMed] [Google Scholar]
- 13. Dutt S., Wilch C., Gersthagen T., Talbiersky P., Bravo-Rodriguez K., Hanni M., Sánchez-García E., Ochsenfeld C., Klärner F. G., Schrader T. (2013) Molecular tweezers with varying anions: a comparative study. J. Org. Chem. 78, 6721–6734 [DOI] [PubMed] [Google Scholar]
- 14. Loo J. A. (1997) Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 16, 1–23 [DOI] [PubMed] [Google Scholar]
- 15. Xie Y., Zhang J., Yin S., Loo J. A. (2006) Top-down ESI-ECD-FT-ICR mass spectrometry localizes noncovalent protein-ligand binding sites. J. Am. Chem. Soc. 128, 14432–14433 [DOI] [PubMed] [Google Scholar]
- 16. Yin S., Loo J. A. (2010) Elucidating the site of protein-ATP binding by top-down mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 899–907 [DOI] [PubMed] [Google Scholar]
- 17. Yin S., Loo J. A. (2011) Top-Down Mass Spectrometry of Supercharged Native Protein-Ligand Complexes. Int. J. Mass Spectrom 300, 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sinha S., Lopes D. H., Du Z., Pang E. S., Shanmugam A., Lomakin A., Talbiersky P., Tennstaedt A., McDaniel K., Bakshi R., Kuo P. Y., Ehrmann M., Benedek G. B., Loo J. A., Klärner F. G., Schrader T., Wang C., Bitan G. (2011) Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 133, 16958–16969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bitan G., Teplow D. B. (2004) Rapid photochemical cross-linking—a new tool for studies of metastable, amyloidogenic protein assemblies. Acc. Chem. Res. 37, 357–364 [DOI] [PubMed] [Google Scholar]
- 20. Bitan G., Lomakin A., Teplow D. B. (2001) Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 276, 35176–35184 [DOI] [PubMed] [Google Scholar]
- 21. Prabhudesai S., Sinha S., Attar A., Kotagiri A., Fitzmaurice A. G., Lakshmanan R., Ivanova M. I., Loo J. A., Klärner F. G., Schrader T., Stahl M., Bitan G., Bronstein J. M. (2012) A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 9, 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lapidus L. J., Eaton W. A., Hofrichter J. (2002) Measuring dynamic flexibility of the coil state of a helix-forming peptide. J. Mol. Biol. 319, 19–25 [DOI] [PubMed] [Google Scholar]
- 23. Szabo A., Schulten K., Schulten Z. (1980) 1st Passage Time Approach to Diffusion Controlled Reactions. J. Chem. Phys. 72, 4350–4357 [Google Scholar]
- 24. Lapidus L. J., Steinbach P. J., Eaton W. A., Szabo A., Hofrichter J. (2002) Effects of chain stiffness on the dynamics of loop formation in polypeptides. Appendix: Testing a 1-dimensional diffusion model for peptide dynamics. J. Phys. Chem. 106, 11628–11640 [Google Scholar]
- 25. Lapidus L. J., Eaton W. A., Hofrichter J. (2001) Dynamics of intramolecular contact formation in polypeptides: Distance dependence of quenching rates in a room-temperature glass. Phys. Rev. Lett. 87, 4. [DOI] [PubMed] [Google Scholar]
- 26. Wang W., Perovic I., Chittuluru J., Kaganovich A., Nguyen L. T., Liao J., Auclair J. R., Johnson D., Landeru A., Simorellis A. K., Ju S., Cookson M. R., Asturias F. J., Agar J. N., Webb B. N., Kang C., Ringe D., Petsko G. A., Pochapsky T. C., Hoang Q. Q. (2011) A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 108, 17797–17802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fauvet B., Mbefo M. K., Fares M. B., Desobry C., Michael S., Ardah M. T., Tsika E., Coune P., Prudent M., Lion N., Eliezer D., Moore D. J., Schneider B., Aebischer P., El-Agnaf O. M., Masliah E., Lashuel H. A. (2012) α-Synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J. Biol. Chem. 287, 15345–15364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sinha S., Du Z., Maiti P., Klärner F. G., Schrader T., Wang C., Bitan G. (2012) Comparison of three amyloid assembly inhibitors: the sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem. Neurosci. 3, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bier D., Rose R., Bravo-Rodriguez K., Bartel M., Ramirez-Anguita J. M., Dutt S., Wilch C., Klärner F. G., Sanchez-Garcia E., Schrader T., Ottmann C. (2013) Molecular tweezers modulate 14–3-3 protein-protein interactions. Nat. Chem. 5, 234–239 [DOI] [PubMed] [Google Scholar]
- 30. Mao A. H., Crick S. L., Vitalis A., Chicoine C. L., Pappu R. V. (2010) Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A. 107, 8183–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Das R. K., Pappu R. V. (2013) Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U. S. A. 110, 13392–13397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y., Wedemeyer W. J., Lapidus L. J. (2010) A General Polymer Model of Unfolded Proteins under Folding Conditions. J. Phys. Chem. 114, 15969–15975 [DOI] [PubMed] [Google Scholar]