

Background: Coordinated synaptic and nuclear signaling is required for long lasting changes in neuronal morphology.
Results: Afadin undergoes activity-dependent bi-directional shuttling to synapses and the nucleus resulting in dendritic spine remodeling and histone modifications.
Conclusion: Afadin is required for coordinated signaling at synapses and the nucleus.
Significance: Bi-directional trafficking of afadin is required for coordinated synaptic and nuclear signaling in response to activity-dependent stimulation.
Keywords: Cell Biology, Chromatin Histone Modification, Neurobiology, Nucleus, Signaling, Synapses, Synaptic Plasticity, AF-6, Dendritic Spine, Small GTPase
Abstract
The ability of a neuron to transduce extracellular signals into long lasting changes in neuronal morphology is central to its normal function. Increasing evidence shows that coordinated regulation of synaptic and nuclear signaling in response to NMDA receptor activation is crucial for long term memory, synaptic tagging, and epigenetic signaling. Although mechanisms have been proposed for synapse-to-nuclear communication, it is unclear how signaling is coordinated at both subcompartments. Here, we show that activation of NMDA receptors induces the bi-directional and concomitant shuttling of the scaffold protein afadin from the cytosol to the nucleus and synapses. Activity-dependent afadin nuclear translocation peaked 2 h post-stimulation, was independent of protein synthesis, and occurred concurrently with dendritic spine remodeling. Moreover, activity-dependent afadin nuclear translocation coincides with phosphorylation of histone H3 at serine 10 (H3S10p), a marker of epigenetic modification. Critically, blocking afadin nuclear accumulation attenuated activity-dependent dendritic spine remodeling and H3 phosphorylation. Collectively, these data support a novel model of neuronal nuclear signaling whereby dual-residency proteins undergo activity-dependent bi-directional shuttling from the cytosol to synapses and the nucleus, coordinately regulating dendritic spine remodeling and histone modifications.
Introduction
The ability of a neuron to transduce activity-dependent signals into long lasting changes in neuronal morphology is central to its normal function. Although changes in neuronal morphology can occur via local signaling at synapses, regulation of gene transcription is required to make these alterations into long lasting effects (1). Two main mechanisms have been proposed to allow signals generated at synapses and along the dendrite to communicate with the nucleus and thus regulate transcription. First, rapid synaptonuclear signaling is thought to depend on the propagation of action potentials and subsequent calcium influx into the nucleus (2). Second, synaptic proteins are thought to shuttle to the nucleus in response to activity-dependent stimuli. Several proteins that display dual synaptic and nuclear localization have been shown to translocate to the nucleus following stimulation (1, 3). However, the spatiotemporal relationships between synaptic, cytosolic, and nuclear populations of these synaptonuclear proteins and the functional consequences of their translocation are not well understood.
It has been proposed that upon nuclear accumulation, these synaptonuclear proteins participate in nuclear events that result in gene expression changes. This includes the post-translational modification of histone protein, the core proteins required for the packaging of tightly coiled chromatin (4). The phosphorylation or acetylation of histones is associated with the initiation of gene transcription (4), and they are thought of as essential transcriptional regulatory mechanisms (5, 6). Increasing evidence suggests that phosphorylation of the histone H3 protein occurs in response to activity-dependent stimuli and is required for cognition (5–8). However, the mechanism(s) by which activity-dependent signaling can result in histone H3 phosphorylation is not well understood.
In cortical neurons, the PDZ domain-containing scaffold protein afadin (also known as AF-6) controls spine morphology downstream of several synaptic membrane proteins (9–11). In hippocampal and non-neuronal cells, afadin has been localized to the nucleus (12, 13) suggesting that it may signal in this subcompartment. Moreover, our previous data have demonstrated that afadin is a mobile protein that changes its subcellular localization in response to various stimuli (9–11). Here, we report that afadin is present at both extranuclear sites and within the nucleus of cortical neurons and that NMDA receptor activation results in a time-dependent accumulation of afadin within the nucleus. This accumulation is independent of gene transcription or protein synthesis. Interestingly, afadin concurrently accumulates at dendritic spines with a simultaneous decrease in its content in dendrites, suggesting a bi-directional trafficking of this protein from the cytosol to both synapses and the nucleus. Moreover, we observed a time-dependent increase in phosphorylation of histone H3 at serine 10 (H3S10p) and its direct upstream target, p90 ribosomal S6 kinase (p90RSK) (5), in cells positive for afadin nuclear accumulation. Remarkably, blocking nuclear accumulation of afadin attenuated activity-dependent spine remodeling and phosphorylation of both histone H3 and p90RSK in a time-specific manner. Collectively, these data support a novel model of neuronal nuclear signaling whereby dual-residency proteins undergo activity-dependent bi-directional and concomitant shuttling from the cytosol to synapses and the nucleus, coordinately regulating dendritic spine remodeling and histone modifications.
MATERIALS AND METHODS
Reagents and Plasmid Constructs
The following antibodies were purchased: GFP mouse monoclonal (MAB3580), NeuN mouse monoclonal (clone A60; MAB377), phospho-histone H3 serine 10 mouse (H3S10p) monoclonal (clone 3H10; 05-806), and Myc rabbit polyclonal (06-549) were from Millipore; GFP chicken polyclonal (ab13972) and histone 3 (total) rabbit polyclonal were from Abcam; Myc mouse monoclonal (9E10; Developmental Studies Hybridoma Bank); l4/s-afadin rabbit polyclonal (AF-6; A0224), l-afadin rabbit polyclonal (A0224), and β-actin mouse monoclonal were from Sigma; phospho-p90RSK (Thr-359/Ser-363) rabbit polyclonal (9344) was from Cell Signaling Technologies; DAPI was from Invitrogen. Plasmids used in this study, Myc-l-afadin, Myc-s-afadin, Myc-afadin-ΔNT, or Myc-afadin-NT, have been previously described (11).
Neuronal Culture and Transfections
Medium and high density cortical neuron cultures were prepared from Sprague-Dawley rat E18 embryos as described previously (14). Briefly, neurons were plated onto coverslips coated with poly-d-lysine (0.2 mg/ml, Sigma) and maintained in feeding media (Neurobasal media supplemented with B27 (Invitrogen) and 0.5 mm glutamine). 200 μm dl-aminophosphonovalerate (Ascent Scientific) was added to the media 4 days later. Cortical neurons were transfected at days in vitro (DIV) 23 using Lipofectamine 2000 following the manufacturer's recommendations (14). Transfections were allowed to continue for 2 days.
Neuronal Treatments
To induce an activity-dependent stimulus, we activated synaptic NMDA receptors on cultured cortical pyramidal neurons by activating NMDA receptors with the co-agonist glycine and acutely unmasking receptors chronically inhibited with aminophosphonovalerate (14, 15). Briefly, cells were preincubated in artificial cerebrospinal fluid (in mm: 125 NaCl, 2.5 KCl, 26.2 NaHCO3, 1 NaH2PO4, 11 glucose, 5 Hepes, 2.5 CaCl2, and 1.25 MgCl2) with 200 μm aminophosphonovalerate for 30 min at 37 °C. Cells were then transferred into treatment medium (artificial cerebrospinal fluid without MgCl2, plus 10 μm glycine, 100 μm picrotoxin, and 1 μm strychnine) for 30 min, before being returned to artificial cerebrospinal fluid. Inhibitors were incubated 30 min prior to treatment with the concentrations indicated in the text. Following treatment(s), cells were processed for immunocytochemistry or biochemistry.
Immunocytohistochemistry
Neurons were fixed in either 4% formaldehyde, 4% sucrose/PBS for 10 min or in 4% formaldehyde, 4% sucrose/PBS followed by a 10-min fix with methanol pre-chilled to −20 °C. Coverslips were then permeabilized and blocked simultaneously in PBS containing 2% normal goat serum and 0.2% Triton X-100 for 1 h at room temperature. Primary antibodies were added in PBS containing 2% normal goat serum for 2 h at room temperature or overnight at 4 °C, followed by three 10-min washes in PBS. Secondary antibodies were incubated for 1 h at room temperature, also in 2% normal goat serum in PBS. Three further washes (15 min each) were performed before coverslips were mounted using ProLong antifade reagent (Invitrogen).
Quantitative Analysis of Nuclear Immunofluorescence
Micrographs were acquired essentially as described previously (14). Confocal images of single- and double-stained neurons were obtained with a Zeiss LSM5 Pascal confocal microscope. Z series images of neurons were taken using the 63× oil immersion objective (N.A. 1.4; Zeiss). The acquisition parameters were kept the same for all scans. Regions were drawn around nuclei, as delineated by NeuN or DAPI staining, and saved as regions of interest. These regions of interest were then applied to a corresponding image of afadin staining from which the mean average intensity was collected to determine nuclear immunoreactivity levels. Images were selected by examining a z stack series of images through the nucleus and choosing a central/representative plane. Orthogonal images were produced from z stack images of using MetaMorph. Immunoreactivity levels of phospho-histone H3 or -p90RSK were analyzed by collecting mean average intensity from regions determined by DAPI staining. One- or two-way ANOVAs were used to compare means between three or more groups, followed by Tukey's B post hoc test for multiple comparisons. Statistical analyses were performed in GraphPad.
Quantitative Analysis of Synaptic and Dendritic Immunofluorescence
Synaptic and dendritic localization of afadin was quantified using MetaMorph (14). Images were acquired as described above. The background corresponding to areas without cells was subtracted to generate a “background-subtracted” image. Images were then thresholded equally to include clusters with intensity at least 2-fold above the adjacent dendrite. Regions along dendrites were outlined using the “Parameters” utility, and the total gray values (immunofluorescence integrated intensity) of each cluster, or all clusters within a region, were measured automatically. Quantification was performed as detailed above.
Quantitative Analysis of Spine Morphologies
Two-dimensional maximum projection reconstructions of images were generated, and morphometric analysis (spine number, area, and breadth) was done using MetaMorph software (Universal Imaging) (14). Cultures that were directly compared were stained simultaneously and imaged with the same acquisition parameters. For each condition, 8–16 neurons each from at least three separate experiments were used, and at least two dendrites from each neuron were analyzed. Experiments were done blind to conditions and on sister cultures. To examine the morphologies of dendritic spines, individual spines on dendrites were manually traced, and spine dimensions were measured by MetaMorph. One- or two-way ANOVAs were used to compare means between three or more groups, followed by Tukey's B post hoc test for multiple comparisons. Statistical analyses were performed in GraphPad.
Biochemistry Cell Fractionation
Subcellular fractions were prepared using the Proteo-Extract kit (EMD Biosciences) following the manufacturer's recommendations. Lysates were subjected to Western blotting; membranes were probed with the appropriate antibodies.
RESULTS
Afadin Localizes to the Nucleus of Cortical Neurons
We have previously reported that afadin has a critical role in regulating synapse structure and function, in response to several stimuli (9–11, 16). Two isoforms of afadin are present within neural tissue l- and s-afadin (Fig. 1A). Afadin is expressed at sites of cell-to-cell contact in a variety of cell types, including pyramidal neurons (16, 17). Interestingly s-afadin but not l-afadin has been reported to localize to the nucleus non-neuronal cells, whereas l-afadin has been reported to localize in the nucleus of hippocampal neurons (12, 13). Therefore, to determine which isoform could be found in the nucleus of cortical neurons, we performed a series of immunocytohistochemical and biochemical studies. Immunostaining of cultured cortical neurons with an antibody that detects both l- and s-afadin revealed punctate staining along dendrites as well as in the nucleus (Fig. 1B). Importantly, l/s-afadin colocalized with the neuronal nuclear marker, NeuN though the X, Y, and Z planes (Fig. 1B). Using an antibody against l-afadin, we found that the longer isoform also displayed a similar distribution with puncta along the dendrite and staining within the nucleus (data not shown). We further confirmed the presence of both l- and s-afadin in the nucleus of cortical neurons by Western blotting of subcellular fractions of cultured neuron homogenates (Fig. 1B).
FIGURE 1.

Afadin nuclear localization is dependent on its N-terminal domain. A, schematic diagram depicting the structure and important domains of l- and s-afadin and truncated constructs. B, confocal image of cortical neuron (DIV 25) immunostained for endogenous l/s-afadin. Red arrow indicates nuclear afadin; yellow open arrowheads indicate dendritic afadin. Orthogonal projections show afadin colocalized with the nuclear marker NeuN. C, Western blot of neuronal subcellular fractionations confirmed l-/s-afadin was present in multiple subcellular compartments. D, representative images of Myc-l-afadin, Myc-s-afadin, Myc-afadin-ΔNT, or Myc-afadin-NT expression in neurons. E, cultured cortical neurons (DIV 25) expressing enhanced GFP and either Myc-l-afadin or Myc-afadin-NT constructs; red arrow highlights restricted localization of Myc-afadin-NT to the nucleus. Inset image shows Myc-afadin-NT localization to the nucleus. F, Myc-afadin-ΔNT, but not Myc-afadin-NT, is found at spines. Scale bars, 5 μm.
To further validate the nuclear presence of both l- and s-afadin, we overexpressed Myc-tagged l- or s-afadin in cultured cortical neurons. Both Myc-l- and Myc-s-afadin colocalized with NeuN indicating a nuclear localization for both isoforms (Fig. 1C). The presence of two nuclear localization sequences has been described to be present within the first 350 amino acids of afadin (12). Thus, to confirm whether the N-terminal portion of afadin was required for its nuclear localization, we ectopically expressed constructs encoding either amino acids 1–350 (afadin N terminus (NT)) or afadin lacking the N terminus (ΔNT) (amino acids 351–1829) (Fig. 1A) (11). When afadin-NT (Myc-afadin-NT) was ectopically expressed in cortical neurons, it almost exclusively localized within the nucleus and was not observed along the dendrite or at synapses (Fig. 1, D–F). Conversely, exogenous l-afadin lacking the N terminus (Myc-afadin-ΔNT) was not present in the nucleus but localized to synapses (Fig. 1, D–F), demonstrating the requirement of the N terminus of afadin for its nuclear localization. Collectively, these data revealed that both l- and s-afadin were present in the nucleus of cortical neurons and that the nuclear localization of these isoforms is dependent on the N-terminal region of the protein.
Afadin Accumulations within the Nucleus of Cortical Neurons Following Activity-dependent Stimulation
Our previous studies have demonstrated that afadin clusters at synapses in response to several stimuli (9, 10), including chemical activation of NMDA receptors (11). As several other proteins have been shown to shuttle to the nucleus of neurons following stimulation (18–20), we reasoned that afadin may also translocate to the nucleus of neurons in response to NMDA receptor activation (11, 15). To investigate this possibility, we performed an activity-dependent stimulation (15) of cortical neurons and examined afadin nuclear content at 0 (control), 30, 120, or 240 min from the start of treatment. Afadin nuclear content was not different from the control levels 30 min post-treatment. However, a significant increase in the nuclear localization of afadin was seen 120 and 240 min post-treatment (relative afadin intensities (arbitrary units (a.u.)) are as follows: 0 min, 1.16 ± 0.03; 30 min, 1.19 ± 0.02; 120 min, 1.39 ± 0.05; 240 min, 1.3 ± 0.05; *, p < 0.05; **, p < 0.01; Fig. 2, A and B). Interestingly, an increase in afadin clustering could be observed within the cell somas, which were not associated with the nucleus 120 and 240 min post-treatment (Fig. 2A). We have observed a similar clustering of afadin following the stimulated clustering of N-cadherin adhesion junctions (10), and thus we believe this to be indicative of afadin accumulation at adherent junctions surrounding the cell soma. Taken together, these data indicate that afadin is trafficked to the nucleus of cortical neurons in response to activity-dependent stimulation.
FIGURE 2.

Activity-dependent afadin nuclear accumulation. A, representative images of afadin nuclear accumulation following treatment. Red dashed lines indicate the nucleus. B, time course of afadin nuclear accumulation in all neurons 30, 120, and 240 min after activity-dependent simulation or control (0 min)-treated cells. Activity-dependent afadin nuclear localization peaked 120 min post-activation (*, p < 0.05; **, p < 0.01, ANOVA). C, representative immunofluorescence images of cortical neurons staining for afadin, subjected to NMDA receptor activation (120 min) or not, in the presence or absence of 20 μm ActD. Nuclei were identified by DAPI staining, and are indicated by the red dashed line in lower panels. D, quantification of C demonstrated that nuclear localization of afadin increases 120 min post-activation in the presence or absence of ActD (***, p < 0.001, two-way ANOVA). E, endogenous afadin staining in cortical neurons following NMDA receptor activation, in the presence or absence of 0.5 μm CycHx. The nucleus, identified by DAPI, is indicated by the red dashed line in lower panels. F, quantification of E revealed increased afadin nuclear accumulation 120 min post-activation, regardless of CycHx treatment (***, p < 0.001, two-way ANOVA). APVwd, activity-dependent stimulation.
Previously, it has been postulated that the accumulation of proteins within the nucleus following activity-dependent stimulation may be misinterpreted (1). Indeed, observed protein accumulation may actually result from the induction of new gene transcription and subsequent protein synthesis, rather than translocation from more distal cellular regions (1). Therefore, we sought to determine whether afadin nuclear accumulation was dependent on gene transcription or protein synthesis. We first repeated our activation protocol in the presence or absence of pretreatment, for 30 min, with actinomycin D (ActD), a transcription inhibitor. Afadin nuclear content was not altered under basal (control) conditions, and a significant increase in afadin nuclear accumulation was observed 120 min post-treatment, in the presence or absence of ActD pretreatment (relative afadin intensities (a.u.) are as follows: 0 min, 1.12 ± 0.01; 0 min + ActD, 1.13 ± 0.03; 120 min, 1.37 ± 0.02; 120 min + ActD, 1.34 ± 0.02; ***, p < 0.001; Fig. 2, C and D). We next stimulated cells in the presence or absence of pretreatment, for 30 min, with the translation inhibitor cycloheximide (CycHx). At 120 min post-activation, afadin nuclear content was again significantly increased regardless of the presence or absence of CycHx (relative afadin intensities (a.u.) are as follows: 0 min, 1.0 ± 0.08; 0 min + CycHx, 0.96 ± 0.07; 120 min, 1.59 ± 0.13; 120 min + CycHx, 1.46 ± 0.11; ***, p < 0.001; Fig. 2, E and F). Taken together, these results indicate that afadin nuclear accumulation occurs independently of gene transcription and new protein synthesis, an important criterion for demonstrating nuclear translocation of a cytoplasmic protein (1), and thus the increase in afadin nuclear content is likely due to the translocation of the protein from extranuclear locations following NMDA receptor activation.
Activity-dependent Bi-directional Shuttling of Cytosolic Afadin to Discrete Nuclear and Synaptic Sites
To examine the source of afadin that shuttles to the nucleus in response to activity-dependent stimulation, we performed Western blotting on neuronal cell fractions generated from stimulated cortical neurons. Consistent with our immunocytohistochemical data, we observed an increase in both l- and s-afadin in nuclear fractions 120 and 240 min following NMDA receptor activation (*, p < 0.05; Fig. 3A). Interestingly, we also detected an increase of l/s-afadin 120 and 240 min after treatment in the membrane fraction (*, p < 0.05; Fig. 3B). This is consistent with our previous study demonstrating that afadin clusters to synapses in response to activity-dependent stimulation (11). Conversely, examination of the cytosolic fraction revealed a congruent decrease in the presence of l/s-afadin in the cytosol fraction 120 and 240 min following treatment (*, p < 0.05; Fig. 3C). Together, these results suggest that following the activation of NMDA receptors, a pool of afadin located within the cytosol is trafficked to the nucleus and membrane.
FIGURE 3.
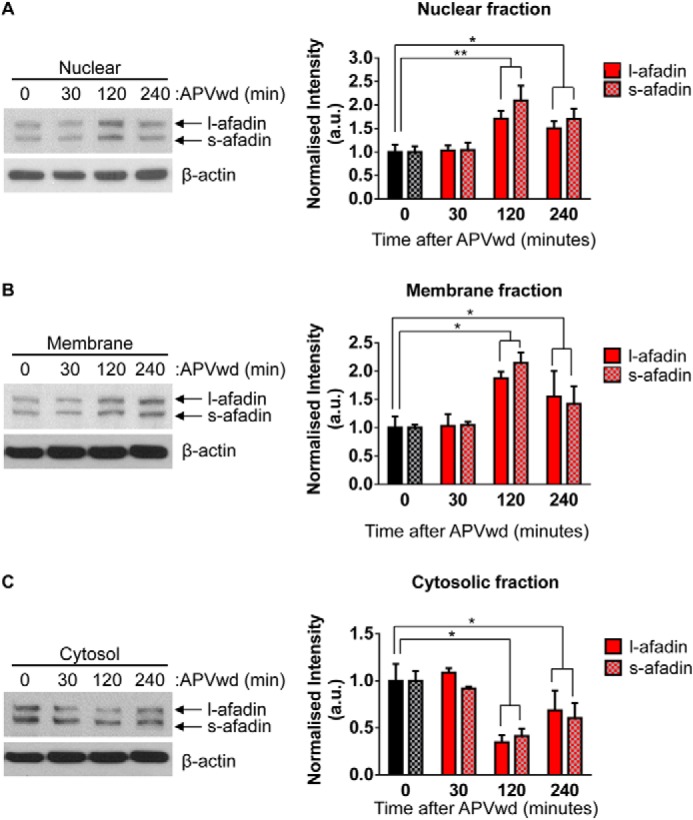
Altered afadin presence in distinct subcompartments following activity-dependent stimulation. A, assessment of l- and s-afadin presence in nuclear (A), membrane (B). and cytosol (C) of cortical neurons before and after NMDA receptor stimulation by Western blotting. Quantification reveals a significant increase in l/s-afadin presence in nuclear and membrane fractions 120 and 240 min post-treatment (A and B; *, p < 0.05; **, p < 0.01, two-way ANOVA). Conversely, a significant decrease in both l- and s-afadin levels was observed in cytosol fractions 120 and 240 min following activity-dependent stimulation (C; *, p < 0.05, two-way ANOVA). APVwd, activity-dependent stimulation.
Although our biochemical data indicate that afadin is capable of being trafficked to both synaptic and nuclear compartments, it does not allow us to assess whether this can occur within the same cell and whether afadin within the cytosol is the source of the mobile protein. To test this, we measured afadin content at synapses and within the dendritic shaft of neurons that displayed increased nuclear accumulation of afadin at 120 and 240 min following treatment (data not shown) (Fig. 4A). Consistent with our biochemical data, measurements of synaptic afadin immunofluorescence in this subpopulation of neurons revealed a significant increase in synaptic afadin puncta size and number 120 and 240 min post-treatment (integrated intensities in spines (a.u.) are as follows: 0 min, 11.5 ± 1.1; 30 min, 12.8 ± 0.8, 120 min, 15.9 ± 1.2; 240 min, 15.1 ± 1.1; afadin puncta per 10 μm as follows: 0 min, 5.1 ± 0.68; 30 min, 5.8 ± 0.76; 120 min, 9.6 ± 0.88; 240 min, 7.8 ± 0.65; *, p < 0.05; ***, p < 0.001; Fig. 4, A–C). Remarkably, as afadin content was increased at synapses and in the nucleus, afadin content decreased in dendrites within the same a time-dependent manner (integrated intensities in dendrite (a.u.) are as follows: 0 min, 23.9 ± 2.2; 30 min, 20.7 ± 0.8, 120 min, 11.7 ± 0.9; 240 min, 10.32 ± 0.6; afadin puncta per 10 μm as follows: 0 min, 3.0 ± 0.47; 30 min, 2.7 ± 0.37; 120 min, 1.4 ± 0.0.17; 240 min, 1.9 ± 0.15; **, p < 0.01; ***, p < 0.001; Fig. 4, D and E). Line scans of afadin distribution within the cytosol and at synapses further demonstrate a loss of the protein from the dendritic shaft and an increase at synapses in cells also showing an increase in nuclear afadin content 120 and 240 min post-treatment (Fig. 4F). Taken together, these data indicate that concomitant with an increase at synapses and within the nucleus, there is a decrease of afadin content in dendrites. This suggests that a mobile pool of cytosolic afadin is capable of being bi-directionally trafficked to both synaptic and nuclear compartments within the same neuron in response to activity-dependent stimulation.
FIGURE 4.

Bi-directional translocation of afadin following activity-dependent stimulation. A, representative images of afadin nuclear and dendritic localization in neurons with increased nuclear content following NMDA receptor activation. Insets show magnified regions of dendrites outlined by yellow boxes; images were pseudo-colored or binarized to demonstrate endogenous afadin puncta in dendrites and synapses of control and treated neurons. B and C, quantification of afadin puncta linear density at synapses or within dendrites following activity-dependent stimulation. At 120 and 240 min post-treatment, a significant increase in afadin puncta linear density at synapses (B) concurrent with a decrease of afadin puncta in dendrites (C) is observed (*, p < 0.05; **, p < 0.01; ***, p < 0.001, ANOVA). D and E, quantification of afadin puncta intensity at synapses and dendrites following activity-dependent stimulation reveals an increase in afadin puncta intensity at synapses (D) and concomitant decrease in dendrites (E) (*, p < 0.05; ***, p < 0.001, ANOVA). F, high magnification images of endogenous afadin following NMDA receptor activation. Yellow dotted lines outline the dendritic shaft; red lines represent line scans taken through dendritic spines and shaft. Graphical representation of line scans of endogenous afadin localization following treatment was produced by averaging values of three line scans (6 μm) performed at three different sections of the dendrite, of equal width, from six cells per condition. Values were then averaged and plotted; region where line scans pass through dendrites (dendritic shaft) is indicated. Scale bars, 10 and 5 μm (A) and 1 μm (F). APVwd, activity-dependent stimulation.
Nuclear Accumulation of Endogenous Afadin Is Blocked by Afadin N-terminal Domain
As Myc-afadin-NT localized exclusively to the nucleus, we speculated that it would act as a dominant-negative and attenuating afadin nuclear translocation. We first confirmed that exogenous afadin-NT (N terminus of afadin) was present in the nucleus; Myc-afadin-NT colocalized with the nuclear marker DAPI through the X, Y, and Z planes (Fig. 5A). When we examined afadin nuclear immunofluorescence under basal conditions, no differences were observed between NT-expressing and nonexpressing cells (Fig. 5, B and C). Moreover, expression of the Myc-afadin-NT was not expressed outside of the nucleus and did not alter the distribution of the endogenous protein outside the nucleus (Figs. 1, D and E, and 5, D–F) indicating that synaptic afadin was unaffected. As expected, neurons not expressing Myc-afadin-NT displayed a significant increase in nuclear afadin content 120 min post-treatment. However, a significantly reduced accumulation of nuclear afadin was observed in afadin-NT-expressing neurons 120 min post-treatment (relative afadin intensities (a.u.) are as follows: 0 min, 1.0 ± 0.04; 0 min + afadin-NT, 1.1 ± 0.05; 120 min, 1.49 ± 0.08; 120 min + afadin-NT, 1.17 ± 0.08; *, p < 0.05; ***, p > 0.001; Fig. 4, B and C). These results suggest that overexpression of Myc-afadin-NT domain does not preclude localization of endogenous afadin to the nucleus or synapses, under basal conditions, but is sufficient to attenuate activity-dependent translocation of afadin into the nucleus.
FIGURE 5.
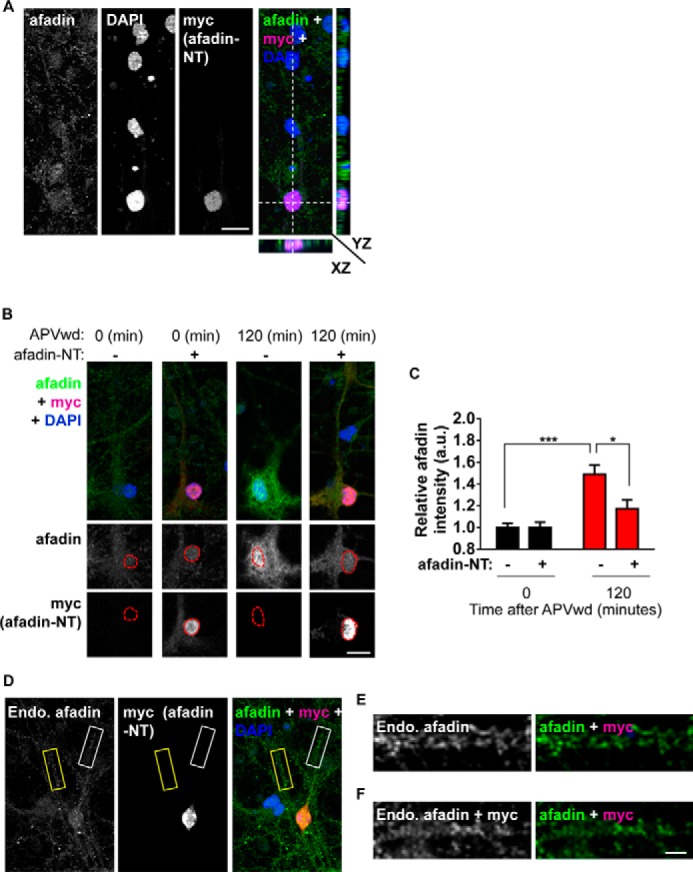
Myc-afadin-NT blocks activity-dependent afadin nuclear accumulation. A, confocal image of cortical neuron costained for endogenous l/s-afadin, DAPI, and Myc (afadin-NT). Orthogonal projections of XZ and YZ planes reveal that afadin-NT colocalizes with DAPI, demonstrating restricted localization of afadin-NT to the nucleus. B, representative images of control (0 min) or NMDA receptor-activated (120 min) neurons expressing, or not, Myc-afadin-NT. C, quantification of afadin nuclear content in B (*, p < 0.05; ***, p < 0.001, two-way ANOVA). Scale bars, 15 μm. D, representative image of cortical neurons expressing, or not, Myc-afadin-NT. Yellow box indicates a dendrite from a cell not expressing Myc-afadin-NT; white box indicates a dendrite from a Myc-afadin-NT positive cell. E, high magnification image of dendrite from a non-Myc-afadin-NT positive cell, outlined by a yellow box in D. D, high magnification image of dendrite from a Myc-afadin-NT positive cell, outlined by a white box in D. Scale bar, 15 μm (A and B) and 5 μm (F). APVwd, activity-dependent stimulation.
Nuclear Translocation of Afadin Contributes to Activity-dependent Spine Remodeling
One potential function of protein nuclear translocation is to transduce extracellular stimuli into nuclear signals that yield long term changes in neuronal structure (3, 21). Indeed, coordination of both local signaling and gene expression-dependent mechanisms is thought to be required for activity-dependent remodeling of dendritic spines. We have previously reported that activity-dependent stimulations induce a time-dependent alteration of spine morphologies (11, 15). Moreover, we have also demonstrated that afadin is required for the maintenance of dendritic spine morphologies (16) and can play an important role in mediating the effects of extracellular signals, including activity-dependent stimuli, on the remodeling of dendritic spine morphologies (9–11, 16). As afadin nuclear translocation occurs in a time frame consistent with the activity-dependent spine remodeling (11), we asked whether this translocation was also required for activity-dependent remodeling of spines. Expression of Myc-afadin-NT did not alter spine linear density or the spine area under basal conditions (Fig. 6, A–C). Activity-dependent stimulation for 120 min resulted in increased spine linear density and area in control cells (***, p < 0.001; Fig. 6, A–C). However, in neurons expressing Myc-afadin-NT, activity-dependent remodeling of spines was significantly attenuated (spines/10 μm are as follows: control 0 min, 5.55 ± 0.4; control 120 min, 10.1 ± 0.4; Myc-afadin-NT 0 min, 5.32 ± 0.3; Myc-afadin-NT 120 min, 6.9 ± 0.3; n = 11; spine area μm2 is as follows: control 0 min, 0.71 ± 0.02; control 120 min, 0.89 ± 0.03; Myc-afadin-NT 0 min, 0.69 ± 0.02; Myc-afadin-NT 120 min, 0.79 ± 0.03; *, p < 0.05; ***, p < 0.001; Fig. 6, A–C). Notably, Myc-afadin-NT expression did not completely block activity-dependent remodeling of spines, as the area of dendritic spines in neurons expressing afadin-NT was significantly different compared with both control and control 120 min-treated cells following activity-dependent stimulation (*, p < 0.05; Fig. 6C), indicating that multiple pathways, possibly including local synaptic signaling of afadin (11), are required for long term changes in synapse structure. Collectively, these data indicate that activity-dependent translocation of afadin plays an important role in mediating long lasting changes in neuronal structure, in response to synaptic stimuli.
FIGURE 6.
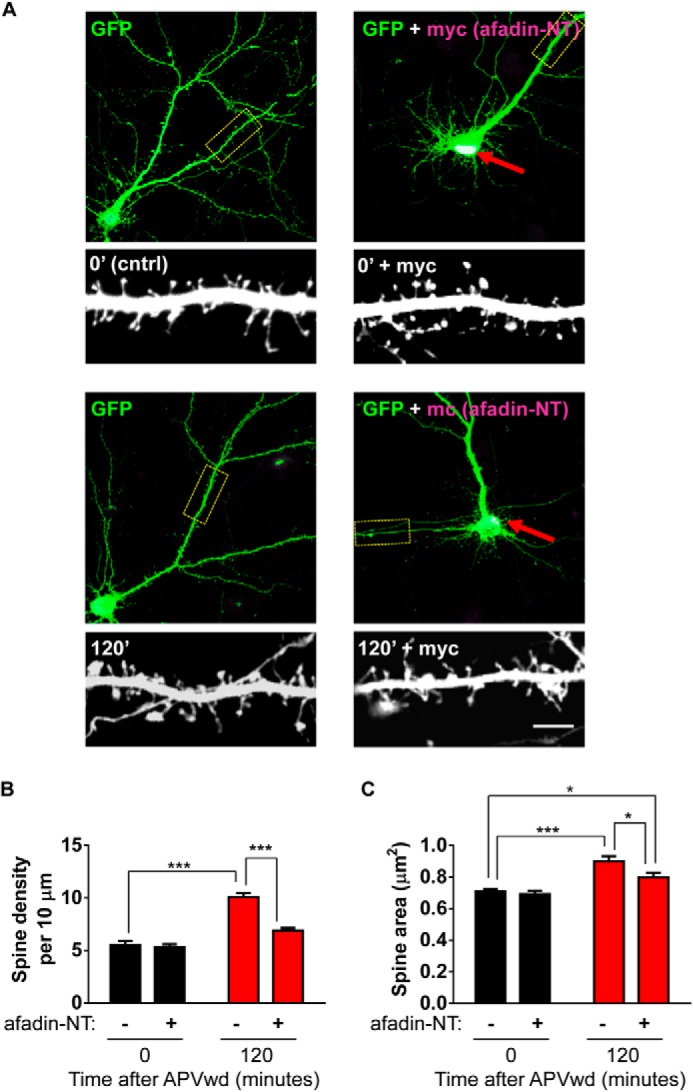
Afadin nuclear accumulation contributes to activity-dependent spine remodeling. A, representative images of treated cortical neurons expressing enhanced GFP with or without Myc-afadin-NT; red arrows indicate restricted nuclear expression of Myc-afadin-NT. Insets are representative of high magnification images of secondary dendrites and dendritic spines. B and C, quantification of spine morphology and linear density. B, NMDA receptor activation (120 min) results in an increase in dendritic spine linear density; this effect is attenuated in neurons expressing Myc-afadin-NT (***, p < 0.001, two-way ANOVA). C, examination of dendritic spine area reveals an increase in spine area following treatment. In neurons expressing Myc-afadin-NT, activity-dependent stimulation increased spine area compared with control but was significantly reduced compared with control stimulated cells (*, p < 0.05; ***, p < 0.001, two-way ANOVA). Scale bars, 5 μm. APVwd, activity-dependent stimulation.
Activity-dependent Histone Modification Coincides with Afadin Nuclear Accumulation
The mechanisms that underlie long lasting remodeling of dendritic spines likely require the coordination of multiple factors, including transcriptional regulation. Indeed, a large number of genes (>45) have been shown to be regulated in an activity- and time-dependent manner (22). As the nuclear accumulation of afadin supports the activity-dependent remodeling of dendritic spines, we hypothesized that afadin translocation to the nucleus may be involved in the regulation of nuclear events. We first sought to determine whether histone modifications are altered following activation of NMDA receptors. We thus examined the phosphorylation of the histone H3 protein at serine 10 (H3S10p) in cortical neurons. Importantly, this modification has been shown to be sufficient to induce a change in chromatin from a condensed heterochromatin state to a euchromatin state more amenable to gene transcription and associated with transcriptional activation (4, 6, 23). Moreover, it has previously been shown that H3S10p can be induced by activity-dependent stimuli in striatal neurons and in hippocampal neurons following behavioral testing (5, 7, 8). Additionally, a variety of neurotransmitters, such as glutamate, dopamine, and acetylcholine, can elicit neuronal responses resulting in increased H3S10p (7, 23, 24); similarly, acute cocaine administration in rats induces greater phosphorylation on histone H3 (25). Following activation of NMDA receptors, we observed an increase in H3S10p levels 30 and 120 min post-treatment in all neurons (relative H3S10p intensities (a.u.) are as follows: 0 min, 1.1 ± 0.08; 30 min, 1.51 ± 0.1; 120 min, 1.79 ± 0.17; 240 min, 1.42 ± 0.18; *, p < 0.05; Fig. 7, A and B), indicating that maximal afadin nuclear accumulation occurred concurrently with a significant increase in H3S10p levels. We next tested whether preventing activity-induced translocation of afadin by overexpression of Myc-afadin-NT could also attenuate the induction of H3S10p 120 min post-activation. Indeed, 120 min post-activation, neurons expressing Myc-afadin-NT displayed reduced H3S10p levels compared with non-Myc-afadin-NT expressing cells (relative H3S10p intensities (a.u.) are as follows: 0 min, 1.06 ± 0.03; 0 min + afadin-NT, 1.01 ± 0.1; 120 min, 1.4 ± 0.05; 120 min + afadin-NT, 0.92 ± 0.06; *, p < 0.05; Fig. 7, C and D). Importantly an increase in H3S10p could be observed at 30 min of treatment in both, but only in nonexpressing cells after 120 and 240 min within the same cell populations (Fig. 7, E and F; **, p < 0.01; ***, p < 0.001).
FIGURE 7.

Activity-dependent phosphorylation of histone H3 at serine 10 requires afadin nuclear translocation. A, representative high magnification images of cortical neurons costained for H3S10p and afadin following activity-dependent stimulation. Red dashed lines outline the nucleus (DAPI) in lower panels. B, quantification of H3S10p in afadin-positive cells revealed an increase in H3 phosphorylation 30, 120, and 240 min post-stimulation; neurons also display an increase in afadin nuclear content 120 min post-treatment (*, p < 0.05, ANOVA). C, representative high magnification images of neurons overexpressing, or not, Myc-afadin-NT and costained for H3S10p, following NMDA receptor activation. Red dashed lines outline the nucleus (DAPI) in lower panels. D, quantification of H3S10p in C revealed that in cells expressing Myc-afadin-NT H3 phosphorylation levels are significantly decreased compared with nonexpressing cells (*, p < 0.05; ANOVA). E, low magnification images of H3S10p and Myc-stained cortical neurons following activity-dependent stimulation for 0, 30, 120, or 240 min. Cortical neurons (DIV 25) were transfected with Myc-afadin-NT or not and probed with H3S10p. Red dashed boxes indicate cells expressing Myc-afadin-NT, and yellow dashed boxes enclose cells not expressing Myc-afadin-NT. H3S10p (images are pseudo-colored) increases in a time-dependent manner after activity-dependent stimulation in non-Myc-afadin-NT expressing cells. In Myc-afadin-NT expressing cells, H3S10p levels are significantly increased after 30 min but not at 120 or 240 min. F, quantification of relative H3S10p intensity in stimulated nontransfected and Myc-afadin-NT expressing cells shown in yellow boxes (nontransfected) or red boxes (transfected) in E. (**, p < 0.01; ***, p < 0.001, two-way ANOVA.) Scale bar, 15 μm. APVwd, activity-dependent stimulation.
To investigate the mechanism underlying the phosphorylation of histone H3 in response to activity-dependent stimulation, we examined the phosphorylation of the p90RSK protein. This protein directly phosphorylated histone H3 at serine 10 and is itself phosphorylated directly by ERK1/2 and following NMDA receptor activation and learning (5, 26, 27). Following activity-dependent stimulation, a time-dependent increase in p-p90RSK levels at Thr-359/Ser-363 was observed in the nucleus of cortical neurons (relative p-p90RSK (Thr-359/Ser-363) intensities (a.u.) are as follows: 0 min, 0.98 ± 0.07; 30 min, 1.82 ± 0.22; 120 min, 1.55 ± 0.15; 240 min, 1.39 ± 0.07; *, p < 0.05; **, p < 0.01; ***, p < 0.001; Fig. 8, A and B). Consistent with our data for H3S10p, overexpression of afadin-NT attenuated p-p09RSK(Thr-359/Ser-363) nuclear levels compared with nonexpressing cells, 120 min post-treatment (relative p-p90RSK (Thr-359/Ser-363) intensities (a.u.) are as follows: 0 min, 1.00 ± 0.21; 0 min + afadin-NT, 1.18 ± 0.11; 120 min, 1.92 ± 0.26; 120 min + afadin-NT, 0.93 ± 0.15; *, p < 0.05; **, p < 0.01; Fig. 8, C and D). Taken together, these data suggest that activity-dependent stimulation increases H3S10p in a time-dependent manner, potentially via the direct actions of p90RSK, in cortical neurons. Interestingly, our data suggest that phosphorylation of histone H3 and p90RSK specifically at 120 min post-stimulation may require the increased presence of afadin within the nucleus.
FIGURE 8.

Afadin nuclear shuttling is required for long term phosphorylation of nuclear p90RSK following activity-dependent stimulation. A, representative high magnification images of cortical neurons costained for p-p90RSK (Thr-359/Ser-363) and NeuN; red dashed lines outline the nucleus (NeuN) in lower panels. B, quantification of p-p90RSK (Thr-359/Ser-363) following activity-dependent simulation revealed an increase in p90RSK phosphorylation 30, 120, and 240 min post-stimulation (*, p < 0.05; **, p < 0.01; ***, p < 0.001, ANOVA). C, representative high magnification images of neurons overexpressing, or not, Myc-afadin-NT and costained for p-p90RSK (Thr-359/Ser-363), following NMDA receptor activation. Red dashed lines outline the nucleus (DAPI) in lower panels. D, quantification of p-p90RSK (Thr-359/Ser-363) in C revealed that in cells expressing Myc-afadin-NT H3 phosphorylation levels are significantly decreased compared with nonexpressing cells (*, p < 0.05; **, p < 0.01, ANOVA). APVwd, activity-dependent stimulation.
DISCUSSION
In this study, we have demonstrated that NMDA receptor activation results in a time-dependent accumulation of afadin in the nuclei of cortical neurons. In addition, neurons that displayed afadin nuclear accumulation also exhibited an increase of afadin presence at synapses. Importantly, this synaptic and nuclear accumulation occurred within the same time frame, suggesting a bi-directional shuttling of afadin as opposed to a singular synapse-to-nucleus movement. Interestingly, this bi-directional shutting was paralleled by a reduction in afadin content in dendrites, indicating that a mobile population of afadin located in the cytosol was the source protein being bi-directionally trafficked into synapses and nuclei. We have previously shown that afadin signaling at synapses contributes to activity-dependent spine morphogenic activity (9–11); however, here we observed that preventing afadin nuclear entry abrogated activity-dependent spine remodeling. We further showed that phosphorylation of histone H3 and of p90RSK occurred following activity-dependent stimulation and that blocking afadin nuclear entry attenuated phosphorylation of both proteins in a time-specific manner. Collectively, these data indicate that the activity-dependent nuclear accumulation of afadin results in histone H3 phosphorylation and is required for long lasting changes in neuronal morphology (Fig. 9).
FIGURE 9.
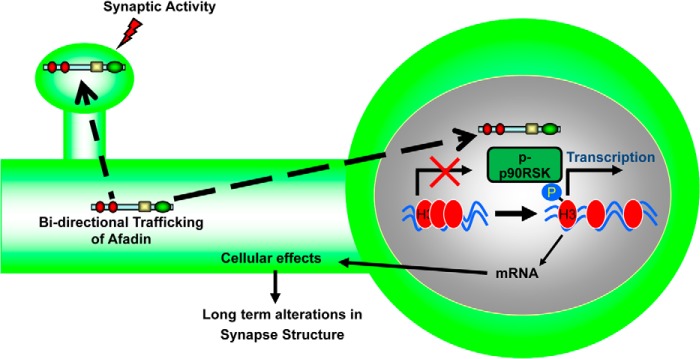
Model of afadin nuclear shuttling. Following activity-dependent stimulation, a mobile pool of afadin located in the cytosol is bi-directionally trafficked to both synapses as well to the nucleus in a time-dependent manner. In the nucleus, afadin's presence is required for the late phosphorylation of p90RSK that can directly phosphorylate histone H3 at serine 10. This in turn contributes to long term alterations in synapse structure.
The molecular mechanisms whereby neurons communicate signals generated at dendrites and synapses to the nucleus are not well understood (1, 3, 21). Several models have been proposed. Signals initiated at the synapse by neuronal activity can be rapidly propagated to the nucleus by calcium signaling and can trigger new gene transcription within a short time frame (21, 22). Shuttling of proteins directly from synapses to the nucleus is another modality by which activity-dependent signals are communicated to the nucleus. Protein-mediated nuclear signaling is a slower process that occurs over the course of minutes to hours (1, 3). Although extensive evidence supports the idea of synapse-to-nucleus protein shuttling, several important questions remain unanswered, including the lack of unequivocal evidence for translocation of synaptic proteins from individual synapses to the nucleus (1). Furthermore, it is possible that additional modalities might also exist for communication between the synaptic, cytosolic, and nuclear compartments.
An interesting observation from this study is that the N terminus of afadin (afadin-NT) was sufficient to block the accumulation of afadin in the nucleus following activity-dependent stimulation. It is important to note that two nuclear localization sequences have been identified within the first 350 amino acids of the protein (12). Indeed, the restricted localization of afadin-NT within the nucleus is consistent with that fact that the afadin-NT construct encodes the first 350 amino acids and thus contains the putative nuclear localization sequences. Thus, one explanation is that afadin-NT acts as a dominant-negative mutant that competes for binding sites or transport mechanisms and therefore attenuates the ability of endogenous afadin to translocate into the nuclear compartment.
The bi-directional protein trafficking processes, as described in this study, could simultaneously mediate local synaptic and global nucleus-mediated changes. Simultaneous translocation to synapses and nucleus of one protein species, in response to one specific stimulus, would be a simple mechanism to achieve a coordinated cellular response to specific stimuli. It should be noted that inhibiting afadin accumulation within the nucleus, by overexpression of afadin-NT, was not sufficient to completely block activity-dependent spine remodeling. Even in the presence of ectopically expressed Myc-afadin-NT, the average spine area is significantly increased compared with control and NMDA receptor-activated control cells. Interestingly, our previous data have shown that disrupting afadin's PDZ binding domain is sufficient to completely block activity-dependent increases in spine area (11). At the synapse, afadin likely mediates an immediate, local, and highly regulated response to synaptic activation. Indeed, synaptic afadin is rapidly regulated by a variety of stimuli, including NMDA receptor activation, Rap1 activation, N-cadherin-mediated adhesion, and estrogens (9–11), providing the ability to locally control its spine morphogenic activity. These direct spine morphogenic effects of afadin are mediated by afadin's PDZ domain-dependent interactions with proteins like kalirin-7 (10, 11). Conversely, the spatiotemporal integration of nuclear afadin likely controls global and long lasting changes in neuronal structure and possibly function. Thus, these data suggest that multiple pathways, including local signaling at synapses, potentially mediated by afadin, are required for long lasting changes in both dendritic spine linear density and morphology.
Multiple studies have demonstrated that histone H3 can undergo post-translational modifications in response to both activity-dependent stimuli and in vivo in behavioral situations (5, 7, 8). Phosphorylation of histone H3 at serine 10 has been shown to be sufficient to induce a change in chromatin from a condensed heterochromatin state to a euchromatin state more amenable to gene transcription (4, 6). A variety of neurotransmitters, such as glutamate, dopamine, and acetylcholine, can elicit neuronal responses that result in increased H3S10p (7, 23, 24). Moreover, acute cocaine administration in rats induces greater phosphorylation on histone H3 (25). However, the signaling events that mediate activity-dependent histone modifications are not well understood. The p90RSK protein has been previously shown to be phosphorylated with a response to activity-dependent stimulation and learning (26, 27), and it has also been shown to translocate to the nucleus and directly phosphorylate histone H3 at serine 10 (5). In this study, we observed that H3S10p at 120 min post-activation was dependent on afadin nuclear translocation. In contrast, the increased histone H3 phosphorylation noted at 30 min post-activation persisted even when nuclear afadin accumulation was attenuated. Moreover, we observe that p90RSK phosphorylation occurred within the nucleus in a time-dependent manner, and at 120 min, it is dependent on afadin nuclear translocation. Interestingly, it has recently been shown that uncaging of glutamate on seven spines is sufficient to increase phospho-ERK1/2 levels in the nucleus of neurons, up to 120 min (20). Critically, p90RSK is directly phosphorylated by ERK1/2 (5); an intriguing possibility is that in the nucleus afadin could function as a scaffold to assemble transcription factors or histone-modifying proteins such as p90RSK, bringing these proteins together as a signaling complex, although future studies will need to test this hypothesis directly. Nevertheless, our data indicate that at 120 min afadin nuclear accumulation is permissive for the phosphorylation of p90RSK, which in turn can directly phosphorylate histone H3 at serine 10. Taken together, these data suggest a functional role for afadin in the nucleus but also provide support for the idea that activity-dependent nucleosomal events are mediated by multiple signaling pathways operating along different time scales.
Several proteins with dual nuclear and synaptic localization have been identified; many of these proteins accumulate in the nucleus within minutes of stimulation (19, 24), whereas others, like afadin, show increased accumulation over the course of hours (28, 29). The functional significance and cellular consequences of the spatiotemporal nuclear integration of dual residency synaptic proteins are not well understood. Here, we show that afadin nuclear accumulation is required for spine morphogenesis. Previous reports support a role for neuronal activity-dependent nuclear translocation of other synaptic proteins in transcription initiation and repression, synapse elimination, and altering behavior (28, 30). Furthermore, both long term potentiation and long term depression are shown to induce nuclear translocation of some synaptic proteins (18–20). Future studies will aim to examine the roles of afadin in these processes.
Collectively, our data suggest a novel model whereby the concomitant translocation of proteins from the dendritic cytoplasm to synapses and the nucleus occurs in response to synaptic activity to control neuronal plasticity. Moreover, the ability to influence histone modification adds to the growing repertoire of afadin cellular functions, such as regulation of synapse structure and function (9–11, 16). Recently, afadin has also been reported to regulate the accumulation of PINK1/parkin to mitochondria, indicating a potential role in the pathophysiology of Parkinson disease (31). Therefore, our findings have implications for the understanding cytonuclear shuttling in general and add to a growing understanding of activity-dependent nuclear signaling pathways and the roles they play in synaptic plasticity.
This work was supported, in whole or in part, by National Institutes of Health Grant DA013141-11A1 (to R. J. M.), Grants MH071316 and MH097216 from NIMH (to P. P.), and Ruth L. Kirschstein NRSA 1F31MH087043 (to J. E. V.), 1F31MH085362 (to K. A. J.), and 1F31NS076201 (to R. H.). This work was also supported by grants from the Royal Society United Kingdom, Brain and Behavior Foundation (formally National Alliance for Research on Schizophrenia and Depression (NARSAD)), Psychiatric Research Trust, and American Heart Association (to D. P. S.), National Science Foundation Grant DGE-0824162 (to I. R.), and Autism Speaks, NARSAD, Brain Research Foundation.
- l
- long
- s
- short
- ANOVA
- analysis of variance
- DIV
- days in vitro
- a.u.
- arbitrary unit
- ActD
- actinomycin D
- CycHx
- cycloheximide
- NT
- N terminus.
REFERENCES
- 1. Jordan B. A., Kreutz M. R. (2009) Nucleocytoplasmic protein shuttling: The direct route in synapse-to-nucleus signaling. Trends Neurosci. 32, 392–401 [DOI] [PubMed] [Google Scholar]
- 2. Greer P. L., Greenberg M. E. (2008) From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860 [DOI] [PubMed] [Google Scholar]
- 3. Ch'ng T. H., Martin K. C. (2011) Synapse-to-nucleus signaling. Curr. Opin. Neurobiol. 21, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berger S. L. (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 5. Riccio A. (2010) Dynamic epigenetic regulation in neurons: Enzymes, stimuli and signaling pathways. Nat. Neurosci. 13, 1330–1337 [DOI] [PubMed] [Google Scholar]
- 6. Maze I., Noh K. M., Allis C. D. (2013) Histone regulation in the CNS: Basic principles of epigenetic plasticity. Neuropsychopharmacology 38, 3–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brami-Cherrier K., Lavaur J., Pagès C., Arthur J. S., Caboche J. (2007) Glutamate induces histone H3 phosphorylation but not acetylation in striatal neurons: Role of mitogen- and stress-activated kinase-1. J. Neurochem. 101, 697–708 [DOI] [PubMed] [Google Scholar]
- 8. Lubin F. D., Sweatt J. D. (2007) The IκB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 55, 942–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Srivastava D. P., Woolfrey K. M., Woolfrey K., Jones K. A., Shum C. Y., Lash L. L., Swanson G. T., Penzes P. (2008) Rapid enhancement of two-step wiring plasticity by estrogen and NMDA receptor activity. Proc. Natl. Acad. Sci. U.S.A. 105, 14650–14655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie Z., Photowala H., Cahill M. E., Srivastava D. P., Woolfrey K. M., Shum C. Y., Huganir R. L., Penzes P. (2008) Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. J. Neurosci. 28, 6079–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xie Z., Huganir R. L., Penzes P. (2005) Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron 48, 605–618 [DOI] [PubMed] [Google Scholar]
- 12. Buchert M., Poon C., King J. A., Baechi T., D'Abaco G., Hollande F., Hovens C. M. (2007) AF6/s-afadin is a dual residency protein and localizes to a novel subnuclear compartment. J. Cell. Physiol. 210, 212–223 [DOI] [PubMed] [Google Scholar]
- 13. Nishioka H., Mizoguchi A., Nakanishi H., Mandai K., Takahashi K., Kimura K., Satoh-Moriya A., Takai Y. (2000) Localization of l-afadin at puncta adhaerentia-like junctions between the mossy fiber terminals and the dendritic trunks of pyramidal cells in the adult mouse hippocampus. J. Comp. Neurol. 424, 297–306 [DOI] [PubMed] [Google Scholar]
- 14. Srivastava D. P., Woolfrey K. M., Penzes P. (2011) Analysis of dendritic spine morphology in cultured CNS neurons. J. Vis. Exp. 53, e2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie Z., Srivastava D. P., Photowala H., Kai L., Cahill M. E., Woolfrey K. M., Shum C. Y., Surmeier D. J., Penzes P. (2007) Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56, 640–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srivastava D. P., Copits B. A., Xie Z., Huda R., Jones K. A., Mukherji S., Cahill M. E., VanLeeuwen J. E., Woolfrey K. M., Rafalovich I., Swanson G. T., Penzes P. (2012) Afadin is required for maintenance of dendritic structure and excitatory tone. J. Biol. Chem. 287, 35964–35974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Majima T., Ogita H., Yamada T., Amano H., Togashi H., Sakisaka T., Tanaka-Okamoto M., Ishizaki H., Miyoshi J., Takai Y. (2009) Involvement of afadin in the formation and remodeling of synapses in the hippocampus. Biochem. Biophys. Res. Commun. 385, 539–544 [DOI] [PubMed] [Google Scholar]
- 18. Ch'ng T. H., Uzgil B., Lin P., Avliyakulov N. K., O'Dell T. J., Martin K. C. (2012) Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150, 207–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jordan B. A., Fernholz B. D., Khatri L., Ziff E. B. (2007) Activity-dependent AIDA-1 nuclear signaling regulates nucleolar numbers and protein synthesis in neurons. Nat. Neurosci. 10, 427–435 [DOI] [PubMed] [Google Scholar]
- 20. Zhai S., Ark E. D., Parra-Bueno P., Yasuda R. (2013) Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science 342, 1107–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohen S., Greenberg M. E. (2008) Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol. 24, 183–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saha R. N., Wissink E. M., Bailey E. R., Zhao M., Fargo D. C., Hwang J. Y., Daigle K. R., Fenn J. D., Adelman K., Dudek S. M. (2011) Rapid activity-induced transcription of Arc and other IEGs relies on poised RNA polymerase II. Nat. Neurosci. 14, 848–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crosio C., Heitz E., Allis C. D., Borrelli E., Sassone-Corsi P. (2003) Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J. Cell Sci. 116, 4905–4914 [DOI] [PubMed] [Google Scholar]
- 24. Stipanovich A., Valjent E., Matamales M., Nishi A., Ahn J. H., Maroteaux M., Bertran-Gonzalez J., Brami-Cherrier K., Enslen H., Corbillé A. G., Filhol O., Nairn A. C., Greengard P., Hervé D., Girault J. A. (2008) A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature 453, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar A., Choi K. H., Renthal W., Tsankova N. M., Theobald D. E., Truong H. T., Russo S. J., Laplant Q., Sasaki T. S., Whistler K. N., Neve R. L., Self D. W., Nestler E. J. (2005) Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48, 303–314 [DOI] [PubMed] [Google Scholar]
- 26. Sananbenesi F., Fischer A., Schrick C., Spiess J., Radulovic J. (2002) Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: Interactions between Erk-1/2 and Elk-1. Mol. Cell. Neurosci. 21, 463–476 [DOI] [PubMed] [Google Scholar]
- 27. Thomas G. M., Rumbaugh G. R., Harrar D. B., Huganir R. L. (2005) Ribosomal S6 kinase 2 interacts with and phosphorylates PDZ domain-containing proteins and regulates AMPA receptor transmission. Proc. Natl. Acad. Sci. U.S.A. 102, 15006–15011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dieterich D. C., Karpova A., Mikhaylova M., Zdobnova I., König I., Landwehr M., Kreutz M., Smalla K. H., Richter K., Landgraf P., Reissner C., Boeckers T. M., Zuschratter W., Spilker C., Seidenbecher C. I., Garner C. C., Gundelfinger E. D., Kreutz M. R. (2008) Caldendrin-Jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 6, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Proepper C., Johannsen S., Liebau S., Dahl J., Vaida B., Bockmann J., Kreutz M. R., Gundelfinger E. D., Boeckers T. M. (2007) Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J. 26, 1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abe K., Takeichi M. (2007) NMDA-receptor activation induces calpain-mediated β-catenin cleavages for triggering gene expression. Neuron 53, 387–397 [DOI] [PubMed] [Google Scholar]
- 31. Haskin J., Szargel R., Shani V., Mekies L. N., Rott R., Lim G. G., Lim K. L., Bandopadhyay R., Wolosker H., Engelender S. (2013) AF-6 is a positive modulator of the PINK1/parkin pathway and is deficient in Parkinson's disease. Hum. Mol. Genet. 22, 2083–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]