

Background: Nuclear import of protein kinase Cδ is required for DNA-damage-induced apoptosis.
Results: c-Src and c-Abl phosphorylate PKCδ to regulate nuclear import. Tyrosine kinase inhibitors block nuclear translocation of PKCδ and suppress apoptosis.
Conclusion: Tyrosine kinase inhibitors can regulate the pro-apoptotic function of protein kinase Cδ.
Significance: Tyrosine kinase inhibitors may improve the quality of life in cancer patients receiving radiation therapy.
Keywords: Apoptosis, Nonreceptor Tyrosine Kinase, Protein Kinase C (PKC), Radiation Biology, Salivary Gland
Abstract
Radiation therapy for head and neck cancer can result in extensive damage to normal adjacent tissues such as the salivary gland and oral mucosa. We have shown previously that tyrosine phosphorylation at Tyr-64 and Tyr-155 activates PKCδ in response to apoptotic stimuli by facilitating its nuclear import. Here we have identified the tyrosine kinases that mediate activation of PKCδ in apoptotic cells and have explored the use of tyrosine kinase inhibitors for suppression of irradiation-induced apoptosis. We identify the damage-inducible kinase, c-Abl, as the PKCδ Tyr-155 kinase and c-Src as the Tyr-64 kinase. Depletion of c-Abl or c-Src with shRNA decreased irradiation- and etoposide-induced apoptosis, suggesting that inhibitors of these kinases may be useful therapeutically. Pretreatment with dasatinib, a broad spectrum tyrosine kinase inhibitor, blocked phosphorylation of PKCδ at both Tyr-64 and Tyr-155. Expression of “gate-keeper” mutants of c-Abl or c-Src that are active in the presence of dasatinib restored phosphorylation of PKCδ at Tyr-155 and Tyr-64, respectively. Imatinib, a c-Abl-selective inhibitor, also specifically blocked PKCδ Tyr-155 phosphorylation. Dasatinib and imatinib both blocked binding of PKCδ to importin-α and nuclear import, demonstrating that tyrosine kinase inhibitors can inhibit nuclear accumulation of PKCδ. Likewise, pretreatment with dasatinib also suppressed etoposide and radiation induced apoptosis in vitro. In vivo, pre-treatment of mice with dasatinib blocked radiation-induced apoptosis in the salivary gland by >60%. These data suggest that tyrosine kinase inhibitors may be useful prophylactically for protection of nontumor tissues in patients undergoing radiotherapy of the head and neck.
Introduction
Ionizing radiation (IR)2 therapy in patients with head and neck cancer can result in moderate to severe damage to the salivary glands, resulting in loss of saliva, chronic oral infections, and severe discomfort (1). Other nontumor tissues in the oral cavity, such as the taste buds and oral mucosa, can also be unintended targets of IR damage; however, damage to these tissues generally resolves in the months after therapy (2). In contrast, damage to the salivary gland is permanent because it primarily affects salivary acinar cells which are postmitotic (1). Protein kinase Cδ (PKCδ) is essential for the apoptotic response of nontransformed cells to cell damaging agents (3–7), and our studies have defined PKCδ as a critical regulator of IR-induced apoptosis in salivary epithelial cells (8–13). Salivary epithelial cells from PKCδ−/− mice are resistant to multiple apoptotic stimuli in vitro, whereas PKCδ−/− mice are protected from IR-induced damage to the salivary gland and thymus in vivo and have a delay in mammary gland involution, a process driven by apoptosis (8, 14–16).
PKCδ is ubiquitously expressed and regulates a variety of cell functions in addition to apoptosis, including cell survival, migration, and proliferation (17). The ability of PKCδ to control diverse cellular functions is due in part to tight regulation of its subcellular localization (17–19). In resting cells PKCδ largely resides in the cytoplasm; however, upon DNA damage a series of highly regulated events results in its nuclear import and activation of downstream apoptotic pathways (9–12). We reported previously that tyrosine phosphorylation of PKCδ is rate-limiting for this process because phosphorylation at Tyr-64 and Tyr-155 results in a conformational change that facilitates importin-α binding to a C-terminal nuclear localization signal and nuclear import (10–12). Candidate tyrosine kinases for phosphorylation of PKCδ include c-Abl, which plays a prominent role in DNA repair, especially double-stranded break repair induced by DNA-damaging agents, and members of the Src family kinases (SFKs), known to control proliferation and cell migration (20–24).
In our current studies we have identified the tyrosine kinases that mediate activation of PKCδ in apoptotic cells and have explored the use of TKIs (tyrosine kinase inhibitors) for protection of the salivary gland in patients undergoing radiotherapy for head and neck cancer. We show that phosphorylation of PKCδ at Tyr-64 and Tyr-155, nuclear accumulation of PKCδ, and apoptosis can be specifically inhibited by pretreatment with TKIs. Our studies suggest that suppression of tyrosine phosphorylation of PKCδ with TKIs may be a useful therapeutic strategy for protection of salivary gland function in patients undergoing head and neck irradiation.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
Culture of the ParC5 cell line has been described previously (25). ParC5 cells were stably transduced with a nontargeting lentiviral shRNA or lentiviral shRNAs against c-Abl (TRCN0000023354 and TRCN0000034456; Open Biosystems, Pittsburg, PA) or c-Src (TRCN0000023596 and TRCN0000023597; Open Biosystems). ParC5 cells were transfected at 30–40% confluence using FuGENE 6 (11988387001; Roche Applied Science), according to the manufacturer's instructions. 293T cells were cultured in DMEM/high glucose medium (SH30243.02; Thermo Scientific) supplemented with 10% FBS (F2442; Sigma). 293T cells were transfected using FuGENE 6.
Plasmids and Site-directed Mutagenesis
pGFP-PKCδ, pY64F-PKCδ, and pY155F-PKCδ have been described previously (11). The pBABE-WT-Src and pBABE-SrcT341 vectors were a generous gift from Dr. Rebecca Schweppe (University of Colorado Anschutz Medical Campus). The pBABE-WT-Abl vector was generated by ligating a PCR product digested with EcoRI and BamHI where the primers 5′-TATGGAGCCATGGGGCAGCAGCCT-3′ and 5′-TATGAATTCCTACCTCCGGACAATGTC-3′ (Integrated DNA Technologies, Coralville, IA) were used to amplify off of pcDNA-Abl-WT.
The pBABE-AblT315I vector was generated using the QuikChange site-directed mutagenesis kit (200518-5; Stratagene) with primers 5′-CCATAGGTCATGAACTCAATGATTATGTAGAATGGTG-3′ and 5′-CACCATTCTACATAATCATTGAGTTCATGACCTATGG-3′ (Integrated DNA Technologies).
Immunoprecipitation and Immunoblotting
293T cells were transfected with pGFP-PKCδ and treated with 5 mm hydrogen peroxide (H2O2) (H1009; Sigma) either with or without the pretreatment with 20 nm dasatinib (Sprycel) or 1 μm imatinib (Gleevec) (University of Colorado Anschutz Medical Campus Pharmacy). Immediately following treatments, cells were lysed with buffer A (50 mm Tris, pH 7.4, 1% Triton X-100, 100 mm NaCl, 5 mm EDTA, 1× Complete Protease Inhibitor (11697498001; Roche Applied Science), and 1× phosphatase inhibitor (04906837001; Roche Applied Science). Protein concentrations were measured using the DC Protein Assay kit (500-0111; Bio-Rad). For immunoprecipitation, total protein (1.0 mg) was mixed with anti-GFP (green fluorescent protein) antibody (ab290; Abcam) or control rabbit IgG (sc-2027; Santa Cruz Biotechnology) for 16 h at 4 °C. Immunocomplexes were bound to protein A-Sepharose beads (P6649; Sigma) for 1 h at 4 °C. The immunocomplexes were then washed using three 15-min washes in buffer A prior to SDS-PAGE. The immunoblots were probed with antibodies to importin-α (610486; BD Transduction Laboratories) and anti-GFP (632280; Clontech). Immunoblots from other experiments were probed with antibodies to phospho-PKCδ (Tyr-64) (A8171; Assay Biotech, Sunnyvale, CA), phospho-PKCδ (Tyr-155) (sc-233770-R; Santa Cruz Biotechnology), phospho-PKCδ (Tyr-311) (2055; Cell Signaling), actin (ab49900; Abcam), PKCδ (sc-937 and sc-213; Santa Cruz Biotechnology), phospho-c-Abl (Tyr-412) (NB100-92665; Novus Biological, Littleton, CO), c-Abl (sc-23; Santa Cruz Biotechnology), phospho-c-Src family kinase (2110; Cell Signaling), and c-Src (2108; Cell Signaling). In some instances the signal was quantified by densitometry and expressed as ratio of pY64/total PKCδ or pY155/total PKCδ.
Fluorescent Microscopy
ParC5 cells were grown on glass coverslips and transfected with pGFP-PKCδ. Following treatment with H2O2, coverslips were first rinsed with 1× PBS (3 × 10 min) then fixed with 2% paraformaldehyde for 15 min. Coverslips containing fixed cells were mounted on slides using Vectashield with DAPI mounting medium (H-1200; Vector Laboratories, Burlingame, CA). Subcellular localization of GFP-PKCδ was analyzed by fluorescent microscopy. For quantification of GFP-PKCδ localization, fixed cells were scanned using Olympus hardware and software (Center Valley, PA), and nuclear localization was quantified as the percentage of total cells with predominantly nuclear localized GFP. More than 200 cells were counted for each variable per experiment.
Analysis of Apoptosis in Vitro
Active caspase-3 was detected with the Caspase-3 Cellular Activity Assay kit PLUS (BML-ALK7030001; BIOMOL, Farmingdale, NY), which uses N-acetyl-Asp-Glu-Val-Asp-p-nitroaniline (Ac-DEVD.FMK-pNA) as a substrate, according to the manufacturer's instructions.
Analysis of Apoptosis in Vivo
C57BL/6 female mice were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were maintained at the University of Colorado, Anschutz Medical Campus, in accordance with Laboratory Animal Care guidelines and protocols and with approval of the University of Colorado Denver Institutional Animal Use and Care Committee. Six- to 8-week-old female mice were left untreated or pretreated with dasatinib (20 mg/kg) via oral gavage 1 h prior to irradiation. Mice were anesthetized as described, and the head and neck region was irradiated using a cesium-137 source, while the remainder of the body was shielded with lead (8). Three h after irradiation, mice treated with dasatinib received a second dose of dasatinib (20 mg/kg). Mice were sacrificed 24 h following irradiation, and salivary glands were removed, fixed in 10% neutral buffered formalin, and embedded in paraffin for immunohistochemistry. Five-μm sections were cut from the paraffin-embedded tissue for immunohistochemistry for detection of activated caspase-3 and counterstained with hematoxylin. For quantification of caspase-3 staining, sections were scanned Olympus hardware and software. Active caspase-3-positive cells in five random 40× fields were quantified for each mouse (n = 7 mice per condition). The data are expressed as the percentage caspase-3-positive cells/total epithelial cells.
RESULTS
c-Abl and c-Src Phosphorylate PKCδ at Tyr-155 and Tyr-64, Respectively
We have shown previously that PKCδ is phosphorylated at Tyr-64 and Tyr-155 in response to H2O2 and DNA-damaging agents and that tyrosine phosphorylation of PKCδ is required for nuclear translocation and activation of its pro-apoptotic function (Refs. 11, 12 and Fig. 1A). Because tyrosine phosphorylation of PKCδ may be a therapeutic target for regulation of apoptosis, we sought to identify the tyrosine kinases that phosphorylate PKCδ at these sites. Both SFKs and c-Abl have been shown previously to phosphorylate PKCδ, and c-Src has been identified as the PKCδ Tyr-311 kinase (21–23, 26, 27). To investigate the contribution of these tyrosine kinases to activation and nuclear import of PKCδ following apoptotic signals, we treated salivary gland acinar cells (ParC5) with H2O2 and assayed activation of c-Abl and SFKs using antibodies that specifically recognize the activated form of the tyrosine kinase. As seen in Fig. 1B, activation of both c-Abl and SFKs is evident by 5 min and persists for at least 40 min. Notably, tyrosine kinase activation parallels the kinetics of phosphorylation of PKCδ at Tyr-64 and Tyr-155 (Ref. 12 and Fig. 2, A and C).
FIGURE 1.
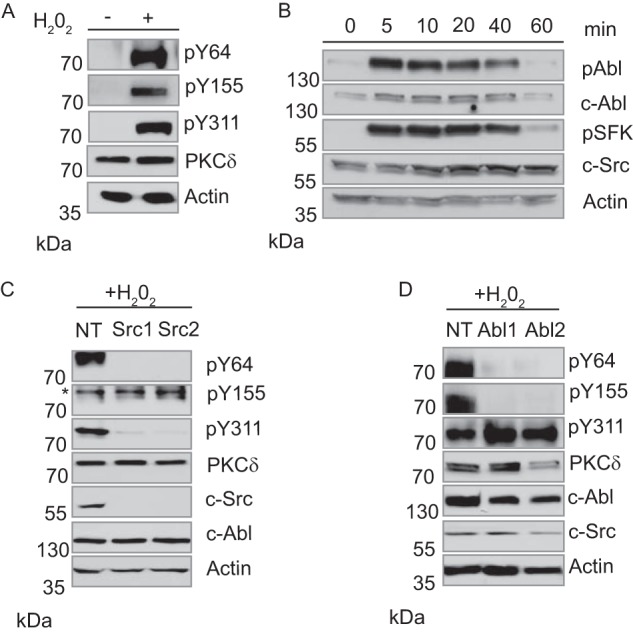
c-Src and c-Abl phosphorylate PKCδ in response to H2O2. A, ParC5 cells were left untreated or treated with 5 mm H2O2 for 10 min. Whole cell lysates were separated by SDS-PAGE and probed using phospho-specific antibodies against PKCδ pY64, pY155, and pY311. To determine loading, membranes were stripped and probed for total PKCδ and actin. B, ParC5 cells were treated with 5 mm H2O2 for the indicated times. Whole cell lysates were resolved by SDS-PAGE and probed using a phospho-specific antibody against the c-Abl activation site (pY412) or the conserved SFK activation site (pY416). Membranes were stripped and re-probed for total c-Abl and c-Src. C and D, ParC5 cells stably expressing either an nontargeting shRNA or two unique shRNAs against c-Src (C) or c-Abl (D) were treated with 5 mm H2O2 for 10 min. Whole cell lysates were resolved using SDS-PAGE and analyzed for PKCδ pY64, pY155, and pY311. Blots were stripped and probed for total PKCδ and actin. Efficiency and specificity of knockdown were determined by probing for c-Src and c-Abl. In C an asterisk denotes the band representing PKCδ pY155. MW, molecular mass.
FIGURE 2.
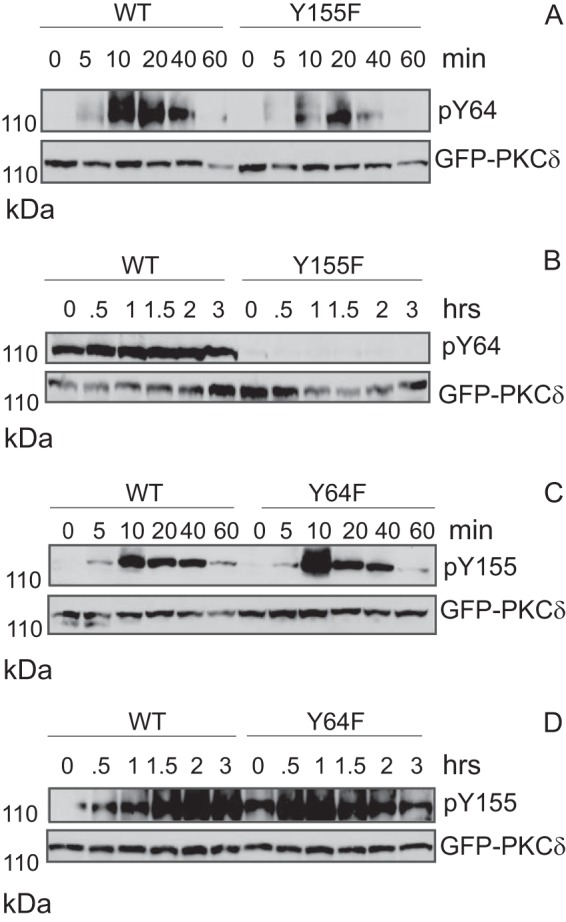
Phosphorylation of PKCδ at Tyr-155 facilitates phosphorylation at Tyr-64. A and B, 293T cells were transfected with either pGFP-PKCδWT or pGFP-PKCδY155F. Transfected cells were treated with 5 mm H2O2 (A) or 50 μm etoposide (B) for the indicated times. Whole cell lysates were separated using SDS-PAGE and analyzed for PKCδ pY64. Membranes were stripped and probed for total GFP-PKCδ. C and D, 293T cells were transfected with either pGFP-PKCδWT or pGFP-PKCδY64F. Transfected cells were treated with 5 mm H2O2 (C) or 50 μm etoposide (D) for the indicated times. Whole cell lysates were separated using SDS-PAGE and analyzed for PKCδ pY155. Membranes were stripped and probed for total GFP-PKCδ. MW, molecular mass.
To demonstrate a specific role for c-Abl and c-Src in PKCδ-dependent apoptosis we asked whether depletion of either tyrosine kinase with shRNA blocks phosphorylation of PKCδ at Tyr-64 or Tyr-155. In ParC5 cells stably expressing a nontargeting shRNA, treatment with H2O2 results in phosphorylation of PKCδ on Tyr-64, Tyr-155, and Tyr-311, whereas in cells expressing shRNA to c-Src, phosphorylation of Tyr-64 and Tyr-311, but not Tyr-155, is dramatically reduced (Fig. 1C). This confirms previous studies that have identified Tyr-311 as a c-Src site and suggests that Tyr-64 is also phosphorylated by c-Src (28). In contrast, depletion of c-Abl with shRNA leads to a significant reduction in phosphorylation of Tyr-155 (Fig. 1D). Unexpectedly, phosphorylation of PKCδ at Tyr-64 was also reduced in cells depleted of c-Abl (Fig. 1D). Although it is possible that both c-Src and c-Abl can phosphorylate PKCδ on Tyr-64, alternatively, phosphorylation at Tyr-64 by c-Src may be dependent upon prior phosphorylation of Tyr-155 by c-Abl. To test this directly, we transfected ParC5 cells with pGFP-PKCδWT, or pGFP-PKCδY155F, a mutant that cannot be phosphorylated at Tyr-155, and assayed phosphorylation of PKCδ at Tyr-64 (Fig. 2A). In cells treated with H2O2, phosphorylation of PKCδ at Tyr-64 was diminished and delayed in the context of the PKCδ Y155F mutant (Fig. 2A), suggesting that under these conditions, phosphorylation at Tyr-155 facilitates but is not required for phosphorylation of PKCδ at Tyr-64. However, in cells treated with etoposide, phosphorylation of the PKCδ Y155F mutant at Tyr-64 was not detectable (Fig. 2B). In a reciprocal experiment where cells were transfected with pGFP-PKCδY64F and probed for phosphorylation of Tyr-155, no decrease in pY155 was detected in cells treated with either agent (Fig. 2, C and D). These data support a model in which phosphorylation of PKCδ at Tyr-155 by c-Abl increases availability of the Tyr-64 site for phosphorylation by c-Src, presumably through a conformational change in the kinase which exposes the Tyr-64 site. The dependence upon phosphorylation at PKCδ Tyr-155 for phosphorylation at Tyr-64 is much more apparent in the context of etoposide than H2O2. Notably, H2O2 alone can induce oxidation-related conformational changes in proteins containing C1 domains. Because Tyr-155 in PKCδ is adjacent to the C1 domain, oxidation by H2O2 could result in some exposure and phosphorylation of Tyr-64 even in the absence of phosphorylation of PKCδ at Tyr-155 (28).
Tyrosine Kinase Inhibitors Block Phosphorylation of PKCδ at Tyr-64 and Tyr-155
TKIs encompass a large family of drugs that are used clinically for the treatment of neoplastic diseases (27). Our studies suggest that these drugs may also be useful for protection of nontumor tissue in patients undergoing IR treatment. To address this, we first explored whether TKIs could suppress tyrosine phosphorylation of PKCδ. ParC5 cells were treated with H2O2 alone or following pretreatment with dasatinib, a broad spectrum TKI that inhibits both c-Abl and SFKs (29, 30). Treatment with dasatinib plus H2O2 suppresses activation of c-Src and c-Abl dramatically and blocks phosphorylation of PKCδ at Tyr-64 and Tyr-155, as well as the previously described c-Src site, Tyr-311 (Fig. 3A and Ref. 26). Notably, we consistently observed a reduction (average decrease 45%) in total Src protein with Src activation (see Fig. 3, A, B, C, right, and D).
FIGURE 3.

Dasatinib and imatinib can inhibit phosphorylation of PKCδ at Tyr-64 and Tyr-155. A and D, ParC5 cells were left untreated or treated with 5 mm H2O2 for 10 min with or without pretreatment with 20 nm dasatinib (A) or 1 μm imatinib (D) for 30 min. Whole cell lysates were resolved using SDS-PAGE and analyzed for PKCδ pY64, pY155, and pY311. Inhibition of c-Src and c-Abl was determined by probing for their respective activation sites pY416 (c-Src) and pY412 (c-Abl). Blots were stripped and probed for total PKCδ, total c-Src, total c-Abl, and actin. B and C, 293T cells were transfected with either wild type c-Src (B), wild type c-Abl (C), or the gatekeeper mutation for c-Src T341I (B) or c-Abl T315I (C). Transfected cells were treated as in A. Whole cell lysates were resolved using SDS-PAGE and analyzed for PKCδ pY64, pY155, and pY311. For B and C, an asterisk denotes the band representing PKCδ pY155. Blots were stripped and probed for total PKCδ, total c-Src, total c-Abl, and actin. For D, an asterisk distinguishes the band representing c-Abl pY412 from a lower background band.
To confirm that inhibition of tyrosine phosphorylation indeed results from suppression of c-Src and c-Abl activation in dasatinib-treated cells, we repeated this experiment in cells transfected with plasmids encoding gatekeeper mutants for c-Src (T341I) (Fig. 3C) or c-Abl (T315I) (Fig. 3B). These mutants are resistant to inhibition by dasatinib (31). Expression of c-Src T341I, but not WT c-Src, rescued phosphorylation of Tyr-64 (and Tyr-311) in cells treated with dasatinib and H2O2, verifying our previous finding that Tyr-64 is a c-Src site. Quantification by densitometry (n = 3 experiments) shows that phosphorylation at Tyr-64 was reduced 70% in cells transfected with WT Src and treated with H2O2 plus dasatinib, compared with H2O2 alone. Conversely, in cells transfected with Src T341I and treated with H2O2 plus dasatinib, phosphorylation at Tyr-64 was 171% of that seen in cells treated with H2O2 alone (Fig. 3C). Phosphorylation at Tyr-155 was not rescued by c-Src T341I, but was rescued by expression of the c-Abl gatekeeper mutant, c-Abl T315I (Fig. 3B), consistent with this site being phosphorylated by c-Abl. Phosphorylation at Tyr-155 was reduced 50% in cells transfected with WT Abl and treated with H2O2 plus dasatinib, compared with H2O2 alone. Expression of Abl T341I completely restored phosphorylation at Tyr-155 in cells treated with H2O2 plus dasatinib (Fig. 3B). Interestingly, expression of either gatekeeper mutant resulted in an increase in basal phosphorylation at their respective activation sites (pY412 for c-Abl and pY416 for c-Src) (Fig. 3, B, 3C, and Ref. 31). Finally, we show that in ParC5 cells pretreated with the c-Abl-selective inhibitor imatinib, phosphorylation of PKCδ at Tyr-155 is also abolished (Fig. 3D and Ref. 30). The dose of imatinib used had very minimal effects on c-Src activation by H2O2 and did not inhibit phosphorylation of the c-Src site, Tyr-311. However, imatinib did reduce phosphorylation of Tyr-64, again consistent with our previous data that suggests co-operativity between phosphorylation at Tyr-64 and Tyr-155.
Tyrosine Kinase Inhibition Suppresses Nuclear Localization of PKCδ
Phosphorylation of PKCδ on Tyr-64 and Tyr-155 regulates its nuclear import in response to apoptotic agents by facilitating interaction with importin-α (12). Based on our observation that treatment with TKIs blocks Tyr-64 and Tyr-155 phosphorylation, we predicted that TKIs would prevent nuclear translocation of PKCδ. To address this, we asked whether treatment with dasatinib results in exclusion of PKCδ from the nucleus in cells treated with H2O2. ParC5 cells were transfected with pGFP-PKCδ and treated with H2O2 alone or in combination with dasatinib, and cells with nuclear PKCδ were quantified. Nuclear localization of GFP-PKCδ was observed in 27% of ParC5 cells treated with H2O2 for 30 min and 56% of cells treated for 60 min. Nuclear accumulation of GFP-PKCδ was substantially reduced in cells pretreated with dasatinib prior to the addition of H2O2 (15% at 30 min) and (25% at 60 min) (Fig. 4A). We have shown previously that tyrosine phosphorylation results in a conformational change in the kinase, exposing the binding site for importin-α (12). To determine whether dasatinib suppression of nuclear accumulation of GFP-PKCδ is because of reduced importin-α binding, GFP-PKCδ was immunoprecipitated and probed for bound importin-α. Whereas treatment with H2O2 increased importin-α binding to PKCδ, this was dramatically reduced in cells pretreated with dasatinib (Fig. 4B, left). Likewise, pretreatment with the c-Abl-selective inhibitor, imatinib, also substantially reduced H2O2 induced binding of importin-α (Fig. 4B, right).
FIGURE 4.

Dasatinib and imatinib prevent nuclear translocation of PKCδ. A, ParC5 cells were transfected with pGFP-PKCδWT. Transfected cells were left untreated or treated with 5 mm H2O2 for the indicated times with or without pretreatment with 20 nm dasatinib 30 min. Treated cells were fixed in 2% paraformaldehyde, and GFP or DAPI fluorescence was determined using fluorescence microscopy. Shown are representative images for GFP (top) DAPI (middle), and merged (bottom) taken at ×40 magnification. Quantification of fluorescence is shown on the right and represents the average values from two separate experiments plus the S.D. (error bars; >200 cells quantified per condition); * indicates a significant difference compared with cells treated with H2O2 alone (p < 0.05, t test). Black bars represent data from cells treated with H2O2 alone; white bars represent data from cells treated with the combination of H2O2 and dasatinib. B, 293T cells were transfected with pGFP-PKCδWT and treated with H2O2 with or without pretreatment with 20 nM dasatinib (left) or 1 micromolar imatinb (right) for 30 min. GFP-PKCδ was immunoprecipitated (IP) from whole cell lysates (WCL) using an anti-GFP antibody or a rabbit control IgG. Immunocomplexes were separated by SDS-PAGE and analyzed by immunoblotting for importin-α. Immunoprecipitation efficiency was determined by probing with an anti-PKCδ antibody. Whole cell lysate was probed for GFP-PKCδ and importin-α. The blots for the control IgG and GFP lanes were taken from the same experiment. MW, molecular mass.
The studies above suggest that PKCδ-dependent apoptotic signaling, and possibly apoptosis, can be targeted by inhibition of specific tyrosine kinases through the use of specific TKIs. To explore this, we first utilized ParC5 cells that express two unique shRNAs against c-Src, c-Abl, or a scrambled control shRNA. ParC5 cells depleted of c-Src or c-Abl were treated with IR or the DNA damaging agent etoposide, and activation of caspase-3 was assayed (Fig. 5, A–D). When ParC5 cells depleted of c-Src or c-Abl were treated with etoposide there was up to a 50% reduction in caspase-3 activation compared with cells expressing the scrambled control shRNA (Fig. 5, A and B). Similar results were seen when c-Src- or c-Abl-depleted cells were exposed to IR (Fig. 5, C and D). These studies confirm and extend previous work from our laboratory by demonstrating that c-Src and c-Abl play an essential role in DNA damage-induced apoptosis through phosphorylation of PKCδ at Tyr-64 and Tyr-155 (11, 12).
FIGURE 5.

Inhibition of c-Src or c-Abl blocks apoptosis in vitro. ParC5 cells stably expressing nontargeting shRNA (NT) or two unique shRNAs against c-Src (A and C) or c-Abl (B and D) were treated with 50 μm etoposide (A and B) and harvested 6 h later. C and D, cells were treated with 10 Gy of γ-irradiation and harvested 18 h later. Also shown is an immunoblot to demonstrate knockdown of c-Src and c-Abl. E, ParC5 cells were left untreated (NT) or treated with up to 10 Gy of γ-irradiation with or without pretreatment with 20 nm dasatinib for 30 min. Cells were harvested 18 h later. Caspase-3 activity was assayed as described under “Experimental Procedures.” The data represent the specific activity of caspase-3 and are the average of duplicate measurements from triplicate samples from a representative experiment plus the S.D. (error bars). Each experiment was repeated a minimum of three times. For A–D, asterisks indicate a significant difference compared with cells expressing the nontargeting shRNA (*, p < 0.05, t test; **, p < 0.005, t test). Black bars represent data from untreated cells, whereas white bars represent data from cells treated with etoposide (A and B) or radiation (C and D). For E,* indicates a significant difference compared with cells treated with IR only (p < 0.05, t test). Black bars represent data from irradiated cells, whereas white bars represent data from cells pretreated with dasatinib, prior to radiation.
To determine the potential use of TKIs to protect against salivary gland damage, we asked whether treatment with dasatinib could suppress apoptosis either in vitro or in mice exposed to head and neck IR. In vitro pretreatment with dasatinib suppressed apoptosis by 80% in cells treated with 10 Gy γ-irradiation (Fig. 5E). For the in vivo analysis, dasatinib was administered via oral gavage 60 min before and again 3 h after exposure to IR. Dasatinib treatment reduced IR-induced apoptosis by >60% in the parotid salivary gland, as measured by analysis of caspase-3 activation (Fig. 6). These data demonstrate a potential novel use for TKIs as a prophylactic strategy for protection of the salivary gland and possibly other oral tissues in head and neck cancer patients receiving radiation therapy.
FIGURE 6.
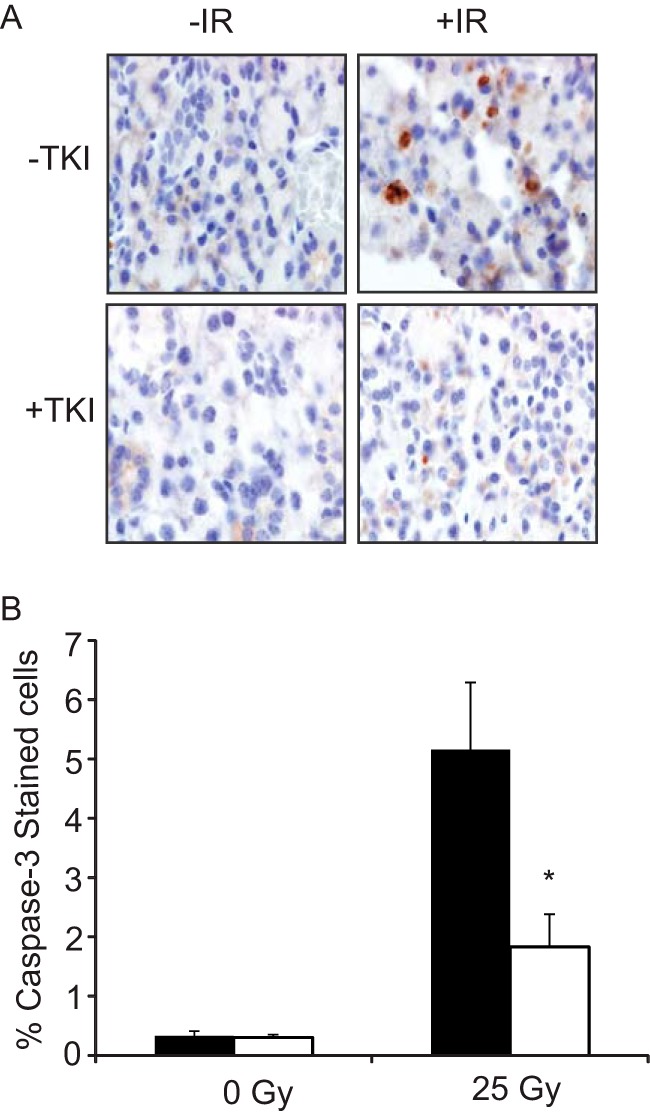
Dasatinib inhibits radiation-induced apoptosis in vivo. A, mice were irradiated to the head and neck with 25 Gy of γ-irradiation with or without the application of dasatinib (20 mg/kg) by oral gavage 1 h before and 3 h after IR. Salivary glands were removed, sectioned, and stained for activated caspase-3 as described under “Experimental Procedures.” Shown is a representative image for each condition (magnification, ×60). B, positively stained cells were quantified and expressed as the percentage of caspase-3-positive/total cells; >3000 cells were counted per condition. Data shown represent the average value plus the S.E. (error bars) (n = 7); * indicates a significant difference compared with mice treated with IR only (p < 0.05, t test).
DISCUSSION
Radiation therapy for head and neck cancer patients can result in extensive and permanent damage to adjacent healthy tissues including the salivary gland. Therapeutics designed to protect the salivary gland, such as the free radical scavenger amifostine, have limited efficacy and significant toxicity (32). Our previous studies have identified tyrosine phosphorylation of PKCδ as a rate-limited step in IR-induced apoptosis in the salivary gland (11). Here we show that TKIs effective against c-Src and c-Abl are able to block multiple key regulatory steps necessary for PKCδ nuclear localization leading to suppression of apoptosis both in vitro and in vivo. Our data suggest that some TKIs, currently used in the clinic for treatment of cancer, may also be useful for protection of nontumor tissues in patients undergoing radiotherapy of the head and neck.
Our studies demonstrate a role for the tyrosine kinases c-Src and c-Abl in tyrosine phosphorylation and in turn activation of PKCδ in response to apoptotic stimuli. Specifically, we define c-Abl as the Tyr-155 kinase. Phosphorylation of PKCδ at Tyr-155 is blocked by the c-Abl inhibitors dasatinib and imatinib and by c-Abl depletion using shRNA. Additionally, expression of the c-Abl gatekeeper mutant (T315I) can specifically restore phosphorylation at Tyr-155 in the presence of dasatinib. Our data support and extend previous studies that have reported a functional relationship between c-Abl and PKCδ in response to genotoxic and oxidative stress (33–35). Both Yuan et al. (34) and Sun et al. (33) have shown that IR and H2O2 induce phosphorylation of PKCδ by c-Abl, although the specific tyrosine phosphorylated was not identified. Intriguingly, a recent report showed that hyperglycemia induced apoptosis of neural progenitor cells occurs through a PKCδ·c-Abl-dependent mechanism that involves tyrosine phosphorylation of PKCδ and nuclear translocation of the PKCδ·c-Abl complex (36).
Previous reports have demonstrated a role for members of the Src family of non-receptor tyrosine kinases in the regulation of PKCδ (21, 37, 38). For instance, phosphorylation of PKCδ by c-Src has been shown to control its degradation and activation (23, 39, 40). c-Src and PKCδ also mediate signal transduction through growth factor receptors, including platelet-derived growth factor receptor, extracellular growth factor receptor, and the insulin receptor (24, 41). In addition, c-Src dependent tyrosine phosphorylation has been shown to regulate the pro-apoptotic function of PKCδ in neuronal cells and in response to ceramide and chemotherapeutic drugs (21, 38, 42). Our studies strongly support a role for c-Src as the PKCδ Tyr-64 kinase. We show that stable depletion of c-Src using shRNA blocks Tyr-64 phosphorylation and that expression of the Src T341I gatekeeper mutant specifically restores Tyr-64 phosphorylation in the presence of dasatinib (Figs. 1C and 3B). Unexpectedly, inhibition or depletion of c-Abl can also partially reduce phosphorylation at Tyr-64 independent of suppression of Src. This is explained at least in part by our findings that Tyr-155 phosphorylation by c-Abl facilitates phosphorylation of Tyr-64. However, c-Abl may also be able to directly phosphorylate the Tyr-64 site as some phosphorylation of Tyr-64 is recovered in dasatinib-treated cells that express the c-Abl gatekeeper mutation, under conditions where c-Src activity is inhibited (Fig. 3B).
We have shown previously that phosphorylation of Tyr-64 and Tyr-155 promotes a conformational change in PKCδ leading to its association with importin-α and subsequent nuclear translocation (12). Our current data suggest a hierarchical relationship between Tyr-155 and Tyr-64, where phosphorylation of Tyr-155 by c-Abl serves as a permissive signal for phosphorylation of Tyr-64. These data support a model in which phosphorylation of PKCδ at Tyr-155 presumably leads to a structural change that makes Tyr-64 more accessible to phosphorylation by c-Src. Notably, phosphorylation of the PKCδ Y155F mutant at Tyr-64 was completely lost in cells treated with etoposide, but only reduced in cells treated with H2O2 (Fig. 2, A and B). This is consistent with a partial loss of protein structure in the presence of H2O2 but not etoposide (28, 43). Sequential phosphorylation of PKCδ by c-Abl and c-Src is likely to be critical to cellular homeostasis. Because c-Abl is a damage-induced tyrosine kinase, this assures that pro-apoptotic signaling by PKCδ is coordinated with activation of other cell death signals. Furthermore, this model explains how c-Src, which regulates many survival/proliferation signals, may also play a role in apoptosis. Based on this relationship, the kinase activity of c-Abl and/or c-Src provides a logical target for the disruption of the pro-apoptotic function of PKCδ.
TKIs have been developed against many kinases required for cancer cell proliferation, including members of the Src family and c-Abl (27). Here we explored the novel use of these inhibitors as prophylactic agents to prevent IR-induced cell death within the salivary gland. We show that pretreatment with dasatinib, which inhibits SFKs and c-Abl, is sufficient to suppress activation and nuclear translocation of PKCδ. Further, a 20-mg/kg dose in our mouse model equates to a serum concentration that is easily obtained in patient populations (44, 45). Expectedly, pretreatment with the c-Abl-selective inhibitor imatinib also blocked nuclear translocation, as evidenced by reduced binding of importin-α to PKCδ following H2O2 treatment. Importantly, treatment of mice with dasatinib significantly reduced IR-induced salivary gland apoptosis. Taken together our in vitro and in in vivo data suggest that TKIs can offer radioprotection within the salivary gland by inhibiting the activation of PKCδ. These findings support our previous studies which showed that IR-induced apoptosis in the salivary gland is significantly reduced in PKCδ−/− mice (46) and recent studies by Ovitt and co-workers which demonstrate that depletion of PKCδ within the mouse salivary gland using nanoparticle delivered PKCδ siRNA protects against IR-induced loss of salivary gland function (47). Pabla et al. have also recently shown that PKCδ inhibition leads to a reduction in nephrotoxicity caused by cisplatin with no effect on the efficaciousness of cisplatin (38).
Our findings provide a rationale for future clinical trials to investigate TKIs as a radioprotective therapy to preserve salivary function in patients being treated for head and neck cancer. In the context of IR treatment, our studies suggest that short term dosing with a TKI may be sufficient. Most patients receive fractionated IR over the course of a few months, thus treatment with TKIs would likely be limited to that time frame. An important concern with all radioprotective treatments is that they do not promote tumor growth or hamper tumor therapy. Although few TKIs have been tested in patients with head and neck cancer, phase I/II clinical trials with dasatinib showed no enhancement of tumor growth or progression (48). At the same time this study failed to demonstrate activity of dasatinib as a single agent against head and neck cancer. However, a report by Lin et al. shows that IR can be combined with dasatinib to further sensitize head and neck squamous cell carcinoma cells (49). Therefore, it can be suggested that dasatinib will protect salivary glands while if anything promoting apoptosis of tumor tissue in patients receiving IR treatment for head and neck cancer. Finally, our studies may have more far reaching implications for protection of other nontumor tissues in patients undergoing IR or some types of chemotherapy.
Acknowledgments
We thank David Jones, Hannah Edlin, Angela Ohm, Vadym Zaberezhnyy, and Andrew Lewis for intellectual contributions and technical help.
This work was supported, in whole or in part, by National Institutes of Health Grants 5R01DE015648 (to M. E. R.), 1R01CA180175 (to J. D.), R01CA138482 (to S. M. A.), and P30CA046934 (Cancer Center Support; DNA Sequencing, Tissue Culture, Histology and Radiation Cores).
- IR
- ionizing radiation
- Gy
- Gray
- H2O2
- hydrogen peroxide
- SFK
- Src family kinase
- TKI
- tyrosine kinase inhibitor.
REFERENCES
- 1. Vissink A., Mitchell J. B., Baum B. J., Limesand K. H., Jensen S. B., Fox P. C., Elting L. S., Langendijk J. A., Coppes R. P., Reyland M. E. (2010) Clinical management of salivary gland hypofunction and xerostomia in head-and-neck cancer patients: successes and barriers. Int. J. Radiat. Oncol. Biol. Phys. 78, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vissink A., Jansma J., Spijkervet F. K., Burlage F. R., Coppes R. P. (2003) Oral sequelae of head and neck radiotherapy. Crit. Rev. Oral Biol. Med. 14, 199–212 [DOI] [PubMed] [Google Scholar]
- 3. Yoshida K., Yamaguchi T., Shinagawa H., Taira N., Nakayama K. I., Miki Y. (2006) Protein kinase Cδ activates topoisomerase IIα to induce apoptotic cell death in response to DNA damage. Mol. Cell. Biol. 26, 3414–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haughian J. M., Jackson T. A., Koterwas D. M., Bradford A. P. (2006) Endometrial cancer cell survival and apoptosis is regulated by protein kinase C α and δ. Endocr. Relat. Cancer 13, 1251–1267 [DOI] [PubMed] [Google Scholar]
- 5. Yamanouchi D., Kato K., Ryer E. J., Zhang F., Liu B. (2010) Protein kinase Cδ mediates arterial injury responses through regulation of vascular smooth muscle cell apoptosis. Cardiovasc. Res. 85, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bright R., Raval A. P., Dembner J. M., Pérez-Pinzón M. A., Steinberg G. K., Yenari M. A., Mochly-Rosen D. (2004) Protein kinase Cδ mediates cerebral reperfusion injury in vivo. J. Neurosci. 24, 6880–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lasfer M., Davenne L., Vadrot N., Alexia C., Sadji-Ouatas Z., Bringuier A. F., Feldmann G., Pessayre D., Reyl-Desmars F. (2006) Protein kinase PKCδ and c-Abl are required for mitochondrial apoptosis induction by genotoxic stress in the absence of p53, p73 and Fas receptor. FEBS Lett. 580, 2547–2552 [DOI] [PubMed] [Google Scholar]
- 8. Humphries M. J., Limesand K. H., Schneider J. C., Nakayama K. I., Anderson S. M., Reyland M. E. (2006) Suppression of apoptosis in the protein kinase Cδ-null mouse in vivo. J. Biol. Chem. 281, 9728–9737 [DOI] [PubMed] [Google Scholar]
- 9. DeVries-Seimon T. A., Ohm A. M., Humphries M. J., Reyland M. E. (2007) Induction of apoptosis is driven by nuclear retention of protein kinase Cδ. J. Biol. Chem. 282, 22307–22314 [DOI] [PubMed] [Google Scholar]
- 10. DeVries T. A., Neville M. C., Reyland M. E. (2002) Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 21, 6050–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Humphries M. J., Ohm A. M., Schaack J., Adwan T. S., Reyland M. E. (2008) Tyrosine phosphorylation regulates nuclear translocation of PKCδ. Oncogene 27, 3045–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adwan T. S., Ohm A. M., Jones D. N., Humphries M. J., Reyland M. E. (2011) Regulated binding of importin-α to protein kinase Cδ in response to apoptotic signals facilitates nuclear import. J. Biol. Chem. 286, 35716–35724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reyland M. E., Anderson S. M., Matassa A. A., Barzen K. A., Quissell D. O. (1999) Protein kinase Cδ is essential for etoposide-induced apoptosis in salivary gland acinar cells. J. Biol. Chem. 274, 19115–19123 [DOI] [PubMed] [Google Scholar]
- 14. Allen-Petersen B. L., Carter C. J., Ohm A. M., Reyland M. E. (2014) Protein kinase Cδ is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene 13, 1306–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leitges M., Mayr M., Braun U., Mayr U., Li C., Pfister G., Ghaffari-Tabrizi N., Baier G., Hu Y., Xu Q. (2001) Exacerbated vein graft arteriosclerosis in protein kinase Cδ-null mice. J. Clin. Invest. 108, 1505–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Banninger G. P., Cha S., Said M. S., Pauley K. M., Carter C. J., Onate M., Pauley B. A., Anderson S. M., Reyland M. E. (2011) Loss of PKCδ results in characteristics of Sjögren's syndrome including salivary gland dysfunction. Oral. Dis. 17, 601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reyland M. E. (2009) Protein kinase C isoforms: multi-functional regulators of cell life and death. Front. Biosci. 14, 2386–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H., Xiao L., Kazanietz M. G. (2011) p23/Tmp21 associates with protein kinase Cδ (PKCδ) and modulates its apoptotic function. J. Biol. Chem. 286, 15821–15831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gomel R., Xiang C., Finniss S., Lee H. K., Lu W., Okhrimenko H., Brodie C. (2007) The localization of protein kinase Cδ in different subcellular sites affects its proapoptotic and antiapoptotic functions and the activation of distinct downstream signaling pathways. Mol. Cancer Res. 5, 627–639 [DOI] [PubMed] [Google Scholar]
- 20. Chan C. M., Jing X., Pike L. A., Zhou Q., Lim D. J., Sams S. B., Lund G. S., Sharma V., Haugen B. R., Schweppe R. E. (2012) Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin. Cancer Res. 18, 3580–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kajimoto T., Sawamura S., Tohyama Y., Mori Y., Newton A. C. (2010) Protein kinase Cδ-specific activity reporter reveals agonist-evoked nuclear activity controlled by Src family of kinases. J. Biol. Chem. 285, 41896–41910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paugh B. S., Paugh S. W., Bryan L., Kapitonov D., Wilczynska K. M., Gopalan S. M., Rokita H., Milstien S., Spiegel S., Kordula T. (2008) EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCδ, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 22, 455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rybin V. O., Guo J., Gertsberg Z., Elouardighi H., Steinberg S. F. (2007) Protein kinase Cϵ (PKCϵ) and Src control PKCδ activation loop phosphorylation in cardiomyocytes. J. Biol. Chem. 282, 23631–23638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amos S., Martin P. M., Polar G. A., Parsons S. J., Hussaini I. M. (2005) Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cδ/c-Src pathways in glioblastoma cells. J. Biol. Chem. 280, 7729–7738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson S. M., Reyland M. E., Hunter S., Deisher L. M., Barzen K. A., Quissell D. O. (1999) Etoposide-induced activation of c-Jun N-terminal kinase (JNK) correlates with drug-induced apoptosis in salivary gland acinar cells. Cell Death Differ. 6, 454–462 [DOI] [PubMed] [Google Scholar]
- 26. Rybin V. O., Guo J., Gertsberg Z., Feinmark S. J., Steinberg S. F. (2008) Phorbol 12-myristate 13-acetate-dependent protein kinase Cδ–Tyr-311 phosphorylation in cardiomyocyte caveolae. J. Biol. Chem. 283, 17777–17788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levitzki A. (2013) Tyrosine kinase inhibitors: views of selectivity, sensitivity, and clinical performance. Annu. Rev. Pharmacol. Toxicol. 53, 161–185 [DOI] [PubMed] [Google Scholar]
- 28. Lin D., Takemoto D. J. (2005) Oxidative activation of protein kinase Cγ through the C1 domain. Effects on gap junctions. J. Biol. Chem. 280, 13682–13693 [DOI] [PubMed] [Google Scholar]
- 29. Nam S., Kim D., Cheng J. Q., Zhang S., Lee J. H., Buettner R., Mirosevich J., Lee F. Y., Jove R. (2005) Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 65, 9185–9189 [DOI] [PubMed] [Google Scholar]
- 30. Hantschel O., Rix U., Superti-Furga G. (2008) Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk. Lymphoma 49, 615–619 [DOI] [PubMed] [Google Scholar]
- 31. Azam M., Seeliger M. A., Gray N. S., Kuriyan J., Daley G. Q. (2008) Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 15, 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soref C. M., Hacker T. A., Fahl W. E. (2012) A new orally active, aminothiol radioprotector-free of nausea and hypotension side effects at its highest radioprotective doses. Int. J. Radiat. Oncol. Biol. Phys. 82, e701–7 [DOI] [PubMed] [Google Scholar]
- 33. Sun X., Majumder P., Shioya H., Wu F., Kumar S., Weichselbaum R., Kharbanda S., Kufe D. (2000) Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J. Biol. Chem. 275, 17237–17240 [DOI] [PubMed] [Google Scholar]
- 34. Yuan Z. M., Utsugisawa T., Ishiko T., Nakada S., Huang Y., Kharbanda S., Weichselbaum R., Kufe D. (1998) Activation of protein kinase Cδ by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene 16, 1643–1648 [DOI] [PubMed] [Google Scholar]
- 35. Majumder P. K., Mishra N. C., Sun X., Bharti A., Kharbanda S., Saxena S., Kufe D. (2001) Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ. 12, 465–470 [PubMed] [Google Scholar]
- 36. Gonfloni S., Maiani E., Di Bartolomeo C., Diederich M., Cesareni G. (2012) Oxidative stress, DNA damage, and c-Abl signaling: at the crossroad in neurodegenerative diseases? Int. J. Cell Biol. 2012, 683097–683107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chari R., Kim S., Murugappan S., Sanjay A., Daniel J. L., Kunapuli S. P. (2009) Lyn, PKCδ, SHIP-1 interactions regulate GPVI-mediated platelet-dense granule secretion. Blood 114, 3056–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pabla N., Dong G., Jiang M., Huang S., Kumar M. V., Messing R. O., Dong Z. (2011) Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Invest. 121, 2709–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blake R. A., Garcia-Paramio P., Parker P. J., Courtneidge S. A. (1999) Src promotes PKCδ degradation. Cell Growth Differ. 10, 231–241 [PubMed] [Google Scholar]
- 40. Rosenzweig T., Aga-Mizrachi S., Bak A., Sampson S. R. (2004) Src tyrosine kinase regulates insulin-induced activation of protein kinase C (PKC) δ in skeletal muscle. Cell. Signal. 16, 1299–1308 [DOI] [PubMed] [Google Scholar]
- 41. Saito S., Frank G. D., Mifune M., Ohba M., Utsunomiya H., Motley E. D., Inagami T., Eguchi S. (2002) Ligand-independent trans-activation of the platelet-derived growth factor receptor by reactive oxygen species requires protein kinase Cδ and c-Src. J. Biol. Chem. 277, 44695–44700 [DOI] [PubMed] [Google Scholar]
- 42. Kaul S., Anantharam V., Yang Y., Choi C. J., Kanthasamy A., Kanthasamy A. G. (2005) Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cδ in dopaminergic neuronal cells. J. Biol. Chem. 280, 28721–28730 [DOI] [PubMed] [Google Scholar]
- 43. Korichneva I., Hoyos B., Chua R., Levi E., Hammerling U. (2002) Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J. Biol. Chem. 277, 44327–44331 [DOI] [PubMed] [Google Scholar]
- 44. Furmanski B. D., Hu S., Fujita K., Li L., Gibson A. A., Janke L. J., Williams R. T., Schuetz J. D., Sparreboom A., Baker S. D. (2013) Contribution of ABCC4-mediated gastric transport to the absorption and efficacy of dasatinib. Clin. Cancer Res. 19, 4359–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Demetri G. D., Lo Russo P., MacPherson I. R., Wang D., Morgan J. A., Brunton V. G., Paliwal P., Agrawal S., Voi M., Evans T. R. (2009) Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin. Cancer Res. 15, 6232–6240 [DOI] [PubMed] [Google Scholar]
- 46. Arany S., Xu Q., Hernady E., Benoit D. S., Dewhurst S., Ovitt C. E. (2012) Pro-apoptotic gene knockdown mediated by nanocomplexed siRNA reduces radiation damage in primary salivary gland cultures. J. Cell. Biochem. 113, 1955–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arany S., Benoit D. S., Dewhurst S., Ovitt C. E. (2013) Nanoparticle-mediated gene silencing confers radioprotection to salivary glands in vivo. Mol. Ther. 21, 1182–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brooks H. D., Glisson B. S., Bekele B. N., Johnson F. M., Ginsberg L. E., El-Naggar A., Culotta K. S., Takebe N., Wright J., Tran H. T., Papadimitrakopoulou V. A. (2011) Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer 117, 2112–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin Y. C., Wu M. H., Wei T. T., Chuang S. H., Chen K. F., Cheng A. L., Chen C. C. (2012) Degradation of epidermal growth factor receptor mediates dasatinib-induced apoptosis in head and neck squamous cell carcinoma cells. Neoplasia 14, 463–475 [DOI] [PMC free article] [PubMed] [Google Scholar]