

Background: ATP sulfurylase catalyzes the energetically unfavorable formation of adenosine 5′-phosphosulfate in plant sulfur assimilation.
Results: Structural and kinetic analyses identifies key active site residues.
Conclusion: A reaction mechanism involving distortion of nucleotide conformation and stabilizing interactions is proposed.
Significance: These results provide the first molecular insights on a plant ATP sulfurylase and the committed step of plant sulfur assimilation.
Keywords: Enzyme Mechanisms, Enzyme Structure, Plant, Plant Biochemistry, Protein Structure
Abstract
Enzymes of the sulfur assimilation pathway are potential targets for improving nutrient content and environmental stress responses in plants. The committed step in this pathway is catalyzed by ATP sulfurylase, which synthesizes adenosine 5′-phosphosulfate (APS) from sulfate and ATP. To better understand the molecular basis of this energetically unfavorable reaction, the x-ray crystal structure of ATP sulfurylase isoform 1 from soybean (Glycine max ATP sulfurylase) in complex with APS was determined. This structure revealed several highly conserved substrate-binding motifs in the active site and a distinct dimerization interface compared with other ATP sulfurylases but was similar to mammalian 3′-phosphoadenosine 5′-phosphosulfate synthetase. Steady-state kinetic analysis of 20 G. max ATP sulfurylase point mutants suggests a reaction mechanism in which nucleophilic attack by sulfate on the α-phosphate of ATP involves transition state stabilization by Arg-248, Asn-249, His-255, and Arg-349. The structure and kinetic analysis suggest that ATP sulfurylase overcomes the energetic barrier of APS synthesis by distorting nucleotide structure and identifies critical residues for catalysis. Mutations that alter sulfate assimilation in Arabidopsis were mapped to the structure, which provides a molecular basis for understanding their effects on the sulfur assimilation pathway.
Introduction
In plants, the uptake of sulfate and its assimilation provide an essential nutrient for the synthesis of a diverse set of metabolites, including cysteine, methionine, glutathione, iron-sulfur clusters, vitamin cofactors like biotin and thiamin, and multiple specialized metabolites such as glucosinolates (1–8). The central role of sulfur metabolism in plants and its integration with carbon and nitrogen metabolism and the biosynthesis of multiple specialized metabolites have led to efforts to understand this critical pathway for applications in nutritional enhancement for food and feed and for improved plant growth under environmental stresses (2, 9–15). Utilizing this environmental source of sulfur requires the enzymatic conversion of sulfate into chemical forms that are energetically favorable for reduction.
The first enzymatic step of plant sulfur assimilation is the nonreductive adenylation of sulfate by ATP sulfurylase (ATP:sulfate adenylyltransferase; EC 2.7.7.4), which catalyzes the formation of adenosine 5′-phosphosulfate (APS)5 and inorganic pyrophosphate (PPi) from sulfate and ATP (Fig. 1A) (16, 17). Production of APS by ATP sulfurylase, which is energetically unfavorable compared with the reverse reaction, is the committed step in plant sulfur assimilation and yields a high energy phosphosulfate bond that drives subsequent metabolic steps in the pathway (17–19). Reduction of APS to sulfite and then sulfide completes the sulfur assimilation pathway and supports cysteine biosynthesis (1, 4–8). APS can also be phosphorylated to 3′-phosphoadenosine 5′-phosphosulfate (PAPS) by APS kinase to provide a substrate for sulfonation reactions in plant primary and specialized metabolic pathways (20–27).
FIGURE 1.
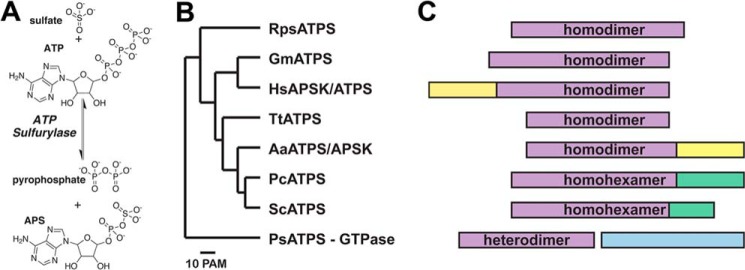
ATP sulfurylase domain structure and oligomerization. A, overall reaction catalyzed by ATP sulfurylase. B, dendrogram of ATP sulfurylase sequences from a bacterial symbiont of hydrothermal vent tubeworm R. pachyptila (RpsATPS), GmATPS, human APSK (HsAPSK/ATPS or PAPS synthetase), T. thermophilus (TtATPS), A. aeolicus (AaATPS/APSK), P. chrysogenum (PcATPS), S. cerevisiae (ScATPS), and P. syringae (PsATPS). The dendrogram was generated from multiple sequence alignment of ATP sulfurylases using the MultAlin webpage (69). C, domain structure of each protein is shown with regions corresponding to ATP sulfurylase, APS kinase, APS kinase-like, and GTPase colored purple, yellow, green, and light blue, respectively.
Multigene families encode ATP sulfurylase in the plant species examined to date, including Arabidopsis thaliana (thale cress), Solanum tuberosum (potato), Brassica juncea (Indian mustard), Glycine max (soybean), and Camellia sinensis (tea plant) (5, 18, 19, 28–34). ATP sulfurylase isoforms are localized to different organelles with the chloroplast form associated with reductive sulfate assimilation and accounting for the highest portion of enzyme activity (35, 36). For example, soybean contains four isoforms (GmATPS1–4), all of which are predicted to localize to the chloroplast (5). Expression of ATP sulfurylase is highest in young leaves, the early stages of seed development, and the elongation zone of roots (19, 35–38). Traditionally, ATP sulfurylase has not been considered to be important for the control of flux through the sulfate assimilation pathway (39). The plastidic forms of ATP sulfurylase, however, are targeted by sulfur deprivation-inducible microRNA miR395, which increases translocation of sulfate from the roots to the shoots (40, 41). Accordingly, disruption of the ATPS1 isoform of ATP sulfurylase in Arabidopsis results in increased accumulation of sulfate in the leaves (41). Thus, ATP sulfurylase contributes significantly to the control of plant sulfur metabolism; however, the molecular basis for its function remains poorly understood.
ATP sulfurylase is also important in a diverse range of organisms for sulfate activation and assimilation; however, the structural organization of the enzyme varies widely in both domain architecture and oligomerization (Fig. 1, B and C). Some prokaryotic forms of ATP sulfurylase, including those from Escherichia coli and Pseudomonas syringae, are heterodimeric proteins composed of a GTPase subunit and a catalytic ATP sulfurylase subunit (42). In other prokaryotes, including the thermophiles Aquifex aeolicus and Thermus thermophilus, the purple sulfur bacterium Allochromatium vinosum, and a bacterial symbiont of the vent tubeworm Riftia pachyptila, ATP sulfurylase functions either as a monofunctional homodimeric enzyme or a bi-functional homodimeric ATP sulfurylase/APS kinase, also known as PAPS synthetase (43–46). In fungi, such as Saccharomyces cerevisiae and Penicillium chrysogenum, ATP sulfurylase is a homohexamer in which each monomer contains an N-terminal ATP sulfurylase domain and a C-terminal APS kinase-like domain (47–49). The APS kinase-like domain can also function as an allosteric regulatory region in response to PAPS, as shown for the P. chrysogenum enzyme (50). The human PAPS synthetase operates as a homodimeric enzyme containing distinct ATP sulfurylase and APS kinase domains (51). Sequence comparisons and biochemical analyses suggest that the plant ATP sulfurylases are most closely related to the human ATP sulfurylase domain of PAPS synthetase and function as homodimeric proteins (19, 52). To date, no structural information is available for an ATP sulfurylase from any plant species to provide insight on how this enzyme functions as the committed step in plant sulfur assimilation.
EXPERIMENTAL PROCEDURES
Materials
All oligonucleotides used for site-directed mutagenesis were synthesized by Integrated DNA Technologies, Inc. Ni2+-nitrilotriacetic acid was from Qiagen. The HiLoad 26/60 Superdex-200 FPLC column was purchased from GE Healthcare. All other reagents were of ACS grade or better and were purchased from Sigma.
Protein Expression, Purification, and Site-directed Mutagenesis
The pET-28a-GmATPSΔ48 bacterial expression construct, which encodes G. max ATP sulfurylase isoform 1 (Glyma10g38760a) lacking the plastid localization sequence (residues 1–48) and with an N-terminal hexahistidine tag, was previously described (19). E. coli Rosetta (DE3) transformed with pET-28a-GmATPSΔ48 were grown at 37 °C in Terrific broth containing kanamycin (50 μg ml−1) and chloramphenicol (34 μg ml−1) until A600 nm ∼0.8. Following induction of protein expression with 1 mm isopropyl 1-thio-β-d-galactopyranoside, the cultures were grown at 20 °C for 8 h. Cells were pelleted by centrifugation (10,000 × g) and resuspended in lysis buffer (50 mm Tris (pH 8.0), 500 mm NaCl, 20 mm imidazole, 10% glycerol, 1% Tween 20, and 1 mm β-mercaptoethanol). After sonication and centrifugation, the supernatant was passed over a Ni2+-nitrilotriacetic acid column equilibrated with lysis buffer. Unbound protein was eluted with wash buffer (lysis buffer without Tween 20), and His-tagged GmATPS was eluted with elution buffer (wash buffer containing 250 mm imidazole). Eluted protein was loaded onto a Superdex-200 FPLC column equilibrated with 25 mm HEPES (pH 7.5), 150 mm NaCl, and 1 mm dl-dithiothreitol. Peak fractions were collected and concentrated to 12.5 mg ml−1 using centrifugal concentrators (Amicon). Purity was assessed by SDS-PAGE. Purified protein was flash-frozen in liquid nitrogen and stored at −80 °C. Protein concentration was determined by the Bradford method (Protein Assay, Bio-Rad) with BSA as standard. Site-directed mutants of GmATPS (F245L, F245A, Q246E, Q246N, Q246A, R248K, N249D, N249A, H252Q, H252N, H252A, H255Q, H255N, H255A, L258V, L258A, H333Q, H333N, H333A, and R349K) were generated using the QuikChange PCR method (Agilent) with expression and purification as described for wild-type GmATPS.
Protein Crystallography
Crystals of GmATPS in complex with APS were grown at 4 °C in hanging drops with a 1:1 ratio of protein and crystallization buffer (0.1 m HEPES (pH 7.25), 18% PEG-8000, and 0.2 m NaCl) supplemented with 5 mm APS. For x-ray data collection, crystals were transferred to a cryoprotectant solution of mother liquor containing 20% ethylene glycol and flash-frozen in liquid nitrogen. X-ray diffraction data (0.5° oscillations; 360 images; 100 K) were collected at SBC beamline 19-ID of the Advanced Photon Source, Argonne National Laboratory. The HKL3000 software suite was used to integrate, merge, and scale diffraction intensities (53). The GmATPS structure was solved by molecular replacement using the structure of the ATP sulfurylase domain of human PAPS synthetase (residues 234–624; 59% sequence similarity; Protein Data Bank code 1XNJ (51)) with ligands and water molecules removed as a search model. Molecular replacement was implemented with PHASER (54) and yielded an asymmetric unit with eight molecules with translation search Z-scores ranging from 18 to 23. The structural model of GmATPS was built in COOT (55) and refined using PHENIX (56). Initial model building and refinement used noncrystallographic symmetry restraints, which were released in later rounds. A translation-libration-screen model, in which each protein chain was treated as a single rigid group, was used throughout refinement. Iterative rounds of model building and refinement were continued until the R-factors converged to those reported in Table 1. Coordinates and structure factors for the GmATPS·APS complex were deposited in the Protein Data Bank (Protein Data Bank code 4MAF).
TABLE 1.
Summary of crystallographic statistics for the GmATPS·APS complex
| Crystal | |
| Space group | C222 |
| Cell dimensions | a = 204.3 Å, b = 230.8 Å, c = 159.2 Å |
| Data collection | |
| Wavelength | 0.979 Å |
| Resolution range (highest shell resolution) | 48.2 to 2.48 Å (2.52 to 2.48 Å) |
| Reflections (total/unique) | 686,045/123,420 |
| Completeness (highest shell) | 93.6% (93.7%) |
| 〈I/σ〉 (highest shell) | 18.0 (2.5) |
| Rsyma (highest shell) | 9.4% (51.5%) |
| Model and refinement | |
| Rcrystb/Rfreec | 17.0/21.8 |
| No. of protein atoms | 25,454 |
| No. of water molecules | 825 |
| No. of ligand atoms | 216 |
| r.m.s.d. bond lengths | 0.008 Å |
| r.m.s.d. bond angles | 1.149° |
| Average B-factor (Å2): protein, waters, and ligands | 49.4, 43.7, and 37.7 |
| Stereochemistry: most favored, allowed, and outlier | 97.8, 2.1, and 0.1% |
a Rsym = Σ|Ih − 〈Ih〉|/ΣIh, where 〈Ih〉 is the average intensity over symmetry.
b Rcryst = Σ|Fo − 〈Fc〉|/ΣFo, where summation is over the data used for refinement.
c Rfree is defined the same as Rcryst but was calculated using 5% of data excluded from refinement.
Steady-state Kinetic Analysis of Wild-type and Mutant GmATPS
Steady-state kinetic analysis was performed using an enzyme-coupled spectrophotometric assay (52). Initial velocities for the reverse (i.e. ATP synthesis) reaction were measured using a Beckman DU800 UV-visible spectrophotometer by observing the rate of change in absorbance of NADP+ at 340 nm (ϵ = 6270 m−1 cm−1) in 0.5-ml systems at 25 °C. Assays contained 50 mm Tris (pH 8.0), 5 mm MgCl2, 1 mm NADP+, 1 mm glucose, 2 units of hexokinase, and 1 unit of glucose-6-phosphate dehydrogenase. Kinetic constants were determined using a matrix of varied substrate concentrations. Assays with wild-type GmATPS and the F245L, F245A, H252N, and H255Q mutants used 2.5–300 μm APS and 2.5–2,000 μm NaPPi. For the Q246N and Q246A mutants, substrate concentrations were 2.5–500 μm APS and 5–2,000 μm NaPPi. For the Q246E mutant, 25–5,000 μm APS and 5–2,000 μm NaPPi were used. Assays of the L258V, L258A, and R248K mutants used 2.5–300 μm APS and 20–5,000 μm NaPPi. Substrate concentrations of 2–500 μm APS and 20–5,000 μm NaPPi were used for assays of the N249A and R248K mutants. The N249D, H255A, and H333Q mutants were assayed using 1–200 μm APS and 2.5–2,000 μm NaPPi. Assays to determine forward reaction (i.e. APS synthesis) kinetics of wild-type and mutant GmATPS contained 50 mm Tris (pH 8.0), 15 mm MgCl2, 100 mm NaCl, 0.2 mm phosphoenolpyruvate, 20 mm KCl, 0.2 mm NADH, 0.05 units of APS kinase, 20 units of pyruvate kinase, and 30 units of lactate dehydrogenase with varied ATP (50–10,000 μm) and Li2SO4 (50–10,000). Calculation of kcat and Km values used SigmaPlot (Systat Software) to fit data to the rate equation for an ordered Bi Bi Reaction 1,
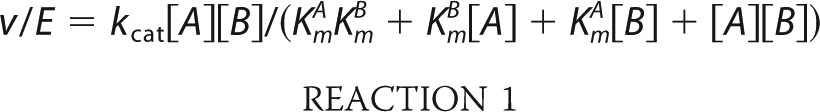 |
Substrate Modeling
Molecular docking experiments were performed by Autodock vina (Version 1.1.2) (57) with standard protocols. Computer docking of substrates into the GmATPS active site used a 30 × 30 × 30 Å grid box with the level of exhaustiveness = 20. For modeling of the forward reaction, ATP and sulfate were manually modeled into the structure of GmATPS as a starting point. The position of sulfate was fixed based on the sulfate moiety in the GmATPS·APS complex structure. Docking of ATP yielded a calculated affinity of −8.6 kcal/mol with r.m.s.d. ranges of 1.35–2.90 Å. For the reverse reaction, the crystallographic position of APS in GmATPS was used, and the initial placement of PPi was based on the crystal structure of yeast ATP sulfurylase in complex with APS and PPi (48). Docking of PPi into the structure yielded a calculated affinity of −4.8 kcal/mol with r.m.s.d. ranges of 0–0.44 Å.
Analysis of Plant Material
Arabidopsis accessions were grown for 5 weeks in controlled environment room under a short day 10-h light/14-h dark cycle at constant temperature of 22°C, 60% relative humidity, and light intensity of 160 microeinsteins s−1 m−2. ATP sulfurylase activity was measured in leaf extracts as the APS- and PPi-dependent formation of ATP and sulfate was determined by HPLC, as described in Kawashima et al. (41).
RESULTS
Overall Three-dimensional Structure of a Plant ATP Sulfurylase
Soybean ATP sulfurylase isoform 1 (GmATPS) was overexpressed in E. coli as an N-terminal His-tagged protein and purified using Ni2+-affinity and size-exclusion chromatography, as described previously (19, 52). Purified GmATPS eluted from the gel filtration column as a dimer of ∼100 kDa formed from two 48-kDa monomers (19, 52).
The x-ray crystal structure of GmATPS was determined at 2.48 Å resolution by molecular replacement (Table 1; Fig. 2). Each monomer of GmATPS consists of two mixed α/β structural domains (Fig. 2A). The N-terminal domain (residues 48–211) is centered on an eight-stranded β-sheet (β1a–β8a) surrounded by four α-helices (α1, α2, α4, and α6). A second β-sheet (β1b–β2b) and two additional α-helices (α3 and α5) form most of the dimerization interface (Fig. 2B). A short loop (residues 212–226) connects the two halves of the monomer. The C-terminal domain (residues 227–447) is formed by a core β-sheet (β1c–β5c) and five α-helices (α7–α11) with a subdomain consisting of a β-sheet (β1d–β3d) and two α-helices (α12–α13) positioned as a lid over the APS-binding site. One helix of the C-terminal domain (α11) also contributes to the dimer interface. The dimeric structure of GmATPS (Fig. 2B) positions the active site of each monomer in opposite orientations by a 2-fold rotation axis with respect to the dimer interface.
FIGURE 2.
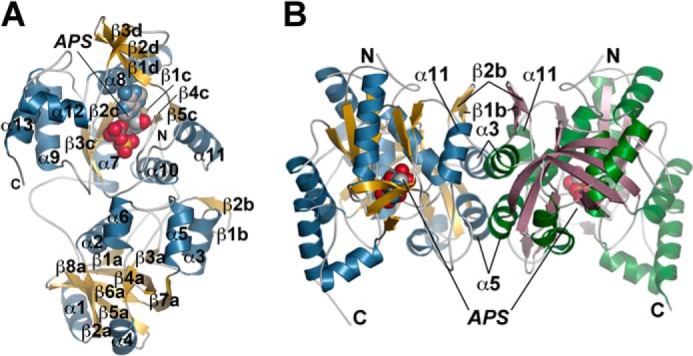
Three-dimensional structure of GmATPS. A, ribbon diagram showing the structure of the GmATPS monomer. The N- and C-terminal residues are indicated. The α-helices and β-strands are colored blue and gold, respectively, and are numbered. APS bound in the active site is shown as a space-filling model. In this orientation, the N-terminal domain corresponds to the lower half of the structure, and the C-terminal domain corresponds to the upper half of the structure. All structural figures were generated using PyMOL (PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC). B, overall structure of the GmATPS dimer. Structural features in one monomer are colored as in A with α-helices and β-strands of the second monomer colored green and rose, respectively. Secondary structure features along the dimer interface are indicated. The position of bound APS in each monomer is shown by the space-filling model.
Structural Comparison with ATP Sulfurylases from Other Species
A search of the Protein Data Bank using DALI (58) showed that the overall fold of the GmATPS monomer was most similar to that of the ATP sulfurylase domain of human PAPS synthetase with a 0.9-Å r.m.s.d. for 390 aligned Cα atoms. This is consistent with the 59% amino sequence similarity between these two eukaryotic ATP sulfurylases (Figs. 1, B and C, and 3). The ATP sulfurylases from T. thermophilus, S. cerevisiae, P. chrysogenum, A. aeolicus, and the Riftia symbiont, which share 26–29% amino acid sequence similarity with GmATPS, displayed larger structural variations of 1.6–2.7-Å r.m.s.d. over 360–390 aligned Cα atoms, although the overall fold was similar. No significant structural or sequence similarity was detected between GmATPS and the catalytic ATP sulfurylase subunit of the P. syringae enzyme.
FIGURE 3.

Sequence comparison of ATP sulfurylases. A multiple sequence alignment of the ATP sulfurylases from soybean (GmATPS; AF452454) human (HsPAPPSS; O43252), the Riftia symbiont (RpsATPS; Q54506), S. cerevisiae (ScATPS, P08563), P. chrysogenum (PcATPS, Q12650), A. aeolicus (AaATPS, O67174), and T. thermophilus (TtATPS, Q5SKH7) is shown with amino acid numbering indicated in parentheses. The α-helices (blue rectangles) and β-strands (yellow rectangles) of GmATPS are depicted above the alignment. Conserved residues are highlighted in orange with residues in white indicating a variation from the conserved sequence. Red highlighting denotes active site residues in GmATPS. The linker region between the N- and C-terminal structural domains is indicated. The light gray boxes indicate regions at the dimer interface of GmATPS; these regions also display large sequence variation across the ATP sulfurylases. Multiple sequence alignment used the MultAlin webpage.
Although the overall structure of the ATP sulfurylase monomer from various organisms is generally conserved, with the exception of the heterodimeric form of the enzyme, the oligomerization of the enzyme from different species is diverse (Fig. 4). As noted above, the dimer interface of GmATPS is formed by a β-sheet (β1b–β2b) and three α-helices (α3, α5, and α11) (Figs. 2B and 4). This organization is also found in the ATP sulfurylase domain of the bifunctional human PAPS synthetase (Fig. 4). The APS kinase domain of the human enzyme is also dimeric with a flexible loop linking the two domains of each bifunctional monomer. Structural and sequence comparison of GmATPS with the Riftia symbiont and T. thermophilus ATP sulfurylases shows that major sequence variation in the features defining the dimerization region of the soybean enzyme (Fig. 3) leads to structural remodeling of the interface between monomers in the two bacterial proteins (Fig. 4). Interestingly, dimerization of the bifunctional A. aeolicus enzyme does not involve the ATP sulfurylase domains but occurs through the APS kinase domain (Fig. 4). Similarly, the hexameric structure of the S. cerevisiae and P. chrysogenum ATP sulfurylases is also mediated through other regions of the monomer and not the catalytic domain (Fig. 4). Ultimately, the diverse sequence changes that have lead to varied oligomeric interaction features do not alter conserved regions of ATP sulfurylase necessary for catalysis (Fig. 3).
FIGURE 4.
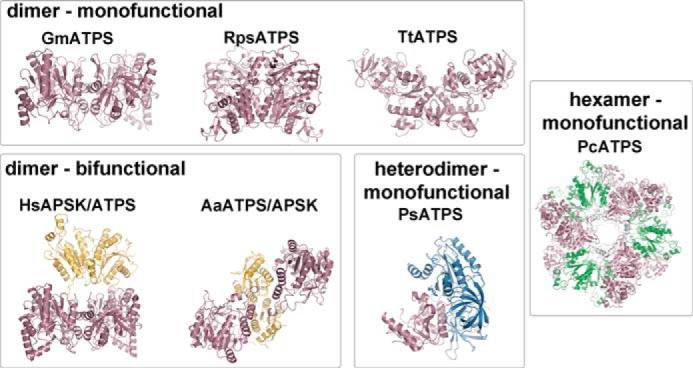
Structural diversity in ATP sulfurylases. The three-dimensional structures of the dimeric monofunctional ATP sulfurylases are from soybean (GmATPS; described here), the Riftia symbiont (RpsATPS), and T. thermophilus (TtATPS); the dimeric bifunctional PAPS synthetases are from human (HsAPSK/ATPS) and A. aeolicus (AaATPS/APSK); the heterodimeric monofunctional ATP sulfurylase from P. syringae (PsATPS); and the homohexameric enzyme from fungi (PcATPS) are shown as ribbon diagrams. The domain structure of each protein is colored by function as follows: ATP sulfurylase, rose; APS kinase, yellow; APS kinase-like, green; and GTPase, blue.
Active Site Structure of GmATPS
In the GmATPS structure, APS binds in the C-terminal domain of each monomer to clearly define the location of the active site (Fig. 2). Unambiguous electron density for APS was observed in each monomer of the asymmetric unit (Fig. 5A). The ligand is bound in an enclosed cavity with a wide channel leading to the surface (Fig. 5B). The channel is positioned above the α-phosphate of APS and is lined with positively charged residues, including Arg-248, His-252, His-255, and Arg-349 and provides a site for binding of the β- and γ-phosphates of ATP and PPi. Residues from the β1c–α8 loop, α10, and the N-terminal part of the β4c–α11 loop contribute to the active site and are highly conserved across the ATP sulfurylases from different organisms (Fig. 3). The enclosed active site suggests a possible movement of the C-terminal subdomain (residues 390–447; β1d–β3d and α12–α13) that forms a lid over the APS-binding site to provide access of adenine nucleotides at the catalytic site. A recent crystal structure of the apoenzyme form of ATP sulfurylase from the purple sulfur bacterium A. vinosum reveals movement of α12 to open the active site (46).
FIGURE 5.
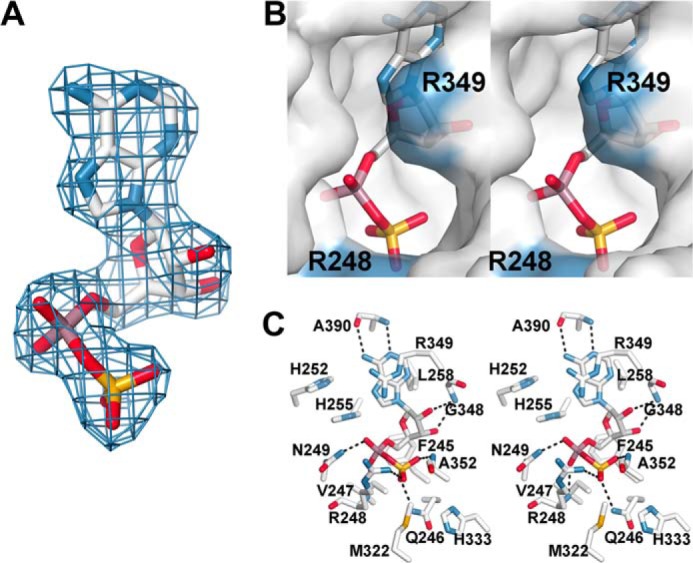
Active site of the GmATPS·APS complex. A, electron density of APS in the GmATPS active site. A final Fo − Fc omit map (1.5σ) is shown. B, stereoview molecular surface rendering of the GmATPS active site. The view is oriented down the putative pyrophosphate-binding site. Surfaces corresponding to Arg-248 and Arg-349 are colored (blue). APS is shown as a stick model. C, stereoview showing protein-ligand interactions in the GmATPS·APS complex. Dotted lines represent hydrogen bonds between active site residues and the ligand.
The GmATPS·APS complex provides a detailed view of interactions in the active site (Fig. 5C). The N1 and side-chain amine of the adenine ring form hydrogen bonds with the backbone nitrogen of Ala-390 (2.9 Å) and the carbonyl of Ala-390 (3.1 Å), respectively. In addition, the adenine ring makes van der Waals contacts with the side chains of Leu-258 and Arg-349. The hydroxyl groups of the ribose hydrogen interact with the backbone nitrogen of Gly-348 (3.1 Å). Extensive interactions between the phosphosulfate-mixed anhydride and active site residues are observed. Backbone nitrogens of Arg-248 (3.1 Å) and Asn-249 (2.6 Å) contact the phosphate group of APS. The side-chain amide of Gln-246 (3.0 Å), the backbone nitrogen of Ala-352 (2.9 Å), and the basic guanidyl group of Arg-248 (3.1 Å) anchor the sulfate-derived portion of APS in the active site. Phe-245, Val-247, Met-322, and His-333 contribute additional van der Waals contacts. The side chains of His-252, His-255, and Arg-349 are positioned in the cavity leading to the surface of the protein and likely contribute to binding the β- and γ-phosphates of ATP in the forward reaction and PPi in the reverse reaction. With the exceptions of Ala-352 and Ala-390, the amino acids forming the catalytic site of GmATPS are invariant across the structurally characterized ATP sulfurylases (Fig. 3).
Functional Analysis of GmATPS Active Site Mutants
Based on the crystal structure of the GmATPS·APS complex, 20 point mutants of GmATPS (F245L, F245A, Q246E, Q246N, Q246A, R248K, N249D, N249A, H252Q, H252N, H252A, H255Q, H255N, H255A, L258V, L258A, H333Q, H333N, H333A, and R349K) were generated to introduce changes in active site residues for subsequent steady-state kinetic analysis. The H252Q, H252A, H255N, H333N, and H333A mutants led to expression of insoluble protein in E. coli; however, all other GmATPS were soluble and purified as dimeric enzymes.
Assays of the APS synthesis reaction catalyzed by wild-type and mutant GmATPS showed that only the F245L, Q246N, Q246A, and H333Q mutants retained sufficient activity for determination of kinetic constants (Table 2). In general, the F245L, Q246N, Q246A, and H333Q mutants displayed modest changes in turnover rates and/or Km values compared with wild type. Additional steady-state kinetic analysis of point mutants in the active site used the reverse (i.e. ATP synthesis) reaction because it provides better assay sensitivity compared with the forward reaction (Table 3).
TABLE 2.
Steady-state kinetic parameters of wild-type and mutant GmATPS for APS synthesis
Reactions were performed as described under “Experimental Procedures.” All parameters are expressed as means ± S.E. for n = 3.
| Protein | V/E | KmATP | V/KmATP | Kmsulfate | V/Kmsulfate |
|---|---|---|---|---|---|
| s−1 | μm | m−1 s−1 | μm | m−1 s−1 | |
| Wild type | 0.512 ± 0.013 | 165 ± 18 | 3,100 | 320 ± 25 | 1,600 |
| F245L | 0.283 ± 0.022 | 2,150 ± 338 | 132 | 306 ± 52 | 93 |
| Q246N | 0.134 ± 0.007 | 880 ± 145 | 155 | 236 ± 57 | 579 |
| Q246A | 0.288 ± 0.010 | 346 ± 30 | 833 | 1,070 ± 103 | 269 |
| H333Q | 0.162 ± 0.005 | 575 ± 51 | 281 | 311 ± 53 | 520 |
TABLE 3.
Steady-state kinetic parameters of wild-type and mutant GmATPS for ATP synthesis
Reactions were performed as described under “Experimental Procedures.” All parameters are expressed as means ± S.E. for n = 3.
| Protein | V/E | KmAPS | V/KmAPS | KmPPi | V/KmPPi |
|---|---|---|---|---|---|
| s−1 | μm | m−1 s−1 | μm | m−1 s−1 | |
| Wild type | 32.0 ± 1.4 | 34.2 ± 5.2 | 936,000 | 45.8 ± 6.8 | 699,000 |
| F245L | 93.8 ± 6.1 | 24.7 ± 5.8 | 3,798,000 | 118 ± 29 | 795,000 |
| F245A | 108 ± 9 | 36.3 ± 5.6 | 2,980,000 | 153 ± 24 | 706,000 |
| Q246E | 10.3 ± 1.7 | 312 ± 26 | 33,000 | 61.1 ± 3.9 | 170,000 |
| Q246N | 60.6 ± 1.8 | 45.0 ± 5.4 | 1,350,000 | 55.6 ± 6.1 | 1,090,000 |
| Q246A | 64.3 ± 4.1 | 40.2 ± 8.5 | 1,600,000 | 117 ± 25 | 373,000 |
| R248K | 19.7 ± 1.4 | 47.0 ± 4.6 | 419,000 | 428 ± 78 | 46,000 |
| N249D | 0.104 ± 0.005 | 6.51 ± 1.79 | 16,000 | 69.4 ± 14.1 | 1,500 |
| N249A | 34.6 ± 4.3 | 43.2 ± 6.1 | 801,000 | 1,078 ± 153 | 32,100 |
| H252N | 40.3 ± 2.5 | 27.2 ± 4.4 | 1,480,000 | 85.0 ± 17.2 | 474,000 |
| H255Q | 63.5 ± 2.8 | 22.6 ± 2.2 | 2,810,000 | 114 ± 12 | 557,000 |
| H255A | 2.45 ± 0.16 | 4.41 ± 0.55 | 556,000 | 39.9 ± 6.2 | 61,900 |
| L258V | 129 ± 8 | 38.5 ± 5.4 | 3,350,000 | 208 ± 26 | 629,000 |
| L258A | 109 ± 5 | 28.2 ± 2.9 | 3,870,000 | 373 ± 34 | 292,000 |
| H333Q | 27.0 ± 1.6 | 12.7 ± 2.6 | 2,130,000 | 22.0 ± 3.0 | 1,130,000 |
| R349K | 3.22 ± 0.11 | 34.6 ± 6.7 | 93,100 | 590 ± 96 | 5,460 |
Mutations in residues making van der Waals contacts with APS (i.e. F245L, F245A, L258V, L258A, and H333Q) generally led to modest <5-fold changes in steady-state kinetic parameters with either APS or sodium pyrophosphate. An 8-fold increase in Km values for pyrophosphate was observed with the L258A mutant.
Changes to amino acids in the sulfate- and phosphate-binding pockets of the active site introduced varied effects. The Q246A and Q246N mutants displayed kcat and Km values comparable with wild-type enzyme, but introduction of a negatively charged side chain in the Q246E mutant resulted in a 28-fold decrease in catalytic efficiency (kcat/Km) for APS and a modest 4-fold decrease in kcat/Km for PPi. Replacement of Arg-248 with a lysine altered catalytic efficiency with APS less than 2-fold but decreased the kcat/Km for PPi by 15-fold. The N249A mutant affected the kinetic parameters of PPi with a 22-fold decrease in catalytic efficiency that resulted largely from an increased Km value for this substrate. The kinetic parameters of this mutant should be considered as estimates because a saturating concentration of NaPPi could not be used. Substitution of an aspartate at this position (N249D) led to a 308-fold decrease in kcat and 59- and 466-fold decreases in kcat/Km for APS and PPi, respectively.
Mutations in the putative PPi-binding site also altered the kinetic parameters of GmATPS. The R349K mutant reduced the turnover rate by 10-fold and lowered the kcat/Km values for APS and PPi by 10- and 127-fold, respectively. Although changes of His-252 largely resulted in insoluble protein, as noted above, the H252N mutant exhibited kinetic parameters comparable with wild-type protein. Substitution of His-255 with a glutamine also did not dramatically alter the kinetic properties of the mutant enzyme; however, the H255A mutant displayed a 15-fold effect on kcat for both substrates and a 10-fold decrease in catalytic efficiency for PPi.
Analysis of Sequence Variation, Sulfate Content, and ATP Sulfurylase Activity in Arabidopsis ATPS1 Haplotypes
In A. thaliana, alteration of ATP sulfurylase expression leads to increased sulfate accumulation (40–41, 59). To investigate the effect of sequence variation in Arabidopsis ATPS1 haplotypes, the ATP sulfurylase activity and sulfate levels of selected representative accessions were analyzed (Table 4). Haplotype data for ATPS1 was derived from the Arabidopsis 1001 Genomes database (60). ATP sulfurylase activity and sulfate levels were determined in leaves of accessions representative of each haplotype. For comparison, ATP sulfurylase activity and sulfate content were measured in A. thaliana Col-0 and atps1 mutant grown at the same conditions. High variation in sulfate levels was observed, with some accessions, such as Dog-4, Vie-0, Goettingen-7, Ciste-2, Naes-2, and Kulturen, accumulating higher sulfate levels than Col-0 (Table 4). These accessions were thus similar in the high sulfate phenotype to atps1, although the accumulation was not as pronounced as in the mutants (Table 4).
TABLE 4.
Summary of variation in Arabidopsis ATPS1 haplotypes and the ATPS activities and sulfate contents of representative accessions
The haplotype information is derived from the 1001 Genome browser. ATP sulfurylase activity and sulfate levels were determined in leaves of accessions representative of each haplotype grown for 5 weeks in a controlled environment room under short days. For comparison, ATP sulfurylase activity and sulfate were measured in A. thaliana Col-0 and atps1 mutant grown at the same conditions. Asterisks mark values significantly (p < 0.05) different from wild-type. ND indicates activity not determined, and NA indicates accession not available or not germinated.
| Mutation | Comment | No. of accessions | Example accession | Sulfate content | ATPS activity |
|---|---|---|---|---|---|
| μmol/g fresh wt | nmol/min/mg protein | ||||
| Wild type | A. thaliana Col-0 | Col-0 | 7.6 ± 1.4 | 145 ± 23 | |
| atps1 | atps1 A. thaliana Col-0 | atps1 | 16 ± 2 | 92 ± 19 | |
| S9R | Transit peptide | 14 | Tsu-0 | 8 ± 1 | ND |
| V43N | Transit peptide | 5 | Dog-4 | 11.5 ± 2.0* | 140 ± 40 |
| G56S | Invariant in A. thaliana Col-0; disordered region | 11 | Ull2–3 | 8.2 ± 2 | 121 ± 10 |
| L122V | Invariant in A. thaliana Col-0; dimer interface | 1 | Vie-0 | 14 ± 1* | 135 ± 10 |
| T150S | Loop region | 36 | Ang-0 | 4.6 ± 0.3* | ND |
| A144T/T150S | Loop region | 2 | TDr-2 | NA | NA |
| N160K | Loop region | 14 | Kn-0 | 7 ± 0.5 | ND |
| S166N | Asn in other A. thaliana isoforms | 8 | Edu-1 | NA | NA |
| E169A | Invariant in A. thaliana Col-0; dimer interface | 6 | Goettingen-7 | 11.3 ± 0.8* | 135 ± 40 |
| T198A | Invariant in A. thaliana Col-0; internal packing | 2 | Ciste-2 | 11.1 ± 2.2* | 140 ± 36 |
| N202Sa | Loop region | 1 | Hov3–2 | NA | NA |
| E312V | Loop region | 1 | Sakata | NA | NA |
| V316F | Internal packing | 1 | T800 | 9.9 ± 2.1 | 247 ± 11* |
| A337S | Invariant in A. thaliana Col-0; loop region | 18 | Bil-7 | 7.6 ± 0.1 | 145 ± 51 |
| G342D | Invariant in A. thaliana Col-0; internal packing | 1 | Naes-2 | 12.3 ± 3* | 72 ± 3* |
| K372R | Arg in other A. thaliana isoforms; dimer interface | 1 | Kulturen | 10.7 ± 1.6* | ND |
a The sequence quality is low in this region; the haplotype might be a sequencing error.
DISCUSSION
ATP sulfurylase plays a critical role in the plant sulfur assimilation pathway by catalyzing its first committed step via the energetically unfavorable formation of APS (Fig. 1A). The resulting phosphosulfate bond of APS contains twice the energy of the pyrophosphate linkage in ATP and provides the driving force for reductive sulfur assimilation in plants (16, 17). To overcome this energetic barrier, plants use a range of metabolic strategies, including the rapid use of the products of ATP sulfurylase in subsequent reactions catalyzed by APS reductase for sulfur assimilation, APS kinase for the generation of PAPS as a sulfonation donor molecule, and removal of PPi by pyrophosphatase (5–8, 19–22, 25, 61, 62).
The diversity in both sequence and structure of ATP sulfurylase (Figs. 1, B and C, 3, and 4) reflects the distant origins of sulfur metabolism in different organisms and its various metabolic purposes. For the reductive assimilation of sulfate from the environment, ATP sulfurylase provides activated molecules for the synthesis of amino acids and specialized metabolites. In some microorganisms, sulfate reduction and sulfide oxidation function in a dissimilatory process to support consumption of sulfate and sulfur compounds for electron transfer processes. Multiple studies of ATP sulfurylases from a variety of organisms highlight the conservation of the catalytic core of the enzyme in terms of both three-dimensional structure and active site residues despite the distant evolutionary relationships of these organisms. Given the retention of the three-dimensional structure of ATP sulfurylase, this activity likely evolved before divergence of the different branches on the tree-of-life (46). These studies also reveal the versatility of the ATP sulfurylase fold as sequence variations that alter the oligomerization interface lead to a range of architectures for this enzyme (Figs. 3 and 4) (43–45, 47–51).
Interestingly, genome analysis of the sulfate utilization genes in the major eukaryotic and prokaryotic lineages suggests complex origins of the enzymes in these pathways and that crystallographic and biochemical studies of ATP sulfurylase have only examined a few of the possible ways to organize the initial steps of sulfur assimilation (3, 62). For example, bioinformatic studies have identified independent and fused forms of ATP sulfurylase, APS kinase, sulfotransferase, pyrophosphatase, APS reductase, and related regulatory domains in various organisms (3, 62). Phylogenetic analysis of sulfur pathway genes by Bradley et al. (62) suggests a model for the evolution of these enzymes that invokes an ancestral gene fusion of ATP sulfurylase and APS kinase in two possible orientations to provide a common evolutionary origin for ATP sulfurylase/APS kinase fusion and another origin for APS kinase/ATP sulfurylase fusion. Subsequent gene fission, gene loss, and lateral gene transfer led to the variety of molecular architectures for these enzymes.
It remains unclear whether ATP sulfurylase architecture is related to the evolution of regulatory structural features. In the heterodimeric enzymes from E. coli and P. syringae, the GTPase subunit allosterically activates the catalytic ATP sulfurylase subunit (42); however, this mechanism seems specialized for certain prokaryotes. Of the ATP sulfurylases structurally related to GmATPS (Figs. 1 and 4), only the P. chrysogenum enzyme has a demonstrated allosteric regulatory mechanism involving PAPS (49, 50). The observed structural transition in response to PAPS levels provides for control of sulfate activation in P. chrysogenum, but it does not appear to be a widely used regulatory feature in other organisms. However, localized structural changes are important for substrate binding.
The enclosed active site of GmATPS (Figs. 2 and 5B) suggests that conformational changes occur to accommodate substrate access and product exit from the enzyme. Substrate recognition in ATP sulfurylases requires several regions around the active site for nucleotide and sulfate/PPi binding and reduced solvent access (Fig. 5C) (43–45, 47–50). Localized structural changes were observed in the apoenzyme structure of the Riftia symbiont ATP sulfurylase, which showed residues corresponding to the β2c–α9 loop of GmATPS rotated away from the active site (43). The recently determined crystal structure of an apoenzyme form of the enzyme from the purple sulfur bacterium A. vinosum reveals movement of α12 to open the active site (46).
Although multiple ATP sulfurylases from different organisms have been crystallized and structurally characterized, functional analysis of these enzymes is limited to the two histidines near the active site that correspond to His-252 and His-255 of GmATPS (63, 64). The HXXH motif was originally identified in nucleotidyltransferases and type I amino acyltransferases (65–67). In these enzymes, the conserved histidines are directly involved in cleavage of the α/β-phosphate bond. Mutagenesis of the HXXH motif in the ATP sulfurylase domain of human and mouse PAPS synthetase led to reduced specific activity (63, 64), but more detailed steady-state kinetic analysis of the effect on function was not performed. To date, no structure-function analysis of other active site residues of an ATP sulfurylase from any other species has been reported.
The structure of GmATPS in complex with APS provides detailed information on the active site and protein-ligand interactions (Figs. 2 and 5). Mutagenesis of GmATPS identifies key active site residues and examines their contributions to catalysis and substrate recognition (Tables 2 and 3). Substitutions in residues making surface contacts with APS (including Phe-245, Leu-258, and His-333) result in minor changes in steady-state kinetic parameters. Mutations in the His-252 and His-255 (i.e. the HXXH motif) of GmATPS suggest that the role of the first histidine may be structural, as some changes in this residue led to production of insoluble protein. Removal of the His-255 side chain in the H255A mutant shows a 15-fold reduction in turnover rate, which suggests the loss of a stabilizing hydrogen bond interaction in catalysis. These results are consistent with the proposal that these two conserved histidines are not directly involved in cleavage of the α/β-phosphodiester bond.
Mutation of Gln-246, which hydrogen bonds with the sulfate group of APS, suggests that this residue is not critical for substrate interaction; however, introduction of a negative charge at this position (i.e. Q246E) weakens affinity for APS, as suggested by the 28-fold decrease in kcat/Km. This likely results from charge repulsion in the APS-binding site of this mutant. Subtle alteration in Arg-248 to a lysine retains interaction with APS but reduces the kcat/Km value for PPi by 15-fold suggesting the loss of interaction with this group during catalysis. Of the GmATPS active site residues, Asn-249 and Arg-349 are critical for efficient catalysis. The N249A mutation resulted in a 22-fold decrease in catalytic efficiency with PPi. Introduction of a negative charge (N249D) caused the largest fold changes in steady-state kinetic parameters of any mutation in the GmATPS active site for both substrates. Substitution of Arg-349 with a lysine reduced turnover rate by up to 10-fold but decreased the kcat/Km value for PPi by 128-fold.
Molecular docking of the forward reaction substrates ATP and sulfate (Fig. 6A) and the reverse reaction substrates APS and PPi (Fig. 6B) in the GmATPS active site suggests that Asn-249 and Arg-349, which are on opposite sides of the active site, have potential roles in catalysis. For the forward reaction, the positions of the sulfate ion and the adenine and ribose groups of ATP were initially placed based on the GmATPS·APS complex. Docking of ATP places the phosphate groups into the opening that leads to solvent from the active site and interactions with Arg-248, Asn-249, His-255, and Arg-349 (Fig. 6A). The distorted U-shape of the ATP is similar to the conformation observed in nucleotidyltransferases (65). This conformation situates the α-phosphate group of ATP for in-line nucleophilic attack by the sulfate group and subsequent formation of APS and release of PPi. Distortion of the phosphate backbone energetically contributes to overcoming the reaction barrier and leads to formation of the high energy mixed anhydride bond of APS (68). For the energetically favorable reverse reaction (i.e. ATP synthesis), positioning of PPi by interactions with Asn-249, His-255, and Arg-349 allows for facile nucleophilic attack on the α-phosphate of APS (Fig. 6B). Modeling of substrates in the GmATPS structure supports the proposed roles for various active site residues. Overall, both the forward and reverse reactions are substitution mechanisms that rely on reactive groups in close contact with both substrates to provide protein-ligand interactions that stabilize a pentavalent transition state and enhance reaction rates. Because the active site residues of GmATPS are invariant across the ATP sulfurylases from other organisms, the functional role of these amino acids is likely conserved.
FIGURE 6.
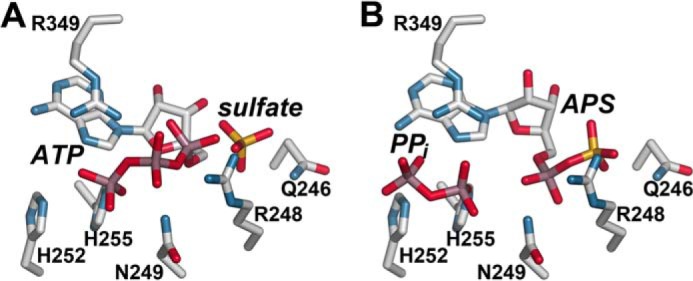
Active site models for the forward and reverse reactions catalyzed by GmATPS. A, model of substrates of the forward reaction in the GmATPS active site. ATP adopts a U-shaped conformation that allows nucleophilic attack by the sulfate ion. B, model of substrates of the reverse reaction in the GmATPS active site. The position of APS is from the GmATPS·APS x-ray crystal structure, and PPi was computationally docked in the active site.
The structural and functional analyses presented here provide the first molecular insights on a plant ATP sulfurylase and the committed step of plant sulfur assimilation. Each method identified several amino acid residues important for the enzyme activity located in the active center. To identify further residues that might be important for ATP sulfurylase function, we exploited natural variation in A. thaliana accessions. The ATP sulfurylase activity in the atps1 mutant is decreased by ∼50%, which together with the strong sulfate phenotype suggests that in Arabidopsis ATPS1 is the major isoform (4). Thus, sequence variation in ATPS1 that alters protein stability and/or proper folding may affect sulfate levels in Arabidopsis leaves.
Examination of ATPS1 sequences in the Arabidopsis 1001 Genome database identified numerous accessions with altered amino acid sequences of the protein (Table 4) (60). Sulfate levels in leaves of accessions representative of most haplotypes were determined. Some accessions (Dog-4, Vie-0, Goettingen-7, Ciste-2, Naes-2, and Kulturen) contained higher sulfate levels than Col-0. These accessions were thus similar in the high sulfate phenotype to atps1, although the accumulation has not been as pronounced as in the mutants (Table 4). Because the differences in sulfate levels may not be connected with the variation in ATPS1, we determined total ATP sulfurylase activity in the accessions. In all the accessions except Naes-2, activity was not different from Col-0 or even higher. The resulting effects of Naes-2 suggest that the amino acid variation in the ATPS1 protein (i.e. G342D) may affect protein function.
The 79% amino acid sequence identity between GmATPS and AtATPS1 allows for the mapping of Arabidopsis haplotype variations onto the GmATPS structure (Fig. 7). No variations were observed in any residues linked to active site structure. Three mutations (S9R, V43N, and G56S) are in the localization sequence and/or region corresponding to the disordered region of GmATPS and were not mapped to the structural model of AtATPS1. Multiple changes (A144T, T150S, N160K, N202S, E312V, and A337S) occur in residues found in loops between secondary structural features (Fig. 7). Because mutations in loop regions are often tolerated, these changes would be predicted not to alter ATP sulfurylase function.
FIGURE 7.
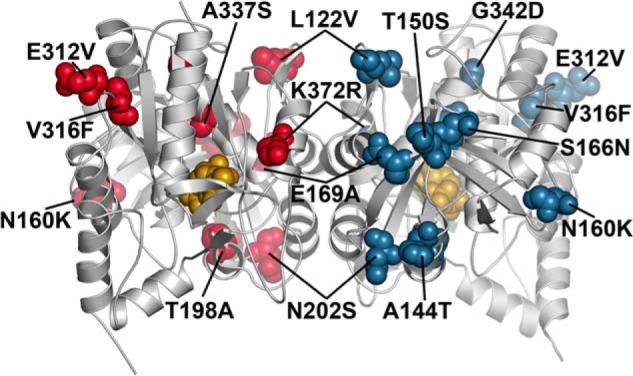
Structural mapping of A. thaliana haplotype differences in ATP sulfurylase. Positions of mutations in Arabidopsis ATPS1 haplotypes were mapped to the corresponding residues of GmATPS. Residues of each monomer are colored as red and blue space-filling models, respectively. The position of APS in the structure is shown as the gold space-filling model. The orientation shown is the same as in Fig. 2B.
In comparison, changes in amino acids found in the dimer interface may affect activity by altering oligomerization and/or disrupting proper protein folding. Three variants (L122V, E169A, and K372R) across the Arabidopsis accessions are found in the dimer interface. Leu-122 of AtATPS1 is invariant in the β1b strand of all ATP sulfurylases from different organisms (Fig. 3). The L122V change may affect protein stability and/or folding. Similarly, the E169A mutation leads to removal of a charged side chain at the start of α7 where conservation of an acidic residue at this position occurs (Fig. 3). This mutation may alter stability and/or structure of AtATPS1. The K372R variant likely does not alter enzyme activity, because an arginine is found at this position in other plant homologs. Based on the positions of the T198A, V316F, and G342D, changes in these residues, all of which alter the size and/or properties of the side chain, may negatively impact protein structure because these residues are internally packed. Indeed, the Naes-2 accession with the G342D variant of ATPS1 shows decreased total ATP sulfurylase activity and sulfate accumulation. Mapping of Arabidopsis haplotype mutations onto the GmATPS crystal structure provides a molecular basis for understanding their effects on sulfur metabolism and for dissecting the natural variation in ATP sulfurylase and the structure/function relationship of the enzyme isoforms.
Acknowledgments
Portions of this research were carried out at the Argonne National Laboratory Structural Biology Center of the Advanced Photon Source, a national user facility operated by the University of Chicago for the Department of Energy Office of Biological and Environmental Research and supported by Grant DE-AC02-06CH11357. Product names are necessary to report factually on available data; however, the United States Department of Agriculture neither guarantees nor warrants the standard of product, and the use of the name by the United States Department of Agriculture implies no approval of the product to the exclusion of others that may be suitable.
This work was supported in part by National Science Foundation Grant MCB-0904215.
The atomic coordinates and structure factors (code 4MAF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- APS
- adenosine 5′-phosphosulfate
- HsAPSK
- human adenosine-5′-phosphosulfate kinase, also known as adenylyl-sulfate kinase, E.C. 2.7.1.25
- ATP sulfurylase
- ATP:sulfate adenylyltransferase, E.C. 2.7.7.4
- GmATPS
- G. max (soybean) ATP sulfurylase
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- PPi
- pyrophosphate
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Leustek T., Martin M. N., Bick J. A., Davies J. P. (2000) Pathways and regulation of sulfur metabolism revealed through molecular and genetic studies. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 141–165 [DOI] [PubMed] [Google Scholar]
- 2. Krishnan H. B. (2005) Engineering soybean for enhanced sulfur amino acid content. Crop Sci. 45, 454–461 [Google Scholar]
- 3. Patron N. J., Durnford D. G., Kopriva S. (2008) Sulfate assimilation in eukaryotes: Fusions, relocations, and lateral transfers. BMC Evol. Biol. 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yi H., Galant A., Ravilious G. E., Preuss M. L., Jez J. M. (2010) Sensing sulfur conditions: Simple to complex protein regulatory mechanisms in plant thiol metabolism. Mol. Plant 3, 269–279 [DOI] [PubMed] [Google Scholar]
- 5. Yi H., Ravilious G. E., Galant A., Krishnan H. B., Jez J. M. (2010) From sulfur to homoglutathione: Thiol metabolism in soybean. Amino Acids 39, 963–978 [DOI] [PubMed] [Google Scholar]
- 6. Takahashi H., Kopriva S., Giordano M., Saito K., Hell R. (2011) Sulfur assimilation in photosynthetic organisms: Molecular functions and regulations of transporters and assimilatory enzymes. Annu. Rev. Plant Biol. 62, 157–184 [DOI] [PubMed] [Google Scholar]
- 7. Kopriva S., Mugford S. G., Baraniecka P., Lee B. R., Matthewman C. A., Koprivova A. (2012) Control of sulfur partitioning between primary and secondary metabolism in Arabidopsis. Front. Plant Sci. 3, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ravilious G. E., Jez J. M. (2012) Structural biology of plant sulfur metabolism: From assimilation to biosynthesis. Nat. Prod. Rep. 29, 1138–1152 [DOI] [PubMed] [Google Scholar]
- 9. Höfgen R., Kreft O., Willmitzer L., Hesse H. (2001) Manipulation of thiol contents in plants. Amino Acids 20, 291–299 [DOI] [PubMed] [Google Scholar]
- 10. Hawkesford M. J., De Kok L. J. (2006) Managing sulphur metabolism in plants. Plant Cell Environ. 29, 382–395 [DOI] [PubMed] [Google Scholar]
- 11. White P. J., Brown P. H. (2010) Plant nutrition for sustainable development and global health. Ann. Bot. 105, 1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ohkama-Ohtsu N., Wasaki J. (2010) Recent progress in plant nutrition research: Cross-talk between nutrients, plant physiology, and soil microorganisms. Plant Cell Physiol. 51, 1255–1264 [DOI] [PubMed] [Google Scholar]
- 13. Shin R., Jez J. M., Basra A., Zhang B., Schachtman D. P. (2011) 14-3-3 proteins fine-tune plant nutrient metabolism. FEBS Lett. 585, 143–147 [DOI] [PubMed] [Google Scholar]
- 14. Aharoni A., Galili G. (2011) Metabolic engineering of the plant primary-secondary metabolism interface. Curr. Opin. Biotechnol. 22, 239–244 [DOI] [PubMed] [Google Scholar]
- 15. Chan K. X., Wirtz M., Phua S. Y., Estavillo G. M., Pogson B. J. (2013) Balancing metabolites in drought: The sulfur assimilation conundrum. Trends Plant Sci. 18, 18–29 [DOI] [PubMed] [Google Scholar]
- 16. Asahi T. (1964) Sulfur metabolism in higher plants: IV. Mechanism of sulfate reduction in chloroplasts. Biochim. Biophys. Acta 82, 58–66 [Google Scholar]
- 17. Osslund T., Chandler C., Segel I. (1982) ATP sulfurylase from higher plants: Purification and preliminary kinetics studies of the cabbage leaf enzyme. Plant Physiol. 70, 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murillo M., Leustek T. (1995) Adenosine-5′-triphosphate sulfurylase from Arabidopsis thaliana and Escherichia coli are functional equivalent but structurally and kinetically divergent: Nucleotide sequence of two adenosine-5′-triphosphate sulfurylase cDNAs from Arabidopsis thaliana and analysis of a recombinant enzyme. Arch. Biochem. Biophys. 323, 195–204 [DOI] [PubMed] [Google Scholar]
- 19. Phartiyal P., Kim W. S., Cahoon R. E., Jez J. M., Krishnan H. B. (2006) Soybean ATP sulfurylase, a homodimeric enzyme involved in sulfur assimilation, is abundantly expressed in roots and induced by cold treatment. Arch. Biochem. Biophys. 450, 20–29 [DOI] [PubMed] [Google Scholar]
- 20. Klein M., Papenbrock J. (2004) The multi-protein family of Arabidopsis sulphotransferases and their relatives in other plant species. J. Exp. Bot. 55, 1809–1820 [DOI] [PubMed] [Google Scholar]
- 21. Mugford S. G., Yoshimoto N., Reichelt M., Wirtz M., Hill L., Mugford S. T., Nakazato Y., Noji M., Takahashi H., Kramell R., Gigolashvili T., Flügge U. I., Wasternack C., Gershenzon J., Hell R., Saito K., Kopriva S. (2009) Disruption of adenosine-5′-phosphosulfate kinase in Arabidopsis reduces levels of sulfated secondary metabolites. Plant Cell 21, 910–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mugford S. G., Matthewman C. A., Hill L., Kopriva S. (2010) Adenosine-5′-phosphosulfate kinase is essential for Arabidopsis viability. FEBS Lett. 584, 119–123 [DOI] [PubMed] [Google Scholar]
- 23. Yatusevich R., Mugford S. G., Matthewman C., Gigolashvili T., Frerigmann H., Delaney S., Koprivova A., Flügge U. I., Kopriva S. (2010) Genes of primary sulfate assimilation are part of the glucosinolate biosynthetic network in Arabidopsis thaliana. Plant J. 62, 1–11 [DOI] [PubMed] [Google Scholar]
- 24. Ravilious G. E., Nguyen A., Francois J. A., Jez J. M. (2012) Structural basis and evolution of redox regulation in plant adenosine-5′-phosphosulfate kinase. Proc. Natl. Acad. Sci. U.S.A. 109, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ravilious G. E., Jez J. M. (2012) Nucleotide binding site communication in Arabidopsis thaliana adenosine 5′-phosphosulfate kinase. J. Biol. Chem. 287, 30385–30394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ravilious G. E., Westfall C. S., Jez J. M. (2013) Redox-linked gating of nucleotide binding by the N-terminal domain of adenosine 5′-phosphosulfate kinase. J. Biol. Chem. 288, 6107–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang M., Ravilious G. E., Hicks L. M., Jez J. M., McCulla R. (2012) Redox switching of adenosine-5′-phosphosulfate kinase with photoactivatable atomic oxygen precursors. J. Am. Chem. Soc. 134, 16979–16982 [DOI] [PubMed] [Google Scholar]
- 28. Leustek T., Murillo M., Cervantes M. (1994) Cloning of a cDNA encoding ATP sulfurylase from Arabidopsis thaliana by functional expression in Saccharomyces cerevisiae. Plant Physiol. 105, 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klonus D., Höfgen R., Willmitzer L., Riesmeier J. W. (1994) Isolation and characterization of two cDNA clones encoding ATP-sulfurylases from potato by complementation of a yeast mutant. Plant J. 6, 105–112 [DOI] [PubMed] [Google Scholar]
- 30. Klonus D., Riesmeier J. W., Willmitzer L. (1995) A cDNA clone for an ATP-sulfurylase from Arabidopsis thaliana. Plant Physiol. 107, 653–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Logan H. M., Cathala N., Grignon C., Davidian J. C. (1996) Cloning of a cDNA encoded by a member of the Arabidopsis thaliana ATP sulfurylase multigene family. J. Biol. Chem. 271, 12227–12233 [DOI] [PubMed] [Google Scholar]
- 32. Heiss S., Schäfer H. J., Haag-Kerwer A., Rausch T. (1999) Cloning sulfur assimilation genes of Brassica juncea L.: Cadmium differentially affects the expression of a putative low-affinity sulfate transporter and isoforms of ATP sulfurylase and APS reductase. Plant Mol. Biol. 39, 847–857 [DOI] [PubMed] [Google Scholar]
- 33. Hatzfeld Y., Lee S., Lee M., Leustek T., Saito K. (2000) Functional characterization of a gene encoding a fourth ATP sulfurylase isoform from Arabidopsis thaliana. Gene 248, 51–58 [DOI] [PubMed] [Google Scholar]
- 34. Zhu L., Deng W. W., Ye A. H., Yu M., Wang Z. X., Jiang C. J. (2008) Cloning of two cDNAs encoding a family of ATP sulfurylase from Camellia sinensis related to selenium or sulfur metabolism and functional expression in Escherichia coli. Plant Physiol. Biochem. 46, 731–738 [DOI] [PubMed] [Google Scholar]
- 35. Renosto F., Patel H. C., Martin R. L., Thomassian C., Zimmerman G., Segel I. H. (1993) ATP sulfurylase from higher plants: kinetic and structural characterization of the chloroplast and cytosol enzymes from spinach leaf. Arch. Biochem. Biophys. 307, 272–285 [DOI] [PubMed] [Google Scholar]
- 36. Rotte C., Leustek T. (2000) Differential subcellular localization and expression of ATP sulfurylase and 5′-adenylylsulfate reductase during ontogenesis of Arabidopsis leaves indicates that cytosolic and plastid forms of ATP sulfurylase may have specialized functions. Plant Physiol. 124, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brunold C., Suter M. (1984) Regulation of sulfate assimilation by nitrogen nutrition in the duckweed Lemna minor L. Plant Physiol. 76, 579–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cacco G., Saccomani M., Ferrari G. (1977) Development of sulfate uptake capacity and ATP-sulfurylase activity during root elongation in maize. Plant Physiol. 60, 582–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vauclare P., Kopriva S., Fell D., Suter M., Sticher L., von Ballmoos P., Krähenbühl U., den Camp R. O., Brunold C. (2002) Flux control of sulphate assimilation in Arabidopsis thaliana: Adenosine 5′-phosphosulphate reductase is more susceptible than ATP sulphurylase to negative control by thiols. Plant J. 31, 729–740 [DOI] [PubMed] [Google Scholar]
- 40. Liang G., Yang F., Yu D. (2010) MicroRNA395 mediates regulation of sulfate accumulation and allocation in Arabidopsis thaliana. Plant J. 62, 1046–1057 [DOI] [PubMed] [Google Scholar]
- 41. Kawashima C. G., Matthewman C. A., Huang S., Lee B. R., Yoshimoto N., Koprivova A., Rubio-Somoza I., Todesco M., Rathjen T., Saito K., Takahashi H., Dalmay T., Kopriva S. (2011) Interplay of SLIM1 and miR395 in the regulation of sulfate assimilation in Arabidopsis. Plant J. 66, 863–876 [DOI] [PubMed] [Google Scholar]
- 42. Mougous J. D., Lee D. H., Hubbard S. C., Schelle M. W., Vocadlo D. J., Berger J. M., Bertozzi C. R. (2006) Molecular basis for G-protein control of the prokaryotic ATP sulfurylase. Mol. Cell 21, 109–122 [DOI] [PubMed] [Google Scholar]
- 43. Beynon J. D., MacRae I. J., Huston S. L., Nelson D. C., Segel I. H., Fisher A. J. (2001) Crystal structure of ATP sulfurylase from the bacterial symbiont of the hydrothermal vent tubeworm Riftia pachyptila. Biochemistry 40, 14509–14517 [DOI] [PubMed] [Google Scholar]
- 44. Taguchi Y., Sugishima M., Fukuyama K. (2004) Crystal structure of a novel zinc-binding ATP sulfurylase from Thermus thermophilus HB8. Biochemistry 43, 4111–4118 [DOI] [PubMed] [Google Scholar]
- 45. Yu Z., Lansdon E. B., Segel I. H., Fisher A. J. (2007) Crystal structure of the biofunctional ATP sulfurylase-APS kinase from the chemolithotrophic thermophile Aquifex aeolicus. J. Mol. Biol. 365, 732–743 [DOI] [PubMed] [Google Scholar]
- 46. Parey K., Demmer U., Warkentin E., Wynen A., Ermler U., Dahl C. (2013) Structural, biochemical, and genetic characterization of dissimilatory ATP sulfurylase from Allochromatium vinosum. PLoS One 8, e74707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ullrich T. C., Blaesse M., Huber R. (2001) Crystal structure of ATP sulfurylase from Saccharomyces cerevisiae, a key enzyme in sulfate activation. EMBO J. 20, 316–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ullrich T. C., Huber R. (2001) The complex structures of ATP sulfurylase with thiosulfate, ADP, and chlorate reveal new insights in inhibitory effects and the catalytic cycle. J. Mol. Biol. 313, 1117–1125 [DOI] [PubMed] [Google Scholar]
- 49. MacRae I. J., Segel I. H., Fisher A. J. (2001) Crystal structure of ATP sulfurylase from Penicillium chrysogenum: Insights into the allosteric regulation of sulfate assimilation. Biochemistry 40, 6795–6804 [DOI] [PubMed] [Google Scholar]
- 50. MacRae I. J., Segel I. H., Fisher A. J. (2002) Allosteric inhibition via R-state destabilization in ATP sulfurylase from Penicillium chrysogenum. Nat. Struct. Biol. 9, 945–949 [DOI] [PubMed] [Google Scholar]
- 51. Harjes S., Bayer P., Scheidig A. J. (2005) The crystal structure of human PAPS synthetase 1 reveals asymmetry in substrate binding. J. Mol. Biol. 347, 623–635 [DOI] [PubMed] [Google Scholar]
- 52. Ravilious G. E., Herrmann J., Goo Lee S., Westfall C. S., Jez J. M. (2013) Kinetic mechanism of a dimeric ATP sulfurylase from plants. Biosci. Rep. 33, e00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 54. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Trott O., Olson A. J. (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holm L., Rosenström P. (2010) Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koprivova A., Giovannetti M., Baraniecka P., Lee B. R., Grondin C., Loudet O., Kopriva S. (2013) Natural variation in the ATPS1 isoform of ATP sulfurylase contributes to the control of sulfate levels in Arabidopsis. Plant Physiol. 163, 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gan X., Stegle O., Behr J., Steffen J. G., Drewe P., Hildebrand K. L., Lyngsoe R., Schultheiss S. J., Osborne E. J., Sreedharan V. T., Kahles A., Bohnert R., Jean G., Derwent P., Kersey P., Belfield E. J., Harberd N. P., Kemen E., Toomajian C., Kover P. X., Clark R. M., Rätsch G., Mott R. (2011) Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Phartiyal P., Kim W. S., Cahoon R. E., Jez J. M., Krishnan H. B. (2008) The role 5′-adenylylsulfate reductase in the sulfur assimilation pathway of soybean: Molecular cloning, kinetic characterization, and gene expression. Phytochemistry 69, 356–364 [DOI] [PubMed] [Google Scholar]
- 62. Bradley M. E., Rest J. S., Li W. H., Schwartz N. B. (2009) Sulfate activation enzymes: phylogeny and association with pyrophosphatase. J. Mol. Evol. 68, 1–13 [DOI] [PubMed] [Google Scholar]
- 63. Deyrup A. T., Singh B., Krishnan S., Lyle S., Schwartz N. B. (1999) Chemical modification and site-directed mutagenesis of conserved HXXH and PP-loop motif arginines and histidines in the murine bifunctional ATP sulfurylase/adenosine 5′-phosphosulfate kinase. J. Biol. Chem. 274, 28929–28936 [DOI] [PubMed] [Google Scholar]
- 64. Venkatachalam K. V., Fuda H., Koonin E. V., Strott C. A. (1999) Site-selected mutagenesis of a conserved nucleotide binding HXGH motif located in the ATP sulfurylase domain of human bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase. J. Biol. Chem. 274, 2601–2604 [DOI] [PubMed] [Google Scholar]
- 65. Perona J. J., Rould M. A., Steitz T. A. (1993) Structural basis for transfer RNA aminoacylation by Escherichia coli glutaminyl-tRNA synthetase. Biochemistry 32, 8758–8771 [DOI] [PubMed] [Google Scholar]
- 66. Park Y. S., Gee P., Sanker S., Schurter E. J., Zuiderweg E. R., Kent C. (1997) Identification of functional conserved residues of CTP:glycerol-3-phosphate cytidylyltransferase: Role of histidines in the conserved HXGH in catalysis. J. Biol. Chem. 272, 15161–15166 [DOI] [PubMed] [Google Scholar]
- 67. Weber C. H., Park Y. S., Sanker S., Kent C., Ludwig M. L. (1999) A prototypical cytidylyltransferase: CTP:glycerol-3-phosphate cytidylyltransferase from Bacillus subtilis. Structure 7, 1113–1124 [DOI] [PubMed] [Google Scholar]
- 68. Parey K., Fritz G., Ermler U., Kroneck P. M. (2013) Conserving energy with sulfate around 100°C–Structure and mechanism of key metal enzymes in hyperthermophilic Archaeoglobus fulgidus. Metallomics 5, 302–317 [DOI] [PubMed] [Google Scholar]
- 69. Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]