

Abstract
Anti-influenza drugs, amantadine and rimantadine, targeting the M2 channel from influenza A virus are no longer effective because of widespread drug resistance. S31N is the predominant and amantadine-resistant M2 mutant, present in almost all of the circulating influenza A strains as well as in the pandemic 2009 H1N1 and the highly pathogenic H5N1 flu strains. Thus, there is an urgent need to develop second-generation M2 inhibitors targeting the S31N mutant. However, the S31N mutant presents a huge challenge to drug discovery, and it has been considered undruggable for several decades. Using structural information, classical medicinal chemistry approaches, and M2-specific biological testing, we discovered benzyl-substituted amantadine derivatives with activity against both S31N and WT, among which 4-(adamantan-1-ylaminomethyl)-benzene-1,3-diol (44) is the most potent dual inhibitor. These inhibitors demonstrate that S31N is a druggable target and provide a new starting point to design novel M2 inhibitors that address the problem of drug-resistant influenza A infections.
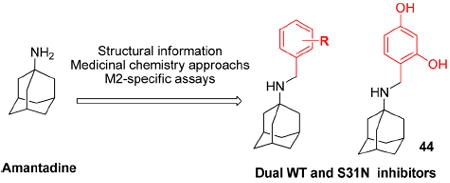
INTRODUCTION
Seasonal influenza infections as well as the emergence of life-threatening strains of influenza are a major worldwide health concern. Every year, influenza epidemics cause numerous deaths and millions of hospitalizations.1,2 Besides the yearly epidemic outbreaks, influenza viruses are even more threatening pathogens due to their potential to cause pandemics, as occurred in 2009 with the emergence and worldwide spread of pandemic H1N1. Available prophylactic vaccines are not completely effective against emerging flu strains, and thus, effective antiviral therapy is an essential component in the fight against influenza A infections. Currently, there are only four antivirals approved for use against influenza virus infections in humans, two targeting the M2 proton channel, amantadine (oral) and rimantadine (oral), and two targeting the neuraminidase, oseltamivir (oral), and zanimivir (inhaled) (Figure1). Resistance to both classes is a problem: resistance to the adamantane class of drugs is now so pervasive that the Centers for Disease Control and Prevention (CDC) has advised against its continued use,2,3 and widespread resistance to oseltamivir has been increasingly reported since 2007–2008.4-8 Even more of a concern are the reports of the emergence of flu strains with dual resistance to both adamantane and neuraminidase classes of antivirals.9
Figure 1.
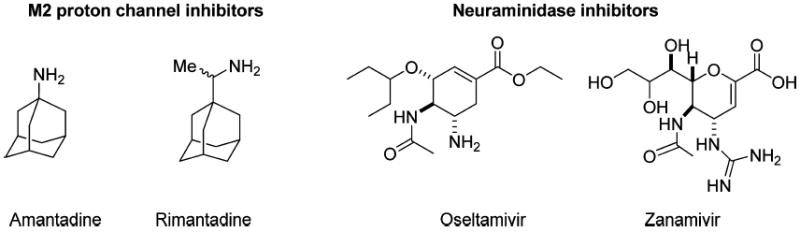
Structures of four approved antiviral drugs for influenza A.
Resistance to M2 proton channel drugs is associated with mutations in the transmembrane domain of the M2 protein. The homotetrameric structure of the M2 channel places constraints on the types of drug-resistant mutations that can be accommodated. A single mutation to the pore-lining residues of the protein results in four changes to the residues lining the pore, where the drug binds. While many mutations can be tolerated with retention of function in vitro,10-12 only a few mutants appear to be tolerated in highly transmissible viruses seen in the wild. V27A, L26F, and S31N have consistently comprised more than 99% of transmissible M2 mutants, among which S31N is now predominant in 98–100% of the transmissible amantadine-resistant H1N1, H5N1, and H3N2 strains isolated from humans, birds, and swine in the past decade.1 As a result, there is an urgent need to develop second-generation novel M2 inhibitors targeting the most prevalent S31N mutant. However, this mutation presents significant challenges to drug discovery because the side chain of Asn31 partially fills the amantadine-binding site and also markedly changes its physical properties.
M2 inhibitors generally consist of a hydrophobic portion (adamantane in the drugs amantadine and rimantadine) connected to a polar group, which is generally a positively charged substituent such as an amine in amantadine or an aminoethyl group in rimantadine (Figure 1). A variety of scaffolds can be substituted for the hydrophobic adamantane group, including fused and spiro-fused multicyclic alkanes, acyclic branched alkanes, and silanes.13,14 These compounds showed excellent activity for WT, and some were highly active against V27A and L26F.14,15 However, none showed significantly better activity vs S31N than had already been seen for amantadine. The polar group has also been the subject of extensive structure–activity relationship (SAR) studies. Again, a variety of substituents were tolerated when the target was WT, but only the simple primary amine found in amantadine gave significant activity against S31N. Thus, after several decades of medicinal chemistry investigations, no progress had been made toward inhibiting this predominant mutant form, suggesting that this variant might be essentially undruggable.16-20
Our journey to discover S31N inhibitors started with modification of amantadine (1) whose potency against S31N drops approximately 12-fold from 16 μM against WT21 to 200 μM22 in a TEVC (two electrode voltage clamp) electrophysiological assay. Amantadine (1) is, however, more active than rimantadine (2) against S31N (199.9 μM vs >2 mM)23 (Figure 2A). We envisioned that the S31N inhibitory activity could be restored if we captured additional interactions within the channel. Recent crystallographic and NMR studies have provided major advances in our understanding of the mode of inhibition of M2 by amantadine and related drugs (Figure 2B and C).15,21,24-33
Figure 2.
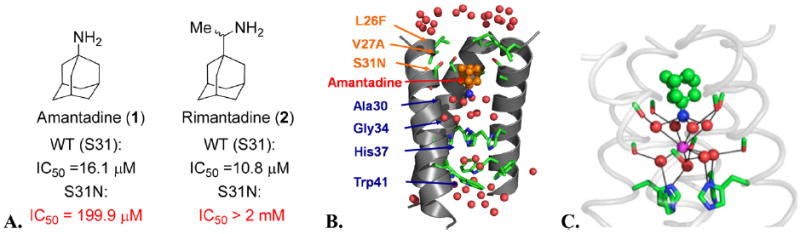
(A) Activity of amantadine and rimantadine against the WT (S31) and S31N mutant in the TEVC assay. (B) The M2 channel-lining residues found in transmissible drug-resistant mutants are shown in orange letters. Amantadine (orange bound drug) causes a break in the aqueous path (water molecules, red balls). One helix has been removed for clarity. (C) Snapshot from a simulation of amantadine with WT.15 Water molecules (red) associate with carbonyl groups (green/red sticks) in a square planar array. This array of water molecules can stabilize the bound ammonium group of amantadine (green and blue bound drug) or a centrally located water molecule (magenta).
The location of the pharmacologically relevant drug binding site in M2 was extensively debated until recently. Our earlier crystallographic structures suggested a pore-binding model,24 while Chou and co-workers proposed a more peripheral site on the surface of the protein based on NMR studies.25 However, last year, Chou and co-workers solved a structure, showing that the drug binds to the site we had originally identified, ending the controversy, and validating our earlier crystallographic structures.31 Specifically, the drug binds in a pocket within the channel lumen and is surrounded by the apolar side chain of Val27, Ala30, the C-beta of Ser31, and the C-alpha of Gly34. The ammonium group projects downward toward the invariant His37 residue but does not actually interact with this residue. Instead, it is stabilized by water molecules, which are in turn stabilized by interactions with backbone carbonyl groups lining the pore.15 The mutation S31N disrupts the size and polarity of the pore in precisely the region that accommodates the adamantane group of amantadine, explaining the decrease in potency of amantadine from 16 μM against WT to 200 μM in a TEVC electrophysiological assay. Moreover, NMR studies showed that S31N was more dynamic and possibly also more hydrated in the pore than the corresponding WT protein.34 This led us to consider strategies in which we either increased the polarity or dimensions of the adamantane core to capture more interactions with the backbone of the channel (Figure 3, approach I). We synthesized over 300 amantadine analogues and other diverse cores, some of which have been published.14,15,22,35-38 While we succeeded in inhibiting V27A and L26F, none of these compounds inhibited S31N.15
Figure 3.
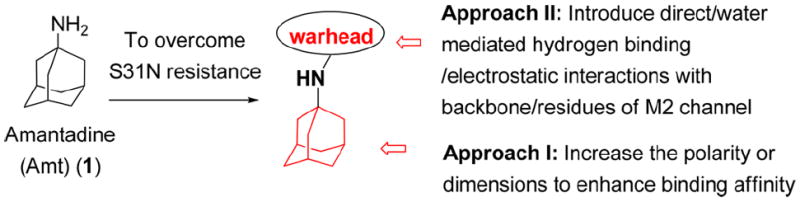
Two strategies to overcome drug-resistance.
We turned our attention to the strategy of introducing additional groups (warhead groups) to the amine, in order to enhance affinity for S31N as well as WT by introducing direct/water mediated hydrogen binding/electrostatic interactions with backbone or the groups lining the pore of the M2 channel (Figure 3, approach II). Examination of the structure of M2’s channel showed there was sufficient space in the channel to accommodate additional substituents to mediate these interactions. However, this line of modification of amantadine (1) was not obvious; primary amines tended to be more potent than secondary amines, and many early substitutions of the primary amines decreased activity toward the WT channel.14 Nevertheless, two reports had shown that aromatic groups could be appended to the amines of 1-adamantanemethylamine (a rimantadine (2) derivative)39 and pinanamine40 and showed activity against M2 channels. We therefore synthesized the most potent inhibitors in these two series, compounds 339 and 4,40 and found that they were active against WT (21% and 91% inhibition for 3 and 4, respectively) (Table 1). However, they showed no activity against S31N (0% inhibition for both) in our electrophysiological TEVC assay. Since amantadine (1) is more potent against S31N (35% inhibition) than rimantadine (2) (13% inhibition) and pinanamine (0% inhibition23) in a TEVC assay, we envisioned that installation of an aromatic group to the amine group of amantadine (5–49) might improve activity against S31N as well as WT. This led to the discovery of benzyl-substituted amantadine derivatives as dual WT and S31N inhibitors, which is described in the present article. These compounds serve as starting points for the design of novel compounds that address the problem of drug-resistant influenza A infections. At the same time of writing this article, a pinanamine bearing an imidazole derivative was reported to show weak inhibitory activity against the S31N M2 channel.41
Table 1.
M2 Inhibitors with an Appended Aromatic Groupa
| Structure |
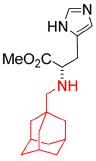
|
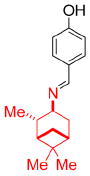
|

|
|---|---|---|---|
| 3 | 4 | 5-49 | |
| Rimantadine-based | Pinanamine-based | Amantadine-based (this work) | |
| S31N % inhibition (TEVC) | 0% | 0% | See the text |
| WT % inhibition (TEVC) | 27 ± 2% | 91 ± 2% |
The activity of the inhibitors was measured using the TEVC technique with full length AM2 protein in the Xenopus oocytes membrane; average of three measurements was reported (see Experimental Section in the Supporting Information).
A significant obstacle to developing inhibitors of M2 is to devise assays that are specific to the inhibition of M2. In establishing an assay for S31N, we found it important to verify that the assay identified true M2 inhibitors, as we found both vesicle proton flux assays and viral inhibition assays are complicated by the fact that many amantadine-like compounds are proton carriers. Proton carriers such as chloroquine are good inhibitors of the virus in vitro but lack efficacy in animal models and human trials.42-44 However, by conducting electrophysiological assays in parallel with antiviral assays it is possible to eliminate many false positives from compounds that acted as proton carriers and delayed acidification of the endosome. We report here the first examples of compounds with specificity for both S31N and WT. Such compounds will be particularly important as mechanistic probes as well as serving as starting points for the design of novel compounds that address the problem of drug-resistant influenza A infections.
CHEMISTRY
The synthetic routes of compounds 5, 8, 9, 11, 12, 15, 17–23, and 29–49 are shown in Scheme 1. A variety of benzyl groups were installed on the N-terminal of 1 either through reductive amination (procedure a or b)45-47 or amide bond formation followed by lithium aluminum hydride (LAH) reduction (procedure c).48-50 Subsequent reactions included deprotection, oxidation of a thioether,51 tetrazole formation,52 or N-methylation, yielding compounds 6, 10, 13, 16, and 27, respectively. Due to a low yield in the reductive animation with 2,4-dihydroxybenzaldehyde, compound 44 was synthesized by reductive amination with 2-hydroxy-4-methoxy-benzaldehyde followed by demethylation with BBr3.53 Compounds 14, 24, 25, 31, and 33 were prepared via similar routes (Scheme 2). The syntheses of the imine analogue, 26,40 and the ether analogue, 28, are shown in Scheme 3.
Scheme 1.
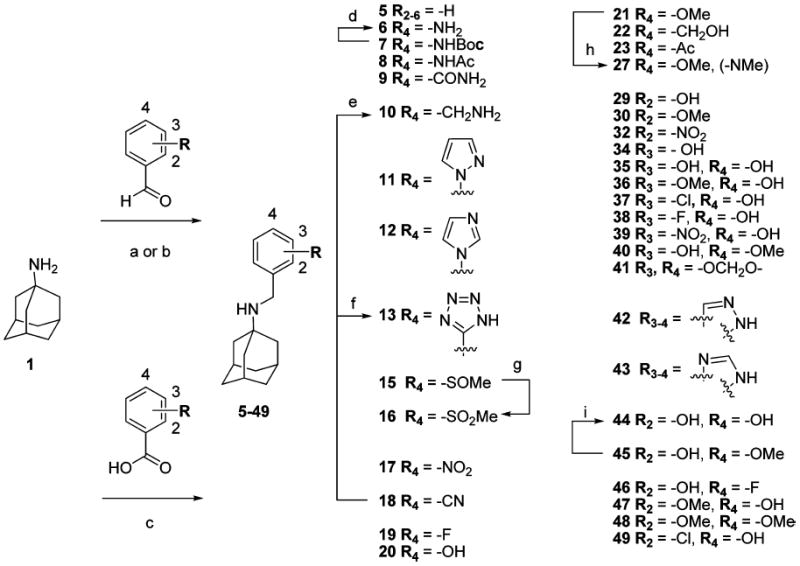
a
aReagents and conditions: (a) NaBH3CN, MeOH, RT, overnight. (b) (i) Ti(OiPr)4; (ii) NaBH4, MeOH. (c) (i) EDCI, HOAT, DIPEA, CH2Cl2, RT; (ii) LAH, THF, reflux. (d) HCl/dioxane, rt. (e) LAH, THF, reflux. (f) NaN3, CH3CN. (g) m-CPBA (1.2 equiv), CH2Cl2, RT. (h) NaH, Mel (1.1 equiv), 0 °C, DMF. (i) BBr3 (1.2 equiv), CH2Cl2, −78 to 0 °C.
Scheme 2.
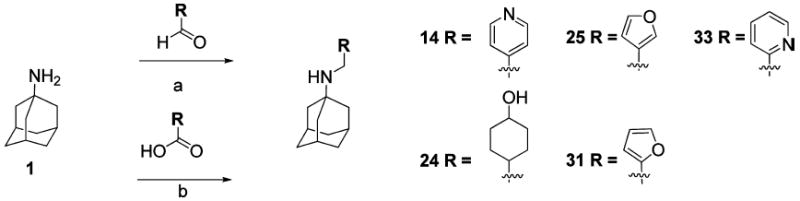
a
aReagents and conditions: (a) NaBH3CN, MeOH, RT. (b) (i) EDCl, HOAT, DIPEA, DMF, RT; (ii) LAH, THF, Reflux.
Scheme 3.
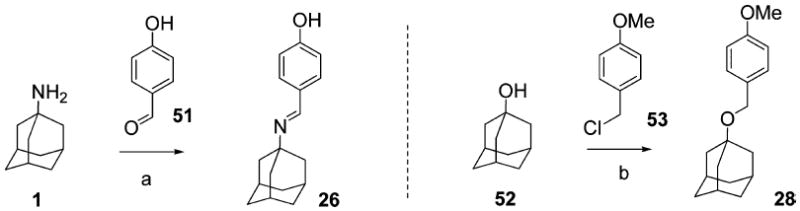
a
aReagents and conditions: (a) piperidine (cat.), benzene, Dean–Stark. (b) NaH, PMBCl (1.1 equiv), RT, DMF.
STRUCTURE–ACTIVITY RELATIONSHIP (SAR) STUDY
The activity of the inhibitors was measured using the TEVC technique with full length M2 protein expressed in Xenopus oocytes.21,54-56 All inhibitors were initially tested at 100 μM; those that inhibited the AM2 channel activity more than amantadine (Amt) (1) (by 35% inhibition) were chosen for the measurement of their IC50. Inhibitors of M2 enter the channel from the outside of the virus through a narrow opening near Val27. Thus, M2-blockers show slow on/off inhibition, with association occurring on the minute to hour time scale (at 1 μM) and dissociation hour to day time scale. Due to the limited stability of oocytes at low pH, we measured the percent inhibition after only 2 min, before equilibrium was reached. Due to the kinetics of inhibition, this provides an apparent kinetic IC50 up to 100-fold higher than the true Kdiss measured with much longer times.57 Nevertheless, the relative potency of the compounds tracks well with their relative potencies measured in other methods. Thus, the low micromolar IC50 values of compounds seen in our primary electrophysiologic assays are numerically larger than their antiviral potency in plaque assays. While the TEVC assay underestimates the true potencies of the compounds, it has the advantage of being remarkably reproducible, allowing us to reliably interpret even small degrees of inhibition. All measurements were made in triplicate, with a typical standard error being between 1% and 3%. Moreover, we observed less than 5% error when making repeat measurements over time and with different batches of oocytes.
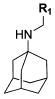
Since the reported nonamantadine-based aryl-containing M2 inhibitors (Table 1)39,40 showed essentially no inhibition against S31N, we started to explore amantadine-based derivatives with an aromatic group appended to the primary amine group, which led to the discovery of a new series of substituted N-benzyl amantadines as promising M2 inhibitors against S31N as well as WT. Beginning with the unsubsituted N-benzyl analogue of amantadine, we examined the effects of substitution at various positions in the ring. Because unsubstituted amantadine is already a relatively hydrophobic drug, we focused the present studies primarily on polar substituents to capture additional hydrogen bonding or electrostatic interactions as well as to maintain desirable drug-like properties. Although the unsubstituted derivative 5 was inactive toward S31N (1% inhibition), it did show significant inhibition against the WT (77% inhibition) at 100 μM drug concentration. We therefore focused on phenyl substituents with a hydrogen-bonding donor/acceptor at 4-position (Table 2) and were gratified to observe that the S31N inhibitory activity did increase gradually for 4-substituted series. Introduction of a 4-amino substituent (compound 6) increased S31N activity from 1% to 10% inhibition but decreased activity against WT from 77% to 31% inhibition. Interestingly, a Boc derivative of the aniline of 6 improved S31N activity to 20% inhibition (compound 7), and acetylation of 6 further boosted S31N activity to 32% inhibition (compound 8), which is similar to the activity of 1 (35% inhibition). However, the closely related benzamide 9 and benzylamine 10 lost considerable potency against both S31N and WT.
Table 2.
SAR of 4-Substituted Adamantan-1-yl-benzyl-aminesa
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1= | -H Amt |

|
|
|
|
|
|
|
|
|
|
| / ID # | 1 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| S31N | |||||||||||
| % inh/ | 35 ± 2/ | 1 ± 1 | 10 ± 1 | 20 ± 2 | 32 ± 2 | 3 ± 1 | 8 ± 1 | 26 ± 3 | 25 ± 2 | 8 ± 1 | 15 ± 1 |
| IC50 | 199.9 μM | ||||||||||
| WT | |||||||||||
| % inh/ | 91 ± 3/ | 77 ± 1 | 31 ± 2 | 13 ± 2 | 3 ± 2 | 7 ± 2 | 3 ± 2 | 4 ± 3 | 4 ± 2 | 6 ± 1 | 6 ± 1 |
| IC50 | 16.1 μM | ||||||||||
| R1= |
|
|
|
|
|
|
|
|
|
|
|
| / ID # | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 |
| S31N | |||||||||||
| % inh/ | 12 ± 1 | 21 ± 2 | 14 ± 1 | 8 ± 2 | 8 ± 2 | 42 ± 2/ | 31 ± 2 | 17 ± 2 | 31 ± 2 | 19 ± 2 | 8 ± 2 |
| IC50 | 166 μM | ||||||||||
| WT | |||||||||||
| % inh/ | 2 ± 2 | 45 ± 2 | 49 ± 2 | 36 ± 2 | 77 ± 1 | 67 ± 2 | 43 ± 1 | 26 ± 2 | 2 ± 2 | 16 ± 1 | 73 ± 2 |
| IC50 | |||||||||||
The activity of the inhibitors was measured using the TEVC technique with full length AM2 protein in the Xenopus oocytes membrane; the average of three measurements was reported (see Experimental Section in the Supporting Information).
Next, heteroaryl substituents at the 4-position were examined. Introduction of a pyrazole 11 or imidazole 12 gave compounds with some activity against S31N (26% and 25% inhibition, respectively) but still less potent than unsubstituted amantadine (1). A tetrazole derivative was even less active than the pyrazole and imidazole derivatives (8% inhibition); therefore, we examined other substituents at the 4-position. Replacement of aniline of 6 with its bioisomer, 4-pyridine 14, slightly increased S31N and WT activity. Other substituents at the 4-position such as sulfone 15, sulfoxide 16, nitro 17, cyano 18, and fluorine 19 showed some improvement in S31N activity compared to that of the parent compound 5 but did not reach the same activity as that of amantadine (1).
The 4-phenolic derivative, 4-(adamantan-1-ylaminomethyl)-phenol (20) was the first compound that showed slightly higher inhibitory activity against S31N (IC50 = 166 μM) than amantadine (1) (IC50 = 199.9 μM). Methylation of the 4-hydroxyl group of 20 reduced S31N inhibitory activity from 42% to 31% inhibition (compound 21), which indicated the importance of the 4-hydroxy group. Methyl ketone 23 and methyl ether 21 showed similar S31N activity (31% inhibition). One carbon homologated derivative of 20 (compound 22) markedly decreased S31N inhibitory activity from 42% to 17% inhibition. Interestingly, 4-cyclohexanol 24, a saturated version of 20, dramatically dropped S31N activity from 42% to 19% inhibition, which demonstrated that the aromaticity is crucial for S31N activity. We also found that the 4-hydroxy of 20 could not be replaced by its bioisomer, 3-furan 25, as this modification led to a marked loss of S31N inhibitory activity from 42% to 8%, respectively.
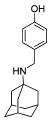
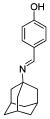
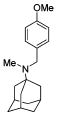
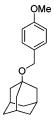
We also explored the SAR of the linkers (Table 3). A secondary amine is preferred over an imine analogue for S31N activity (42% and 21% inhibition for compounds 20 and 26, respectively). The secondary amine linker also showed better S31N activity than either a tertiary amine or an ether linkage (31%, 20%, and 13% inhibition for compounds 21, 27, and 28, respectively). Accordingly, in the following SAR studies, we kept the secondary amine linker constant.
Table 3.
SAR of the Linkers
| Structure |
|
|
|
|
|
|---|---|---|---|---|---|
| 20 | 26 | 21 | 27 | 28 | |
| S31N % inh | 42 ± 2% | 21 ± 1% | 31 ± 2% | 20 ± 3% | 13 ± 1% |
| WT % inh | 67 ± 2% | 81 ± 2% | 43 ± 1% | 34 ± 3% | 51 ± 2% |
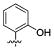
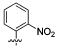
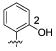
We further examined the SAR of 2- and 3-substituted benzyl amine analogues (Table 4). Two phenol regioisomers of 20, 3-hydroxy 34, and 2-hydroxy 29 displayed much lower S31N inhibitory activity (6% and 18% inhibition, respectively) but improved WT inhibitory activity (88% and 81% inhibition, respectively). As was the case for 4-methyl ether 21 (Table 2), 2-methyl ether 30 also decreased rather than enhanced the S31N inhibitory activity. Similarly, replacement of the 2-hydroxy in 29 with a 2-furan 31, 2-nitro-phenyl 32, or 2-pyridyl 33 group led to a further decrease in activity.
Table 4.
SAR of 2- and 3-Substituted Adamantan-1-yl-benzyl-amines
|
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| R1= | -H Amt |
|
|
|
|
|
|
|
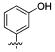
|
| / ID # | 1 | 5 | 20 | 29 | 30 | 31 | 32 | 33 | 34 |
| S31N | |||||||||
| % inh/ | 35 ± 2/ | 1 ± 1 | 42 ± 2/ | 18 ± 2 | 14 ± 1 | 6 ± 2 | 1 ± 1 | 0% | 6 ± 2 |
| IC50 | 199.9 μM | 166 μM | |||||||
| WT | |||||||||
| % inh/ | 91 ± 3/ | 77 ± 1 | 67 ± 2 | 81 ± 2 | 22 ± 2 | 66 ± 3 | 48 ± 1 | 12 ± 1 | 88 ± 1 |
| IC50 | 16.1 μM | ||||||||
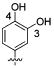
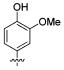
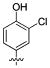
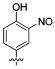
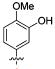
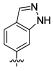
Attention was turned to effects of 3-substituents in the context of the 4-hydroxy 20, which had significant activity against S31N and WT (Table 5). Both electron withdrawing groups (3-Cl, 3-F, 3-NO2) (compounds 37, 38, and 39, respectively) and electron donating groups (3-OH, 3-OMe) (compounds 35 and 36, respectively) reduced potency, with 3-fluoro derivative 38 showing the smallest decrease in potency (31% inhibition). 3-Methoxy analogue 36 is less effective than 3-hydroxy derivative 35, which may be due to steric hindrance. This was supported by a more potent inhibitor 41, in which the methylene at 3-position was constrained into a 5-membered ring. Methylation of the 4-hydroxyl group of 35 (compound 40) maintained both WT and S31N activity and displayed slightly decreased activity than the more constrained 41. We also found that 4-hydroxy cannot be replaced with two cyclic bioisomers, 42 and 43, which displayed much weaker activity against S31N (10% and 12% inhibition, respectively).
Table 5.
SAR of 3,4-Disubstituted Adamantan-1-yl-benzyl-amines
|
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1= | H Amt |
|
|
|
|
|
|
|
|
|
|
| / ID # | 1 | 20 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 |
| S31N | |||||||||||
| % inh/ | 35 ± 2/ | 42 ± 2/ | 23 ± 1 | 3 ± 1 | 16 ± 1 | 31 ± 3 | 13 ± 2 | 20 ± 1 | 29 ± 1 | 10 ± 1 | 12 ± 1 |
| IC50 | 199.9 μM | 166 μM | |||||||||
| WT | |||||||||||
| % inh/ | 91 ± 3/ | 67 ± 2 | 44 ± 3 | 76 ± 2 | 78 ± 1 | 65 ± 1 | 66 ± 1 | 46 ± 3 | 54 ± 2 | 26 ± 1 | 79 ± 1 |
| IC50 | 16.1 μM | ||||||||||
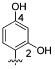
Because 2- and 4-hydroxyl and methoxy analogues provided better inhibition compared to the 3-hydroxy analogue (Table 4), we further examined whether their beneficial effects were additive. Toward this end, we synthesized 2,4-disubstituted benzyl amine derivatives (Table 6). Pleasingly, the 2,4-dihydroxy substituted analogue, 44, markedly enhanced S31N inhibitory activity (67% inhibition at 100 μM, IC50 = 35.2 μM). Compound 44 was approximately 6-fold more potent against S31N than amantadine (1) (IC50 = 199.9 μM), and only 2-fold less potent than amantadine (1) for WT (IC50 = 16 μM) in the TEVC assay. Compound 44 was also active against the WT with an IC50 of 59 μM, approximately 3.7-fold less potent than amantadine (1). The antiviral activity of 44 was also profiled in viral plaque reduction assays (Figure 4). The plaque assays showed that the compounds were more active than in the electrophysiological TEVC assays, which underestimated the affinities due to the brief period of inhibition. Compound 44 had an EC50 of 3.2 μM against S31N and was 7-fold more potent than amantadine (1) (EC50 = 22.5 μM) (Figure 4A). In the plaque assay, compound 44 was also active against the WT virus with complete inhibition of plaque formation at l μM (Figure 4B).
Table 6.
SAR of 2,4-Disubstituted Adamantan-1-yl-benzyl-amine of Selective Compounds
|
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R1= | -H Amt |
|
|
|

|
|
|
|

|
|
| /ID # | 1 | 20 | 29 | 44 | 45 | 46 | 47 | 48 | 49 | |
| S31N | % inh | 35 ± 2 | 42 ± 2 | 18 ± 2 | 67 ± 1 | 66 ± 1 | 37 ± 2 | 12 ± 2 | 11 ± 1 | 14 ± 1 |
| IC50 (μM) | 199.9 μM | 166 μM | 35.2 μM | 43.1 μM | NA | NA | NA | NA | ||
| WT | % inh | 91 ± 3 | 67 ± 2 | 81 ± 2 | 64 ± 1 | 56 ± 1 | 87 ± 2 | 0 | 16 ± 2 | 78 ± 1 |
| IC50 (μM) | 16.1 μM | NA | 59 μM | 79 μM | NA | NA | NA | NA | ||
Figure 4.
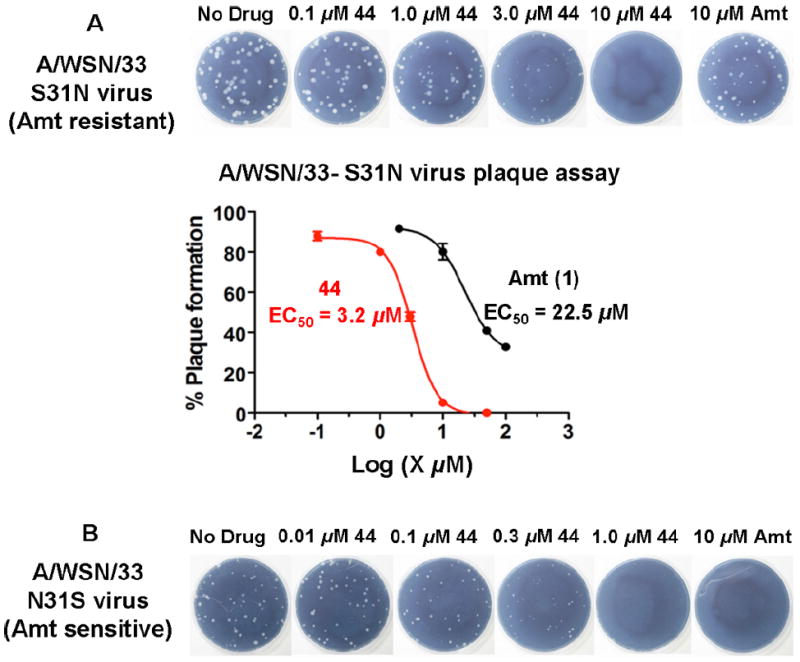
(A) Plaque reduction assay with A/WSN/33-S31N influenza virus. (B) Plaque reduction assay with A/WSN/33-N31S (WT) virus. The inhibitory effect of compound 44 and amantadine on influenza A virus replication was evaluated by plaque formation in MDCK cells as described in the Experimental Section (Supporting Information).
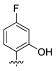
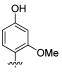
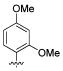
We also found that methylation of the 4-hydroxy 45 maintained the potency for both WT and S31N with IC50 values of 79 μM and 41.3 μM, respectively. The importance of the 2-hydroxy was observed in other 4-substituted analogues. For example, installation of 2-hydroxy to 4-fluoro analogue 19 markedly improved S31N inhibitory activity from 8% to 37% inhibition (compound 46). However, methylation of 2-hydroxy in 44 and 45 (compounds 47 and 48, respectively) dramatically reduced both S31N and WT activities, which might be due to the steric hindrance at this position. 2-Chloro analogue 49 also showed much lower S31N activity than 44, which may be attributed to the fact that chlorine is a weaker hydrogen acceptor than a hydroxyl group. These data might also indicate that a hydrogen bond donating hydroxyl is essential at this position of the ring.
CONCLUSIONS
Using a combination of structural information of M2 proton channel and medicinal chemistry approaches, we report the discovery of promising dual M2 inhibitors for S31N and WT M2 channels, demonstrating that M2 S31N is a druggable target. On the basis of this work, several more potent series of S31N inhibitors have been discovered, one series has already been published,58 and others will be the subject of future reports. Such compounds will be also important as mechanistic probes to address the problem of drug-resistant influenza A infections.
Supplementary Material
Acknowledgments
We acknowledge support from grant U01 A1074571 from the NIH.
ABBREVIATIONS USED
- Amt
amantadine
- S31
serine 31
- S31N
Serine 31 arganine
- WT
wild type
- Cmpd
compound
- TEVC
two-electrode voltage clamp
- % inh
% inhibition
- IC50
half maximal inhibitory concentration
- MDCK
Madin–Darby canine kidney cells
- LAH
lithium aluminum hydride
- HOAT
1-hydroxy-7-azabenzotriazole
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide
- NMR
nuclear magnetic resonance
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- RT
room temperature
- m-MCPA
meta-chloroperoxybenzoic acid
Footnotes
Supporting Information
Details of the two-electrode voltage clamp (TEVC) assay, plaque reduction assay, and synthesis of compounds 5-49. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Bright RA, Medina MJ, Xu XY, Perez-Oronoz G, Wallis TR, Davis XHM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 2.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 3.Cheng PK, Leung TW, Ho EC, Leung PC, Ng AY, Lai MY, Lim WW. Oseltamivir- and amantadine-resistant influenza viruses A (H1N1) Emerg Infect Dis. 2009;15:966–968. doi: 10.3201/eid1506.081357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheu TG, Deyde VM, Okomo-Adhiambo M, et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob Agents Chemother. 2008;52:3284–3292. doi: 10.1128/AAC.00555-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moscona A. Global transmission of oseltamivir-resistant influenza. N Engl J Med. 2009;360:953–956. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- 6.Moore C, Galiano M, Lackenby A, Abdelrahman T, Barnes R, Evans MR, Fegan C, Froude S, Hastings M, Knapper S, Litt E, Price N, Salmon R, Temple M, Davies E. Evidence of person to person transmission of oseltamivir resistant pandemic influenza A(H1N1) 2009 virus in a hematology unit. J Infect Dis. 2010;203:18–24. doi: 10.1093/infdis/jiq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayden FG, deJong MD. Emerging influenza antiviral resistance threats. J Infect Dis. 2011;203:6–10. doi: 10.1093/infdis/jiq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baz M. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N Engl J Med. 2009;361:2296–2297. doi: 10.1056/NEJMc0910060. [DOI] [PubMed] [Google Scholar]
- 9.Sheu TG, Fry AM, Garten RJ, Deyde VM, Shwe T, Bullion L, Peebles PJ, Li Y, Klimov AI, Gubareva LV. Dual resistance to adamantanes and oseltamivir among seasonal influenza A (H1N1) viruses: 2008-2010. J Infect Dis. 2011;203:13–17. doi: 10.1093/infdis/jiq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, DeGrado WF, Pinto LH. Functional studies and modeling of pore-lining residue mutants of the influenza a virus M2 ion channel. Biochemistry. 2010;49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular-basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grambas S, Hay AJ. Maturation of influenza A virus hemagglutinin-Estimates of the pH encountered during transport and its regulation by the M2 protein. Virology. 1992;190:11–18. doi: 10.1016/0042-6822(92)91187-y. [DOI] [PubMed] [Google Scholar]
- 13.Hu W, Zeng S, Li C, Jie Y, Li Z, Chen L. Identification of hits as matrix-2 protein inhibitors through the focused screening of a small primary amine library. J Med Chem. 2010;53:3831–3834. doi: 10.1021/jm901664a. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Ma C, Balannik V, Pinto LH, Lamb RA, Degrado WF. Exploring the requirements for the hydrophobic scaffold and polar amine in inhibitors of M2 from Influenza A virus. ACS Med Chem Lett. 2011;2:307–312. doi: 10.1021/ml100297w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Klein ML, DeGrado WF. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J Am Chem Soc. 2011;133:12834–12841. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurtz S, Luo G, Hahnenberger KM, Brooks C, Gecha O, Ingalls K, Numata K, Krystal M. Growth impairment resulting from expression of influenza virus M2 protein in Saccharomyces cerevisiae: identification of a novel inhibitor of influenza virus. Antimicrob Agents Chemother. 1995;39:2204–2209. doi: 10.1128/aac.39.10.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discovery. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Severson WE, McDowell M, Ananthan S, Chung DH, Rasmussen L, Sosa MI, White EL, Noah J, Jonsson CB. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2) J Biomol Screening. 2008;13:879–887. doi: 10.1177/1087057108323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagoja IM, De Clercq E. Anti-influenza virus agents: Synthesis and mode of action. Med Res Rev. 2008;28:1–38. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das K. Antivirals targeting influenza A virus. J Med Chem. 2012;55:6263–6277. doi: 10.1021/jm300455c. [DOI] [PubMed] [Google Scholar]
- 21.Jing X, Ma C, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duque MD, Ma C, Torres E, Wang J, Naesens L, Juarez-Jimenez J, Camps P, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J Med Chem. 2011;54:2646–2657. doi: 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma C. Unpublished results. [Google Scholar]
- 24.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–593. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cady SD, Luo W, Hu F, Hong M. Structure and function of the influenza A M2 proton channel. Biochemistry. 2009;48:7356–7364. doi: 10.1021/bi9008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J Mol Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Insight into the mechanism of the influenza a proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci U S A. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pielak RM, Oxenoid K, Chou JJ. Structural investigation of rimantadine inhibition of the AM2-BM2 chimera channel of influenza viruses. Structure. 2011;19:1655–1663. doi: 10.1016/j.str.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J Am Chem Soc. 2011;133:4274–4284. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andreas LB, Eddy MT, Chou JJ, Griffin RG. Magic-angle-spinning NMR of the Drug Resistant S31N M2 Proton Transporter from Influenza A. J Am Chem Soc. 2012;134:7215–7218. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci U S A. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, DeGrado WF, Pinto LH. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Fan X, Cristian L. Preparation of Adamantane Derivatives as Antiviral Agents. US 20110065762 A1 U S Pat Appl Publ. 2011
- 37.Wang J, Fan X, Cristian L. Preparation of Adamantane Derivatives as Antiviral Agents. US 20110065766 A1 U S Pat Appl Publ. 2011
- 38.Wang J, Ma C, Wu Y, Lamb RA, Pinto LH, DeGrado WF. Exploring organosilane amines as potent inhibitors and structural probes of influenza a virus M2 proton channel. J Am Chem Soc. 2011;133:13844–13847. doi: 10.1021/ja2050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Xu J, Liu F, Li C, Jie Y, Chen S, Li Z, Liu J, Chen Ling, Zhou G. Heterodimers of histidine and amantadine as inhibitors for wild type and mutant M2 channels of influenza A. Chin J Chem. 2010;28:1417–1423. [Google Scholar]
- 40.Zhao X, Li C, Zeng S, Hu W. Discovery of highly potent agents against influenza A virus. Eur J Med Chem. 2011;46:52–57. doi: 10.1016/j.ejmech.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 41.Zhao X, Jie Y, Rosenberg MR, Wan J, Zeng S, Cui W, Xiao Y, Li Z, Tu Z, Casarotto MG, Hu W. Design and synthesis of pinanamine derivatives as anti-influenza A M2 ion channel inhibitors. Antiviral Res. 2012;96:91–99. doi: 10.1016/j.antiviral.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 42.Di Trani L, Savarino A, Campitelli L, Norelli S, Puzelli S, D’Ostilio D, Vignolo E, Donatelli I, Cassone A. Different pH requirements are associated with divergent inhibitory effects of chloroquine on human and avian influenza A viruses. Virol J. 2007;4:39. doi: 10.1186/1743-422X-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ooi EE, Chew JS, Loh JP, Chua RC. In vitro inhibition of human influenza A virus replication by chloroquine. Virol J. 2006;3:39. doi: 10.1186/1743-422X-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paton NI, Lee L, Xu Y, Ooi EE, Cheung YB, Archuleta S, Wong G, Wilder-Smith A. Chloroquine for influenza prevention: a randomised, double-blind, placebo controlled trial. Lancet Infect Dis. 2011;11:677–6783. doi: 10.1016/S1473-3099(11)70065-2. [DOI] [PubMed] [Google Scholar]
- 45.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures (1) J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 46.Dalpathado DS, Jiang H, Kater MA, Desaire H. Reductive amination of carbohydrates using NaBH(OAc)3. Anal Bioanal Chem. 2005;381:1130–1137. doi: 10.1007/s00216-004-3028-9. [DOI] [PubMed] [Google Scholar]
- 47.Salmi C, Loncle C, Letourneux Y, Brunel JM. Efficient preparation of secondary aminoalcohols through a Ti(IV) reductive amination procedure. Application to the synthesis and antibacterial evaluation of new 3 beta-N-[hydroxyalkyl]aminosteroid derivatives. Tetrahedron. 2008;64:4453–4459. [Google Scholar]
- 48.Webb TR, Eigenbrot C. Conformationally restricted arginine analogues. J Org Chem. 1991;56:3009–3016. [Google Scholar]
- 49.Webb TH, Wilcox CS. Improved synthesis of symmetrical and unsymmetrical 5,11 methanodibenzo[b,f][1,5]diazocines. Readily available nanoscale structural units. J Org Chem. 1990:363–365. [Google Scholar]
- 50.Kihara N, Hashimoto M, Takata T. Redox behavior of ferrocene-containing rotaxane: Transposition of the rotaxane wheel by redox reaction of a ferrocene moiety tethered at the end of the axle. Org Lett. 2004;6:1693–1696. doi: 10.1021/ol049817d. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X, Yu M, Yao J, Zhang Y. Palladium-catalyzed regioselective arylation of arene C-H bond assisted by the removable 2-pyridylsulfinyl group. Synlett. 2012;23:463–467. [Google Scholar]
- 52.Finnegan WG, Hennry RA, Lofquist R. An improved synthesis of 5-substituted tetrazoles. J Am Chem Soc. 1958;80:3908–3911. [Google Scholar]
- 53.Williams AB, Hanson RN. Synthesis of substituted asymmetrical biphenyl amino esters as alpha helix mimetics. Tetrahedron. 2012;68:5406–5414. [Google Scholar]
- 54.Balannik V, Lamb RA, Pinto LH. The oligomeric state of the active BM2 ion channel protein of influenza B virus. J Biol Chem. 2008;283:4895–4904. doi: 10.1074/jbc.M709433200. [DOI] [PubMed] [Google Scholar]
- 55.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 57.Balannik V, Obrdlik P, Inayat S, Steensen C, Wang J, Rausch JM, DeGrado WF, Kelety B, Pinto LH. Solid-supported membrane technology for the investigation of the influenza A virus M2 channel activity. Pflugers Arch. 2010;459:593–605. doi: 10.1007/s00424-009-0760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci U S A. 2013;110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.