

Background: Autolysins ensure the plasticity of bacterial cell walls, and deletion leads to impaired cell clusters.
Results: High resolution structures of Staphylococcus aureus amidase AmiA shed light on peptidoglycan binding and cleavage.
Conclusion: AmiA distinguishes peptidoglycan mostly by the peptide, and cleavage is facilitated by a zinc-activated water molecule.
Significance: These structures will inform strategies to develop new therapeutics against MRSA.
Keywords: Drug Design, Enzyme Mechanisms, Peptidoglycan, Staphylococcus aureus, X-ray Crystallography, Zinc, MRSA, Amidase, Autolysin, Complex Structure
Abstract
The bifunctional major autolysin AtlA of Staphylococcus aureus cleaves the bacterium's peptidoglycan network (PGN) at two distinct sites during cell division. Deletion of the enzyme results in large cell clusters with disordered division patterns, indicating that AtlA could be a promising target for the development of new antibiotics. One of the two functions of AtlA is performed by the N-acetylmuramyl-l-alanine amidase AmiA, which cleaves the bond between the carbohydrate and the peptide moieties of PGN. To establish the structural requirements of PGN recognition and the enzymatic mechanism of cleavage, we solved the crystal structure of the catalytic domain of AmiA (AmiA-cat) in complex with a peptidoglycan-derived ligand at 1.55 Å resolution. The peptide stem is clearly visible in the structure, forming extensive contacts with protein residues by docking into an elongated groove. Less well defined electron density and the analysis of surface features indicate likely positions of the carbohydrate backbone and the pentaglycine bridge. Substrate specificity analysis supports the importance of the pentaglycine bridge for fitting into the binding cleft of AmiA-cat. PGN of S. aureus with l-lysine tethered with d-alanine via a pentaglycine bridge is completely hydrolyzed, whereas PGN of Bacillus subtilis with meso-diaminopimelic acid directly tethered with d-alanine is not hydrolyzed. An active site mutant, H370A, of AmiA-cat was completely inactive, providing further support for the proposed catalytic mechanism of AmiA. The structure reported here is not only the first of any bacterial amidase in which both the PGN component and the water molecule that carries out the nucleophilic attack on the carbonyl carbon of the scissile bond are present; it is also the first peptidoglycan amidase complex structure of an important human pathogen.
Introduction
Gram-positive, spherical staphylococci arrange in clusters and colonize the skin or mucous membranes, causing severe infections in infants, the elderly, transplantation patients, and people suffering from immunocompromising diseases, such as AIDS. Staphylococci are among the main causes of hospital-acquired infections (1). Staphylococcus aureus and Staphylococcus epidermidis are the most abundant human pathogens of the Staphylococcus genus. Both species form a biofilm that protects them from the human immune system (2) and antibiotics as well as contributing to persistent infections (3). In the case of S. epidermidis, this multilayered polysaccharide matrix (4) is responsible for infections of patients with implants, such as intravasal catheters, prostheses, or pacemakers (2, 5). This may require implant replacement and cause severe complications for the affected patients (6). The biofilm of S. aureus primarily serves as protection but also contributes to its pathogenicity (7). S. aureus is responsible for a large number of life-threatening infections that can result in diseases, such as endocarditis, meningitis, pneumonia, septicaemia, and toxic shock syndrome (8).
Resistance against S. aureus is on the rise, posing a serious threat to human health. There is therefore an urgent need for the development of new antibiotics to control emerging methicillin-resistant and vancomycin-resistant S. aureus strains (MRSA3 and VRSA, respectively). Worldwide numbers are not available, but with about 132,000 cases in Germany per year (9), hospital-acquired MRSA currently accounts for ∼20% of all staphylococcus infections (10), whereas in the early 1990s, the MRSA fraction was only ∼1% (11). In high risk areas, such as intensive care units, the MRSA infection rate increases up to 37% (12), causing 5,000 deaths and leading to additional costs of ∼380 million € (9) per year in Germany alone. In the United States, annual S. aureus infections have reached 475,000, 275,000 of which are MRSA-related, with $1–10 billion in extra expenses for the health care system and ∼11,000 to ∼19,000 deaths (13, 14). Targeting staphylococcal enzymes, critical for survival and growth of the bacterium, represents an attractive strategy for the development of new antibiotics.
Several hydrolytic enzymes ensure the plasticity of the staphylococcal cell wall by processing the complex PGN network. One of these, the major autolysin AtlA, is composed of two enzymes with hydrolytic activity (an amidase (AmiA) and a glucosaminidase (NAGase)) that cleave PGN at different locations (15). In the precursor AtlA protein, the two catalytic functions (cat) are each linked to targeting repeats (R1–R3) and also connected to a propeptide and a signal peptide (Fig. 1A). Posttranslational processing generates active AmiA and NAGase, which both localize at the septal region (16), where they process staphylococcal PGN during cell growth and division. S. aureus AtlA deletion mutants show a severely impaired phenotype that is unable to proliferate, forming large cell clusters instead (17). These findings demonstrate the essential function of AtlA in the S. aureus life cycle and also highlight a therapeutic potential for specific inhibition of AtlA.
FIGURE 1.
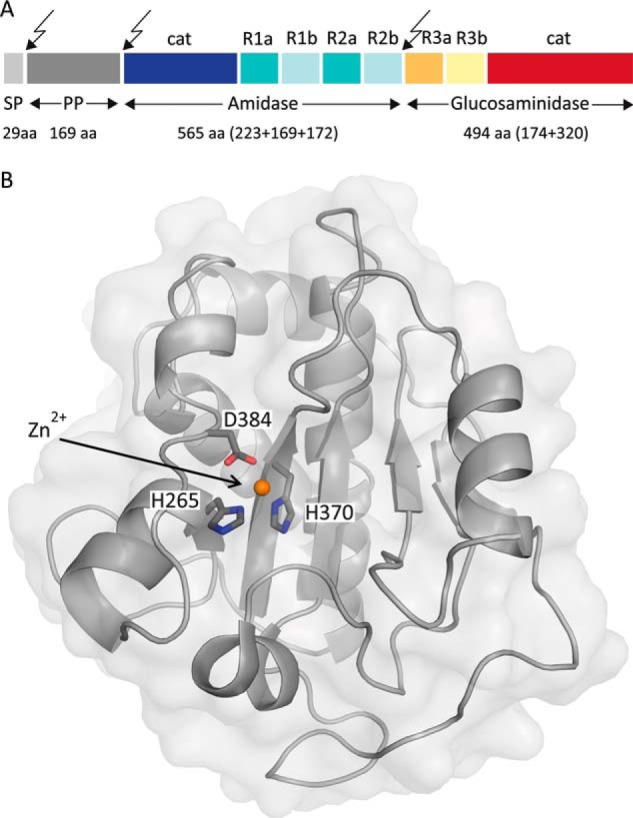
Prepro-AtlA holoenzyme and structure of AmiA-cat. A, domain arrangement of AtlA with sites of post-translational cleavage indicated by arrows. SP, signal peptide; PP, propeptide; cat, catalytic domain; R, repeat domain. This figure was adapted and modified from Ref. 19. B, mixed α/β fold of unliganded AmiA-cat in a schematic representation with a transparent surface. The zinc ion (orange) and its coordinating residues (dark gray) are highlighted.
The staphylococcal Atl domain organization is highly conserved in all of the species, with the amidase being the most conserved domain. It has been shown that the Atl-based phylogenetic tree correlates well with the corresponding 16 S rRNA- and core genome-based tree and represents a useful tool for staphylococcal genus and species typing (18).
The catalytic domain of AmiA (referred to here as AmiA-cat) is a zinc-dependent amidase that cleaves the amide bond between the peptide stem and carbohydrate backbone of PGN (16, 19, 20). The previously proposed mechanism for hydrolysis of the lactyl-alanine bond was based on in silico docking studies of the homologous catalytic domain AmiE from S. epidermidis (19). Further data from structures of a homologous protein originate from Escherichia coli (21). Consequently, detailed structural information on amidase-PGN interaction in Gram-positive bacteria is limited to date.
To determine the specificity of recognition and the mechanism of catalysis of AmiA-cat, we determined crystal structures of the enzyme in the absence (Fig. 1B) and presence (Fig. 2A) of the muramyltetrapeptide MurNAc-l-Ala-d-iGln-l-Lys-NHAc-d-Ala-NH2 (MtetP), a ligand that includes the previously characterized minimal ligand for the S. epidermidis amidase (muramyltripeptide) (19). Both structures were solved to high resolution, and they unambiguously establish the specificity of interaction as well as the reaction mechanism used by this essential cell wall enzyme. Our results form an excellent basis for the design of new antibiotic lead structures.
FIGURE 2.
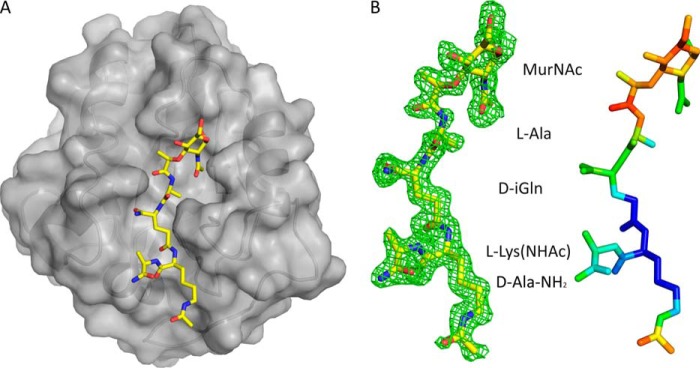
AmiA-cat in complex with MtetP. A, AmiA-cat (semitransparent gray surface) bound MtetP (yellow sticks) in the active site. B, the close-up on MtetP surrounded by omit density illustrates that the ligand is well defined. Still, the B-factor distribution of MtetP displays the elevated flexibility of its MurNAc moiety. The difference omit map is shown at a σ level of 2.0, and the color scale for B-factors ranges from blue (20 Å2) to red (50 Å2).
EXPERIMENTAL PROCEDURES
Molecular Biology
The cDNA coding for AmiA-cat (residues 199–421) was cloned into a pGEX-4–3T vector for expression. The expressed protein contains an N-terminal GST tag fused to AmiA-cat with a six-amino acid thrombin-cleavable linker. Active site mutants were created using site-directed mutagenesis as described in the QuikChange® protocol (22).
Protein Expression and Purification
Proteins were expressed in E. coli BL21 (DE3). After induction, cultures were incubated for 72 h at 20 °C. Harvested cells were then resuspended in buffer (150 mm NaCl, 50 mm Tris, pH 8.0) supplemented with PMSF and Roche Applied Science Complete protease inhibitor mix. Filtered cell lysate was loaded onto a 5-ml GSTrap FF column (GE Healthcare). 100 units of thrombin were added for on-column overnight cleavage at 20 °C and release of the fusion protein. Size exclusion chromatography removed the remaining small impurities and aggregates from the protein. Purity was confirmed by SDS-PAGE and MALDI-MS.
Protein Crystallization
AmiA-cat crystals belong to space group C2 and contain two protomers in the asymmetric unit, giving rise to a solvent content of 41.2%. Crystals were grown using the hanging drop vapor diffusion method at 20 °C. 1 μl of protein solution (11 mg/ml) was mixed with 1 μl of a well solution containing 0.1 m MES/imidazole buffer at pH 6.5 and a mix of sodium formate, ammonium acetate, sodium citrate, racemic sodium/potassium tartrate, and sodium oxamate at concentrations of 0.02 m each as well as 12.5% (w/v) PEG 1000, 12.5% (w/v) PEG 3350, and 12.5% (w/v) 2-methyl-2,4-pentanediol (Molecular Dimensions). Microseeding improved crystal quantity and quality. In order to obtain catalytically inactive enzyme, the AmiA-cat crystals were first incubated for 72 h in well solution supplemented with 20 mm EDTA to remove the active site zinc ion. For complex formation, crystals were next soaked for 60 h in well solution containing 20 mm EDTA and 20 mm MtetP. Crystals yielding a complex belong to space group P21 with four protomers in the asymmetric unit and similar solvent content. All crystals could be directly flash-frozen in liquid nitrogen because the well solutions contained sufficient cryoprotectant.
X-ray Diffraction
All data were collected at 100 K on PILATUS detectors using synchrotron radiation at beamlines X06DA and X06SA of the Swiss Light Source in Villigen, Switzerland.
Structure Determination
Indexing, integrating, and scaling were done with the XDS software package (23). Molecular replacement for all AmiA-cat structures was performed with PHASER (24, 25). Initial phases for AmiA-cat were determined with an AmiE search model (Protein Data Bank accession code 3LAT, 81% identity). The refined AmiA-cat structure was then used to solve the ligand structure by molecular replacement. Model building, refinement, and validation were performed with Coot (26, 27), the CCP4 suite (28–31), Phenix (32), and the MolProbity Web page (33). The coordinate and parameter files for MtetP were obtained from the PRODRG2 server (34). Simulated annealing (Phenix) was performed to remove model bias for the ligand structure, especially in the active site. The ligand was then added manually and modeled according to difference or simulated annealing omit density maps in Coot. The final structures contain almost all of the 223 residues, with the exception of 13–15 poorly ordered N-terminal and 1–4 C-terminal amino acids. Figures were generated with PyMOL (35), and electrostatic potentials were calculated with PBD2PQR and APBS 2.1 (36), implemented in PyMOL.
Purification of Peptidoglycan
PGN was isolated from S. aureus SA113 (37) or Bacillus subtilis ATCC 6051 using the method of de Jonge et al. (38) with some modifications. Briefly, cells were grown to A578 nm of 0.6 and harvested by centrifugation at 3,000 × g for 30 min, boiled with 5% SDS for 30 min, and broken with glass beads. Insoluble PGN was harvested by centrifugation at 30,000 × g for 30 min and washed several times with lukewarm water to remove SDS. Broken cell walls were suspended in 100 mm Tris-HCl, pH 7.2, treated with 10 μg/ml DNase (Sigma) and 50 μg/ml RNase (Sigma) for 2 h, and subsequently treated with 100 μg/ml trypsin for 16 h at 37 °C. To remove wall teichoic acid, the PGN preparations were incubated with 48% hydrofluoric acid for 24 h at 4 °C while stirring. PGN was harvested by centrifugation at 30,000 × g for 30 min and washed several times with water until complete removal of hydrofluoric acid. The final PGN product was lyophilized.
Preparation and HPLC/MS Analysis of PGN
Purified PGN (5 mg) was resuspended in 1 ml of 25 mm sodium phosphate buffer (pH 6.8) and digested with mutanolysin for 16 h at 37 °C. The enzyme reaction was stopped by boiling the sample for 5 min at 95 °C, and insoluble contaminants were removed by centrifugation. 50 μl of AmiA-cat, AmiA-H370A, or AmiA gel filtration buffer as a negative control were added to 100 μl of mutanolysin-digested PGN and incubated overnight at 37 °C while stirring. HPLC separation of digestion products was carried out on a reversed-phase column (Poroshell 120 EC-C18 4.6 × 150 mm, 2.7 μm; Agilent Technologies, Waldbronn, Germany) fitted with a poroshell EC-C18, 4.6-mm guard column using an Agilent 1200 system operating ChemStation software.
HPLC was performed using a linear 150-min gradient from 100% HPLC-buffer A (100 mm sodium phosphate, pH 2.2, and 5% methanol) to 100% HPLC-buffer B (100 mm sodium phosphate, pH 2.8, and 30% methanol) with a column temperature of 52 °C. Detection was made at 205 nm. Samples were prepared by mixing 100 μl of the product with an equal volume of 0.5 m sodium borate buffer (pH 8.0) containing freshly dissolved sodium borohydride (10 mg/ml) and reduced for 30 min at room temperature. The reaction was stopped, and excess borohydride was deactivated by lowering the pH to <3 using 20% phosphoric acid. 100 μl of the prepared sample was injected.
LC/MS analysis of these samples was performed with an Agilent HPLC-electrospray ionization-MS system (LC/MSD Ultra Trap System XCT 6330), using a gradient of A (H2O with 0.1% formic acid) and B (0.06% formic acid in acetonitrile) as follows: 0–10% B:A over 25 min, 10% B to 27 min, and 100% B to 30 min at 0.4 ml min−1 at 40 °C on a Nucleosil 100 C18 3 μm column (100 × 2-mm inner diameter) with a precolumn (10 × 2-mm inner diameter, Dr. Maisch, Ammerbuch, Germany). Detection of m/z values consistent with AmiA-cat digestion products was conducted using Agilent Data Analysis for 6300 Series Ion Trap LC/MS 6.1 version 3.4 software (Bruker-Daltonik GmbH).
RESULTS
Overall Structures of Unliganded and MtetP-bound AmiA-cat
The structures of unliganded and liganded AmiA-cat were determined at high resolution (1.12 and 1.55 Å, respectively, Table 1), allowing us to analyze their salient features with confidence. The proteins adopt a globular, mixed α/β fold that is highly similar to that of the previously reported unliganded AmiE-cat structure (19). Essential features of this fold are seven α-helices that surround a central six-stranded β-sheet (Fig. 1B). The rear of the β-sheet is shielded by two long α-helices, whereas its front is solvent-accessible and forms the bottom of a recessed groove. In the case of unliganded AmiA-cat, one end of this groove accommodates a zinc ion that is required for catalysis. Residues His-265, His-370, and Asp-384 directly coordinate the zinc ion (Fig. 1B), and nearby residues Glu-324 and His-382 are positioned to participate in catalysis. To produce a complex with MtetP, the zinc ion was first removed by treatment of the AmiA-cat crystals with EDTA, followed by extensive soaking with MtetP (see “Experimental Procedures”). Ligand binding was confirmed by a simulated annealing omit density map that displays unbiased electron density for MtetP over the entire tetrapeptide and as far as the N-acetylmuramic acid (MurNAc) moiety of the ligand. The tetrapeptide portion of MtetP is well defined by electron density (Fig. 2B), showing that this part of the ligand binds in an extended conformation to the recessed groove. The lactate moiety of MurNAc linking the peptide to the carbohydrate as well as the cyclic MurNAc moiety are less well ordered, suggesting higher mobility due to fewer defined interactions. These differences in mobility are reflected in the B-factor distribution of the ligand (Fig. 2B). The MtetP-bound AmiA-cat crystals contain four molecules in their asymmetric unit, only two of which contain fully occupied ligand binding sites. The flexible, cyclic MurNAc moiety presented sufficient electron density for model building in both copies. The binding sites of the remaining two molecules are less accessible due to smaller solvent channels. As a result, they contain electron density features that suggest only partially bound ligand.
TABLE 1.
Data collection and refinement statistics for AmiA-cat (Protein Data Bank code 4KNK) and liganded AmiA-cat (Protein Data Bank code 4KNL)
Values for the highest resolution bin are given in parentheses.
| Parameters | AmiA-cat | Liganded AmiA-cat |
|---|---|---|
| Beamline | X06DA (PX III) | X06SA (PX I) |
| Space group | C2 | P21 |
| Cell dimensions (Å) | a = 96.9, b = 81.7, c = 68.7 | a = 67.7, b = 82.8, c = 77.6 |
| α = γ = 90°. β = 128.8° | α = γ = 90°. β = 95.9° | |
| Wavelength (Å) | 1.00000 | 1.00605 |
| Detector | Pilatus 2M | Pilatus 6M |
| Resolution (Å) | 50-1.12 (1.15-1.12) | 50.0-1.55 (1.59-1.55) |
| Measured reflections | 885,445 (14,326) | 442,448 (27,121) |
| Unique reflections | 146,370 (6,937) | 122,742 (8,927) |
| Rmeas (%) | 4.7% (60.3%) | 6.5% (82.3%) |
| Completeness (%) | 92.6% (59.7%) | 99.5% (98.1%) |
| Redundancy | 6.0 (2.0) | 3.6 (3.0) |
| I/σ (I) | 21.1 (1.8) | 13.9 (1.7) |
| CC1/2 (%) | 100 (71.7) | 99.9 (69.7) |
| Resolution (Å) | 40.9-1.12 | 48.2-1.55 |
| Rwork/Rfree | 12.09/13.89 | 17.12/19.81 |
| No. of atoms | 3,894 | 7,321 |
| Protein | 3,318 | 6,531 |
| Water | 535 | 630 |
| MtetP | 102 | |
| Zn2+ | 2 | |
| Others | 39 | 58 |
| B-Factors (Å2) | ||
| Wilson | 13.7 | 24.5 |
| Mean | 16.9 (7.9–41.8) | 25.9 (11.9–69.9) |
| Protein | 15.1 (7.9–38.8 ) | 25.3 (11.9–69.9) |
| Water | 27.9 (9.0–41.8) | 33.1 (14.8–55.9) |
| MtetP | 36.7 (18.0–61.5) | |
| Zn2+ | 12.5 (12.2–12.8) | |
| Others | 27.8 (11.0–38.7) | 37.0 (18.8–53.0) |
| Root mean square deviations | ||
| Bond length (Å) | 0.009 | 0.012 |
| Bond angles (degrees) | 1.389 | 1.380 |
| Ramachandran plot | ||
| Most favorable (%) | 97.4 | 97.6 |
| Allowed (%) | 2.6 | 2.4 |
| Disallowed (%) | 0.0 | 0.0 |
With the exception of the active site region, unliganded and liganded AmiA-cat exhibit no structural differences. The two structures can be superimposed with a root mean square deviation of 0.2 Å (DaliLite pairwise (39)) onto each other, demonstrating that ligand binding does not induce larger structural changes.
Interactions of AmiA-cat with MtetP
Inspection of the structure of AmiA-cat in complex with MtetP reveals a sophisticated interaction network that significantly extends knowledge derived from earlier in silico docking (19). The tetrapeptide backbone of MtetP is anchored into the binding groove along its entire length via several direct or water-mediated hydrogen bonds to protein residues (Fig. 3). In addition, all four amino acid side chains of MtetP (l-Ala, d-iGln, l-Lys-NHAc, and d-Ala-NH2) are also involved in individual contacts with the protein, accounting for specificity. These interactions are detailed in Fig. 3 and summarized below.
FIGURE 3.

Interactions between AmiA-cat and MtetP. A, interactions of AmiA-cat with the MurNAc moiety and l-Ala of MtetP at the active site. The zinc ion from the native structure (orange sphere) is superimposed on the AmiA-cat complex (gray) with MtetP (yellow). B, close-up of the interactions of the peptide moiety of MtetP with AmiA. C, ChemSketch (57) plot of interactions between MtetP and AmiA-cat. Van-der-Waals interactions are depicted as green arcs, and hydrogen bonds are shown as black dashed lines. Coordination of Wat-9 involves His-370, Asp-384, and Wat-10 and a hydrogen bond with the carbonyl oxygen of the scissile amide bond in the ligand, which itself is positioned by interaction with His-382. Wat-10 lies next to Asp-266, Glu-324, Wat-9, and the carbonyl carbon of the scissile bond. MurNAc forms an intramolecular and four further hydrogen bonds with Glu-277, Thr-267, and Asn-269. Hydrophobic interactions of the methyl groups involve Ala-268 and Phe-293, respectively. l-Ala inserts into a small hydrophobic pocket formed by residues Ala-288 and Val-290. The amide side chain of d-iGln forms direct hydrogen bonds with Thr-380 and His-370 as well as two water-mediated hydrogen bonds to Asp-381. The carbonyl oxygen bonds with Gly-313 and Asn-317. l-Lys is stabilized by interactions with Asn-287 as well as Asn-317, whereas the acetylated side chain engages in hydrophobic interactions with Trp-310. A water (Wat-7 (w7)) bridges Nϵ of l-Lys-NHAc and the carbonyl oxygen of d-Ala, which engages additional water-mediated interactions with AmiA-cat residues Asn-287, Ala-314, and Gly-379.
The methyl side chain of the first amino acid of MtetP, l-Ala, inserts into a small hydrophobic pocket formed by residues Ala-288 and Val-290 (Fig. 3, A and C). The methyl group of l-Ala is also only 4.4 Å away from the Cα carbon of the conserved Gly-311. Lack of a side chain at position 311 contributes to specificity because even a medium sized side chains would lead to clashes with the l-Ala group.
The short amide side chain of d-isoglutamine (d-iGln), which is a key determinant of ligand binding (20), projects into a shallow pocket and forms direct hydrogen bonds with the hydroxyl group of Thr-380 and Nϵ of His-370, as well as two water-mediated hydrogen bonds to Asp-381 (Fig. 3, B and C). The side chain of His-382 seals the pocket on one side, whereas Thr-380 is located at the other end.
The side chain of the third amino acid, l-Lys, is acetylated (l-Lys-NHAc) in MtetP to approximate the physiologic state of this residue in PGN, where it is linked to additional glycine residues (see Fig. 7A). Specificity of recognition derives primarily from hydrophobic interactions between the aliphatic side chain of l-Lys-NHAc and the side chain of Trp-310. The entire lysine side chain lies parallel to the large indole ring of Trp-310, with distances below 4 Å (Fig. 3B). In addition, a water molecule (Wat-7) bridges the carbonyl oxygen of d-Ala and Nϵ of l-Lys-NHAc, thus helping to fix the orientation of the lysine side chain. Similarly, the peptide oxygen of Lys-NHAc forms a water-mediated (Wat-1) hydrogen bond with Asn-317.
FIGURE 7.

Schematic and molecular illustration of the PGN structures. A, in S. aureus, the PGN subunit is composed of d-isoglutamine and l-lysine and is cross-linked via a pentaglycine bridge (43). It comprises C-terminally either d-Ala-d-Ala or d-Ala-pentaglycine. B, B. subtilis PGN from vegetative cells (53, 55), however, contains d-isoglutamic acid and amidated meso-DAP and lacks a d-alanine at the meso-DAP α-carboxyl group, and it is directly cross-linked with d-alanine of the next subunit. In addition, B. subtilis PGN has 1,6-anhydro-MurNAc, shown in gray, at its terminus instead of a reducing MurNAc.
The fourth residue, d-Ala-NH2, is engaged in backbone hydrogen bond formation, but its side chain is at least partially exposed to solvent. The terminal amide group of d-Ala-NH2 participates in a network of water-mediated hydrogen bonds that help to fix its orientation with respect to the protein and the preceding l-Lys-NHAc side chain (Fig. 3, B and C). These interactions would still be possible in the context of either a pentaglycine bridge or a free carboxyl terminus.
The carbohydrate moiety of MtetP engages in fewer interactions with AmiA-cat than the peptide moiety, resulting in its higher flexibility. The N-acetyl oxygen of MurNAc forms an intramolecular hydrogen bond with the l-Ala nitrogen and via Wat-4 water-mediated contacts to AmiA-cat. The NAc-methyl group inserts in the hydrophobic portion (Ala-288, Val-290, and Phe-293) of the otherwise hydrophilic binding pocket (Fig. 3, A and C). Only Thr-267, Asn-269, and Glu-277 directly bond with MurNAc. Interestingly, the anomeric carbon is in α-configuration instead of β-configuration.
Active Site
The bound zinc ion in the unliganded structure marks the center of the active site of the enzyme (Fig. 1B). The ion is coordinated by the side chains of His-265, His-370, and Asp-384, with a water (Wat-10) completing its almost perfect tetrahedral coordination sphere. Although the zinc had to be removed to prepare the complex with MtetP, its position can be reliably inferred from superposition. Inspection of the unliganded and liganded structures shows two water molecules (Wat-9 and Wat-10) in each case, at most shifted by 1 Å. Wat-9 is located next to the position of the zinc ion, as well as Wat-10. Coordination of Wat-9 involves His-370, Asp-384, and Wat-10 (Fig. 3A). A fourth hydrogen bond is formed between Wat-9 and the carbonyl group of the scissile amide bond between l-Ala and the lactate of MurNAc in the ligand (Fig. 3, A and C). The high mobility or partial occupancy of Wat-9, indicated by a high B-factor, make a vital role unlikely. However, it may support stabilization of intermediate states during catalysis. The scissile bond is positioned directly adjacent to Wat-10, suggesting that the reaction mechanism proceeds by a Wat-10-mediated nucleophilic attack. Two residues, Asp-266 and Glu-324, lie next to Wat-10 and probably serve to enhance its nucleophilicity, thus favoring an attack on the lactyl amide bond. The carbonyl atom of the scissile amide bond of MtetP is hydrogen-bonded to His-382, which helps to orient the lactic acid moiety in the active site. We note that the His-382 side chain has different orientations in the unliganded and liganded AmiA-cat structures.
Putative Reaction Mechanism
The architecture of the active site and the observed interactions between protein and substrate are very much consistent with a reaction mechanism in which Wat-10 attacks the scissile bond. This water is hydrogen-bonded to the Asp-266 carbonyl and Glu-324 carboxyl groups, which would lead to both hydrogens of Wat-10 facing toward these residues and the free electron pairs of the Wat-10 oxygen facing toward zinc and the bond connecting the lactyl and peptide moieties (Fig. 4A). The likely role of the zinc ion is to polarize the oxygen of Wat-10, rendering it more reactive. The remaining free electron pair of the Wat-10 oxygen would then be able to perform a nucleophilic attack on the scissile bond of MtetP. His-382 could provide a proton for stabilization of the resulting oxyanion, whereas Glu-324 could accept a hydrogen from Wat-10 (Fig. 4B). Stabilization of the tetrahedral intermediate would involve the zinc ion and Nδ of His-382, which could each interact with one of the resulting hydroxyl groups as well as the side chains of Asp-266 and Glu-324. In the next step, the tetrahedral intermediate again forms a carbonyl group but with the peptide moiety as the leaving group rather than the previously attacking water molecule, leading to the separation of the carbohydrate and peptide moieties of PGN (Fig. 4C). At the same time, the N terminus of the new peptide is poised to accept a hydrogen from Glu-324, whereas His-382 can accept a hydrogen atom from the tetrahedral intermediate. Finally, the cleavage products are released from the active site (Fig. 4D).
FIGURE 4.

Proposed reaction mechanism of AmiA. A, Wat-10 (w10) is hydrogen-bonded to Asp-266 and Glu-324, and its free electron pairs face toward the scissile bond. Zn2+ is complexed by His-265, His-370, and Asp-384 and probably renders Wat-10 more reactive, enabling a nucleophilic attack. B, tetrahedral intermediate is stabilized by hydrogen bonds of the resulting hydroxyl groups with Nδ of His-382 as well as Asp-266, Glu-324, and zinc, respectively. C, reformation of a carbonyl group with the peptide moiety as leaving group. His-382 can accept a hydrogen atom from the tetrahedral intermediate, whereas the peptide is poised to accept a hydrogen from Glu-324. D, product release.
Implications for PGN Binding and Cleavage
The MtetP compound is a substrate for AmiA-cat, which cleaves large PGN structures in its physiologic setting. It is possible and indeed likely that additional contacts between PGN components and AmiA-cat exist and that the interactions between PGN and AmiA-cat are somewhat more complex than depicted here. Nevertheless, analysis of surface properties provides at least some clues as to how AmiA-cat would engage components of PGN that extend beyond the muramyltetrapeptide (i.e. the MurNAc-GlcNAc glycan polymer and the pentaglycine bridge) (Fig. 5A).
FIGURE 5.

AmiA-cat binding to PGN. A, electrostatic surface of AmiA-cat with MtetP (yellow), zinc (orange), and modeled PGN components (dark gray, black, and light gray). Uncharged (white surface area), positively charged (blue areas), and negatively charged residues (red surface) are shown. The spacious hydrophilic pocket harboring the zinc ion and active site also accommodates MurNAc. Adjacent GlcNAc rings (dark gray sticks) shift MurNAc slightly when modeled as a polymer. The lower peptide moiety of MtetP binds in the mostly uncharged region of the binding cleft. Two conformations for the pentaglycine bridge (black and light gray sticks, respectively) linked to l-Lys of MtetP were modeled according to uncharged surface area and possible hydrogen bonds. B, a multisequence alignment of bacterial amidases with identical residues colored by conservation from light to dark blue. Residues forming the carbohydrate binding pocket, marked by dark gray boxes, are highly conserved among all compared amidases. Surface-exposed amino acids near the two pentaglycine bridges are marked in black and light gray for the respective conformation models. Conservation, especially among staphylococci is high for both. Alignment was calculated using Clustal Omega (58), and the output was created using Jalview (59). C, conserved residues mapped on the AmiA-cat surface according to the color scheme used in A and B.
A number of residues largely conserved among bacterial amidases (Fig. 5, B and C) define the spacious carbohydrate binding pocket. Thr-267 and Glu-277 together with Met-281 and Phe-293 form the bottom of the pocket, whereas Tyr-280 on one side and Ala-268, Asn-269, and Ser-273 on the other side enclose the carbohydrate and form the lateral edges. Analysis of the complex shows that MurNAc does not engage in many specific contacts on its own. Based on chemical and geometric restraints, we therefore modeled a plausible conformation of GlcNAc-MurNAc-GlcNAc in the carbohydrate pocket (Fig. 5A), which is concurrent with a previously proposed three-dimensional structure of PGN (40) and nicely follows the carbohydrate groove. Steric constraints lead to a minor shift of MurNAc out of the binding pocket in the presence of β-1,4-linked GlcNAc molecules. Hydrogen bonds formed with O1 of MurNAc fall away in this model, whereas hydrophobic interactions and hydrogen bonds of the NAc moiety remain. Nevertheless, only the oxygen atoms O6 of preceding and succeeding GlcNAc may each form additional hydrogen bonds (preceding GlcNAc with His-382 or Wat-5 and succeeding GlcNAc with Gly-276, Glu-277, and Wat-66; data not shown). Physiologically, weaker interactions with MurNAc and the glycan strand make sense because the enzyme achieves its specificity through engagement of the tetrapeptide stem and must dissociate from the PGN after cleavage has occurred.
Investigation of the electrostatic surface of AmiA-cat gives two plausible orientations in terms of uncharged contact area for the pentaglycine bridge linked to l-Lys in PGN (Fig. 5A), although Cϵ and Nϵ would not superimpose with the complex. In both cases, glycines would be able to loosely interact with conserved (Fig. 5B) and uncharged AmiA-cat surface residues. Previous data showed that the presence or absence of the glycine bridge has little effect on catalysis (19, 20), indicating that interactions of the glycines with AmiA-cat contribute little binding energy. Nonetheless, the pentaglycine bridge is most likely important for specificity.
Substrate Specificity
In order to relate the structural data to functional experiments, we investigated wild-type AmiA-cat and an active site mutant AmiA-H370A for its ability to digest PGN from S. aureus as well as B. subtilis. Both PGN structures were first predigested with mutanolysin, which cleaves the glycosidic bond between GlcNAc and MurNAc, to facilitate the digestion. AmiA-cat completely hydrolyzed S. aureus PGN, whereas the AmiA-H370A mutant was inactive (Fig. 6, A–C). Analysis by HPLC shows that the main product generated by AmiA-cat is the disaccharide GlcNAc-MurNAc, with a mass of m/z 496 (Fig. 6, B and F). Interestingly, the intensity of the GlcNAc-MurNAc peak (Fig. 6B) represents the sum of the PGN oligomers seen in Fig. 6A, indicating that >95% of the PGN substrate was digested.
FIGURE 6.

RP-HPLC profile of mutanolysin-digested PGN fragments after incubation with enzyme. A, S. aureus PGN fragments incubated with elution buffer of AmiA-cat as control. Three peaks around 28 min contain monomer species of one peptide stem with carbohydrates, peaks at 58 min comprise species with two peptide stems, peaks at 73 min cover fragments with three peptide stems, and continuing accordingly. B, S. aureus PGN fragments incubated with AmiA-cat result in completely digested polymers with GlcNAc-MurNAc fragments remaining. C, S. aureus PGN fragments incubated with active site mutant AmiA-H370A show no catalytic activity. D, B. subtilis PGN fragments incubated with elution buffer of AmiA-cat result in a pattern similar to A but with specific retention times for B. subtilis. E, B. subtilis PGN fragments incubated with AmiA-cat exhibit no activity of the staphylococcal amidase for B. subtilis PGN. F, mass analysis of the major product shown in B, which corresponds to the disaccharide GlcNAc-MurNAc (m/z 496).
In contrast to S. aureus, B. subtilis PGN could not be hydrolyzed by AmiA-cat (Fig. 6, D and E). The major differences in PGN structure of S. aureus and B. subtilis are illustrated in Fig. 7. B. subtilis incorporates d-isoglutamic acid into the peptide linking the glycan chains (Fig. 7B), whereas S. aureus converts this amino acid to d-iGln (Fig. 7A). It is, however, unlikely that this difference is solely responsible for the inability of AmiA-cat to cleave B. subtilis PGN because the homologous AmiE enzyme still hydrolyzed synthetic substrates composed of MurNAc-l-Ala-d-iGlu-l-Lys, albeit with lower efficiency (20). We consider it more likely that two other alterations in the PGN structures are responsible for the observed difference in activity. The B. subtilis PGN carries a meso-diaminopimelic acid (meso-DAP), which has an amidated ϵ-carboxylate and a free α-carboxylate and also features a directly cross-linked peptide stem (Fig. 7B), without the pentaglycine bridge found in the S. aureus PGN. These two major differences probably prevent a proper fit of the B. subtilis PGN structure into the binding cleft of AmiA-cat.
DISCUSSION
We have determined high resolution structures of the catalytic region of the S. aureus amidase AmiA in its unliganded form and in complex with a compound that includes MurNAc and the tetrapeptide l-Ala-d-iGln-l-Lys-d-Ala. To crystallize the wild-type enzyme in complex with ligand while avoiding cleavage, it proved critical to first remove the catalytic zinc ion through extensive incubation with EDTA and then soak crystals with ligand, whose high concentrations reflect the environment in PGN. Analysis of the two structures provides insights into the parameters that govern specificity as well as the catalytic mechanism.
AmiA-cat folds into a compact structure with a long, exposed ligand binding groove that appears ideally suited to access its specific cleavage sites within the dense, highly cross-linked PGN structure. In addition to the catalytically active AmiA-cat domain, the mature enzyme also contains four repeat domains (Fig. 1A). Analysis of the highly homologous S. epidermidis amidase has shown that these repeats probably anchor the protein to lipoteichoic acid protruding from the staphylococcal cell wall (41). Importantly, the repeats are flexibly linked to the catalytically active domain, allowing cleavage to proceed efficiently (41). The position of the bound MtetP ligand sheds light on the interaction of amidases with more complex, branched PGN fragments. Thus, the structure of liganded AmiA-cat allows us to visualize how the enzyme acts in a physiologic setting. In addition to the groove that accommodates the tetrapeptide of MtetP, AmiA-cat contains surface features that probably allow for binding of MurNAc-GlcNAc polymers at one end and a pentaglycine bridge at the other end of the groove. The peptide composition of PGN in staphylococci is well conserved, typically using a pentaglycine bridge that connects d-Ala of one peptide stem with the Nϵ from l-Lys of another stem (42–44). Variations in S. aureus and among subspecies exist (43, 44) but are rare. Also, they concentrate on the variation of the pentaglycine bridge in terms of length and composition. The number of glycines may vary from four to six, or one glycine may be substituted by a serine or an alanine (43, 44). Although structures of several bacterial amidases have been determined (19, 45–52), the amidase AmiD from E. coli is the only catalytically active amidase for which structural data of an uncleaved ligand-enzyme complex have been available prior to this work (21).
The PGN of E. coli and B. subtilis is highly similar (42) and belongs to the type of variation A1γ (43). It differs in amino acid composition (d-iGln and l-Lys are substituted with d-iGlu and meso-DAP, respectively) as well as direct cross-linkage of amino acids 3 and 4 (no interpeptide bridge) from the PGN of S. aureus (42), which has a PGN of the A3α type of variation (43). Additionally, E. coli and B. subtilis PGN has anhydro-MurNAc at the terminus of the carbohydrate backbone (42, 53), whereas PGN of S. aureus always presents reducing MurNAc (54). Further differences within the E. coli and the B. subtilis A1γ type of variation include deviations in the presence or absence of an unlinked, terminal d-Ala and amidation of meso-DAP carboxyl groups (53, 55).
The inability to digest B. subtilis PGN presumably is a cumulative effect because d-iGlu instead of d-iGln is tolerated (20), and a meso-DAP, which is amidated at its second carboxyl group, itself is flexible enough to be accommodated at the lysine binding site of AmiA-cat. In addition, Asn-317 could partially compensate for a negative charge resulting from the absent, unlinked, terminal d-Ala of B. subtilis PGN. However, the direct cross-linkage is most likely to cause a clash with AmiA-cat due to a high rigidity and bulky side chain compared with a pentaglycine bridge. Since each of these differences is likely tolerated on its own, we suggest that the combined differences account for substrate specificity.
Although the E. coli enzyme AmiD and AmiA both belong to the amidase 2 family and are zinc-dependent, the two enzymes share a sequence identity of only 21%, have different substrates, and exhibit large structural differences (Fig. 8), including differences in the active site shown by a 2.0-Å root mean square deviation of α-carbon atoms (Dali server (56)). For example, the proton donor in AmiD is a lysine instead of a histidine residue (His-382 in AmiA). His-370 and Thr-380 that stabilize d-iGln in AmiA are replaced by a histidine and an arginine, which are better suited to bond with d-iGlu from A1γ-PGN variants. The free carboxylate of meso-DAP or d-Ala could be stabilized by another arginine of AmiD. Interestingly, this is not the case in the two available complex structures of AmiD (cocrystallized with tripeptide l-Ala-d-iGlu-l-Lys (MTP) and soaked with anhydro-MurNAc-l-Ala-d-iGlu-l-Lys (anhydro-MTP)) due to crystal contacts. Also, lysine was used as the third amino acid of the peptide stem instead of meso-DAP. Because anhydro-MurNAc does not occur in S. aureus PGN and the carbohydrate binding pocket of AmiD is smaller than in AmiA, one can draw only limited conclusions for binding and catalysis of staphylococcal PGN from these structures. Moreover, the AmiD complex structure of anhydro-MTP lacks space for both the zinc ion and a water molecule in the active site, which could be activated by the zinc ion and attack the scissile bond. Thus, the structure of AmiA-cat from S. aureus presented here provides new information both about how the ligand is contacted and also about how catalysis is performed.
FIGURE 8.
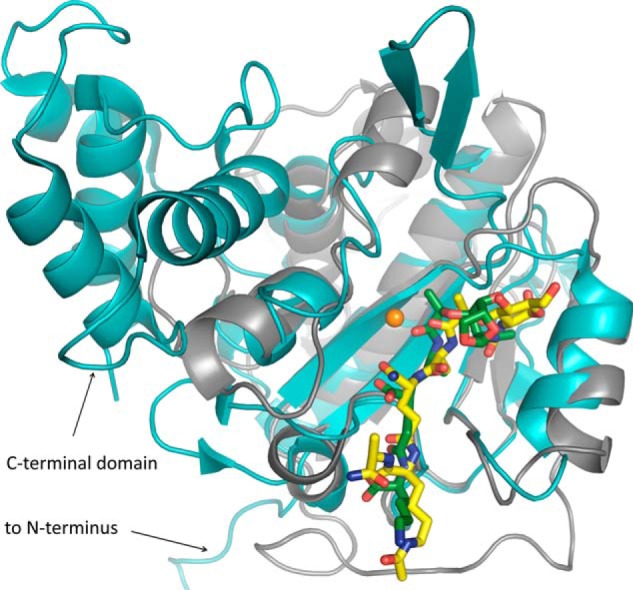
Comparison of the AmiA-cat complex (gray schematic) with AmiD from E. coli (cyan schematic; Protein Data Bank code 3D2Y). Superimposition of the two bacterial amidases reveals a similar fold solely around the binding and active site. AmiD deviates by a 2.0-Å root mean square deviation (DaliLite pairwise) from the AmiA-cat main chain, amino acids in the active and binding sites differ, and AmiD contains additional motifs at its N and C termini. Ligand positioning of anhydro-MTP (green sticks) to AmiD is comparable with the AmiA-cat complex with MtetP (yellow sticks). However, anhydro-MTP has an overall shift in relation to MtetP, and interactions of enzyme with ligand are unalike. Additionally, the MurNAc moiety, including the scissile bond, lies in the direct vicinity of the zinc binding residues and is in the anhydro form, which does not occur in staphylococci. Zn2+ from the unliganded AmiA-cat structure in orange was superimposed.
The presented results also significantly extend prior knowledge obtained from in silico molecular docking of a tripeptide into the highly homologous amidase structure of S. epidermidis, AmiE-cat (19). Although the overall model and the orientation of the ligand agree with our structure, the ligand used for docking AmiE-cat lacked the C-terminal d-Ala and the acetyl group at the l-Lys side chain. The latter resulted in a non-physiologic positive charge due to the protonated Nϵ of the lysine. Although some of the predicted hydrogen bonds and hydrophobic interactions could be validated by our complex structure, the majority of interactions identified in this work were not projected. Furthermore, the docking model did not take the nucleophilic water in the active site into account, causing the substrate to locate closer to the zinc ion. Additionally, a non-physiological zinc ion caused the backbone to shift at residue 63 (Ala-268 in AmiA-cat), making the carbohydrate binding pocket smaller.
Inspection of the liganded AmiA-cat structure directly suggests strategies for inhibition. A competitive inhibitor of AmiA should contain the tetrapeptide stem of MtetP because all four residues make strong interactions with the enzyme. d-Ala, however, contributes least and could therefore be omitted or replaced. The main contact maintained by l-Lys is the hydrophobic interaction with Trp-310. l-Ala and especially d-iGln are conserved in staphylococcal PGN and should be part of a lead structure. Changes in these residues are not tolerated by the enzyme (19) and would therefore not lead to a competitive compound. A reasonable approach would be the introduction of an noncleavable, peptide-mimicking group at the position of the l-Ala-lactyl peptide bond. Suitable bioisosteres are hydroxyethylene or dihydroxyethylene that imitate the tetragonal transition state. Modification of the carbohydrate (e.g. at C1 to form a salt bridge with Glu-277) would also increase affinity for AmiA-cat. On the other hand, it may be advantageous to exchange the sugar moiety and end the lead structure with a non-cleavable lactate analog linked to a cyclic compound because carbohydrate synthesis is expensive.
In conclusion, we have solved the complex structure of AmiA-cat with its ligand, the PGN component MtetP, at high resolution. Inspection of the interactions observed in the complex leads to a plausible model for catalysis and allows us to predict the binding locations of the adjacent GlcNAc molecules and the pentaglycine bridge. We show that the crystallized enzyme is active and that it possesses a narrow specificity that enables it to digest PGN structures from S. aureus but not from B. subtilis. The complex structure offers plausible explanations for these differences in catalytic activity. Our work moreover provides new data on the mechanism of the crucial amidase reaction that may help in the development of therapeutics against MRSA.
Acknowledgments
We thank the team at the Swiss Light Source (Villigen, Switzerland) for beamtime and support. We also acknowledge Andreas Maurer from the Kalbacher laboratory (Tübingen) for the MALDI analysis.
This work was supported by the Collaborative Research Center TRR34.
The atomic coordinates and structure factors (codes 4KNK and 4KNL) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MRSA and VRSA
- methicillin-resistant and vancomycin-resistant S. aureus, respectively
- AtlA
- bifunctional major autolysin of S. aureus
- AmiA
- amidase of S. aureus
- AmiE
- amidase of S. epidermidis
- AmiA-cat and AmiE-cat
- catalytic domain of AmiA and AmiE, respectively
- AmiD
- amidase of E. coli
- NAGase
- glucosaminidase of S. aureus
- PGN
- peptidoglycan network
- MtetP
- muramyl tetrapeptide (MurNAc-l-Ala-d-iGln-l-Lys(NHAc)-d-Ala-NH2)
- MTP
- muramyl tripeptide (l-Ala-d-iGlu-l-Lys)
- anhydro-MTP
- 1,6-anhydro-MTP
- MurNAc
- N-acetylmuramic acid
- d-iGln
- d-isoglutamine
- d-iGlu
- d-isoglutamic acid
- Wat
- water
- meso-DAP
- meso-diaminopimelic acid
- NAc
- N-acetyl.
REFERENCES
- 1. Baron S. (1996) in Medical Microbiology (Baron S., ed) 4th Ed., Chapter 12, University of Texas, Galveston, TX: [PubMed] [Google Scholar]
- 2. O'Gara J. P., Humphreys H. (2001) Staphylococcus epidermidis biofilms: importance and implications. J. Med. Microbiol. 50, 582–587 [DOI] [PubMed] [Google Scholar]
- 3. Götz F. (2002) Staphylococcus and biofilms. Mol. Microbiol. 43, 1367–1378 [DOI] [PubMed] [Google Scholar]
- 4. Heilmann C., Schweitzer O., Gerke C., Vanittanakom N., Mack D., Götz F. (1996) Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol. Microbiol. 20, 1083–1091 [DOI] [PubMed] [Google Scholar]
- 5. Götz F. G. P. (2000) in Infections Associated with Indwelling Medical Devices (Waldvogel F. A., Bisno A. L., eds) pp. 55–88, American Society for Microbiology Press, Washington, D. C [Google Scholar]
- 6. Hall-Stoodley L., Costerton J. W., Stoodley P. (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108 [DOI] [PubMed] [Google Scholar]
- 7. Archer N. K., Mazaitis M. J., Costerton J. W., Leid J. G., Powers M. E., Shirtliff M. E. (2011) Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence 2, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lowy F. D. (1998) Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 [DOI] [PubMed] [Google Scholar]
- 9. Köck R., Becker K., Cookson B., van Gemert-Pijnen J. E., Harbarth S., Kluytmans J., Mielke M., Peters G., Skov R. L., Struelens M. J., Tacconelli E., Navarro Torné A., Witte W., Friedrich A. W. (2010) Methicillin-resistant Staphylococcus aureus (MRSA): burden of disease and control challenges in Europe. Euro Surveill. 15, 19688. [DOI] [PubMed] [Google Scholar]
- 10. European Antimicrobial Resistance Surveillance System (2009) EARSS Annual Report 2008, pp. 55–58, European Centre for Disease Prevention and Control, Bilthoven, The Netherlands [Google Scholar]
- 11. Kresken M., Hafner D., Schmitz F.-J., Wichelhaus T. A. (2009) Resistenzsituation bei klinisch wichtigen Infektionserregern gegenüber Antibiotika in Deutschland und im mitteleuropäischen Raum. Bericht über die Ergebnisse einer multizentrischen Studie der Arbeitsgemeinschaft Empfindlichkeitsprüfungen & Resistenz der Paul-Ehrlich-Gesellschaft für Chemotherapie e.V. aus dem Jahre 2007, p. 9, Antiinfectives Intelligence, Rheinbach, Germany [Google Scholar]
- 12. Kohlenberg A., Schwab F., Geffers C., Behnke M., Rüden H., Gastmeier P. (2008) Time-trends for Gram-negative and multidrug-resistant Gram-positive bacteria associated with nosocomial infections in German intensive care units between 2000 and 2005. Clin. Microbiol. Infect. 14, 93–96 [DOI] [PubMed] [Google Scholar]
- 13. Klein E., Smith D. L., Laxminarayan R. (2007) Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg. Infect. Dis. 13, 1840–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeller J. L., Burke A. E., Glass R. M. (2007) JAMA patient page. MRSA infections. JAMA 298, 1826. [DOI] [PubMed] [Google Scholar]
- 15. Biswas R., Voggu L., Simon U. K., Hentschel P., Thumm G., Götz F. (2006) Activity of the major staphylococcal autolysin Atl. FEMS Microbiol. Lett. 259, 260–268 [DOI] [PubMed] [Google Scholar]
- 16. Schlag M., Biswas R., Krismer B., Kohler T., Zoll S., Yu W., Schwarz H., Peschel A., Götz F. (2010) Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 75, 864–873 [DOI] [PubMed] [Google Scholar]
- 17. Heilmann C., Hussain M., Peters G., Götz F. (1997) Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol. Microbiol. 24, 1013–1024 [DOI] [PubMed] [Google Scholar]
- 18. Albrecht T., Raue S., Rosenstein R., Nieselt K., Götz F. (2012) Phylogeny of the staphylococcal major autolysin and its use in genus and species typing. J. Bacteriol. 194, 2630–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zoll S., Pätzold B., Schlag M., Götz F., Kalbacher H., Stehle T. (2010) Structural basis of cell wall cleavage by a staphylococcal autolysin. PLoS Pathog. 6, e1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lützner N., Pätzold B., Zoll S., Stehle T., Kalbacher H. (2009) Development of a novel fluorescent substrate for Autolysin E, a bacterial type II amidase. Biochem. Biophys. Res. Commun. 380, 554–558 [DOI] [PubMed] [Google Scholar]
- 21. Kerff F., Petrella S., Mercier F., Sauvage E., Herman R., Pennartz A., Zervosen A., Luxen A., Frère J. M., Joris B., Charlier P. (2010) Specific structural features of the N-acetylmuramoyl-l-alanine amidase AmiD from Escherichia coli and mechanistic implications for enzymes of this family. J. Mol. Biol. 397, 249–259 [DOI] [PubMed] [Google Scholar]
- 22. Stratagene (2006) QuikChange® Site-Directed Mutagenesis Kit: Instruction Manual, Stratagene, La Jolla, CA [Google Scholar]
- 23. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 30. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 31. Kantardjieff K. A., Rupp B. (2003) Matthews coefficient probabilities: improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 12, 1865–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 35. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC, New York [Google Scholar]
- 36. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iordanescu S., Surdeanu M. (1976) Two restriction and modification systems in Staphylococcus aureus NCTC8325. J. Gen. Microbiol. 96, 277–281 [DOI] [PubMed] [Google Scholar]
- 38. de Jonge B. L., Chang Y. S., Gage D., Tomasz A. (1992) Peptidoglycan composition in heterogeneous Tn551 mutants of a methicillin-resistant Staphylococcus aureus strain. J. Biol. Chem. 267, 11255–11259 [PubMed] [Google Scholar]
- 39. Hasegawa H., Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348 [DOI] [PubMed] [Google Scholar]
- 40. Meroueh S. O., Bencze K. Z., Hesek D., Lee M., Fisher J. F., Stemmler T. L., Mobashery S. (2006) Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 103, 4404–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zoll S., Schlag M., Shkumatov A. V., Rautenberg M., Svergun D. I., Götz F., Stehle T. (2012) Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J. Bacteriol. 194, 3789–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vollmer W., Joris B., Charlier P., Foster S. (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286 [DOI] [PubMed] [Google Scholar]
- 43. Schleifer K. H., Kandler O. (1972) Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36, 407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Jonge B. L., Chang Y. S., Gage D., Tomasz A. (1992) Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 267, 11248–11254 [PubMed] [Google Scholar]
- 45. Low L. Y., Yang C., Perego M., Osterman A., Liddington R. (2011) Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. J. Biol. Chem. 286, 34391–34403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang D. C., Tan K., Joachimiak A., Bernhardt T. G. (2012) A conformational switch controls cell wall-remodelling enzymes required for bacterial cell division. Mol. Microbiol. 85, 768–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheng X., Zhang X., Pflugrath J. W., Studier F. W. (1994) The structure of bacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 91, 4034–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Low L. Y., Yang C., Perego M., Osterman A., Liddington R. C. (2005) Structure and lytic activity of a Bacillus anthracis prophage endolysin. J. Biol. Chem. 280, 35433–35439 [DOI] [PubMed] [Google Scholar]
- 49. Liepinsh E., Généreux C., Dehareng D., Joris B., Otting G. (2003) NMR structure of Citrobacter freundii AmpD, comparison with bacteriophage T7 lysozyme and homology with PGRP domains. J. Mol. Biol. 327, 833–842 [DOI] [PubMed] [Google Scholar]
- 50. Martínez-Caballero S., Lee M., Artola-Recolons C., Carrasco-López C., Hesek D., Spink E., Lastochkin E., Zhang W., Hellman L. M., Boggess B., Mobashery S., Hermoso J. A. (2013) Reaction products and the x-ray structure of AmpDh2, a virulence determinant of Pseudomonas aeruginosa. J. Am. Chem. Soc. 135, 10318–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carrasco-López C., Rojas-Altuve A., Zhang W., Hesek D., Lee M., Barbe S., André I., Ferrer P., Silva-Martin N., Castro G. R., Martínez-Ripoll M., Mobashery S., Hermoso J. A. (2011) Crystal structures of bacterial peptidoglycan amidase AmpD and an unprecedented activation mechanism. J. Biol. Chem. 286, 31714–31722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prigozhin D. M., Mavrici D., Huizar J. P., Vansell H. J., Alber T. (2013) Structural and biochemical analyses of Mycobacterium tuberculosis N-acetylmuramyl-l-alanine amidase Rv3717 point to a role in peptidoglycan fragment recycling. J. Biol. Chem. 288, 31549–31555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Warth A. D., Strominger J. L. (1971) Structure of the peptidoglycan from vegetative cell walls of Bacillus subtilis. Biochemistry 10, 4349–4358 [DOI] [PubMed] [Google Scholar]
- 54. Boneca I. G., Huang Z. H., Gage D. A., Tomasz A. (2000) Characterization of Staphylococcus aureus cell wall glycan strands, evidence for a new β-N-acetylglucosaminidase activity. J. Biol. Chem. 275, 9910–9918 [DOI] [PubMed] [Google Scholar]
- 55. Atrih A., Bacher G., Allmaier G., Williamson M. P., Foster S. J. (1999) Analysis of peptidoglycan structure from vegetative cells of Bacillus subtilis 168 and role of PBP 5 in peptidoglycan maturation. J. Bacteriol. 181, 3956–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Advanced Chemistry Development, Inc. (2012) ACD/ChemSketch Freeware, version 10.00, Advanced Chemistry Development, Inc., Toronto, Canada [Google Scholar]
- 58. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Jalview Version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]