

Background: Scaffolding of signaling proteins to GPCRs may increase signaling efficiency and spatial fidelity.
Results: Phospholipase C (PLC) β3 binding directly to M3 muscarinic receptor intracellular loops involves a non-canonical PDZ interaction.
Conclusion: M3 muscarinic receptor binding to PLCβ3 optimizes interactions with substrate and G protein activator.
Significance: Scaffolding of PLC enzymes to GPCRs may be important for spatial signal specificity and efficacy.
Keywords: G Protein-coupled Receptors (GPCR), G Proteins, Phosphatidylinositol Signaling, Phospholipase C, Protein-Protein Interactions, Scaffolding
Abstract
Phospholipase Cβ (PLCβ) enzymes are activated by G protein-coupled receptors through receptor-catalyzed guanine nucleotide exchange on Gαβγ heterotrimers containing Gq family G proteins. Here we report evidence for a direct interaction between M3 muscarinic receptor (M3R) and PLCβ3. Both expressed and endogenous M3R interacted with PLCβ in coimmunoprecipitation experiments. Stimulation of M3R with carbachol significantly increased this association. Expression of M3R in CHO cells promoted plasma membrane localization of YFP-PLCβ3. Deletion of the PLCβ3 C terminus or deletion of the PLCβ3 PDZ ligand inhibited coimmunoprecipitation with M3R and M3R-dependent PLCβ3 plasma membrane localization. Purified PLCβ3 bound directly to glutathione S-transferase (GST)-fused M3R intracellular loops 2 and 3 (M3Ri2 and M3Ri3) as well as M3R C terminus (M3R/H8-CT). PLCβ3 binding to M3Ri3 was inhibited when the PDZ ligand was removed. In assays using reconstituted purified components in vitro, M3Ri2, M3Ri3, and M3R/H8-CT potentiated Gαq-dependent but not Gβγ-dependent PLCβ3 activation. Disruption of key residues in M3Ri3N and of the PDZ ligand in PLCβ3 inhibited M3Ri3-mediated potentiation. We propose that the M3 muscarinic receptor maximizes the efficiency of PLCβ3 signaling beyond its canonical role as a guanine nucleotide exchange factor for Gα.
Introduction
G protein-coupled receptors (GPCRs)2 are seven-transmembrane proteins that relay information from extracellular signals. Upon activation by ligand binding, GPCRs catalyze guanosine diphosphate (GDP) dissociation from the Gαβγ heterotrimer substrate; guanosine triphosphate (GTP) binding to Gα follows and leads to functional dissociation of Gβγ from Gα and effector enzyme activation (1, 2). Activation of effector enzymes initiates specific signaling cascades that regulate cell physiology. Hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol and inositol 1,4,5-trisphosphate (IP3) by G protein-responsive phospholipase Cβ (PLCβ) is a principal signal transduction pathway activated by GPCRs (3–5).
Classically, effector activation by G proteins is thought to rely on random collision coupling between activated, diffusible G protein and target effector following receptor activation (1). In recent years, this paradigm has been extensively challenged. Biochemical evidence and studies in live cells have led to an emerging view that some G proteins are precoupled to receptors (6–13). There are also reports of stable complexes between GPCRs and other effectors such as G protein-sensitive inward rectifier potassium channels (14–18). Detailed studies of the Gq-PLCβ signaling system revealed a novel paradigm in G protein signaling, called kinetic scaffolding, where the intrinsic Gq-GTPase stimulating function leads to spatially and temporally focused PLC activation (19, 20).
Phospholipase Cβ is often found in physical complexes with GPCRs through interactions with intermediary scaffolds. One such class of scaffolds is postsynaptic density-95/disc large/ZO-1 (PDZ) domain-containing proteins. PDZ domains are independently folded protein modules that specifically bind and recognize PDZ ligand consensus sequences at the extreme C terminus of target proteins (21, 22). PDZ domain-containing proteins generally have multiple individual PDZ domains and thus can scaffold target proteins together (21–23). All PLCβ isoforms have PDZ ligand motifs at their C terminus with a consensus sequence (X(S/T)X(V/L)-COOH) (21, 22). Examples of PLCβ-interacting PDZ scaffolds are NHERF1, NHERF2, PDZK1, and Shank2 that organize specific signaling complexes with parathyroid hormone receptor PTH1R (24), lysophosphatidic acid receptor LPA2R (25), somatostatin receptor (26), and metabotropic glutamate receptor mGluR1 (27), respectively (21, 22). There is increasing recognition that PDZ-dependent organization is required for receptor-dependent activation of PLCβ (25, 28); however, in a classical collision-coupling model, one would not expect physical scaffolding to be required for GPCR-dependent effector activation.
In the present studies, we investigated a direct interaction between the M3 muscarinic receptor (M3R), a prototypical Gαq-coupled receptor, and its effector enzyme, PLCβ3. We demonstrate here that M3R binds to PLCβ3 and drives plasma membrane enrichment of PLCβ3 in cells. Interaction sites for direct protein-protein binding that alter the efficiency of G protein-dependent PLC activation are also defined. Taken together, the direct binding interaction between M3R and PLCβ3 may represent a regulatory mechanism for PLC signal output beyond receptor-stimulated nucleotide exchange on Gαq.
EXPERIMENTAL PROCEDURES
Materials
n-Dodecyl β-maltoside was purchased from Dojindo Molecular Technologies. Resins for purification were from GE Healthcare (glutathione-Sepharose 4B), Qiagen (Ni-NTA-agarose), and Genscript (protein G-agarose). Antibodies were from Covance (MMS101R, monoclonal HA.11 anti-HA ascites), a generous donation from Dr. J. Wess (anti-M3R directed against its last 18 amino acids (29, 30)), from Sigma (G7781, anti-glutathione S-transferase), from Santa Cruz Biotechnology (sc-385, anti-Gαolf antibody), and from R&D Systems (anti-human PLCβ3 at Lys27–Leu246). For PLCβ3 and PLCβ1 with an intact C terminus, B521 and B517, respectively, were used (31). For Gαq and Gβγ, WO82 (32) and B600 (33), respectively, were used. For Gαs, 584 antiserum was used (33). Lipofectamine 2000 (Invitrogen) was used for all transient transfections according to the manufacturer's instructions. l-α-Phosphatidylinositol 4,5-bisphosphate (brain, porcine) and l-α-phosphatidylethanolamine (liver, bovine) were from Avanti Polar Lipids. [Inositol-2-3H]phosphatidylinositol 4,5-bisphosphate ([3H]PIP2) was from PerkinElmer Life Sciences.
M3R Constructs
3xHA-M3R (human, 1–590) for protein expression was obtained from cDNA.org. 1xHA-M3R (rat, 1–589) in pCD vector (34) was a gift from Dr. J. Wess (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health) and was only used as template to generate fragment constructs of M3R intracellular loops fused with glutathione S-transferase (GST) in pGEX4T2 (GE Healthcare). To allow for purification of GST fusion proteins via a His6 tag, an oligonucleotide dimer encoding the tag was inserted into pGEX4T2 between SalI- and NotI-cut sites. Each M3R loop was defined as follows: i1 (91–103), i2 (164–183), i3 (i3N = 252–389 (10), a gift from Dr. S. Lanier of Medical University of South Carolina; i3M = 352–469; i3C = 389–491), and H8-C terminus (CT) (547–589). Subfragments of M3Ri3N were defined as described in Fig. 6. Each fragment was inserted between EcoRI- and SalI-cut sites in the modified pGEX4T2, resulting in an N-terminal GST tag and a C-terminal His6 tag.
FIGURE 6.

Intracellular loops of M3 muscarinic receptor bind Gαq and Gβγ. A, similar to Fig. 4A, M3R constructs fused to GST were tested for binding to purified Gαq or Gβ1γ2 in a glutathione bead pulldown assay. Results were analyzed by Western blot. Nonspecific recognition for GST fusion proteins by Gαq antibody W082 is indicated by * as shown. Representative Western blots shown were each from three independent experiments. B, within M3Ri3Na (252–322): 1, residues 252–273; 2, residues 274–294; 3, residues 295–322; 4, residues 289–314; 5, residues 262–285; 6, residues 286–309. C, binding site mapping for PLCβ3, Gαq, and Gβγ at M3Ri3N residues 252–322. To map binding sites for PLCβ3 (top), Gαq (middle), or Gβγ (bottom), GST fusion proteins as described in B were used in a pulldown assay. Representative Western blots shown were each from two independent experiments. D, binding of PLCβ3 and different isoforms of G protein α subunits (Gαq, Gαs-long, and Gαolf) to M3Ri3Na fragments 3 and 4 was tested. Results were analyzed by Western blot. Representative Western blots are shown from three independent experiments. Fractions of original input were loaded in the leftmost lanes. Samples were also immunoblotted (IB) with anti-GST antibodies to validate loading equal amounts of protein.
PLCβ3 Constructs
PLCβ3 (human, 1–1234) in pBluescript was moved into pciNeo vector (Promega) for mammalian expression and into pFastbac HTb vector (Invitrogen) for baculoviral insect cell expression between EcoRI and SalI sites. To generate deletion/mutant constructs at the extreme C terminus of human PLCβ3, fragments were generated by PCR flanked by a native 5′ KpnI site (nucleotides 2090–2095) and 3′ SalI site and ligated into vector with a purified N-terminal fragment digested with EcoRI and KpnI. Deletions were made by removing the PLCβ3 PDZ consensus motif NTQL at 1231–1234 (ΔPDZ). A larger deletion from the C terminus (1–886, ΔCT) was made by PCR to generate a fragment flanked by EcoRI and SalI for ligation into pciNeo. YFP-PLCβ3 (rat) full length (FL), pleckstrin homology (PH) (1–147), C2 (712–809), and CT (845–1234) in pEYFP-C1 (Clontech) were generated as described previously (35). To create ΔPDZ versions of PLCβ3 FL and PLCβ3 CT, fragments were generated by PCR flanked by a native 5′ HindIII site (nucleotides 2531–2536) and 3′ EcoRI site for vector ligation. GFP-PLCβ3ΔCT was constructed in pciNeo with a NheI/EcoRI-digested fragment encoding for enhanced GFP from pEGFP-C3 (Clontech) and an EcoRI/SalI-digested PLCβ3ΔCT fragment (human). Primers and sequencing results from regions generated by PCR are available upon request.
Protein Expression and Purification
All buffers for protein purification were ice-cold and supplemented with protease inhibitor mixture (133 μm phenylmethylsulfonyl fluoride, 21 μg/ml 1-chloro-3-tosylamido-7-amino-2-heptanone and l-1-tosylamido-2-phenylethyl chloromethyl ketone, 0.5 μg/ml aprotinin, 0.2 μg/ml leupeptin, 1 μg/ml pepstatin A, 42 μg/ml Nα-p-tosyl-l-arginine methyl ester hydrochloride, 10 μg/ml soybean trypsin inhibitor).
Reported protein concentrations were quantified using an amido black protein assay. Protein purity was estimated by analyzing Coomassie-stained protein gels using band densitometry functions in ImageJ.
Induction and Purification of GST Fusion Proteins
GST-M3R loop fusion proteins (GST-M3Ri1, GST-M3Ri2, GST-M3Ri3N/M/C, and GST-M3RCT) were transformed into BL21(DE3) Escherichia coli. BL21 Rosetta strain was used for some constructs that were otherwise difficult to express. For each protein, up to 3-liter cultures were grown in LB/carbenicillin and induced at A600 ∼0.50 with 100 μm isopropyl β-d-thiogalactopyranoside for 1 h at 37 °C. Cells were pelleted and then resuspended in 1× PBS. The pellets were then frozen with liquid nitrogen for storage at −80 °C. Samples were thawed, resuspended in lysis buffer (50 mm Hepes, pH 7.4, 150 mm NaCl, 5% glycerol, 10 mm β-mercaptoethanol, 15 mm imidazole, and 30 mg of lysozyme/liter of culture) followed by addition of 1 mg of DNase I/liter of culture and 10 mm MgCl2. To enrich for fully translated proteins, purification was achieved by taking advantage of the C-terminal His6. Supernatant was collected after 100,000 × g ultracentrifugation and loaded onto a pre-equilibrated 1-ml column of Ni-NTA resin. The loaded column was washed with 600 mm NaCl, re-equilibrated, and eluted with 100–250 mm imidazole. PD-10 columns (GE Healthcare) were used to equilibrate purified proteins in 50 mm Hepes, pH 7.2, 100 mm NaCl, and 1 mm EDTA. The proteins were snap frozen and stored at −80 °C in aliquots.
For the screening of M3Ri3Na single alanine mutants, glutathione affinity chromatography was used. Protein expression was carried out with 25 ml of BL21 cultures. Cell lysis and supernatant extraction were performed as described above except with a proportional reduction in scale. Each protein extract was applied to pre-equilibrated 50% glutathione-Sepharose slurry (100 μl) for 3 h. Resin washing was performed essentially as described above except with buffers without imidazole addition. The partially purified proteins were stored on ice in 50 mm Hepes, pH 7.2, 100 mm NaCl, and 1 mm EDTA. Protein stoichiometry was quantified by Coomassie staining of 15% SDS-PAGE with the inclusion of pure protein standards of known concentration. The protein amount of each mutant was normalized to the fully translated GST fusion protein band.
Expression and Purification of G Protein Subunits
Expression and purification of G proteins αq or αs/αolf were performed using Ric-8A or Ric-8B affinity chromatography, respectively, as described previously (36).
Expression and Purification of PLCβ3 Proteins
Purification of PLCβ proteins was essentially as described previously (37) with minor modifications. Briefly, His6 N-terminally tagged PLCβ3 protein and variants were expressed in 250 ml of High Five cells infected (at 2 × 106/ml) with freshly amplified baculovirus at a multiplicity of infection of 5. 48 h after infection, a collected cell pellet was resuspended in 50 mm Hepes, pH 8.0, 0.1 mm EGTA, 0.1 mm EDTA, 0.1 mm dithiothreitol, and 100 mm NaCl. The suspension was subjected to four cycles of liquid nitrogen freeze/thaw. The NaCl concentration was adjusted to 1 m (resupplemented with protease inhibitors), and the mixture was then ultracentrifuged at 140,000 × g. The supernatant was diluted 5-fold with buffer A (10 mm Hepes, pH 8.0, 0.1 mm EGTA, 0.1 mm EDTA, 0.5% polyoxyethylene 10-lauryl ether (C12E10), and 10 mm β-mercaptoethanol) and recentrifuged. The resulting supernatant was loaded onto a pre-equilibrated 4-ml Ni-NTA column. The column was washed with 20 column volumes of buffer A supplemented with 15 mm imidazole and 800 mm NaCl. The protein was eluted with buffer A supplemented with 50 mm NaCl and 125 mm imidazole. The yield was ∼40 mg of purified protein/liter of High Five culture. PD-10 columns were used to equilibrate purified proteins in 50 mm Hepes, pH 8.0, 300 mm NaCl, 2 mm dithiothreitol, 0.1 mm EGTA, and 0.1 mm EDTA. No further purification steps were deemed necessary to produce ∼80% pure protein.
Laser Confocal Microscopy
Chinese hamster ovary (CHO) cells were plated on 35-mm dishes at ∼¼ confluence. The following day, they were transiently transfected with YFP/GFP-PLCβ3 constructs either with pciNeo vector or 3xHA-M3R (∼500 ng of each per dish for a total of 1 μg). Prior to live cell imaging, medium was replaced 18 h after transfection with imaging buffer (Hanks' balanced salt solution containing 5.5 mm glucose, 0.56 mm MgCl2, 4.7 mm KCl, 1 mm Na2HPO4, 10 mm Hepes, and 1.2 mm CaCl2, pH 7.4). The following settings were used: for enhanced GFP, excitation, 488 nm; emission, 510 nm; for enhanced YFP, excitation, 515 nm; emission, 527 nm. z stacked (one stack = 1 μm) images were acquired directly from each dish using an Olympus FV1000MP microscope in confocal mode with a LUMPLFL 40× 0.8 numerical aperture W (Olympus) lens. A complete z stack was taken for every field of view from the top to the bottom of cells.
For estimation of the plasma membrane/cytosol ratio of protein distribution, regions of interest were selected from the same z level either along the entire edge of a cell for plasma membrane or from those in the body of the cell excluding the nucleus for cytosol. Mean fluorescence intensities within each region of interest were measured using Olympus Fluoview version 2.0. A z level with the highest fluorescence intensity at the plasma membrane edge was chosen for analysis and for representative images. Because contribution of fluorescence from the cytosol was not subtracted, a ratio value of 1 does not indicate a 1:1 distribution of fluorescence between plasma membrane and cytosol; rather, it represents the baseline value of fluorescence distribution for cytosol-localized proteins. Approximately two to six cells were visible per field of view. Fluorescence ratios were calculated for all cells in every field of view captured for each experimental group. At least 20 independently acquired images from three separate transfections were analyzed for each condition. Rounded cells (height more than 20 μm), indicative of poor health, were discarded from the analysis. Where indicated, a blinded observer (unaware of the experimental conditions behind each image) scored cells with plasma membrane distribution of fluorescence from entire image data sets. For this purpose, the order of images was randomly shuffled.
Each representative micrograph at one z plane is supplemented with a line profile analysis below. Fluorescence intensities were measured along points a to b and plotted as a function of distance AB.
Receptor Immunocytochemistry
CHO cells were grown on a 35-mm dish at ∼⅓ confluence. The following day, each dish of cells was transiently transfected with YFP-PLCβ3 and/or 3xHA-M3R. Empty pciNeo vector was added to equalize between experimental groups to a total plasmid load of 1 μg. Cells were analyzed the next day by confocal microscopy as described above. Immediately prior to live cell imaging, surface 3xHA-M3R in these cells was stained with anti-HA antiserum. Briefly, PBS-washed cells were incubated with anti-HA antibody (1:2000) in Hanks' balanced salt solution with 15 mm Hepes, pH 7.4 for 1 h at room temperature followed by incubation with Alexa Fluor 546 anti-mouse antibody (1:1000) for 30 min. Following each incubation step, the cells were washed three times with 1× PBS. Green and red fluorescence were acquired using separate laser excitations. Merged fluorescent images were generated by ImageJ using the Merge Channels function. The same optical and digital settings for either green or red channels were applied to all experimental groups.
GST Fusion Protein Pulldown Assay
Purified PLCβ3 and variant proteins were mixed with GST-His6 or GST-M3R loops in binding buffer (20 mm Hepes, pH 8.0, 1 mm EDTA, 0.1% polyoxyethylene 10-lauryl ether (C12E10), 2 mm dithiothreitol, and 150 mm NaCl). Equal amounts of GST fusion proteins were added at 300 nm as determined by Coomassie staining. The total reaction volume was 500 μl. Unless otherwise indicated, PLCβ3 was added at 10 nm. For pulldown of G protein subunits, 30 nm purified Gαq, Gαs-long, Gαolf, and Gβ1γ2 were added. Reactions were incubated by rotating for 1 h at 4 °C. Prior to addition, glutathione-Sepharose beads were preblocked in binding buffer supplemented with 0.1% bovine serum albumin (Roche Applied Science) and then made into a 50% slurry. 20 μl of slurry was incubated with each reaction for 2 h at 4 °C. Beads were collected by centrifugation at 100 × g for 2.5 min at 4 °C and washed three times with binding buffer. Bound proteins were eluted from the beads by the addition of 2× loading sample buffer, boiled at 95 °C for 5min, resolved by SDS-PAGE, and detected by Western blot. 8% SDS-PAGE was used to resolve PLCβ3. 12% SDS-PAGE was used to resolve G protein subunits. For internal GST loading controls, 15% SDS-PAGE was used to resolve GST fusion proteins.
Coimmunoprecipitation of Transiently Transfected Proteins
PLCβ3 and variant constructs were cotransfected with 3xHA-M3R in adherent HEK293 cells seeded on poly-d-lysine-coated dishes (1 μg of each construct per dish). In some experiments where indicated, pciNeo empty vector was cotransfected with PLCβ3 in place of 3xHA-M3R as a negative control. 48 h after transfection, cells were washed and then lysed in 0.1% n-dodecyl β-d-maltoside in 1× PBS with protease inhibitors. The lysate was subjected to 100,000 × g ultracentrifugation for 20 min at 4 °C. The supernatant was collected and immunoprecipitated with anti-HA antibody. Where indicated, no antibody addition was used as a negative control. Other controls including immunoselection with HA antibody plus HA-blocking peptide or with myc antibody were performed (data not shown) and yielded similar results as no antibody addition. Protein G-agarose was added to incubate with the lysate for 2 h. The resin was then washed two times in lysis buffer and finally resuspended in 2× loading sample buffer to be boiled. Eluted proteins from the immunoprecipitated resin were analyzed by SDS-PAGE and subsequent immunoblotting. 8% SDS-PAGE was used to resolve PLCβ3 and HA-tagged M3R.
Coimmunoprecipitation of Native M3R-PLC Complex
Rat lungs were disrupted and then homogenized in ice-cold lysis buffer (1% n-dodecyl β-d-maltoside, 20 mm Hepes, pH 7.4, 137 mm NaCl, and 2 mm EDTA with protease inhibitors) with an Ultra Turrax homogenizer at 1-s pulse intervals at 22,000 rpm. Proteins were quantified using an amido black dye assay. 5 mg of total protein was used for each immunoprecipitation. For M3R immunoprecipitation, 1 μg of anti-M3R antibody was used (30). For PLCβ3 immunoprecipitation, 1 μl of B521 antiserum was used (31). After antibody addition, the samples were incubated for 2 h with Dynal protein G beads, washed three times in lysis buffer at 0.1% n-dodecyl β-d-maltoside, and resuspended in sonication buffer (50 mm Hepes, pH 7.2, 3 mm EGTA, 80 mm KCl, and 1 mm DTT). Each sample was further diluted in a solution supplemented with albumin and reconstituted with purified Gαq and phospholipid vesicles containing [3H]PIP2 as described in the next section. The reaction was initiated by the addition of ∼150 nm free calcium. For Gαq preactivation, GDP and aluminum fluoride (a mixture of aluminum chloride and sodium fluoride) were added as described in the next section. The reaction was allowed to proceed for 30 min. Free [3H]IP3 from each sample was measured by liquid scintillation counting (36).
Phospholipase Cβ Activation in Reconstituted Vesicles
PLC substrate was composed of 50 μm phosphatidylethanolamine, 25 μm PIP2, and [inositol-2-3H]PIP2 at 6–8000 cpm/assay. For measuring PLC activity associated with native M3R complex, vesicles consisting of 100 μm phosphatidylethanolamine and 25 μm PIP2 were used. The vesicles were prepared as described previously (38). Gαq was diluted in buffer containing 20 mm Hepes, pH 8.0, 170 mm NaCl, 11 mm CHAPS, 1 mm EDTA, 1 μm GDP, and 1 mm DTT. Final CHAPS concentration was made no higher than 300 μm (39). To activate Gαq, each reaction was supplemented with 10 mm NaF and 30 μm AlCl3 (36). For assays containing Gβγ, 0.15% (w/v) β-octyl glucoside (final assay concentration) was included (36). Purified Gαq-GDP-AlF4− and Gβγ were added at 30 nm unless otherwise indicated. Purified PLCβ proteins were added at 10 ng per reaction (∼1 nm). 1.5 mm CaCl2 (∼150 nm free Ca2+) was added to initiate each reaction, and then the samples were incubated at 30 °C for 45 min. A blank set of samples without CaCl2 addition was also included. Reactions were terminated and then analyzed as described previously (36). All assays for PLC activity were carried out within conditions where IP3 production and time were linearly correlated. For concentration-response curves shown, collected data were analyzed using GraphPad Prism 6.0 and fit using the log(agonist) versus response function with variable Hill slope.
Statistical Analysis
Unless otherwise stated, analysis of variance of at least three independent experiments was performed with Bonferroni post-test using GraphPad Prism 6.0 to determine levels of significance. Error bars indicate S.E. * indicates p < 0.05, and *** indicates p < 0.001 or p < 0.0001.
RESULTS
The M3 Muscarinic Receptor Binds to Phospholipase Cβ3
To determine whether PLCβ3 could form a complex with M3R, an N-terminal HA epitope-tagged M3R was coexpressed in HEK293 cells with PLCβ3, extracted, and immunoprecipitated with an anti-HA antibody. Immunoprecipitations were analyzed by SDS-PAGE and immunoblotting for associated PLCβ3. PLCβ3 was detected in the HA immunoprecipitate only if HA-M3R was expressed (Fig. 1A). To determine whether M3R activation could alter association with PLCβ3, cells expressing HA-M3R and PLCβ3 were treated with carbachol for 5 min followed by cell lysis (Fig. 1A, labeled with *), HA-specific immunoprecipitation, and immunoblotting for PLCβ3. Treatment with carbachol led to a 2–3-fold (2.83 ± 0.65-fold from five experiments, p = 0.009) increase in PLCβ3 association with HA-M3R (Fig. 1A).
FIGURE 1.

M3 muscarinic receptor stably interacts with PLCβ3. A, HEK293 cells were transfected with PLCβ3 and empty vector or PLCβ3 and 3xHA-M3R with and without stimulation with 100 μm carbachol for 5 min (* indicates treatment with carbachol). Cells were lysed, immunoprecipitated (IP), and immunoblotted (IB) for either PLCβ3 or HA as described under “Experimental Procedures.” Representative Western blots shown were from three or more independent experiments. B, M3R or PLCβ3 was immunoprecipitated (IP) from rat lung lysates and assayed for associated PLC activity as described under “Experimental Procedures.” The data were compiled from four independent assays, each with internal triplicates. C, YFP-PLCβ3 or YFP-PLCβ2 was expressed in CHO cells with empty vector or 3xHA-M3R, and cells were analyzed by live cell confocal microscopy as described under “Experimental Procedures.” Line scans from a to b are shown below each image. D, multiple images treated as in C were analyzed as described under “Experimental Procedures,” compiled, and plotted. E, YFP-PLCβ3, 3xHA-M3R, or YFP-PLCβ3+M3R was expressed in CHO cells. Surface M3R was immunostained in red as described under “Experimental Procedures.” Line scans are shown to the right of each image set. Error bars indicate S.E. *, p < 0.05; ***, p < 0.001; N.S., not significant. Ab, antibody.
To demonstrate an M3R-PLCβ protein complex in a native system, proteins were extracted from rat lung tissue (enriched in native M3R and PLCβ3) and immunoprecipitated with anti-M3R antibody (29, 30). No agonists were added. Detection of PLCβ3 in these immunoprecipitates by immunoblotting is difficult because of the relatively low abundance of the two proteins in native tissues. As a sensitive approach to detecting PLC, immunoprecipitated samples were assayed for associated PLC enzymatic activity. With M3R immunoprecipitation, there was significantly more associated PLC activity compared with that in the absence of M3R antibody (Fig. 1B). This M3R-bound PLC is likely a PLCβ isoform because it was activated by purified Gαq-GDP-AlF4−. The proportion of Gαq-GDP-AlF4−-activatable PLC associated with M3R was ∼⅓ of the total PLCβ3 that could be precipitated directly with a PLCβ3-specific antibody. This suggests that, as expected, not all of the PLCβ in tissues is associated with M3R.
M3R Promotes PLCβ3 Localization to the Plasma Membrane
To determine whether expression of M3R could alter the subcellular localization of PLCβ3, M3R was coexpressed with YFP-PLCβ3 in CHO cells, which do not express muscarinic receptors. The distribution of YFP-PLCβ3 fluorescence at the cell periphery containing the plasma membrane (PM) relative to the cytosol was analyzed. Previous data have shown that YFP-PLCβ3 is cytosolic (35) consistent with the data in Fig. 1C. M3R coexpression significantly increased PM fluorescence relative to cytosolic fluorescence of YFP-PLCβ3 (Fig. 1, C and D; in blinded analysis, 5 of 100 cells had YFP-PLCβ3 PM localization; when M3R was coexpressed, 102 of 177 cells showed YFP PM localization). In contrast, YFP-PLCβ2 fluorescence remained entirely cytosolic with M3R expression. This supports the notion that PLCβ3 binds to M3R and suggests that one function of this interaction is to localize PLCβ3 to the plasma membrane near its substrate. Because an agonist was not included in these experiments, the data imply that PLCβ3 can be prebound to the receptor prior to activation.
To confirm colocalization of YFP-PLCβ3 with M3R at the PM, cells were transfected with HA-M3R, YFP-PLCβ3, or both (Fig. 1E). Cells were stained with anti-HA antibody and imaged for YFP and anti-HA in separate channels. In the absence of M3R, YFP-PLCβ3 (Fig. 1E, top panels) was cytosolic, and M3R alone was at the PM (Fig. 1E, middle panels). When expressed together, a significant proportion of the YFP-PLCβ3 fluorescence colocalized at the PM with HA-M3R (Fig. 1E, bottom panels), consistent with the idea that PLCβ3 localization to the PM is due to association with M3R.
M3R Interactions with PLCβ3 Involve the PLCβ3 C Terminus and the PLCβ3 PDZ Ligand
To identify regions within PLCβ3 (Fig. 2A) involved in M3R-directed membrane enrichment, we analyzed the PM localization of three PLCβ3 domains expressed as fusions with YFP: YFP-PLCβ3 PH domain, YFP-PLCβ3 C2 domain, and YFP-PLCβ3 CT. Of the three fragments tested, only PLCβ3 CT (845–1234) showed an enrichment of PM fluorescence with M3R coexpression (Fig. 2, B and C). PLCβ3 CT alone also bound somewhat to membranes (Fig. 2, B and C), consistent with previous findings (35), suggesting that this domain has an intrinsic affinity for membranes that is masked in the holoenzyme.
FIGURE 2.

Plasma membrane localization of PLCβ3 C terminus depends on M3R expression and binding. A, primary structure of PLCβ with PH domain (residues 1–147) followed by four EF hands, X and Y catalytic cores, C2 domain (residues 712–809), and CT (residues 845–1234). B, YFP-PLCβ3 fragment constructs were expressed in CHO cells. Either empty vector or 3xHA-M3R was cotransfected. Live cells were analyzed by confocal microscopy as described under “Experimental Procedures.” Line scans from a to b are shown below each image. C, multiple images treated as in B were analyzed as described under “Experimental Procedures,” compiled, and plotted. D, HEK293 cells were cotransfected with YFP-PLCβ3 CT (residues 845–1234) and 3xHA-M3R. Cells were lysed and immunoprecipitated (IP) with or without anti-HA specific antibody. Input lysate (rightmost lanes) and immunoprecipitated (left) samples were immunoblotted (IB) for either PLCβ3 or HA as described under “Experimental Procedures.” Representative Western blots shown were from two independent experiments. Error bars represent S.E. ***, p < 0.001; N.S., not significant.
The PLCβ3 CT also bound to M3R. Cells were transfected with HA-M3R and YFP-PLCβ3 CT followed by extraction and precipitation with or without inclusion of an anti-HA antibody (Fig. 2D). PLCβ3 CT was significantly enriched in samples precipitated with the anti-HA antibody. These results indicate that M3R can interact with the C terminus of PLCβ3.
To determine whether the C terminus of PLCβ3 is required for interaction with M3R, M3R was coexpressed with GFP-PLCβ3 (1–886) (PLCβ3ΔCT) with the C terminus deleted. PLCβ3ΔCT localization was cytosolic and did not change with M3R expression (Fig. 3, A, left panel, and B). Association of PLCβ3ΔCT with M3R was also assessed by coimmunoprecipitation. In contrast to full-length PLCβ3, PLCβΔCT was not enriched in anti-HA immunoprecipitates (Fig. 3C). Thus, the C terminus of PLCβ3 is required for interaction with M3R.
FIGURE 3.

Enrichment of PLCβ3 at plasma membrane by M3R and full binding of PLCβ3 to M3R require the PDZ ligand at the extreme C terminus of PLCβ3. A, deletion of PDZ ligand (NTQL, residues 1231–1234) in PLCβ3 resulted in loss of M3R-mediated enrichment of PLCβ3 at plasma membrane. YFP-PLCβ3 mutant constructs (ΔCT, 1–886; ΔPDZ, 1–1230) were expressed in CHO cells. Either empty vector or 3xHA-M3R was cotransfected. Live cells were analyzed by confocal microscopy as described under “Experimental Procedures.” Line scans from a to b are shown below each image. B, multiple images treated as in A were analyzed as described under “Experimental Procedures,” compiled, and plotted. Additional quantitation yielded the following results: PLCβ3ΔCT without M3R expression, 0 of 42 exhibited PM localization; PLCβ3ΔCT with M3R, 2 of 34 cells exhibited PM localization; PLCβ3ΔPDZ without M3R, 2 of 45 exhibited PM localization; PLCβ3ΔPDZ with M3R coexpression; 0 of 54 exhibited PM localization. C, PLCβ3ΔCT (1–886) or PLCβ3 full length was cotransfected in HEK293 cells with 3xHA-M3R. Cells were lysed and immunoprecipitated (IP) and immunoblotted (IB) for either PLCβ3 N terminus (NT) or HA as described under “Experimental Procedures.” Representative Western blots shown were from three independent experiments. D, untagged PLCβ3 full length, YFP-PLCβ3 full length, or YFP-PLCβ3ΔPDZ (1–1231) was cotransfected in HEK293 cells with 3xHA-M3R. Cells were lysed and immunoprecipitated (IP) and immunoblotted (IB) for either PLCβ3 N terminus (NT) or HA as in C. Representative Western blots shown were from four independent experiments. Error bars represent S.E.
PLCβ3 contains a type I PDZ ligand (1231–1234) at its C terminus. To determine whether the PDZ ligand in PLCβ3 is important for interactions with M3R, YFP-PLCβ3ΔPDZ with deletion of the C-terminal four-amino acid PDZ ligand (Fig. 3A, right panels) was transfected with and without M3R. In the absence of the PDZ ligand, no M3R-dependent recruitment to the PM was observed. YFP-PLCβ3ΔPDZ was also transfected with HA-M3R, and interactions were examined by coimmunoprecipitation (Fig. 3D). Although the background binding was higher for this construct, there was no significant coprecipitation of YFP-PLCβ3ΔPDZ with HA-M3R. These results demonstrate a critical role for the PDZ ligand of PLCβ3 in binding to M3R and driving M3R-dependent PM localization of PLCβ3.
PLCβ3 Binds Directly to Intracellular Loops of M3R
The M3 muscarinic receptor does not contain a PDZ ligand on its C-terminal tail nor does it contain a PDZ domain, so a canonical PDZ scaffold could not mediate the interaction described here. To determine whether PLCβ3 can bind to M3R through direct protein-protein interaction and to characterize the intracellular surface of M3R involved in PLCβ3 interactions, the intracellular loops of M3R were expressed and purified as fusions with GST. Purified PLCβ3 bound directly to M3R intracellular loop 2 (M3Ri2), various subfragments of loop 3 (M3Ri3), and the full C-terminal tail (M3R/H8-CT) (Fig. 4A). The full C-terminal tail immediately distal to TM7 is composed of helix 8 (H8) parallel to the membrane followed by the remainder of the CT. Neither H8 nor CT alone bound to PLCβ3, suggesting that binding requires both domains or that both domains are required for proper folding of this region.
FIGURE 4.
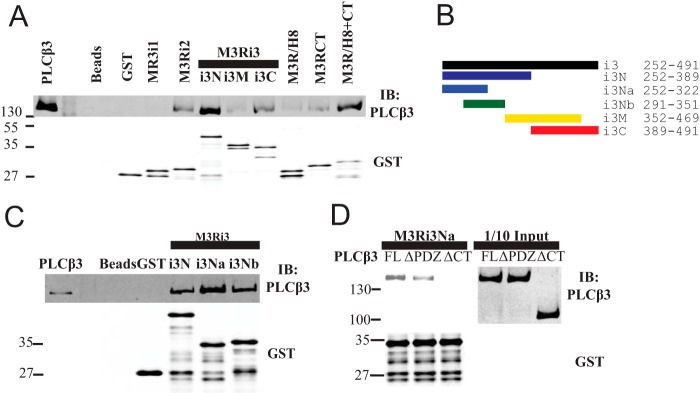
Intracellular loops of M3 muscarinic receptor bind PLCβ3. A, fragments from the intracellular surface of M3R were expressed as GST fusion proteins demarcated as follows: M3Ri1, 92–104; M3Ri2, 165–184; M3Ri3N, 252–389; M3Ri3M, 352–469; M3Ri3C, 389–491; M3R/H8, 547–560; M3R/CT, 564–590; and M3R/H8-CT, 547–590. Each fragment was tested for binding to purified PLCβ3 as described under “Experimental Procedures.” Representative Western blots shown were from three independent experiments. B, M3Ri3 loop subfragments. C, the indicated fragments from B were tested for binding to purified PLCβ3. Representative Western blots shown were from three independent experiments. D, M3Ri3Na was tested for binding to various purified PLCβ3 proteins (FL; ΔPDZ, 1–1230; ΔCT, 1–886; each at 3 nm). Representative Western blots shown were from three independent experiments. IB, immunoblot.
Binding of PLCβ3 to M3R Third Intracellular Loop
The M3Ri3 is large relative to other typical GPCR intracellular loops (240 amino acids) and was thus divided into subfragments for analysis. Within M3Ri3, the strongest apparent binding was to M3Ri3N. To further define the interaction regions within this domain, two overlapping fragments of M3Ri3N (depicted in Fig. 4B) were expressed as GST fusion proteins and tested for binding to purified PLCβ3. Although PLCβ3 binding was observed at all M3Ri3 N-terminal fragments, the strongest binding was at residues 252–322 (Fig. 4C). Because M3Ri3N had weaker binding than the M3Ri3Na subfragment, the larger construct could contain an element inhibitory toward binding of PLCβ3.
To determine whether the PLCβ3 C terminus and PDZ ligand are involved in direct interactions with this region, M3Ri3Na was tested for binding to purified PLCβ3, PLCβ3ΔCT, and PLCβ3ΔPDZ. Complete disruption of binding was achieved with deletion of the entire PLCβ3 CT (Fig. 4D). Removal of PDZ ligand weakened but did not eliminate the interaction (Fig. 4D). Thus, results using purified proteins essentially recapitulate the data from M3R-dependent localization and coimmunoprecipitation experiments. This direct binding to M3R involves multiple intracellular loop regions and requires the C terminus of PLCβ3 and its PDZ ligand.
Intracellular Loops of M3 Muscarinic Receptor Specifically Enhance Gαq Signaling
To determine how binding of M3R intracellular loops influences PLCβ3 signaling, the effects of M3R loop fragments on G protein-dependent PLCβ3 activation were assayed in a purified reconstituted system. Gαq-stimulated PLCβ3 activity was assayed using phospholipid vesicles containing PIP2 substrate and purified Gαq fully activated by GDP-AlF4− (38). Strikingly, M3Ri2, M3Ri3N, and M3R/H8-CT potentiated PLCβ3 activation by Gαq-GDP-AlF4− in a concentration-dependent manner with different efficacies and potencies (Fig. 5A). M3Ri1 and M3Ri3M, which do not bind to PLCβ3, did not have any significant effect (Fig. 5A). Conversely, M3Ri3C bound to PLCβ3 but did not potentiate Gαq activation, indicating that binding to PLCβ3 is not necessarily sufficient to potentiate its activation by Gαq. PLC activity in the presence of calcium alone or its activation by Gβγ was not altered by these M3R constructs, indicating a specific effect on Gαq-dependent PLC activation (Fig. 5B). The effects of M3Ri2, M3Ri3Na, and M3R/H8-CT on the potency and efficacy of Gαq-dependent PLCβ3 activation was tested under conditions where the concentration of Gαq was varied with a fixed concentration of receptor loop fragment (Fig. 5C). All of the loops increased the potency of Gαq as marked by leftward shifts in the [Gαq]-PLC activity curves as well as increases in PLC activity at the maximum [Gαq] tested of 100 nm (Fig. 5C). To determine whether the potentiation of Gαq-dependent PLC activation is specific to PLCβ3, Gαq-dependent PLCβ2 activation was examined for M3Ri2, M3Ri3Na, and M3R/H8-CT (Fig. 5D). Although some potentiation of PLCβ2 activation was observed by the fragments, it was significantly lower than that observed for PLCβ3.
FIGURE 5.
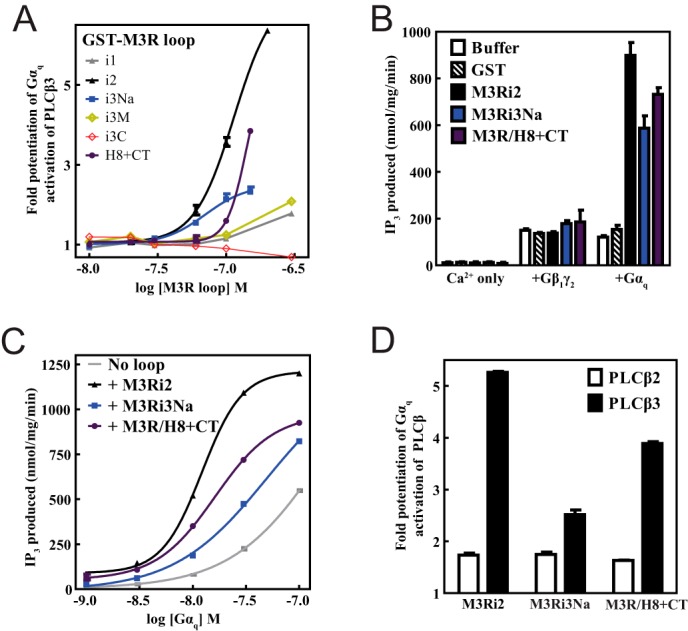
Intracellular loops of M3 muscarinic receptor specifically enhance efficiency of Gαq-dependent activation of PLC. A, [3H]IP3 release from [3H]PIP2-labeled vesicles due to PLCβ3 activation by Gαq-GDP-AlF4− was measured as a function of M3R loop concentration as described under “Experimental Procedures.” B, IP3 release from [3H]PIP2-labeled vesicles due to PLCβ3 activation was measured in the presence of calcium only, Gβ1γ2, or Gαq-GDP-AlF4−. Buffer (in which all fusion proteins were suspended) or a 300 nm concentration of GST, M3Ri2, M3Ri3Na, or M3R/H8-CT was added. C, PLCβ3 activation was measured as a function of [Gαq-Mg-GDP-AlF4−]. A representative plot is shown. D, IP3 release from [3H]PIP2-labeled vesicles due to activation of PLCβ3 (filled) and PLCβ2 (empty) was measured in the presence of Gαq-GDP-AlF4−. For C and D, M3Ri2, M3Ri3Na, and M3R/H8-CT were included at 200 nm. All assay results are representative of at least three independent experiments that contained internal triplicates per experimental condition. Error bars represent S.E.
Binding of M3R Fragments to G Proteins
Some component of the potentiation of Gαq-dependent PLC activation may be derived from an ability of the receptor fragments to bind G proteins. The ability of the M3R fragments to bind Gαq and Gβγ was examined in the GST fusion protein binding assay (Fig. 6A). M3Ri2, M3Ri3N, and M3R/H8-CT directly interacted with purified Gαq-GDP, whereas varying degrees of Gβγ binding were observed for all M3R fragments except M3Ri1. Binding of Gαq to M3Ri3M and M3Ri3C could not be determined due to interference from the GST fusion proteins running at the same molecular weight as Gαq. M3Ri3N was further dissected into six subfragments from residues 252–322 of M3Ri3Na (Fig. 6B) to discriminate the binding determinants for G proteins and PLCβ3. M3R fragments 1, 2, and 5 did not bind PLCβ3, whereas fragments 3 and 4 bound PLCβ3 almost as well as M3Ri3Na 252–322 (Fig. 6C). The binding was substantially reduced in fragment 6 (Fig. 6C), indicating that residues 310–314 of M3R confer M3i3N the ability to bind PLCβ3. In contrast, all six M3Ri3N constructs bound Gαq to varying degrees, whereas Gβγ binding to M3R fragments 3 and 4 was consistent with previous reports (10) (Fig. 6C). In particular, M3Ri3Na fragment 2 bound Gαq but not PLCβ3 (Fig. 6C). Fragments that bound Gαq, Gβγ, and PLCβ3 did not bind to Gαs or Gαolf (Fig. 6D), supporting the specificity of the interaction.
M3R Fragment Binding to PLCβ3 Is Required for Potentiation of Gαq-dependent PLCβ3 Activation
To confirm that binding of PLCβ3 is required for potentiation by M3R loops, we examined the ability of M3Ri3Na subfragments 2, 3, and 4 (from Fig. 6, B and C) to support potentiation of Gαq-dependent PLC activation (Fig. 7A). Fragment 3, which binds to PLCβ3, supported potentiation of PLC activation. Fragment 2, which did not bind PLCβ3, did not support potentiation of Gαq-dependent PLCβ3 activation even though Gαq binding remained intact (Fig. 6C). This suggests that within M3Ri3N PLCβ3 binding is necessary for potentiating PLCβ3 activation in the presence of Gαq-GDP-AlF4−. Surprisingly, fragment 4, which overlaps with fragment 3 and binds Gαq and PLCβ3, did not potentiate Gαq-dependent activation. These data suggest that binding of PLCβ3 to the M3R loop is required but is not sufficient to support potentiation of PLC activation.
FIGURE 7.

Residues 309–314 of M3Ri3N and PDZ ligand of PLCβ3 are determinants for M3Ri3N-mediated potentiation of PLCβ3 activation. A, to identify M3Ri3Na residues that contribute to the potentiation of Gαq-dependent PLCβ3 activation, constructs M3Ri3Na, M3Ri3Na 2, M3Ri3Na 3, and M3Ri3Na 4 were tested. The phospholipase C assay and data analysis were performed as in Fig. 5. A representative plot from four independent experiments is shown. B, M3R-mediated potentiation of PLCβ3 activation was compared between PLCβ3 variants (PLCβ3 FL, 1–1234; PLCβ3ΔPDZ, 1–1230) at 30 and 60 nm M3Ri3Na. *, p < 0.05, paired Student's t test from seven independent experiments. Coomassie staining of purified PLCβ3 proteins (1.5 μg each) is shown on the right. C, PLCβ3 activation by Ca2+ only or Gαq was tested for purified PLCβ3 constructs. The data were compiled from seven independent assays. D, within M3Ri3Na 252–322, alanine-scanning mutagenesis was performed across residues 309RCHFWF314. To facilitate screening for mutations that affect binding, alanine mutant proteins were partially purified by GST-glutathione affinity chromatography. A representative Western blot from three independent experiments is shown. Coomassie staining of each loaded GST fusion protein is shown on the bottommost panel. E, each M3Ri3Na variant was tested at 30 nm. * denotes significantly different (p < 0.05) relative to i3WT-PLCβ3 FL. Data were compiled from at least four independent experiments. Coomassie staining of M3Ri3Na WT (i3wt), M3Ri3Na F312A (i3FA), and M3Ri3Na W313A (i3WA) proteins (1.5 μg each) that were purified using His6-nickel affinity is shown on the left. Error bars represent S.E. IB, immunoblot.
Next we examined how deletion of the PLCβ3 PDZ ligand may affect potentiation. Deletion of the PLCβ3 PDZ ligand decreased but did not eliminate binding to M3Ri3Na (Fig. 4D). There was a small but significant difference between PLCβ3 and PLCβ3ΔPDZ with respect to M3Ri3Na-dependent potentiation of Gαq activation (Fig. 7B). Removal of PLCβ3 PDZ ligand did not affect PLCβ3 activation by Ca2+ or by Gαq in the absence of added M3R fragment (Fig. 7C).
Analysis of PLCβ3 binding to M3Ri3Na fragments 3, 4, and 6 suggested that residues 310–314 were critical binding determinants for PLCβ3. These amino acids were substituted individually with alanine as we sought to identify key residues for PLCβ3 binding (Fig. 7D). Although pulldown of purified PLCβ3 was decreased in all alanine mutants, mutants W313A and H311A/W313A exhibited the most severe binding defects.
Having identified PLCβ3 binding determinants at i3N of M3R (Fig. 7D) and M3R binding determinants on PLCβ3 (Fig. 4D), we examined how these domains may interact in modifying potentiation of Gαq-dependent PLCβ3 activation. The interaction between the ΔPDZ mutation in PLC and the F312A and W313A mutations in M3Ri3Na were investigated using purified components. The W313A and F312A mutations in M3Ri3Na inhibited their ability to potentiate activation of PLCβ3ΔPDZ by Gαq (Fig. 7E). The decrease in potentiation trended with the degree of decreased binding. The W313A mutation inhibited potentiation to a greater extent than F312A (although these were not statistically different from each other). Overall these data support the idea that these specific protein-protein interactions between M3R and PLC regulate Gαq-dependent activation of PLC.
DISCUSSION
Here we demonstrate that PLCβ3 binds directly to the M3 muscarinic receptor intracellular surface and that this binding can alter Gαq-dependent PLC activation. Our model is depicted in Fig. 8. In the inactive state, M3R binds to the C terminus of PLCβ3, resulting in displacement of the C terminus from the remainder of the PLC enzyme. This places PLCβ3 in close spatial proximity to both its substrate, PIP2, and its activator, Gαq. Upon receptor activation, Gαq, which is either prebound or recruited to the receptor, can efficiently activate the M3R-bound PLC. Additionally, M3R activation recruits PLCβ3 to the receptor by either direct binding to the receptor or Gαq-GTP or a cooperative interaction of both.
FIGURE 8.
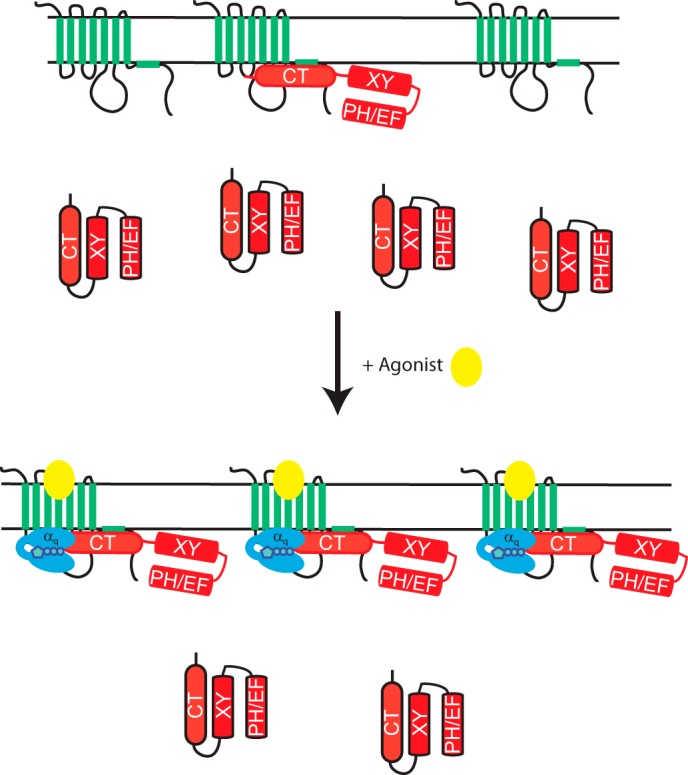
M3R interaction with PLCβ3 determines PLCβ3 signaling efficiency. M3R binding to PLCβ3 localizes PLCβ3 at the plasma membrane via the C-terminal tail of PLCβ3 and the intracellular loops and C-terminal tail of the receptor. This may result in an unfolding of the PLCβ3 enzyme to result in a more optimal interaction with G protein and substrate PIP2. Upon receptor activation, more PLCβ3 is recruited from the cytosol to the receptor. Gαq either prescaffolded to the receptor (not depicted) or recruited by collision coupling can then interact with the scaffolded PLC. PLC recruitment after activation could be due to direct interactions with activated Gαq, M3R, or both. EF, EF hands; XY, X and Y catalytic cores.
We propose that this interaction serves a number of functions. 1) Binding to M3R localizes PLCβ to the plasma membrane where it has local access to its PIP2 substrate. This would allow for spatial regulation of the effector reaction to the vicinity of the receptor. 2) Binding to the M3R increases signaling efficiency by increasing the effective local concentrations of activated Gαq and PLCβ3. 3) Binding of PLCβ3 to M3R increases its intrinsic capacity to be activated by Gαq via an allosteric mechanism.
Direct interactions between Gαq and the M3R C terminus have been suggested to account for the ability of M3R to prevent the lateral mobility of Gαq and alter M3R signaling efficiency (6). Additionally, agonist-independent cross-links between M3Ri2 and Gαq have been identified, suggesting the existence of preformed M3R-Gαq complexes (29). Our data showing direct interactions between Gαq and M3R intracellular loops and C terminus are consistent with these data.
Another mechanism to control signaling efficiency by muscarinic receptors is through the GTPase-accelerating function of PLC thought to kinetically scaffold Gq to a GPCR (19, 20). Here we show a direct physical scaffolding of the effector PLCβ to a GPCR that may contribute to signaling efficiency. It is also possible that this physical scaffolding relates to kinetic scaffolding, but this remains to be determined.
About half of Gq-coupled receptors have a C-terminal PDZ ligand motif (5, 22, 41). The M3 muscarinic receptor does not have a C-terminal PDZ ligand motif that would allow a PDZ scaffold to mediate an interaction with PLCβ (42). Although the M3R intracellular surface bears no homology to canonical PDZ-binding domains, M3Ri3 directly binds PLCβ3 in a PDZ-dependent fashion. Thus, this M3R-PLCβ3 interaction represents a non-canonical version of the highly organized PLC signaling that many Gq-coupled receptors may require to properly regulate phosphoinositide metabolism (5).
Previous studies of PLCβ plasma membrane binding suggest that the C-terminal domain of PLCβ isoforms may participate in an ionic interaction with the negatively charged inner surface of the PM to drive partial membrane association (35, 43–46). The data here, in combination with others, show that PLCβ3 does not associate with the plasma membrane unless M3R is coexpressed (Fig. 1, C and D) (35). Additionally, the PLCβ3 C terminus alone can interact with membranes (Fig. 2B) (35), suggesting that this membrane binding determinant is masked in the full-length enzyme. Binding of the C terminus by M3R could expose these determinants to promote interactions with the plasma membrane. Because M3R expression enhances PM localization of the isolated PLCβ3 CT, a direct tethering to the receptor must underlie part of the mechanism and could work in combination with unmasking from the folded holoenzyme (Fig. 8).
A surprising result was that fragments of M3R altered the efficacy and potency of Gαq-dependent PLC activation, implying a role of the M3R-PLC binding interaction in something other than simple scaffolding. The distal C-terminal domain of PLCβ3 has been suggested to coordinate interactions with the membrane and the N terminus of Gαq so that optimum PLCβ3 activation could be attained (45, 46). An intact PLCβ3 C-terminal domain is coincidentally required for PLCβ3 binding to M3R (Figs. 3, C and D, and 4D) and its potentiation of Gαq-dependent PLCβ3 activation (Fig. 7, B and E). Receptor binding could impart another level of allosteric control on the PLCβ C terminus.
The recent crystal structure of the β2-adrenergic receptor complex with Gs suggests that there is little space for other interactions with a monomeric receptor during G protein activation (47), but such interactions could be imagined in the context of higher order GPCR dimers or oligomers. Biochemical evidence using disulfide cross-linking showed that M3R could form homodimers (48). Recent studies using resonance energy transfer techniques (49, 50) suggest that M3R may actually exist as a dynamic mixture of dimers and rhombic tetramers.
Although we identified residues 294–322 of M3Ri3 as a PLCβ3-binding element necessary for potentiating Gαq-dependent PLCβ3 activation, this element is within an apparently expendable region of M3R 274–469 for carbachol-stimulated total inositol phosphate production (40, 51). There are a few possible explanations for this. PLCβ3 binding by M3Ri3 may reflect a spatially and/or temporally restricted event that is difficult to detect in global inositol phosphate assays, or multiple contacts may be involved in the interaction, and disruption of one of them is insufficient to completely disrupt binding of PLC to M3R and dramatically alter signaling efficiency in cells.
The M3R intracellular surface itself is likely conformationally flexible, and activation of M3R led to increased binding of PLCβ to the receptor. PLCβ3 and Gαq bound more strongly to M3Ri3Na (252–322) compared with the larger fragment M3Ri3N (252–389) (Fig. 4C). One could envision a model where receptor activation coordinates how binding sites on each intracellular loop become exposed to conformationally maximize Gαq-PLC coupling.
In summary, our studies define a novel PDZ-dependent interaction between the M3 muscarinic receptor and its signaling effector, PLCβ3. Receptor expression drives PLCβ3 localization at the plasma membrane and enhances the efficiency of Gαq signaling. These results translate into a mechanism for how the GPCR-effector interactions could fine-tune signaling beyond stimulating guanine nucleotide exchange.
Acknowledgments
We thank Drs. Jianxin Hu and Jürgen Wess (National Institute of Diabetes and Digestive and Kidney Diseases) for generously providing anti-M3R antibody and Nancy Ward for assistance with insect cell culture and virus production.
This work was supported, in whole or in part, by National Institutes of Health Grant GM053536 (to A. V. S.). This work was also supported by American Heart Association Predoctoral Fellowship 10PRE3490032 (to W. K.).
- GPCR
- G protein-coupled receptor
- CT
- C terminus
- FL
- full length
- H8
- helix 8
- i1
- i2, i3, intracellular loops
- IP3
- inositol 1,4,5-trisphosphate
- PDZ
- postsynaptic density-95/disc large/ZO-1
- PH
- pleckstrin homology
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PLC
- phospholipase C
- PM
- plasma membrane
- M3R
- M3 muscarinic receptor
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 2. Oldham W. M., Hamm H. E. (2006) Structural basis of function in heterotrimeric G proteins. Q. Rev. Biophys. 39, 117–166 [DOI] [PubMed] [Google Scholar]
- 3. Sternweis P. C., Smrcka A. V. (1992) Regulation of phospholipase C by G proteins. Trends Biochem. Sci. 17, 502–506 [DOI] [PubMed] [Google Scholar]
- 4. Harden T. K., Waldo G. L., Hicks S. N., Sondek J. (2011) Mechanism of activation and inactivation of Gq/phospholipase C-β signaling nodes. Chem. Rev. 111, 6120–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kadamur G., Ross E. M. (2013) Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154 [DOI] [PubMed] [Google Scholar]
- 6. Qin K., Dong C., Wu G., Lambert N. A. (2011) Inactive-state preassembly of Gq-coupled receptors and Gq heterotrimers. Nat. Chem. Biol. 7, 740–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galés C., Rebois R. V., Hogue M., Trieu P., Breit A., Hébert T. E., Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184 [DOI] [PubMed] [Google Scholar]
- 8. Galés C., Van Durm J. J., Schaak S., Pontier S., Percherancier Y., Audet M., Paris H., Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 [DOI] [PubMed] [Google Scholar]
- 9. Nobles M., Benians A., Tinker A. (2005) Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc. Natl. Acad. Sci. U.S.A. 102, 18706–18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu G., Bogatkevich G. S., Mukhin Y. V., Benovic J. L., Hildebrandt J. D., Lanier S. M. (2000) Identification of Gβγ binding sites in the third intracellular loop of the M3-muscarinic receptor and their role in receptor regulation. J. Biol. Chem. 275, 9026–9034 [DOI] [PubMed] [Google Scholar]
- 11. Wu G., Benovic J. L., Hildebrandt J. D., Lanier S. M. (1998) Receptor docking sites for G-protein βγ subunits. Implications for signal regulation. J. Biol. Chem. 273, 7197–7200 [DOI] [PubMed] [Google Scholar]
- 12. Hein P., Bünemann M. (2009) Coupling mode of receptors and G proteins. Naunyn Schmiedebergs Arch. Pharmacol. 379, 435–443 [DOI] [PubMed] [Google Scholar]
- 13. Daulat A. M., Maurice P., Froment C., Guillaume J. L., Broussard C., Monsarrat B., Delagrange P., Jockers R. (2007) Purification and identification of G protein-coupled receptor protein complexes under native conditions. Mol. Cell. Proteomics 6, 835–844 [DOI] [PubMed] [Google Scholar]
- 14. Lavine N., Ethier N., Oak J. N., Pei L., Liu F., Trieu P., Rebois R. V., Bouvier M., Hebert T. E., Van Tol H. H. (2002) G protein-coupled receptors form stable complexes with inwardly rectifying potassium channels and adenylyl cyclase. J. Biol. Chem. 277, 46010–46019 [DOI] [PubMed] [Google Scholar]
- 15. Benians A., Leaney J. L., Milligan G., Tinker A. (2003) The dynamics of formation and action of the ternary complex revealed in living cells using a G-protein-gated K+ channel as a biosensor. J. Biol. Chem. 278, 10851–10858 [DOI] [PubMed] [Google Scholar]
- 16. David M., Richer M., Mamarbachi A. M., Villeneuve L. R., Dupré D. J., Hebert T. E. (2006) Interactions between GABA-B1 receptors and Kir 3 inwardly rectifying potassium channels. Cell. Signal. 18, 2172–2181 [DOI] [PubMed] [Google Scholar]
- 17. Rebois R. V., Robitaille M., Galés C., Dupré D. J., Baragli A., Trieu P., Ethier N., Bouvier M., Hébert T. E. (2006) Heterotrimeric G proteins form stable complexes with adenylyl cyclase and Kir3.1 channels in living cells. J. Cell Sci. 119, 2807–2818 [DOI] [PubMed] [Google Scholar]
- 18. Fowler C. E., Aryal P., Suen K. F., Slesinger P. A. (2007) Evidence for association of GABAB receptors with Kir3 channels and regulators of G protein signalling (RGS4) proteins. J. Physiol. 580, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ross E. M. (2008) Coordinating speed and amplitude in G-protein signaling. Curr. Biol. 18, R777–R783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waldo G. L., Ricks T. K., Hicks S. N., Cheever M. L., Kawano T., Tsuboi K., Wang X., Montell C., Kozasa T., Sondek J., Harden T. K. (2010) Kinetic scaffolding mediated by a phospholipase C-β and Gq signaling complex. Science 330, 974–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suh P. G., Hwang J. I., Ryu S. H., Donowitz M., Kim J. H. (2001) The roles of PDZ-containing proteins in PLC-β-mediated signaling. Biochem. Biophys. Res. Commun. 288, 1–7 [DOI] [PubMed] [Google Scholar]
- 22. Kim J. K., Lim S., Kim J., Kim S., Kim J. H., Ryu S. H., Suh P. G. (2011) Subtype-specific roles of phospholipase C-β via differential interactions with PDZ domain proteins. Adv. Enzyme Regul. 51, 138–151 [DOI] [PubMed] [Google Scholar]
- 23. van Huizen R., Miller K., Chen D. M., Li Y., Lai Z. C., Raab R. W., Stark W. S., Shortridge R. D., Li M. (1998) Two distantly positioned PDZ domains mediate multivalent INAD-phospholipase C interactions essential for G protein-coupled signaling. EMBO J. 17, 2285–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahon M. J., Segre G. V. (2004) Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase Cβ, and actin increases intracellular calcium in opossum kidney cells. J. Biol. Chem. 279, 23550–23558 [DOI] [PubMed] [Google Scholar]
- 25. Oh Y. S., Jo N. W., Choi J. W., Kim H. S., Seo S. W., Kang K. O., Hwang J. I., Heo K., Kim S. H., Kim Y. H., Kim I. H., Kim J. H., Banno Y., Ryu S. H., Suh P. G. (2004) NHERF2 specifically interacts with LPA2 receptor and defines the specificity and efficiency of receptor-mediated phospholipase C-β3 activation. Mol. Cell. Biol. 24, 5069–5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J. K., Kwon O., Kim J., Kim E. K., Park H. K., Lee J. E., Kim K. L., Choi J. W., Lim S., Seok H., Lee-Kwon W., Choi J. H., Kang B. H., Kim S., Ryu S. H., Suh P. G. (2012) PDZ domain-containing 1 (PDZK1) protein regulates phospholipase C-β3 (PLC-β3)-specific activation of somatostatin by forming a ternary complex with PLC-β3 and somatostatin receptors. J. Biol. Chem. 287, 21012–21024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hwang J. I., Kim H. S., Lee J. R., Kim E., Ryu S. H., Suh P. G. (2005) The interaction of phospholipase C-β3 with Shank2 regulates mGluR-mediated calcium signal. J. Biol. Chem. 280, 12467–12473 [DOI] [PubMed] [Google Scholar]
- 28. Choi J. W., Lim S., Oh Y. S., Kim E. K., Kim S. H., Kim Y. H., Heo K., Kim J., Kim J. K., Yang Y. R., Ryu S. H., Suh P. G. (2010) Subtype-specific role of phospholipase C-β in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell. Signal. 22, 1153–1161 [DOI] [PubMed] [Google Scholar]
- 29. Hu J., Wang Y., Zhang X., Lloyd J. R., Li J. H., Karpiak J., Costanzi S., Wess J. (2010) Structural basis of G protein-coupled receptor-G protein interactions. Nat. Chem. Biol. 6, 541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamada M., Miyakawa T., Duttaroy A., Yamanaka A., Moriguchi T., Makita R., Ogawa M., Chou C. J., Xia B., Crawley J. N., Felder C. C., Deng C. X., Wess J. (2001) Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature 410, 207–212 [DOI] [PubMed] [Google Scholar]
- 31. Smrcka A. V., Sternweis P. C. (1993) Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C β by G protein α and βγ subunits. J. Biol. Chem. 268, 9667–9674 [PubMed] [Google Scholar]
- 32. Pang I. H., Sternweis P. C. (1990) Purification of unique alpha subunits of GTP-binding regulatory proteins (G proteins) by affinity chromatography with immobilized βγ subunits. J. Biol. Chem. 265, 18707–18712 [PubMed] [Google Scholar]
- 33. Mumby S. M., Gilman A. G. (1991) Synthetic peptide antisera with determined specificity for G protein α or β subunits. Methods Enzymol. 195, 215–233 [DOI] [PubMed] [Google Scholar]
- 34. Schöneberg T., Liu J., Wess J. (1995) Plasma membrane localization and functional rescue of truncated forms of a G protein-coupled receptor. J. Biol. Chem. 270, 18000–18006 [DOI] [PubMed] [Google Scholar]
- 35. Adjobo-Hermans M. J., Crosby K. C., Putyrski M., Bhageloe A., van Weeren L., Schultz C., Goedhart J., Gadella T. W., Jr. (2013) PLCβ isoforms differ in their subcellular location and their CT-domain dependent interaction with Gαq. Cell. Signal. 25, 255–263 [DOI] [PubMed] [Google Scholar]
- 36. Chan P., Gabay M., Wright F. A., Kan W., Oner S. S., Lanier S. M., Smrcka A. V., Blumer J. B., Tall G. G. (2011) Purification of heterotrimeric G protein α subunits by GST-Ric-8 association: primary characterization of purified Gαolf. J. Biol. Chem. 286, 2625–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonacci T. M., Ghosh M., Malik S., Smrcka A. V. (2005) Regulatory interactions between the amino terminus of G-protein βγ subunits and the catalytic domain of phospholipase Cβ2. J. Biol. Chem. 280, 10174–10181 [DOI] [PubMed] [Google Scholar]
- 38. Lehmann D. M., Yuan C., Smrcka A. V. (2007) Analysis and pharmacological targeting of phospholipase C β interactions with G proteins. Methods Enzymol. 434, 29–48 [DOI] [PubMed] [Google Scholar]
- 39. Philip F., Kadamur G., Silos R. G., Woodson J., Ross E. M. (2010) Synergistic activation of phospholipase C-β3 by Gαq and Gβγ describes a simple two-state coincidence detector. Curr. Biol. 20, 1327–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zeng F. Y., Soldner A., Schöneberg T., Wess J. (1999) Conserved extracellular cysteine pair in the M3 muscarinic acetylcholine receptor is essential for proper receptor cell surface localization but not for G protein coupling. J. Neurochem. 72, 2404–2414 [DOI] [PubMed] [Google Scholar]
- 41. Ranganathan R., Ross E. M. (1997) PDZ domain proteins: scaffolds for signaling complexes. Curr. Biol. 7, R770–R773 [DOI] [PubMed] [Google Scholar]
- 42. Klenk C., Vetter T., Zürn A., Vilardaga J. P., Friedman P. A., Wang B., Lohse M. J. (2010) Formation of a ternary complex among NHERF1, β-arrestin, and parathyroid hormone receptor. J. Biol. Chem. 285, 30355–30362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu D., Jiang H., Katz A., Simon M. I. (1993) Identification of critical regions on phospholipase C-β 1 required for activation by G-proteins. J. Biol. Chem. 268, 3704–3709 [PubMed] [Google Scholar]
- 44. Singer A. U., Waldo G. L., Harden T. K., Sondek J. (2002) A unique fold of phospholipase C-β mediates dimerization and interaction with G αq. Nat. Struct. Biol. 9, 32–36 [DOI] [PubMed] [Google Scholar]
- 45. Lyon A. M., Dutta S., Boguth C. A., Skiniotis G., Tesmer J. J. (2013) Full-length Gαq-phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat. Struct. Mol. Biol. 20, 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lyon A. M., Tesmer J. J. (2013) Structural insights into phospholipase C-β function. Mol. Pharmacol. 84, 488–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zeng F. Y., Wess J. (1999) Identification and molecular characterization of M3 muscarinic receptor dimers. J. Biol. Chem. 274, 19487–19497 [DOI] [PubMed] [Google Scholar]
- 49. McMillin S. M., Heusel M., Liu T., Costanzi S., Wess J. (2011) Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 286, 28584–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patowary S., Alvarez-Curto E., Xu T. R., Holz J. D., Oliver J. A., Milligan G., Raicu V. (2013) The muscarinic M3 acetylcholine receptor exists as two differently sized complexes at the plasma membrane. Biochem. J. 452, 303–312 [DOI] [PubMed] [Google Scholar]
- 51. Li B., Scarselli M., Knudsen C. D., Kim S. K., Jacobson K. A., McMillin S. M., Wess J. (2007) Rapid identification of functionally critical amino acids in a G protein-coupled receptor. Nat. Methods 4, 169–174 [DOI] [PubMed] [Google Scholar]