

Background: Steroid receptor coactivator-3 (SRC-3) is frequently overexpressed in human urinary bladder cancer.
Results: SRC-3 promotes urinary bladder cancer (UBC) cells proliferation through coactivating HIF1α and up-regulating the expression of genes involved in the glycolytic pathway.
Conclusion: SRC-3 plays an important role in UBC development through enhancing glycolysis.
Significance: Targeting SRC-3 or enzymes in glycolytic pathway could be an attractive approach in UBC therapy.
Keywords: Cancer, Cell Proliferation, Glycolysis, Transcription Coactivators, Tumor Metabolism, HIF1α, SRC-3, Urinary Bladder Cancer
Abstract
Cancer cell proliferation is a metabolically demanding process, requiring high glycolysis, which is known as “Warburg effect,” to support anabolic growth. Steroid receptor coactivator-3 (SRC-3), a steroid receptor coactivator, is overexpressed and/or amplified in multiple cancer types, including non-steroid targeted cancers, such as urinary bladder cancer (UBC). However, whether SRC-3 regulates the metabolic reprogramming for cancer cell growth is unknown. Here, we reported that overexpression of SRC-3 accelerated UBC cell growth, accompanied by the increased expression of genes involved in glycolysis. Knockdown of SRC-3 reduced the UBC cell glycolytic rate under hypoxia, decreased tumor growth in nude mice, with reduction of proliferating cell nuclear antigen and lactate dehydrogenase expression levels. We further revealed that SRC-3 could interact with hypoxia inducible factor 1α (HIF1α), which is a key transcription factor required for glycolysis, and coactivate its transcriptional activity. SRC-3 was recruited to the promoters of HIF1α-target genes, such as glut1 and pgk1. The positive correlation of expression levels between SRC-3 and Glut1 proteins was demonstrated in human UBC patient samples. Inhibition of glycolysis through targeting HK2 or LDHA decelerated SRC-3 overexpression-induced cell growth. In summary, overexpression of SRC-3 promoted glycolysis in bladder cancer cells through HIF1α to facilitate tumorigenesis, which may be an intriguing drug target for bladder cancer therapy.
Introduction
The dysregulated metabolic pathway is one of the hallmarks of cancer (1, 2). As one of them, enhanced glycolysis has been known as the Warburg effect for almost a century (1–3). Cancers are composed of a highly proliferative cell population group. In addition to higher energy demand, cancer cell proliferation requires a wide variety of biomolecules and glucose serving as an important precursor for biosynthesis of those molecules, including nucleic acids and lipids. Up-regulated expression of glucose transporters and enzymes in the glycolytic pathway combined with decelerated tricarboxylic acid cycle, and elevated levels of lactate were often observed in cancer cells.
Hypoxia inducible factor 1α (HIF1α)3 is one of the master regulators of glycolysis. Under normoxic conditions, HIF1α is ubiquitinated by the von Hippel-Lindau protein and degraded through the proteasomal pathway. However, under hypoxic conditions the interaction between HIF1α and von Hippel-Lindau is interrupted, and consequently, the HIF1α protein is not degraded. The stabilized HIF1α dimerizes with HIF1β and binds to hypoxia-responsive elements (HREs) in promoter regions of HIF1α target genes. Glycolytic enzymes HK2, PGK1, and LDHA are all up-regulated HIF1α target genes. Due to consistent low oxygen availability to most solid cancer tissues, these glycolytic enzymes were continuously up-regulated in cancer cells (4–7). Therefore, specifically targeting the HIF1α-mediated metabolic pathway in cancer cells has been postulated as a valuable therapeutic strategy (8–10).
Urinary bladder cancer (UBC) is one of the most common cancers worldwide (11). The incidence of UBC has been increasing steadily. In the developed countries, UBC takes the fourth leading incidence among cancers in men. However, the annual expense on UBC per patient ranks first among all cancers (12). Identification of gene mutations (FGFR3 and p53) and chromosome alterations (gain of 9p and 9q) in UBC specimens has shed some light on understanding UBC development (13, 14). But lacking specific molecular mechanisms underlying the cancer development including initiation, promotion, and progression of UBC hampered the successful treatment of UBC patients.
The steroid receptor coactivator-3 (SRC-3) was originally identified as an amplified gene on chromosome 20q in 30–60% of human breast cancer specimens (15). Multiple lines of evidence indicate that overexpression of SRC-3 is a common event in cancers derived from steroid-targeted tissues such as breast, prostate, and ovary (16). Specific ovrerexpression of SRC-3 in mammary epithelial cells induces breast cancer in mice; therefore SRC-3 has been defined as a bona fide oncogene (17). However, up-regulated expression of SRC-3 had also been found in cancers derived from non-steroid targeted tissues including the lung, liver, and bladder (18–20). In addition, analyses of a cohort of 163 human UBC found that 7.0% of the specimens contain SRC-3 gene amplification and 32.5% show overexpression of the SRC-3 protein (20). Immunohistochemistry analyses showed that SRC-3 overexpression in UBC specimens was not correlated with estrogen receptor, androgen receptor, and progesterone receptor, suggesting SRC-3 may function through mechanisms other than the steroid receptor coactivator (20). However, the exact role of SRC-3 in UBC remains unclear.
In this study, we first found that overexpression of SRC-3 is correlated with elevated levels of HIF1α target genes in human UBC samples. We further demonstrated that overexpressed SRC-3 in UBC cells not only interacts with HIF1α directly but also enhances the expression of HIF1α target genes. Finally, we proved that SRC-3 expression is essential for hypoxia-induced glycolysis and tumorigenesis of UBC.
EXPERIMENTAL PROCEDURES
Tissue Samples, Cell Lines, Plasmids, and Reagents
The tumor and adjacent normal tissue samples were obtained from UBC patients in Drum Tower Hospital at Nanjing University Medical School, using appropriate informed consent obtained after institutional review board approval. Human UBC cell lines, including T24, 5637, BIU87, and SCaBER, were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI1640 medium containing 10% FBS (Hyclone). SRC-3 expression plasmid and shSRC-3 (targeting sequence 5′-AACACTGCACTAGGATTATTG-3′) plasmid were gifts from Dr. Chundong Yu (19). HRE-Luc was a kind gift from Dr. Inna Serganova (21). PGK1-Luc plasmid was generated from human genomic DNA by PCR using primers: forward, 5′-GAAGATCTTCACGGGTAACAGGACTACAGGT-3′; and reverse, 5′-CCCAAGCTTGGGAGAGAGGTCGGTGATTCGGT-3′. To generate the HRE deletion PGK1-Luc plasmid (ΔHRE-PGK1-Luc), we used primers: A2, 5′-AGGGTACTAGTCCGCGAACCGCAA-3′; and B1, 5′-GGTTCGCGGACTAGTACCCTCGCAGA-3′. Puromycin, neomycin, MTT, 2-deoxyglucose (DG), sodium oxamate, and deferoxamine (DFO) were purchased from Sigma. 2-DG and sodium oxamate were added into media in 12.5 mg/ml and 80 mm to decrease cancer cell glycolysis.
Generation of Stable SRC-3 Knockdown and Overexpression Cells, Cell Proliferation, and Soft Agar Assays
To generate stable SRC-3 knockdown and mock cells, T24 and 5637 cells were transfected with pSUPER-shSRC-3 and pSUPER-shGFP plasmids, respectively, followed by selection of 1 μg/ml of puromycin for 10 days. The clones with SRC-3 overexpression were obtained by transfection of SRC-3 expression plasmid, followed by selection of 800 μg/ml of neomycin for 21 days. Western blotting was performed to determine knockdown and overexpression efficiency. Then, cell proliferation of SRC-3 knockdown cells and mock cells were measured by MTT assay. Briefly, 1,000 cells per well were seeded into 96-well plates, followed by MTT assay. UBC cell lines were transfected with 50 nm siRNA to HK2 (5′-CACGATGAAATTGAACCTGGTtt-3′), LDHA (5′-TTGTTGATGTCATCGAAAGtt-3′), and HIF1α (5′-UGUAGUAGCUGCAU GAUCGTtt-3′) at 40 to 60% confluence using Lipofectamine 2000 (Invitrogen). The soft agar assays were carried out as previously described (22). 1,000 cells were suspended as a single-cell suspension in 0.35% agarose in RPMI1640 containing 10% FBS on 0.6% agarose in the same media in 6-well plates. After 21 days incubation, colonies were stained overnight with 0.5 mg/ml of p-iodonitrotetrazolium violet (Sigma). Colonies >100 μm in diameter were counted.
Quantitative RT-PCR
SYBR Green-based real-time reverse transcription-PCR (RT-PCR) was done using an ABI PlusOne PCR instrument. Total RNAs were isolated using TRIzol (Invitrogen) and reverse transcription was performed using gDNA Erase and PrimeScript RT reagent kits (TAKARA Biotechnology, Dalian, China). For real-time PCR, cDNA reversed from 1 μg of total RNA was used. Primer sequences are shown below: src-3 (forward, 5′-GGGACTAAGCAACAGGTGTTT-3′; reverse, 5′-TTTGGCCCACCCATACTTGAG-3′); glut1 (forward, 5′-GATTGGCTCCTTCTCTGTGG-3′; reverse, 5′-TCAAAGGACTTGCCCAGTTT-3′); hk2 (forward, 5′-GAGCCACCACTCACCCTACT-3′; reverse, 5′-CCAGGCATTCGGCAATGTG-3′); pgk1 (forward, 5′-CATACCTGCTGGCTGGATGG-3′; reverse, 5′-CCCACAGGACCATTCCACAC-3′); pgm (forward, 5′-AGCATTCCGTATTTCCAGCAG-3′; reverse, 5′-GCCAGTTGGGGTCTCATACAAA-3′); ldha (forward, 5′-AAGCGGTTGCAATCTGGATTCAG-3′; reverse, 5′-GGTGAACTCCCAGCCTTTCC-3′); mct4 (forward, 5′-CAGTTCGAGGTGCTCATGG-3′; reverse, 5′-ATGTAGACGTGGGTCGCATC-3′); β-actin (forward, 5′-AGCGAGCATCCCCCAAAGTT-3′; reverse, 5′-GGGCACGAAGGCTCATCATT-3′).
Immunoprecipitation, GST Pull-down, and Western Blotting Assays
Immunoprecipitation assay was described previously (23). Briefly, transfected cells were lysed in lysis buffer (20 mm Tris-HCl, pH 8.0, 125 mm NaCl, 0.5% Nonidet P-40, 2 mm EDTA, 0.2 mm NaF, 0.2 mm Na3VO4, protease inhibitor mixture) for 30 min, followed by centrifugation at 12,000 × g for 20 min at 4 °C to remove debris. The lysates were incubated with 1.0 μg of anti-FLAG (Sigma) antibody for 16 h at 4 °C, followed by precipitation of protein A/G-agarose beads. As for GST pull-down, bacterial GST expression vector with SRC-3 domains (bHLH/PAS, S/T, RID, CID, and HAT) were transformed into Escherichia coli BL21(DE3), and the induced fusion protein was expression by 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C. After bacteria were split under the ultrasonic method, GST fusion proteins were purified and added to HEK293T cell lysate with overexpression of SREBP-1c N-terminal for the protein-protein interaction incubation. 20 μg of whole protein extract were fractionated by PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane, probed overnight with the antibodies against SRC-3 (BD Biosciences and Cell Signaling), Bcl-2, LDHA, HIF1α (Bioworld, China), HA, Cyclin D1, and PCNA (Santa Cruz, CA), and Glut1 (Proteintech, IL). β-Actin antibody (Abmax, China) was used as a loading control. The Western blot was visualized using the enhanced chemiluminescence kit (Pierce).
Luciferase Assay
1 × 105 293T or T24 cells were transfected using Lipofectamine 2000 with 0.2 μg of the following plasmids: HRE-Luc, PGK1-Luc, ΔPGK1-Luc, pCMV-3xFLAG-SRC-3, and pCS2–4HA-HIF1α. Renilla luciferase plasmid was co-transfected as a transfection efficiency control. 24 h later, cells were harvested and luciferase activity was determined using the Dual-Luciferase Reporter Assay system (Promega). 100 μm DFO was added into medium 24 h before the luciferase reporter assay.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as previously described (24). In brief, the UBC cells were treated with either normoxia (21% O2) or hypoxia (1% O2) for 24 h and chromatin was immunoprecipitated using anti-HIF1α (Novus Biologicals), anti-SRC-3 (C-20; Santa Cruz Biotechnology Inc.) antibodies, or nonspecific IgG (Santa Cruz). ChIP DNA were purified and amplified by PCR with specific primers for the HRE regions in the promoters of pgk1 and glut1 genes. The PCR products were separated on 1.0% agarose gel and visualized under UV light. Primers used for the ChIP assay were: glut1 (forward, 5′-CTGACTCAGCAAAACCTTTCCT-3′; reverse, 5′-AGGTGCCAGGCGGATGCTGGA-3′) and pgk1 (forward, 5′-TAAGTCGGGAAGGTTCCTTG-3′; reverse, 5′-GGCTTGCAGAATGCGGAACA-3′). Bands were quantified by using ImageJ software (NIH, Bethesda, MD).
Glucose, Lactate, Media pH, and ATP Production Measurements
1 × 105 cells were incubated into a 35-mm dish, the cells were either treated with hypoxic (1% O2) or normoxic (21% O2) conditions for 36 h. The medium was collected by centrifugation to remove the cells, and glucose consumption was measured using the Glucose (HK) Assay Kit (Sigma). Lactate levels and pH values in the culture media were measured using the Lactate Assay kit (BioVision) and pH meter. To determine ATP levels, UBC were trypsinized and washed by Dulbecco's phosphate-buffered saline three times at 4 °C and intracellular ATP was assessed using an ATP bioluminescence assay kit (ZhongkeLiangma Bioengineering, China), per the manufacturer's instructions.
Immunohistochemistry
Twenty human UBC tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Sections (5-μm thick) were stained with antibodies against SRC-3 (1:800; BD Biosciences) and Glut1 (1:3000; H-43, Santa Cruz) in accordance with the previously described procedure (25). Nuclear SRC-3 and membrane/cytoplasmic Glut1 expression were quantified using a 10-value intensity score (0–9) and the percentage (0–100%) of the extent of reactivity by two pathologists. The whole field in each slide was examined for final quantification. An immunohistochemical expression score was obtained by multiplying the intensity and reactivity extension values, and these expression scores were used to determine expression levels.
Tumor Xenograft
All animal experiments were approved by the Animal Care and Use Committee of the Model Animal Research Center, Nanjing University, China. The cells were resuspended in 50% Matrigel (BD Biosciences) in PBS (1 × 107 cells/ml). Athymic mice (6-week-old; Model Animal Research Center) were injected subcutaneously with 200 μl of shSRC-3 or shCTRL T24 cells on the flank. Tumor size was measured every 7 days using a caliper and calculated using the formula: volume = (L × W2)/2, in which L is the widest diameter of the tumor and W is the diameter perpendicular to L. At the end point, the tumors were harvested for weight.
Statistics Analysis
SPSS software (version 17.0) was used for statistical analysis. Student's t test (two-tailed) or Mann-Whitney U tests were performed as appropriate to compare the intergroup. p < 0.05 would be considered statistically significant.
RESULTS
Overexpression of SRC-3 in UBC Cells Enhances Aerobic Glycolysis
To determine the roles of SRC-3 in the development of bladder cancer, we first analyzed SRC-3 expression levels in a panel of human UBC cell lines, and 11 human UBC specimens with their adjacent normal bladder tissues. As shown in Fig. 1A, various levels of SRC-3 were readily detectable in all four tested human UBC cell lines. Compared to that in the adjacent normal bladder tissues, SRC-3 protein levels were much higher in 63.6% (7/11) of UBC specimens. To test whether SRC-3 is overexpressed in UBC samples at the mRNA level, we analyzed a public GEO profile GSE3167 dataset (26), showing that src-3 is significantly increased in tumor samples (Fig. 1B). Moreover, in nine pairs of UBC samples, in which we tested SRC-3 protein levels, we found that SRC-3 is overexpressed in 5 of 9 samples at the mRNA level by qRT-PCR (Fig. 1B), which is correlated with its protein levels in UBC samples (Fig. 1A). Next, we transfected a plasmid overexpressing SRC-3 to BIU87 UBC cells, which have relatively lower SRC-3 expression, and found that, compared to mock cells (CTRL), both stable transfectants (#1 and #2) not only gained a higher proliferative capacity (Fig. 1, C and D), but also formed more colonies (>2-fold) in soft agar assays (Fig. 1E). Western blots showed that two proliferation markers, PCNA and Cyclin D1, were increased in both stable transfectants (Fig. 1D). Consistently, the knockdown of SRC-3 in T24 and 5637 cells decreased both bladder cancer cell proliferation, and colony formation ability in soft agar (Fig. 1, F, I and H, K). Moreover, the protein levels of PCNA and Cyclin D1 were down-regulated in SRC-3 knockdown stable cell lines (Fig. 1, G and J). These data suggest that SRC-3 overexpression may confer a growth advantage to bladder cancer cells.
FIGURE 1.

Overexpressed SRC-3 promoted bladder cancer cell proliferation. A, Western blot analysis of SRC-3 protein expression in four bladder cancer cell lines and 11 pairs of human UBC samples. N, non-tumorous bladder tissue adjacent to UBC; T, tumor tissue. B, overexpression of SRC-3 in human UBC samples at the mRNA level. Compare the mRNA level of src-3 between normal (n = 14) and UBC samples (Tumor, n = 46) in the GEO profile dataset (GSE3167). Quantitative RT-PCR assay detected at the src-3 level in 9 human UBC samples. C, cell growth curve in SRC-3 overexpressed and control cells by the MTT assay over 4 days. D, Western blot analysis of SRC-3, Cyclin D1, and PCNA protein levels in SRC-3 overexpressed and control cells. E, colony formation capacity in soft agar of SRC-3 overexpressed and control cells. CTRL, BIU87 cells transfected empty vector; #1 and #2, two different clones from BIU87 cells stably transfected with the SRC-3 expression plasmid. F–K, SRC-3 deficiency in human UBC T24 (F-H) and 5637 (I–K) cells impairs cell proliferation and colony formation capacity. F and I, cell growth curve in SRC-3 knockdown (shSRC-3) and control (shCTRL) cells by MTT assay. G and J, Western blot analysis of SRC-3, Cyclin D1, and PCNA protein levels in SRC-3 knockdown and control cells. H and K, colony formation capacity in soft agar of the SRC-3 knockdown and control cells. **, p < 0.01; ***, p < 0.001.
Metabolic pathways in cancer cells are usually reprogrammed favoring glycolysis to provide a source of substrates for the synthesis of amino acid, lipids, and nucleic acids that are essential for proliferation (2). We next investigated whether the SRC-3 expression level change would affect glucose metabolism in these cells. Under normoxic condition, overexpression of SRC-3 significantly increased glucose consumption and lactate production in BIU87 cells (Fig. 2A). In addition, hypoxia induced a higher glycolytic rate in all cells, as expected. Notably, SRC-3 overexpression increased both glucose consumption and lactate production further, compared with control cells (p < 0.05, Fig. 2A). Furthermore, we observed that overexpression of SRC-3 also resulted in elevated mRNA expression levels of the glycolytic pathway-related genes, including glut1, hk2, pgk1, pgm, ldha, and mct4, under both normoxic and hypoxic conditions (Fig. 2B). For example, the Glut1 mRNA transcript increased 1.8-fold in SRC-3 #1 cells and 1.6-fold in SRC-3 #2 cells under normoxia conditions; whereas the increases under hypoxia conditions were 2.9-fold in SRC-3 #1 cells and 3.5-fold in SRC-3 #2 cells, respectively, compared with control cells. Data from Western blot analyses further confirmed the induction of Glut1 and LDHA protein levels in SRC-3 transfectants (Fig. 2C).
FIGURE 2.

Ectopic expression of SRC-3 enhanced bladder cancer cell aerobic glycolysis. A, glucose consumption and lactate production in SRC-3 overexpressed and control cells under normoxia and hypoxia conditions. The mRNA and protein expression levels of glycolytic genes in the SRC-3 overexpressed and control cells were detected by quantitative RT-PCR assay (B) and Western blot assay (C). CTRL, BIU87 cells transfected with empty vector; #1 and #2, two stable transfectants of SRC-3. *, p < 0.05; **, p < 0.001.
SRC-3 Deficiency Impairs Glycolysis in Bladder Cancer Cells under Hypoxic Conditions
To confirm whether SRC-3 is essentially required for glycolysis in UBC cells, we tested it in stable SRC-3-knockdown cells (shSRC-3) and control shRNA-transduced mock cells (shCTRL) in T24 and 5637 cells. Under hypoxic conditions, knockdown of SRC-3 decreased glucose consumption (by 49.8 and 20.5% in T24 and 5637, respectively), lactate production (by 36.1 and 24.3% in T24 and 5637, respectively), and the intracellular ATP level (by 38.4 and 34.3% in T24 and 5637, respectively) as well as increased the pH value of culture medium (by 4.4 and 2.1% in T24 and 5637, respectively), compared to parental cells. However, no significant changes were observed between knockdown cells and parental cells under normoxic conditions (Fig. 3, A and B). We also assessed the expression levels of glycolytic genes in shSRC-3 and shCTRL T24 cells under both normoxic and hypoxic conditions. Consistently, SRC-3 knockdown significantly suppressed mRNA levels of glut1, hk2, pgk1, pgm, ldha, and mct4 genes under hypoxia conditions (Fig. 3C). However, the inhibitory effects by depletion of SRC-3 were minimal under normoxia (Fig. 3C). Western blotting confirmed that down-regulation of Glut1 and LDHA proteins in the shSRC-3 cells was only detectable under hypoxia (Fig. 3D). These data indicate that SRC-3 is required for glycolysis of cancer cells only under hypoxic conditions.
FIGURE 3.

Knockdown of SRC-3 decreased glycolysis in bladder cancer cell under hypoxia. Glucose consumption, lactate production, medium pH value, and ATP production in SRC-3 knockdown and control cells under normoxia and hypoxia were detected in both T24 (A) and 5637 (B) cells. The mRNA and protein expression levels of glycolytic genes in SRC-3 knockdown and shCTRL T24 cells were detected by quantitative RT-PCR assay (B) and Western blot assay (C). shSRC-3 and shCTRL, SRC-3-targeting shRNA and control shRNA stably transduced cells. *, p < 0.05.; **, p < 0.01; ***, p < 0.001.
Suppression of SRC-3 Inhibits Tumorigenicity in Nude Mice
We further evaluated the tumorigenicity of SRC-3 knockdown cells in nude mice. shSRC-3 and shCTRL cells were subcutaneously inoculated on the flanks of animals. Tumor size was measured weekly over 7 weeks for the tumor growth study. Calculated tumor volume over time was plotted (Fig. 4A). shCTRL cells exhibited significantly faster growth starting at 3 weeks post-tumor cell injection, compared with the shSRC-3 cells (p < 0.05). At the end point of the experiment, the average tumor weight of the shCTRL xenografts was 70-fold higher than that of the shSRC-3 xenografts (Fig. 4B). Decreased protein expression levels of SRC-3, PCNA (proliferation marker), and LDHA in shSRC-3 xenografts were detected by Western blot (Fig. 4C). In summary, SRC-3 plays an essential role in UBC tumor cell proliferation, metabolism, and tumorigenicity through regulating the glycolytic pathway.
FIGURE 4.

Knockdown of SRC-3 abrogated tumorigenicity of T24 bladder cancer cell in nude mice. A, in vivo tumor growth curve of SRC-3 knockdown and control xenografts over 7 weeks. Tumor size was measured by caliper and tumor volume was calculated by the formula: volume = (L × W2)/2. B, the average tumor weight of the SRC-3 knockdown (shSRC-3, n = 8) and control (shCTRL, n = 6) xenografts at the end point. C, Western blotting assay of SRC-3, PCNA, and LDHA protein expression levels in SRC-3 knockdown and control xenografts. Data, mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
SRC-3 Coactivates HIF1α Transcriptional Activity in UBC
Because HIF1α is a master transcription factor for glycolysis, we explored whether SRC-3 functions as a HIF1α coactivator. The co-immunoprecipitation assay demonstrated that SRC-3 could interact with HIF1α under normoxic conditions, but more HIF1α·SRC-3 protein complex was detected under hypoxic conditions, probably due to stabilization of HIF1α (Fig. 5A). Furthermore, a GST pull-down assay identified that SRC-3 directly interacts with HIF1α through the N-terminal bHLH/PAS domain in vitro (Fig. 5, B and C). DFO, which inhibits the enzymatic activity of prolyl hydroxylases, was used to stabilize HIF1α under normoxic conditions (27). The increased HIF1α protein level by DFO treatment was confirmed by Western blot analysis (Fig. 5D, inset). The luciferase reporter assay based on the transcriptional activity of HIF1α revealed that DFO treatment or SRC-3 overexpression alone increased HRE-luc reporter activities by 2.2- and 3.6-fold, respectively. Combined SRC-3 overexpression and DFO treatment strikingly increased HIF1α-dependent transcriptional activity by 19.3-fold, indicating the synergistic regulation of SRC-3 and HIF1α (p < 0.001; Fig. 5D). In addition, significantly enhanced HIF1α transcriptional activities were proportional to the levels of SRC-3 co-transfected with the HIF1α expression vector (Fig. 5E). Next, we tested whether the effect of SRC-3 on HIF1α transcription activity was HRE-dependent. Reporters using the pgk1 promoter either with the HRE (PGK1-Luc) or without the HRE (ΔPGK1-Luc) were constructed. Loss of the HRE led to a ΔPGK1-Luc reporter that could not respond to DFO treatment, SRC-3 overexpression (Fig. 5F), and even HIF1α overexpression (Fig. 5G), suggesting that HRE is essential in SRC-3-regulated pgk1 gene expression. On the other hand, knockdown of SRC-3 by RNA interference abrogated the HIF1α transcriptional activity (Fig. 5H). Most importantly, the ChIP assay showed that both SRC-3 and HIF1α were recruited to the endogenous glut1 promoter and exogenous pgk1 promoter (Fig. 5I). These data collectively demonstrated that under hypoxic conditions SRC-3 coactivates HIF1α through direct protein binding, and enhances the transcriptional activity of HIF1α in cancer cells.
FIGURE 5.

SRC-3 coactivated HIF1α in vitro. A, ectopic SRC-3 protein interacted with HIF1α in T24 cells. T24 cells were transiently transfected with FLAG-tagged SRC-3. Co-immunoprecipitation (IP) assay was carried out in cells treated under either normoxia or hypoxia conditions. The precipitant by the IgG antibody was used as a negative control. B, schematic representation of five fragments of SRC-3 representing different domains. C, five fragments of SRC-3 were generated in E. coli BL21(DE3) as GST fusion proteins. The purified GST fused proteins were incubated with HEK293T lysate, which contained the ectopic expression of HA-HIF1α. Western blot (WB) was performed to detect the pulled down HIF1α by HA antibody. D–H, luciferase activity assay: D, in the HRE-Luc-transfected 293T cells co-transfected with SRC-3 expression plasmid, with or without DFO treatment, ***, p < 0.001, compared empty vector transfection group; ###, p < 0.001 compared with the SRC-3 transfection group; E, in HRE-Luc 293T cells with co-transfection of SRC-3 and HIF1α expression plasmids with the indicated amount of plasmids, ***, p < 0.001; F, in the PGK1-reporter 293T cells co-transfected with the SRC-3 expression plasmid, with or without DFO treatment, ***, p < 0.001, compared with the empty vector transfection group; ###, p < 0.001, compared with the SRC-3 transfection group; G, in the PGK1-reporter 293T cells with co-transfection of SRC-3 and HIF1α expression plasmids. PGK1-Luc, wild type PGK1 reporter; ΔPGK1-Luc, PGK1 reporter without HRE; followed by transfection with or without HIF1α. H, in HRE-Luc T24 cells transduced with control (−) or SRC-3 shRNA (+), with or without HIF1α overexpression, ***, p < 0.001, compared with the empty vector transfection group; ###, p < 0.001, compared with the group with SRC-3 knockdown and HIF1α overexpression. I, presence of HIF1α and SRC-3 on the endogenous glut1 promoter and pgk1 promoter from the pgk1-luc reporter detected by the ChIP assay. Parental T24 cells (left panel) and T24 wells transfected with the pgk1-luc reporter plasmid (right) were incubated under normoxia (N) or hypoxia (H), respectively. The band intensity was shown under each blot, which was quantified by ImageJ software. The precipitant by IgG antibody was used as a negative control. RLU, relative light units.
Inhibition of the Glycolysis Pathway Suppresses SRC-3-induced Cell Proliferation
To further determine whether SRC-3 plays a pivotal role in cancer cell proliferation via downstream targets of HIF1α in the glycolytic pathway, we knocked down two key glycolytic enzymes, HK2 and LDHA, by siRNA in UBC cells with or without SRC-3 overexpression (Fig. 6A). For instance, in SRC-3 #1 BIU87 cells, suppression of HK2 and LDHA decreased cancer cell proliferation by 21.7 and 34.4%, respectively (Fig. 6, B and C). However, in the parental BIU87 cells, knockdown of HK2 and LDHA slightly reduced proliferation of the cells with endogenous SRC-3 expression by only 12.2 and 22.6%, respectively (Fig. 6, B and C). In addition, treatment with 2-DG (a hexokinase inhibitor) and sodium oxamate (a competitive inhibitor of the LDH enzyme) inhibited cancer cell proliferation by 37.2 and 53.5%, respectively, in the CTRL cells; whereas, in the presence of overexpressed SRC-3, the same treatments reduced cell proliferation remarkably by 50.2 and 60%, respectively (Fig. 6, D and E). Moreover, we impaired glycolysis by knockdown of HIF1α, the proliferation was inhibited by 35.0 and 10.1% in BIU87 #1 and CTRL, respectively (Fig. 6, F and G). Our data indicate that SRC-3 modulates cancer cell proliferation by up-regulating the glycolytic pathway.
FIGURE 6.
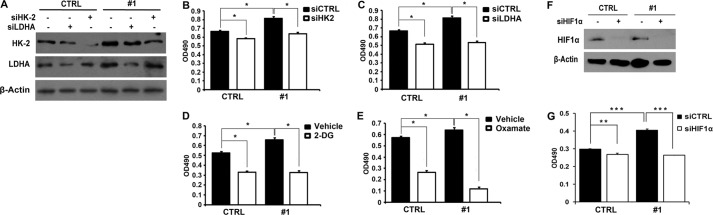
Knockdown of glycolytic genes or HIF1α reduced SRC-3 overexpression accelerated bladder cancer cell proliferation. A, efficiency of RNA interference targeting HK-2 (siHK-2) and LDHA (siLDHA) in SRC-3 overexpressed (#1) and control cells determined by Western blot analysis. B–E, cell proliferation in SRC-3 overexpressed and control cells with the treatment of siHK-2 (B), siLDHA (C), 2-DG (12.5 mg/ml) (D), and sodium oxamate (80 mm) (E). F, efficiency of RNA interference-targeting HIF1α (siHIF1α) in SRC-3 overexpressed (#1) and control cells determined by Western blot analysis. G, cell proliferation in SRC-3 overexpressed (#1) and control (CTRL) cells with treatment of siHIF1α were detected by MTT assay. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
SRC-3 Has Positive Correlation with HIF1α-regulated Genes in the Glycolytic Pathway and Proliferation in Human UBC Specimens
The correlation between the expression levels of SRC-3 and HIF1α-regulated glycolytic genes in 20 human UBC samples was also examined. We observed that the level of SRC-3 nuclear staining was positively associated with the level of Glut1 membrane staining. Immunohistochemistry staining of SRC-3 and Glut1 in two representative UBC patients was shown in Fig. 7A; SRC-3 and Glut1 were not detectable in case 1 and a strong nuclear staining of SRC-3 coincided with a strong membrane staining of Glut1 in case 2. The intensities of SRC-3 nuclear staining and Glut1 membrane staining were quantified (Fig. 7B) and high positive correlation between these two factors was identified by Pearson correlation analysis (r = 0.721; p < 0.001). In addition, 46 human UBC samples from the public GEO profile dataset (GSE3167; 26) were analyzed. SRC-3 was not only positively correlated with two well established HIF1α target genes, pgk1 (r = 0.2721, p = 0.0354) and vegfa (r = 0.4666, p = 0.0002), but also with proliferation marker pcna (r = 0.4567; p = 0.0002; Fig. 7C). Taken together, the correlation between SRC-3 and HIF1α-regulated glycolytic genes was established in clinical UBC specimens, indicating its role in UBC cell proliferation via HIF1α to augment the Warburg effect.
FIGURE 7.

Correlation between expression levels of SRC, HIF1α target genes, and proliferative marker gene, pcna, in clinical human UBC samples. A, two representative UBC specimens for immunohistochemistry staining of SRC-3 and Glut1 in adjacent sections. Case 1, SRC-3 negative and Glut1 negative; case 2, positive nuclear staining of SRC-3 and positive membrane staining of Glut1. Bar, 100 μm. B, correlation between expression levels of SRC-3 and Glut1 in a cohort of human UBC specimens (n = 20, Spearman rank correlation coefficient). C and D, linear regression of src-3, HIF1α target genes (pgk1 and vegfa), and proliferative marker gene (pcna), using UBC samples from the GEO profiles database (GSE3167, n = 46).
DISCUSSION
Deregulation of glucose metabolism is frequently found in cancer cells and known as one of the 10 hallmarks of cancer (1, 28). Because HIF1α is one of the major transcription factors to up-regulate the expression of glycolytic genes, understanding the mechanism regulating HIF1α activity in glycolysis is important for developing more effective and targeted therapies. In this study, we have showed that SRC-3 regulates glucose metabolism through its interaction with HIF1α and coactivation of the transcriptional activity of HIF1α to facilitate cancer cell growth. Overexpression of SRC-3 increases aerobic glycolysis in BIU87 bladder cancer cells, accompanied with elevated expression of genes in the glycolytic pathway (Fig. 2). SRC-3 inhibition suppressed glycolysis under hypoxic conditions in T24 and 5637 UBC cells and T24 xenografts (Figs. 3 and 4), indicating the role of SRC-3 in reprogramming cancer cell metabolism. Furthermore, correlated overexpression of both SRC-3 and enzymes involved in the glycolytic pathway in human UBC specimens confirms the in vitro results. Altogether, these data provide a mechanism whereby SRC-3 functions as an oncogene by regulating glycolytic metabolism.
SRC-3 was characterized as a coactivator for steroid hormone receptors, such as estrogen receptor, progesterone receptor, and androgen receptor (16, 29–31), and overexpression of SRC-3 has been reported in multiple steroid hormone-targeted cancers, including prostate and breast cancers. But none of the samples in a cohort of 163 UBC with amplification and overexpression of SRC-3 showed IHC positive staining of androgen receptor or progesterone receptor, nor any correlation between the expression of SRC-3 and estrogen receptor (20). Multiple lines of evidence from recent reports indicate that SRC-3 may also activate transcription factors such as AP-1 and PEA3 (25, 32–35). Very recently, SRC-3 was reported to coactivate the E2F1 transcriptional factor and drive cell cycle proteins in bladder cancer cells (36). Consistent with these findings, we demonstrated that SRC-3 could also coactivate the HIF1α transcription factor and up-regulate genes in the glycolytic pathway. This suggests that SRC-3 is indeed a master coregulator (37) and activates a much wider spectrum of transcription factors than assumed originally. Therefore, amplification and/or overexpression of SRC-3 in cancer cells made this coactivator readily available for activation of multiple transcription factors and orchestrating the expression of genes in different pathways. Considering that the strong nuclear staining of SRC-3 has been reported as a poor prognostic marker for UBC patients (20), we propose that any strategies specifically targeting SRC-3 could be a promising therapeutic approach in UBC treatment.
HIF1α is degraded through the proteasome pathway under normoxic conditions, but stabilized during hypoxia and consequently promotes the transcription of a battery of genes including those in the glycolytic pathway, such as hk2, pgk1, and ldha (38, 39). Our data revealed that overexpression of SRC-3 coactivates HIF1α in UBC cells to induce expression of glycolytic genes under both normoxic and hypoxic conditions (Fig. 2). However, the loss of SRC-3 affected glycolysis only in hypoxia, but not in normoxia (Fig. 3). Given that several cofactors, such as CBP and p300, have been implicated in HIF1α activation (40–42), we postulate that these coactivators may compensate for the effects from SRC-3 depletion in T24 cancer cells under normoxia.
Cancer cells are more addicted to glycolysis, a pathway providing sufficient metabolic intermediates to support anabolic processes in the highly proliferative cancer cells (43, 44). Therefore, suppression of the intracellular glucose metabolism could be a targeted approach to suppress cancer cell proliferation. Multiple lines of evidence from recent reports revealed this approach is not only reasonable but also feasible. For instance, a specific LDH inhibitor, oxamate, increased sensitivity of breast cancer cells to taxol (45). Animals treated with 2-DG and Mito-CP, a mitochondria-targeted drug, induced significant inhibition of tumor growth without any significant side effects (10). More interestingly, tissue-specific knock-out of HK2 in different animal models demonstrated that KRas-driven lung cancer and ErbB2-driven breast cancer developments were inhibited (46). SRC-3 amplification and overexpression have been detected in 7 and 35% of UBC patients, respectively. In the present report, we demonstrated that SRC-3 overexpression increases the glycolytic rate in bladder cancer cells through up-regulating the expression of genes in the glycolytic pathway. In addition, both knockdown and pharmacologic inhibition of HK2 and LDHA suppressed SRC-3 overexpression-induced cell proliferation, suggesting that SRC-3 overexpressing UBC cells are more addicted to the glycolysis pathway. Because systematic deletion of HK2 does not show any significant adverse physiological consequence (46), targeting any key enzymes in the glycolytic pathway could be an attractive and safe therapeutic approach for solid tumors.
In summary, our data have demonstrated that SRC-3 plays an important role in cell proliferation during tumor progression by enhancing glycolysis through up-regulation of the enzymes in the glycolytic pathway. Whether SRC-3 affects bladder cancer initiation, invasion, and relapse should be further explored.
Acknowledgments
We thank Dr. C. Yu and Dr. I. Serganova for providing plasmids. We also thank Caihong Luo and Mingyang Jiang for helpful technical support.
The work was supported by grants from Ministry of Science and Technology (2011CB944104, 2010CB945101), the National Natural Science Foundation (81172009, 81372168), Doctoral Fund of Ministry of Education of China (20110091120028), and Fundamental Research Funds for the Central Universities (090314340001) (to J. Y.).
- HIF1α
- hypoxia inducible factor 1α
- UBC
- urinary bladder cancer
- SRC-3
- steroid receptor coactivator-3
- HRE
- hypoxia-responsive elements
- DFO
- deferoxamine
- LDHA
- lactate dehydrogenase
- DG
- deoxyglucose
- PCNA
- proliferating cell nuclear antigen
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hsu P. P., Sabatini D. M. (2008) Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707 [DOI] [PubMed] [Google Scholar]
- 4. Denko N. C. (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713 [DOI] [PubMed] [Google Scholar]
- 5. Semenza G. L. (2007) HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 39, 231–234 [DOI] [PubMed] [Google Scholar]
- 6. Robey I. F., Lien A. D., Welsh S. J., Baggett B. K., Gillies R. J. (2005) Hypoxia-inducible factor-1α and the glycolytic phenotype in tumors. Neoplasia 7, 324–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu H., Forbes R. A., Verma A. (2002) Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 277, 23111–23115 [DOI] [PubMed] [Google Scholar]
- 8. Gerlinger M., Santos C. R., Spencer-Dene B., Martinez P., Endesfelder D., Burrell R. A., Vetter M., Jiang M., Saunders R. E., Kelly G., Dykema K., Rioux-Leclercq N., Stamp G., Patard J. J., Larkin J., Howell M., Swanton C. (2012) Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J. Pathol. 227, 146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Le A., Cooper C. R., Gouw A. M., Dinavahi R., Maitra A., Deck L. M., Royer R. E., Vander Jagt D. L., Semenza G. L., Dang C. V. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng G., Zielonka J., Dranka B. P., McAllister D., Mackinnon A. C., Jr., Joseph J., Kalyanaraman B. (2012) Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 72, 2634–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. (2011) Global cancer statistics. CA Cancer J. Clin. 61, 69–90 [DOI] [PubMed] [Google Scholar]
- 12. Botteman M. F., Pashos C. L., Redaelli A., Laskin B., Hauser R. (2003) The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics 21, 1315–1330 [DOI] [PubMed] [Google Scholar]
- 13. Wu X. R. (2005) Urothelial tumorigenesis: a tale of divergent pathways. Nat. Rev. Cancer 5, 713–725 [DOI] [PubMed] [Google Scholar]
- 14. Castillo-Martin M., Domingo-Domenech J., Karni-Schmidt O., Matos T., Cordon-Cardo C. (2010) Molecular pathways of urothelial development and bladder tumorigenesis. Urol. Oncol. 28, 401–408 [DOI] [PubMed] [Google Scholar]
- 15. Anzick S. L., Kononen J., Walker R. L., Azorsa D. O., Tanner M. M., Guan X. Y., Sauter G., Kallioniemi O. P., Trent J. M., Meltzer P. S. (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277, 965–968 [DOI] [PubMed] [Google Scholar]
- 16. Yan J., Tsai S. Y., Tsai M. J. (2006) SRC-3/AIB1: transcriptional coactivator in oncogenesis. Acta Pharmacol. Sin. 27, 387–394 [DOI] [PubMed] [Google Scholar]
- 17. Torres-Arzayus M. I., Font de Mora J., Yuan J., Vazquez F., Bronson R., Rue M., Sellers W. R., Brown M. (2004) High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 6, 263–274 [DOI] [PubMed] [Google Scholar]
- 18. Long W., Foulds C. E., Qin J., Liu J., Ding C., Lonard D. M., Solis L. M., Wistuba I. I., Qin J., Tsai S. Y., Tsai M. J., O'Malley B. W. (2012) ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Invest. 122, 1869–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y., Chen Q., Li W., Su X., Chen T., Liu Y., Zhao Y., Yu C. (2010) Overexpression of transcriptional coactivator AIB1 promotes hepatocellular carcinoma progression by enhancing cell proliferation and invasiveness. Oncogene 29, 3386–3397 [DOI] [PubMed] [Google Scholar]
- 20. Luo J. H., Xie D., Liu M. Z., Chen W., Liu Y. D., Wu G. Q., Kung H. F., Zeng Y. X., Guan X. Y. (2008) Protein expression and amplification of AIB1 in human urothelial carcinoma of the bladder and overexpression of AIB1 is a new independent prognostic marker of patient survival. Int. J. Cancer 122, 2554–2561 [DOI] [PubMed] [Google Scholar]
- 21. Brader P., Riedl C. C., Woo Y., Ponomarev V., Zanzonico P., Wen B., Cai S., Hricak H., Fong Y., Blasberg R., Serganova I. (2007) Imaging of hypoxia-driven gene expression in an orthotopic liver tumor model. Mol. Cancer Ther. 6, 2900–2908 [DOI] [PubMed] [Google Scholar]
- 22. Yan J., Yu Y., Wang N., Chang Y., Ying H., Liu W., He J., Li S., Jiang W., Li Y., Liu H., Wang H., Xu Y. (2004) LFIRE-1/HFREP-1, a liver-specific gene, is frequently downregulated and has growth suppressor activity in hepatocellular carcinoma. Oncogene 23, 1939–1949 [DOI] [PubMed] [Google Scholar]
- 23. van de Sluis B., Mao X., Zhai Y., Groot A. J., Vermeulen J. F., van der Wall E., van Diest P. J., Hofker M. H., Wijmenga C., Klomp L. W., Cho K. R., Fearon E. R., Vooijs M., Burstein E. (2010) COMMD1 disrupts HIF-1α/β dimerization and inhibits human tumor cell invasion. J. Clin. Invest. 120, 2119–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shi W. F., Leong M., Cho E., Farrell J., Chen H. C., Tian J., Zhang D. (2009) Repressive effects of resveratrol on androgen receptor transcriptional activity. PLoS One 4, e7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan J., Erdem H., Li R., Cai Y., Ayala G., Ittmann M., Yu-Lee L. Y., Tsai S. Y., Tsai M. J. (2008) Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 68, 5460–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dyrskjøt L., Kruhøffer M., Thykjaer T., Marcussen N., Jensen J. L., Møller K., Ørntoft T. F. (2004) Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 64, 4040–4048 [DOI] [PubMed] [Google Scholar]
- 27. Porreca E., Ucchino S., Di Febbo C., Di Bartolomeo N., Angelucci D., Napolitano A. M., Mezzetti A., Cuccurullo F. (1994) Antiproliferative effect of desferrioxamine on vascular smooth muscle cells in vitro and in vivo. Arterioscler. Thromb. 14, 299–304 [DOI] [PubMed] [Google Scholar]
- 28. Ward P. S., Thompson C. B. (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou J. X., Zhong Z., Shi X. B., Tepper C. G., deVere White R. W., Kung H. J., Chen H. (2006) ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. Prostate 66, 1474–1486 [DOI] [PubMed] [Google Scholar]
- 30. Lonard D. M., Tsai S. Y., O'Malley B. W. (2004) Selective estrogen receptor modulators 4-hydroxytamoxifen and raloxifene impact the stability and function of SRC-1 and SRC-3 coactivator proteins. Mol. Cell. Biol. 24, 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han S. J., DeMayo F. J., Xu J., Tsai S. Y., Tsai M. J., O'Malley B. W. (2006) Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol. Endocrinol. 20, 45–55 [DOI] [PubMed] [Google Scholar]
- 32. Qin L., Liao L., Redmond A., Young L., Yuan Y., Chen H., O'Malley B. W., Xu J. (2008) The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol. Cell. Biol. 28, 5937–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Louie M. C., Zou J. X., Rabinovich A., Chen H. W. (2004) ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol. Cell. Biol. 24, 5157–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mussi P., Yu C., O'Malley B. W., Xu J. (2006) Stimulation of steroid receptor coactivator-3 (SRC-3) gene overexpression by a positive regulatory loop of E2F1 and SRC-3. Mol. Endocrinol. 20, 3105–3119 [DOI] [PubMed] [Google Scholar]
- 35. Yan J., Yu C. T., Ozen M., Ittmann M., Tsai S. Y., Tsai M. J. (2006) Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Res. 66, 11039–11046 [DOI] [PubMed] [Google Scholar]
- 36. Tong Z. T., Wei J. H., Zhang J. X., Liang C. Z., Liao B., Lu J., Fan S., Chen Z. H., Zhang F., Ma H. H., Qian W. C., Kong L. L., Fang Y., Chen W., Xie D., Luo J. H. (2013) AIB1 predicts bladder cancer outcome and promotes bladder cancer cell proliferation through AKT and E2F1. Br. J. Cancer 108, 1470–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lanz R. B., Bulynko Y., Malovannaya A., Labhart P., Wang L., Li W., Qin J., Harper M., O'Malley B. W. (2010) Global characterization of transcriptional impact of the SRC-3 coregulator. Mol. Endocrinol. 24, 859–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tickoo S. K., Milowsky M. I., Dhar N., Dudas M. E., Gallagher D. J., Al-Ahmadie H., Gopalan A., Fine S. W., Ishill N., Bajorin D. F., Reuter V. E. (2011) Hypoxia-inducible factor and mammalian target of rapamycin pathway markers in urothelial carcinoma of the bladder: possible therapeutic implications. BJU Int. 107, 844–849 [DOI] [PubMed] [Google Scholar]
- 39. Ord J. J., Agrawal S., Thamboo T. P., Roberts I., Campo L., Turley H., Han C., Fawcett D. W., Kulkarni R. P., Cranston D., Harris A. L. (2007) An investigation into the prognostic significance of necrosis and hypoxia in high grade and invasive bladder cancer. J. Urol. 178, 677–682 [DOI] [PubMed] [Google Scholar]
- 40. Datta K., Li J., Bhattacharya R., Gasparian L., Wang E., Mukhopadhyay D. (2004) Protein kinase Cζ transactivates hypoxia-inducible factor α by promoting its association with p300 in renal cancer. Cancer Res. 64, 456–462 [DOI] [PubMed] [Google Scholar]
- 41. Semenza G. L. (2002) Physiology meets biophysics: visualizing the interaction of hypoxia-inducible factor 1α with p300 and CBP. Proc. Natl. Acad. Sci. U.S.A. 99, 11570–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dames S. A., Martinez-Yamout M., De Guzman R. N., Dyson H. J., Wright P. E. (2002) Structural basis for Hif-1α/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. U.S.A. 99, 5271–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vander Heiden M. G., Lunt S. Y., Dayton T. L., Fiske B. P., Israelsen W. J., Mattaini K. R., Vokes N. I., Stephanopoulos G., Cantley L. C., Metallo C. M., Locasale J. W. (2011) Metabolic pathway alterations that support cell proliferation. Cold Spring Harbor Symp. Quant. Biol. 76, 325–334 [DOI] [PubMed] [Google Scholar]
- 44. Schulze A., Harris A. L. (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373 [DOI] [PubMed] [Google Scholar]
- 45. Zhou M., Zhao Y., Ding Y., Liu H., Liu Z., Fodstad O., Riker A. I., Kamarajugadda S., Lu J., Owen L. B., Ledoux S. P., Tan M. (2010) Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 9, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patra K. C., Wang Q., Bhaskar P. T., Miller L., Wang Z., Wheaton W., Chandel N., Laakso M., Muller W. J., Allen E. L., Jha A. K., Smolen G. A., Clasquin M. F., Robey R. B., Hay N. (2013) Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228 [DOI] [PMC free article] [PubMed] [Google Scholar]