

Background: Epigenetic mechanisms of regulating expression of glycosyltransferase genes are largely unknown.
Results: Novel epigenetic factors were identified that specifically regulate a brain-specific glycosyltransferase gene, GnT-IX (Mgat5b).
Conclusion: GnT-IX is epigenetically regulated by a combination of specific chromatin modifiers (HDAC11, OGT, and TET3) and a transcriptional factor NeuroD1.
Significance: This is the first study showing that a glycosyltransferase gene is regulated by another glycosyltransferase, OGT, in combination with histone modifications.
Keywords: Epigenetics, Glycobiology, Glycosyltransferases, Histone Deacetylase, O-GlcNAc, Transcription, GnT-IX, NeuroD1, TET3
Abstract
Expression of glycosyltransferase genes is essential for glycosylation. However, the detailed mechanisms of how glycosyltransferase gene expression is regulated in a specific tissue or during disease progression are poorly understood. In particular, epigenetic studies of glycosyltransferase genes are limited, although epigenetic mechanisms, such as histone and DNA modifications, are central to establish tissue-specific gene expression. We previously found that epigenetic histone activation is essential for brain-specific expression of N-acetylglucosaminyltransferase-IX (GnT-IX, also designated GnT-Vb), but the mechanism of brain-specific chromatin activation around GnT-IX gene (Mgat5b) has not been clarified. To reveal the mechanisms regulating the chromatin surrounding GnT-IX, we have investigated the epigenetic factors that are specifically involved with the mouse GnT-IX locus by comparing their involvement with other glycosyltransferase loci. We first found that a histone deacetylase (HDAC) inhibitor enhanced the expression of GnT-IX but not of other glycosyltransferases tested. By overexpression and knockdown of a series of HDACs, we found that HDAC11 silenced GnT-IX. We also identified the O-GlcNAc transferase (OGT) and ten-eleven translocation-3 (TET3) complex as a specific chromatin activator of GnT-IX gene. Moreover, chromatin immunoprecipitation (ChIP) analysis in combination with OGT or TET3 knockdown showed that this OGT-TET3 complex facilitates the binding of a potent transactivator, NeuroD1, to the GnT-IX promoter, suggesting that epigenetic chromatin activation by the OGT-TET3 complex is a prerequisite for the efficient binding of NeuroD1. These results reveal a new epigenetic mechanism of brain-specific GnT-IX expression regulated by defined chromatin modifiers, providing new insights into the tissue-specific expression of glycosyltransferases.
Introduction
Glycosylation is the most abundant post-translational modification in mammals and frequently takes place in a tissue-specific manner (1). Such restricted expression of glycans is essential for maintaining various physiological functions in multicellular organisms. For instance, in the nervous system, neural specific glycans such as polysialic acid and human natural killer-1 (HNK-1) are required for higher order brain functions, including learning/memory or the formation of neural networks (2, 3). Tissue-specific glycosylation depends largely on expression of glycosyltransferases, which are responsible for glycan biosynthesis. However, it is still not clear how particular glycosyltransferase genes are expressed in specific tissues or how their expression is disturbed during disease progression.
We have been investigating the functions and gene regulation of several glycosyltransferases, especially glycan-branching enzymes acting on glycoprotein, such as N-acetylglucosaminyltransferases (GnT-III,2 -V, and -IX(Vb)) or fucosyltransferase-8 (Fut8) (4). Among them, we and others recently found that GnT-IX (encoded by Mgat5b gene), a brain-specific glycosyltransferase, is involved in remyelination in vivo (5–7). GnT-IX forms a β1,6-branched structure on O-mannose glycans (8, 9), and the branched O-mannose glycans in activated astrocytes inhibit remyelination after myelin injury (5), indicating that the brain-specific expression of GnT-IX contributes to its function in brain. We also examined the regulatory mechanism of brain-specific GnT-IX expression and found that epigenetic histone activation but not DNA hypomethylation in the GnT-IX promoter region is pivotal for its neural specific expression (10). Two transcription factors, NeuroD1 and CTCF (CCCTC-binding factor), were also identified as transactivators for the GnT-IX promoter, but neural specific chromatin activation is likely to be hierarchically dominant because the chromatin activation state is critical for the binding of these transcription factors to the GnT-IX promoter (10). However, it remains to be clarified how chromatin at the GnT-IX gene is activated or repressed in a tissue- and gene-specific manner.
Epigenetic marks, such as histone modification and DNA methylation, are central to the tissue- and cell type-specific gene program (11). Covalent histone modifications (methylation, acetylation, O-GlcNAcylation, etc.) are dynamically regulated by an interplay of adding and removing enzyme pairs, which can influence gene transcription either through direct change of chromatin structure or through binding of effector molecules. For instance, acetylation of histone tails at specific lysine residues is catalyzed by histone acetylases (HATs), whereas acetylation is removed by histone deacetylases (HDACs). These actions are highly correlated with gene activation and silencing, respectively (12). Conversely, methylation of cytosine (5mC) in a gene promoter, which correlates strongly with transcriptional silencing, had been considered to be a static modification (13). However, the recent discovery that ten-eleven translocation (TET) family enzymes (TET1–3) can convert 5mC to 5-hydroxymethyl cytosine (5hmC) led to the hypothesis that TET-mediated hydroxylation of 5mC could be part of a DNA demethylation pathway (14–16). Although TET1 and TET2 were reported to play an important role in ES cell lineage specification (17, 18), depletion of TET3 causes impairment of neural development in Xenopus (19), suggesting that TET proteins, especially TET3, might be involved in tissue-specific gene transcription. Moreover, recent studies have revealed that TET proteins bind tightly with the cytosolic/nuclear glycosyl enzyme, O-GlcNAc transferase (OGT), at transcriptional start sites to regulate gene transcription (20–22). O-GlcNAcylation by OGT on histones or histone-modifying enzymes was recently added to the growing list of epigenetic modifications (23); therefore, it has been revealed that these two emerging epigenetic factors (TET and OGT) can functionally cross-talk. Few studies have linked these epigenetic modifications to glycan expression (24); however, recent works in which an epigenetic inhibitor had a great impact on the cellular N-glycome or composition of neural glycolipid (25–27) have suggested that many glycan-related genes can be regulated by epigenetic mechanisms.
In this study, we investigated which chromatin modifiers are involved in epigenetic regulation of the mouse GnT-IX gene. To this end, we focused on enzymes involved in histone acetylation and O-GlcNAcylation. We identified one HDAC, HDAC11, and the OGT-TET3 complex as regulators of GnT-IX transcription. Meanwhile, the gene expression of other glycosyltransferases (GnT-III and -V and Fut8) was not affected by HDAC11 or the OGT-TET3 complex. Moreover, we found that recruitment of OGT-TET3 facilitates NeuroD1-dependent up-regulation of GnT-IX expression. These results suggest that a set of specific epigenetic factors (TET3-OGT and HDAC11) and a transcription factor (NeuroD1), which are highly expressed in brain, play key roles in the brain-restricted expression of GnT-IX.
EXPERIMENTAL PROCEDURES
Materials
The following antibodies were used: anti-H3K9Ac (07-352, Millipore), anti-acetyl H3 (06-599, Millipore), anti-actin (AC-40, Sigma), anti-FLAG (M2, Sigma), anti-OGT (sc-32921, Santa Cruz Biotechnology), anti-O-GlcNAc (RL2, Thermo Scientific), anti-NeuroD1 (4373, Cell Signaling Technology), anti-histone H3 (4620, Cell Signaling Technology), and normal rabbit IgG (2729, Cell Signaling Technology). siRNAs were purchased from Qiagen: control siRNA, 1027280; mouse HDAC1, SI00173523; mouse HDAC2, SI00173544; mouse HDAC3, SI00192787; mouse HDAC11, SI01063867; mouse TET1, SI00962983; mouse TET2, SI04399878; mouse TET3, SI00968555; and mouse OGT, SI00235172. Primer-probe sets for mRNA quantification were purchased from Life Technologies: control ribosomal RNA, 4308329; GnT-III, Mm00447798_s1; GnT-V, Mm00455036_m1; GnT-IX, Mm00556891_m1; Fut8, Mm00489789_m1; HDAC1, Mm02391771_g1; HDAC2, Mm00515108_m1; HDAC3, Mm00515916_m1; HDAC11, Mm01183513_m1; and TET3, Mm00805756_m1. Trichostatin A was kindly provided by Dr. Minoru Yoshida (Chemical Genetics Laboratory, RIKEN).
Cell Culture and Transfection
Neuro2A cells were cultured in DMEM supplemented with 10% fetal bovine serum. For plasmid transfection, cells plated on a 10-cm (or 6-cm) dish were transfected with 4 μg (or 1 μg) of plasmid using 10 μl (or 2.5 μl) of Lipofectamine 2000 (Life Technologies). For siRNA transfection, cells on a 10-cm (or 6-cm) dish were transfected with 200 pmol (or 80 pmol) of siRNA using 20 μl (or 8 μl) of Lipofectamine 2000. Stable HDAC transfectants were selected in the presence of antibiotics (750 μg/ml G418 for HDAC2 and 7.5 μg/m blasticidin for the other transfectants). In some experiments, cells were treated with trichostatin A (TSA) at 0.1 μm for 24 h.
Isolation of Primary Neurons and an Oligodendrocyte (OL)-rich Fraction
All the animal experiments were approved by the Animal Experiment Committee of RIKEN. Isolation and culture of primary neurons from embryonic C57BL/6 mouse brains were carried out as described previously (5) with slight modification. After plating neurons, Cytosine arabinofranoside (5 μm at final concentration, Sigma) was added to the medium at 2 days in vitro to remove proliferating cells. For isolation of the OL-rich fraction, brains from C57BL/6 mice (8–10 weeks old) were collected and minced. The tissue pieces were incubated in 10 ml of PBS containing 90 units of papain, 2 mg of l-cysteine, and 0.01% DNase at 37 °C for 40 min. The supernatant was passed through a 70-μm cell strainer and mixed with 2 ml of FBS. The precipitate was resuspended with 8 ml of PBS, and the resultant supernatant was passed through a 70-μm cell strainer. The passed solution was mixed with the first passage. This procedure was repeated three more times. The collected solution was centrifuged at 500 × g for 10 min, and the pellet was resuspended with 30 ml of PBS containing 30% (v/v) Percoll. After centrifugation at 20,000 × g for 30 min, the layer between the white and red layers was collected. 2 volumes of PBS were added, and the mixture was centrifuged at 700 × g for 12 min. The pellet was resuspended with 15 ml of PBS and centrifuged at 500 × g for 8 min. This wash step was repeated two more times, with the final pellet representing the OL-rich fraction.
RNA Extraction, Reverse Transcription, and Real-time PCR
Total RNA from cultured cells or mouse tissues was extracted using TRI Reagent (Molecular Research Center, Inc.) according to the manufacturer's protocol. 1 μg of total RNA was reverse-transcribed using Superscript III (Life Technologies). For real-time PCR, cDNA was mixed with TaqMan Universal PCR master mix (Life Technologies) and amplified using an ABI PRISM 7900HT. The levels of mRNA were normalized to the corresponding ribosomal RNA levels.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were carried out using a SimpleChIP enzymatic chromatin IP kit (Cell Signaling Technology) according to the manufacturer's protocol. Approximately 2 × 107 cells were used as starting material. 2% of the chromatin was reserved as input sample. After immunoprecipitation, real-time PCR was performed to quantify the amounts of the precipitated DNA. The data are shown as values relative to the corresponding control IgG sample (indicated as Fold enrichment in figures) or as values relative to the corresponding 2% input sample (indicated as % of input in figures). For ChIP assays of HDAC11-FLAG, anti-FLAG M2-agarose affinity gel (Sigma) was used. For tissue ChIP, adult male C57BL/6 mice (10–15 weeks old) were deeply anesthetized and then sacrificed. Organs were collected and immediately minced on ice. Tissue pieces (100 mg) were incubated in 1% formaldehyde, PBS for 10 min at room temperature. Glycine was then added (125 mm final concentration) followed by incubation for 5 min. After washing twice with PBS, the tissues were homogenized in 4 ml of buffer A (supplied in the above SimpleChIP kit) using a Potter homogenizer. Subsequent procedures were carried out as for cultured cells. Primers and probes for real-time PCR are listed in Table 1.
TABLE 1.
Primers and probes for ChIP analysis
FAM, 6-carboxyfluorescein; MGB, minor groove binder.
| Primer (probe) name | Sequence |
|---|---|
| GnT-III primers | CAAGCACTTTGGCCAGCTCC and GCGCCTCTTTCGTGTGTATG |
| GnT-III probe | 5′-FAM-ACACTCGCGCGTCCGATTGC-MGB-3′ |
| GnT-V primers | GGGAATTTGCGGAACGTCCG and TCCCGGATCGACCTACCCAA |
| GnT-V probe | 5′-FAM-TCCCTTTGCCTTGGAGAGC-MGB-3′ |
| GnT-IX primers | TAGGAAGTTGCATGGTCCAG and CCGGGCTCCCGCGCGGCGCT |
| GnT-IX probe | 5′-FAM-TCAGAGGGAACAATGCGGCG-MGB-3′ |
| Fut8 primers | TCACCCGGCTGAGTCCTCA and GAGTGCGAACTGGCCAATCA |
| Fut8 probe | 5′-FAM-TCGGCACGGGAAGCTCATCT-MGB-3′ |
Plasmids
The construction of a plasmid encoding Neurod1 was described previously (10). Hdac cDNAs except Hdac2 were cloned by PCR using reverse-transcribed total RNA from mouse tissue (Hdac1, -3, -4, -5, and -6 from testis; Hdac7, -10, and -11 from skeletal muscle; Hdac8 and -9 from brain). The amplified fragments were then ligated to pCR4Blunt-TOPO (Life Technologies). All the cDNAs except Hdac2 were subcloned into pcDNA6/Myc-His A (Life Technologies). Hdac1 cDNA was ligated into the EcoRI site. The other Hdac cDNAs in pCR4Blunt-TOPO were released with NotI-SpeI and then ligated to the NotI-XbaI sites of pcDNA6/Myc-His A. Tet3 cDNA was amplified by PCR using reverse-transcribed total RNA from mouse brain. The amplified fragment was digested and directly ligated to pcDNA6/Myc-His A using EcoRI-EcoRV. For HDAC11-FLAG and TET3-FLAG constructs, PCR-amplified cDNA fragments that lacked their respective stop codons were digested and ligated into p3×FLAG-CMV14 (Sigma) using EcoRI-BamHI and EcoRI-XbaI sites, respectively. pcDNA3/mouse HDAC2 was kindly provided by Dr. Akihiro Ito (Chemical Genetics Laboratory, RIKEN). Primers used are listed in Table 2.
TABLE 2.
Primers for plasmid construction
Underlined sequences indicate restriction enzyme sites.
| Primer name | Sequence |
|---|---|
| HDAC1 | GAGCAAGATGGCGCAGACTCAGGG and AGACCTTGCTCAGGCCAACTTGACC |
| HDAC3 | AGCACCATGGCCAAGACCGTGGCGT and GGGGACACAGCATCCATGCTGCTCT |
| HDAC4 | AAGGCACTGACGCTGCTAGCAATGA and GCTTCGGGCTACAGTGGTGGTTCCT |
| HDAC5 | GAGCCGGCATGAACTCTCCCAACGA and GGGATGGGGGCCAGGGTGTCACAG |
| HDAC6 | TGAGGCTCTTCAGCCATGACCTCCA and GGGATTTAGTGTGAGTGGGGCATGT |
| HDAC7 | GGCTCTCAGCCCCAACCCATGGAC and GAGGAGAGCCACATTTGAAGCGCGA |
| HDAC8 | GTTGGAAGTGGGAGATGGAGATGCC and GCTGCCCAGCTCTGGGAACCTGATC |
| HDAC9 | GGGAGCAGCTCTTGGCTCAGCAAAG and TCAGAGACATAAGCTGATAGGAAATAA |
| HDAC10 | GTGACCACGGTGTAGCTATGGGCAC and TTATTATGGTGGAGGAGCAGCCACCT |
| HDAC11 | TGCGGGATGCCTCACGCAACACAGC and TGAGTAGCCGTCAAGGCACAGCACA |
| TET3 | ATGGAATTCTTCCAGGTCAGATGGACTCAGGGC and GCTGATATCTGGCACCTAGATCCAGCGGCTGTA |
| HDAC11-FLAG | AGGGAATTCTGCGGGATGCCTCACGCAAC and TAGGGATCCAGGCACAGCACAGGAAAGCA |
| TET3-FLAG | ATGGAATTCTTCCAGGTCAGATGGACTCAGGGC and TGGTCTAGAGATCCAGCGGCTGTAGGGGC |
Western Blot
Proteins were separated by 4–20% gradient SDS-PAGE using the Laemmli buffer system and then transferred to PVDF or nitrocellulose membranes. After blocking with 5% nonfat dried milk in TBS containing 0.05% Tween 20, the membranes were incubated with primary antibodies followed by HRP-conjugated secondary antibodies. Proteins were detected with SuperSignal West Dura extended duration substrate (Thermo Scientific) using an ImageQuant LAS-4000mini (GE Healthcare).
Immunoprecipitation (IP)
Cells were lysed and sonicated in buffer containing 20 mm Tris-HCl, pH 7.4, 0.3 m NaCl, 0.5% Nonidet P-40, and a protease inhibitor cocktail (Roche Applied Science). After ultracentrifugation at 100,000 × g for 15 min, the resultant supernatant was incubated with antibody for 30 min on ice. Protein G-Sepharose 4 fast flow (GE Healthcare) was then added, and the mixture was incubated for 2 h at 4 °C with gentle rotation. For IP of FLAG-tagged TET3, anti-FLAG M2-agarose affinity gel (Sigma) was used. After washing with excess volumes of TBS containing 0.1% Nonidet P-40, the bound proteins were eluted by boiling with Laemmli sample buffer.
RESULTS
Histone Acetylation Selectively Up-regulates GnT-IX
We previously reported that histone acetylation is a critical factor for GnT-IX expression (10). To investigate whether glycosyltransferase genes are generally regulated by epigenetic histone acetylation, we first treated Neuro2A cells with a global HDAC inhibitor, TSA, that forcibly up-regulates the level of histone acetylation (28). We analyzed the expression of four glycosyltransferase genes that we divided into two groups, tissue-specific genes with the highest expression in brain (GnT-III and GnT-IX) and ubiquitously expressed genes (GnT-V and Fut8) (29, 30), respectively. As a result, only the expression of GnT-IX was drastically up-regulated by TSA treatment (Fig. 1A). To check the levels of histone acetylation around the transcriptional start sites of these genes, ChIP analysis was performed with anti-acetylated histone H3 (H3Ac) antibody. Consistent with the mRNA levels, only the GnT-IX gene had increased levels of H3Ac in TSA-treated cells (Fig. 1B). Although TSA is a pan-HDAC inhibitor, its effect on induction of histone acetylation and transcription is not global but is restricted to certain genes (31, 32), which is consistent with the varying effects of TSA on the different glycosyltransferase genes (Fig. 1B). The specific up-regulation of GnT-IX by TSA shows that GnT-IX can be selectively silenced by HDAC(s).
FIGURE 1.
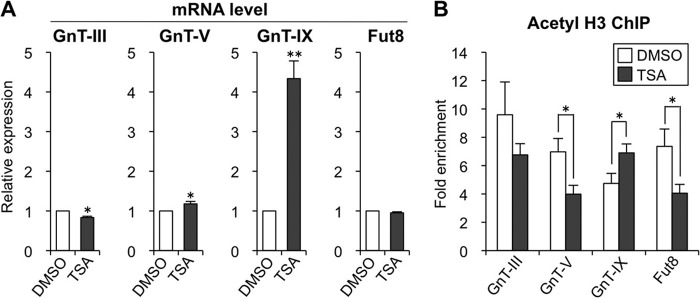
Histone acetylation selectively up-regulates GnT-IX. A, Neuro2A cells were treated with DMSO (vehicle), 0.1 μm TSA for 24 h, and the mRNA levels of GnT-III, -V, and -IX and Fut8 were quantified and normalized to those of rRNA. The mRNA/rRNA levels of TSA-treated samples relative to those of DMSO-treated samples are shown (n = 3). B, Neuro2A cells were treated with DMSO (vehicle), 0.1 μm TSA for 24 h, and the levels of H3Ac around the transcriptional start sites of GnT-III, -V, and -IX and Fut8 genes were analyzed by ChIP assay (n = 4). The precipitated DNA was analyzed by real-time PCR, and the amounts of DNA relative to those with control IgG are shown as -fold enrichment. All graphs show means ± S.E., *, p < 0.05, **, p < 0.01, Student's t test.
HDAC11 Silences the GnT-IX Gene
Next, we explored how GnT-IX gene is epigenetically silenced by HDAC(s). In mammalian cells, 11 HDACs (HDAC1-HDAC11) are expressed (33), but the precise function of each HDAC remains to be elucidated. Moreover, activity of all 11 HDACs is potentially inhibited by TSA. To examine which HDAC is involved in silencing GnT-IX, each HDAC enzyme was overexpressed in Neuro2A cells, and the mRNA levels of the glycosyltransferases were quantified. GnT-IX expression was significantly silenced by overexpression of several HDACs, such as HDAC1, -2, -3, and -11 (Fig. 2A), whereas other glycosyltransferases were moderately or slightly down-regulated, suggesting that GnT-IX gene is highly susceptible to epigenetic silencing by particular HDACs. To further examine whether these HDACs are involved in GnT-IX silencing, HDAC1, -2, -3, and -11 were knocked down by siRNA (Fig. 2, B and C). Depletion of HDAC11 resulted in the highest up-regulation of GnT-IX gene, whereas the ubiquitously expressed Fut8 gene was not up-regulated by knockdown of these HDACs, suggesting that HDAC11 might be a specific factor for epigenetic down-regulation of GnT-IX expression. ChIP analysis showed that knockdown of HDAC11 caused increased histone acetylation in the GnT-IX gene but not in the Fut8 gene (Fig. 2D), which is consistent with the up-regulation of GnT-IX mRNA by HDAC11 knockdown, suggesting that HDAC11 deacetylates histones surrounding the GnT-IX gene. In addition, ChIP analysis of exogenously expressed HDAC11-FLAG demonstrated that HDAC11 is highly recruited to the GnT-IX gene as compared with the Fut8 gene locus (Fig. 2E).
FIGURE 2.

HDAC11 silences GnT-IX gene. A, total RNAs were extracted from Neuro2A cells (control) or transfectants stably expressing HDAC1–11. The mRNA levels of GnT-III, -V, and -IX and Fut8 were quantified and normalized to those of rRNA. The mRNA/rRNA levels of HDAC-expressing samples relative to those of control samples are shown (n = 3, *, p < 0.05, Tukey-Kramer's test). B, Neuro2A cells were treated with control siRNA (siCont), HDAC1, HDAC2, HDAC3, or HDAC11 siRNA for 24 h. Total RNAs were extracted, and the mRNA levels of GnT-IX and Fut8 were quantified and normalized to those of rRNA. The mRNA/rRNA levels of HDAC knockdown samples relative to those of control samples are shown (n = 3, **, p < 0.01, Tukey-Kramer's test). C, Neuro2A cells were treated with control siRNA, HDAC1, HDAC2, HDAC3, or HDAC11 siRNA for 24 h. Total RNAs were extracted, and the mRNA levels of Hdac1, Hdac2, Hdac3, and Hdac11 were quantified and normalized to those of rRNA. The mRNA/rRNA levels of HDAC knockdown samples relative to those of control samples are shown (n = 3). D, Neuro2A cells were treated with control siRNA or HDAC11 siRNA for 24 h, and then the levels of H3K9Ac around the transcriptional start sites of GnT-IX and Fut8 genes were analyzed by ChIP assays (n = 3, *, p < 0.05, Student's t test). E, HDAC11 tagged with 3×FLAG at its C terminus was overexpressed in Neuro2A cells. Recruitment of HDAC11-FLAG to the genomic region around the transcriptional start sites of GnT-IX and Fut8 genes was analyzed by ChIP assays with anti-FLAG antibody (M2)-conjugated beads. The precipitated DNA was analyzed by real-time PCR, and the amount of DNA from the transfectant (+) relative to that from mock-treated cells (−) is shown (n = 3, *, p < 0.05, Student's t test). F, mRNA levels of GnT-IX and Hdac11 were quantified and normalized to those of rRNA in mouse primary neurons (6 days in vitro) or in the oligodendrocyte-rich fraction (OL). The mRNA/rRNA levels are shown as relative values to those in whole brain from 20-week-old male mice (n = 3, *, p < 0.05, **, p < 0.01, Student's t test). All graphs show means ± S.E.
Hdac11 is known to be highly expressed in brain, kidney, heart, and muscle (34), and in brain HDAC11 was reported to regulate oligodendrocyte-specific gene expression (35). Therefore, we assumed that HDAC11 would be involved in silencing of GnT-IX in a specific brain cell type as well as in non-brain tissues. We, therefore, isolated primary neurons and an OL-rich fraction from mouse brain. Quantitative PCR results showed that GnT-IX mRNA is highly expressed in neurons as compared with OLs, whereas HDAC11 shows a higher level of expression in OLs as compared with neurons (Fig. 2F). This negative correlation suggests that HDAC11 may be implicated in cell type-specific silencing of GnT-IX in the brain. Collectively, these data suggest that among HDAC family members, HDAC11 may be a specific suppressor for GnT-IX but not for other glycosyltransferases.
The TET3-OGT Complex Is an Epigenetic Activator Specific for GnT-IX Gene
Next, we investigated the chromatin activation mechanism of GnT-IX gene. Recently, a novel chromatin activation mechanism was reported in which two epigenetic factors, OGT and TET, form a functional complex (20–22). TET molecule can recruit OGT to chromatin, and this in turn results in O-GlcNAcylation of surrounding chromatin molecules, including histones and host cell factor-1 (HCF-1, a component of the H3K4 methyltransferase complex), leading to gene activation (20–22). Considering the fact that OGT is highly expressed in brain as compared with other tissues (36, 37) and that TET-generated 5hmc is most abundant in brain (38–40), we assumed that this OGT-TET machinery could be involved in chromatin activation of GnT-IX gene in brain. To see whether the OGT-TET complex is actually involved in activation of the glycosyltransferase genes, TET family proteins (TET1–3) were knocked down in Neuro2A cells by siRNA. As a result, we found that mRNA levels of GnT-V and -IX but not of GnT-III and Fut8 were down-regulated by TET3 knockdown (Fig. 3).
FIGURE 3.
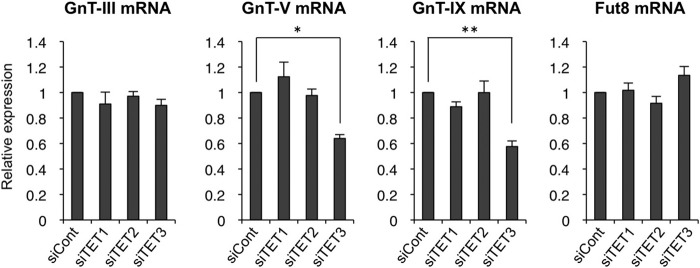
TET3 is involved in GnT-V and -IX expression. Neuro2A cells were treated with control siRNA (siCont), TET1, TET2, or TET3 siRNA for 24 h. Total RNAs were extracted, and the mRNA levels of GnT-III, -V, and -IX and Fut8 were quantified and normalized to those of rRNA. The mRNA/rRNA levels of TET knockdown samples relative to those of control samples are shown. All graphs show means ± S.E. (n = 3, *, p < 0.05, **, p < 0.01, Tukey-Kramer's test).
Next, we checked to see whether the OGT-TET3 complex activates GnT-IX transcription. IP experiments clearly showed that exogenously expressed TET3-FLAG can co-precipitate endogenous OGT and vice versa (Fig. 4A), which is consistent with previous studies using other cell systems (20, 21, 41). If OGT interacts with TET3 for its recruitment to chromatin and gene activation, TET3 depletion would dissociate OGT from chromatin particularly from GnT-V and GnT-IX genes. ChIP assays with anti-OGT antibody showed that OGT was indeed dissociated from GnT-IX gene chromatin by TET3 depletion, together with a reduced level of O-GlcNAc (Fig. 4, B and C), suggesting that OGT and TET3 molecules act as a complex on the GnT-IX gene for epigenetic activation. Surprisingly, however, such dissociation of OGT was not observed for GnT-V gene, suggesting that TET3 regulates GnT-V expression independently of OGT or in an indirect manner. Consistent with this possibility, knockdown of OGT by siRNA revealed that only GnT-IX but not other glycosyltransferases was down-regulated (Fig. 4, D and E). Together, these results indicated that the OGT-TET3 complex may be specifically required for expression of GnT-IX but not for other glycosyltransferases tested.
FIGURE 4.

The TET3-OGT complex is an epigenetic activator specific for GnT-IX gene. A, Neuro2A cells were transfected with the TET3-3×FLAG expression vector or the empty vector (Mock). The cell lysates (Input) were subjected to IP with anti-OGT or anti-FLAG antibody and then Western blotted with an anti-OGT or anti-FLAG antibody. B, Neuro2A cells were treated with control siRNA (siCont) or TET3 siRNA for 24 h. Total RNAs were extracted, and the mRNA levels of TET3 were quantified and normalized to those of rRNA. The mRNA/rRNA level of TET3 knockdown sample relative to that of control sample is shown (n = 3, *, p < 0.05, Student's t test). C, Neuro2A cells were treated with control siRNA or TET3 siRNA for 24 h, and then ChIP assays were performed with anti-OGT or anti-O-GlcNAc (RL2) antibody. Genomic regions around the transcriptional start sites of GnT-III, -V, and -IX and Fut8 genes were analyzed by real-time PCR. The amounts of DNA relative to those with control IgG are shown (left, n = 5; right, n = 3, *, p < 0.05, Student's t test). D, Neuro2A cells were treated with control siRNA or OGT siRNA, and the cell lysates were Western blotted with an anti-OGT, anti-actin, or anti-O-GlcNAc antibody. E, Neuro2A cells were treated with control siRNA or OGT siRNA for 24 h. Total RNAs were extracted, and the mRNA levels of GnT-III, -V, and -IX and Fut8 were quantified and normalized to those of rRNA. The mRNA/rRNA levels of OGT knockdown samples relative to those of control samples are shown (n = 4, *, p < 0.05, **, p < 0.01, Student's t test). All graphs show means ± S.E.
The OGT-TET3 Complex Recruits a Brain-specific Transcription Factor, NeuroD1, to the GnT-IX Promoter
We previously identified NeuroD1, a neuronal basic helix-loop-helix transcription factor (42), as a strong transactivator for the GnT-IX promoter (10). Overexpression of NeuroD1 markedly up-regulates GnT-IX transcription in Neuro2A cells, whereas NeuroD1 fails to activate GnT-IX in a non-neural cell line (3T3-L1), in which the GnT-IX chromatin is highly repressed (10). This suggests that epigenetic chromatin activation is a prerequisite for NeuroD1 binding to the GnT-IX promoter. Therefore, we expected that chromatin activation of GnT-IX by OGT-TET3 would enhance NeuroD1 recruitment. Actually, binding of NeuroD1 to the GnT-IX promoter and subsequent activation of GnT-IX transcription were both reduced by depletion of OGT or TET3 (Fig. 5, A–C). These results suggest that the OGT-TET3 complex is required for the efficient binding of NeuroD1 to the GnT-IX promoter to drive it. We also examined in vivo binding of NeuroD1 and OGT to the GnT-IX promoter. Although we failed to detect the binding of TET3 by ChIP assays with commercially available antibodies, we found that binding of endogenous NeuroD1 and OGT to the GnT-IX promoter was stronger in brain than in other tissues (Fig. 5D), which is consistent with the highest GnT-IX expression level being in the brain. This robust binding of NeuroD1 and OGT may be due to their high expression in brain (Fig. 5E). These results suggest that the OGT-TET3-dependent binding of NeuroD1 to the GnT-IX promoter may also be required for the brain-specific transcription of GnT-IX gene in vivo.
FIGURE 5.

Transactivation of GnT-IX by NeuroD1 is promoted by the OGT-TET3 complex. A, Neuro2A cells were treated with control siRNA (siCont), OGT siRNA, or TET3 siRNA for 24 h and then replated on new dishes. After 24 h, the cells were transfected with the NeuroD1 expression vector followed by a further 24-h culture. Then, ChIP assays were performed with an anti-NeuroD1 antibody. A genomic region around the transcriptional start site of GnT-IX gene, including the NeuroD1 binding site, was analyzed by real-time PCR. The amounts of DNA relative to those of 2% input samples are shown (n = 3, *, p < 0.05, Tukey-Kramer's test). B and C, Neuro2A cells were transfected with siRNA and expression plasmid as in the case of A. Total RNAs were extracted, and the mRNA levels of GnT-IX were quantified and normalized to those of rRNA. The mRNA/rRNA levels relative to those of control siRNA-treated mock sample are shown (n = 3, *, p < 0.05, Tukey-Kramer's test) (B). Cellular proteins extracted after 24 h of NeuroD1-transfection were Western blotted with an anti-NeuroD1 or anti-actin antibody (C). D, chromatin was prepared from brain, liver, or kidney of adult mice, and ChIP assays were performed with anti-NeuroD1 or anti-OGT antibody. A genomic region around the transcriptional start site of GnT-IX gene, including the NeuroD1 binding site, was analyzed by real-time PCR. The amounts of DNA relative to those of 2% input samples are shown (n = 3, **, p < 0.01, Tukey-Kramer's test). E, homogenates of brain, liver, or kidney from adult mice were Western blotted with an anti-NeuroD1, anti-OGT, or anti-histone H3 antibody. All graphs show means ± S.E.
DISCUSSION
We have identified for the first time epigenetic factors involved in the regulation of GnT-IX gene, an epigenetic suppressor HDAC11, and an activator, the OGT-TET3 complex. The other glycosyltransferases tested were not regulated by these factors, which suggests that these chromatin modifiers selectively act on GnT-IX among the glycosyltransferase genes. In addition, our overexpression and knockdown data showed that the other HDACs (HDAC1–10) and TETs (TET1 and TET2) are not or are less involved in GnT-IX regulation, showing the specificity of HDAC11 and OGT-TET3 molecules for GnT-IX gene. Previous studies have revealed that TET proteins are essential for OGT recruitment to a CpG island in a gene promoter, leading to efficient O-GlcNAcylation of chromatin proteins (20–22, 41). Our data indicate that the OGT-TET3 complex is a prerequisite for the efficient binding of the transactivator, NeuroD1, to the GnT-IX promoter to drive it. Taking an overall view of our data, we present a stepwise model for the epigenetic regulation of GnT-IX gene (Fig. 6).
FIGURE 6.
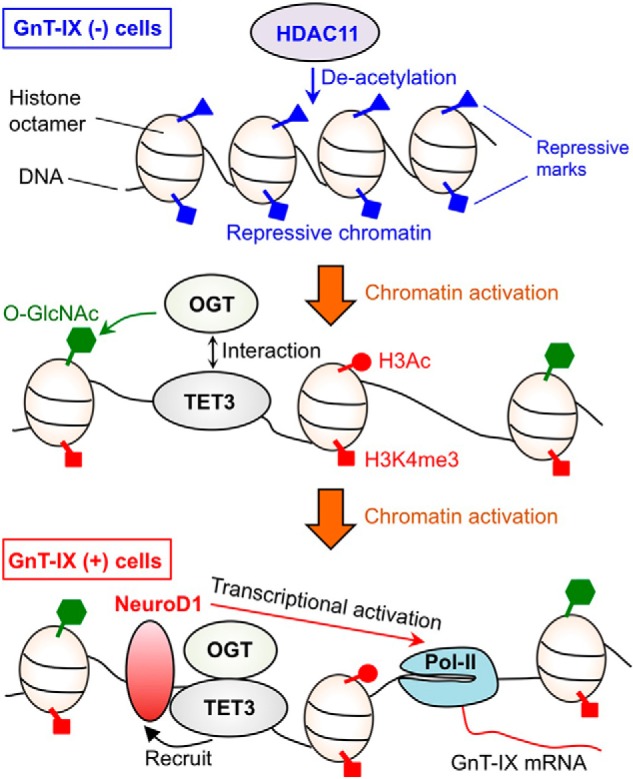
Schematic model for the epigenetic regulation of GnT-IX. In non-GnT-IX-expressing cells, chromatin around the GnT-IX gene is repressed by chromatin modifiers, including HDAC11. When GnT-IX chromatin is activated, TET3 recruits OGT. This OGT-TET3 complex further facilitates the recruitment of NeuroD1 to GnT-IX promoter, which in turn activates GnT-IX transcription. Pol-II, RNA polymerase II.
Recently, we reported that the branched O-mannose glycans produced by GnT-IX inhibit the development of OLs after in vivo induced myelin injury through enhanced astrocyte activation (5). Meanwhile, it was reported that HDAC11 knockdown or pharmacological suppression of HDAC activity also caused impaired development of oligodendrocytes (35, 43). This suggests that HDAC11 regulates OL development through down-regulation of its target genes. The expression of GnT-IX in OLs in vivo is presumed to be silenced by HDAC11 due to the high expression of HDAC11 in these cells (Fig. 2F); therefore, cell lineage-specific expression of GnT-IX might be one of the downstream target genes for HDAC11 during regulation of OL development in the brain. Although the function of HDAC11 still remains largely unclear, it would be interesting to identify the direct target genes of HDAC11 in the brain. Considering that HDACs lack intrinsic DNA binding activity (33), the enrichment of HDAC11 at the GnT-IX gene (Fig. 2E) is probably maintained by the formation of a complex with another sequence-specific DNA-binding protein(s). Our next goal will be to clarify how HDAC11 acts specifically on the GnT-IX locus and not on other glycosyltransferases.
O-GlcNAcylation by OGT is an emerging epigenetic mark because of the recent findings that O-GlcNAc on histone and histone modifiers can regulate gene transcription (23, 44–46). Our present study showed that OGT is involved in epigenetic activation of GnT-IX gene, probably through interaction with TET3 on chromatin. Although GnT-IX gene is likely to be activated by OGT-TET3, probably through promoted O-GlcNAcylation of histone or HCF-1 on the gene promoter, as is generally seen in other genes (20–22), this complex might also facilitate O-GlcNAcylation of NeuroD1 for GnT-IX activation because NeuroD1 was also reported to be modified by OGT (47). O-GlcNAc modification exerts a wide variety of functions depending on the OGT substrate (48). In our case, OGT, a sugar transferase, positively regulates the transcription of another sugar transferase, GnT-IX, which is connected by epigenetics. Meanwhile, OGT is considered as a master sensor for nutrients as it uses UDP-GlcNAc as the high energy donor substrate, which is the end point of the hexosamine biosynthetic pathway (48). The biosynthesis of UDP-GlcNAc integrates the metabolic pathways of glucose, an amino acid (glutamine), a fatty acid (acetyl-CoA), and a nucleotide (UTP). Therefore, nutrient flux to the cells would affect the levels of both cellular UDP-GlcNAc and cellular O-GlcNAc modification. Notably, other glycosyltransferases, including GnT-IX, also use UDP-GlcNAc as the donor substrate. Therefore, nutrient flux and the subsequent metabolic pathway could regulate GnT-IX activity by both donor-substrate use and OGT-mediated gene transcription. We recently proposed that cellular glycosylation forms a functional cycle called the “glycan cycle” comprising nutrient flux, biosynthesis of sugar nucleotide, sugar transfer, biological action, degradation, and recycling (49). We still do not fully understand this cycle at the molecular level, but we believe that the present study provides new insights into how epigenetic regulation of glycogenes is integrated into the glycan cycle.
This work was supported by RIKEN (Systems Glycobiology Research Project) (to N. T.) and Japan Society for the Promotion of Science (JSPS) (KAKENHI Grant 23770163).
- GnT
- N-acetylglucosaminyltransferase
- HDAC
- histone deacetylase
- OGT
- O-GlcNAc transferase
- OL
- oligodendrocyte
- TET
- ten-eleven translocation
- TSA
- trichostatin A
- H3Ac
- acetylated histone H3
- DMSO
- dimethyl sulfoxide
- IP
- immunoprecipitation.
REFERENCES
- 1. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamamoto S., Oka S., Inoue M., Shimuta M., Manabe T., Takahashi H., Miyamoto M., Asano M., Sakagami J., Sudo K., Iwakura Y., Ono K., Kawasaki T. (2002) Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J. Biol. Chem. 277, 27227–27231 [DOI] [PubMed] [Google Scholar]
- 3. Weinhold B., Seidenfaden R., Röckle I., Mühlenhoff M., Schertzinger F., Conzelmann S., Marth J. D., Gerardy-Schahn R., Hildebrandt H. (2005) Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J. Biol. Chem. 280, 42971–42977 [DOI] [PubMed] [Google Scholar]
- 4. Taniguchi N., Miyoshi E., Gu J., Honke K., Matsumoto A. (2006) Decoding sugar functions by identifying target glycoproteins. Curr. Opin. Struct. Biol. 16, 561–566 [DOI] [PubMed] [Google Scholar]
- 5. Kanekiyo K., Inamori K., Kitazume S., Sato K., Maeda J., Higuchi M., Kizuka Y., Korekane H., Matsuo I., Honke K., Taniguchi N. (2013) Loss of branched O-mannosyl glycans in astrocytes accelerates remyelination. J. Neurosci. 33, 10037–10047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inamori K., Endo T., Ide Y., Fujii S., Gu J., Honke K., Taniguchi N. (2003) Molecular cloning and characterization of human GnT-IX, a novel β1,6-N-acetylglucosaminyltransferase that is specifically expressed in the brain. J. Biol. Chem. 278, 43102–43109 [DOI] [PubMed] [Google Scholar]
- 7. Kaneko M., Alvarez-Manilla G., Kamar M., Lee I., Lee J. K., Troupe K., Zhang W. j., Osawa M., Pierce M. (2003) A novel β(1,6)-N-acetylglucosaminyltransferase V (GnT-VB). FEBS Lett. 554, 515–519 [DOI] [PubMed] [Google Scholar]
- 8. Inamori K., Endo T., Gu J., Matsuo I., Ito Y., Fujii S., Iwasaki H., Narimatsu H., Miyoshi E., Honke K., Taniguchi N. (2004) N-Acetylglucosaminyltransferase IX acts on the GlcNAc β1,2-Manα1-Ser/Thr moiety, forming a 2,6-branched structure in brain O-mannosyl glycan. J. Biol. Chem. 279, 2337–2340 [DOI] [PubMed] [Google Scholar]
- 9. Lee J. K., Matthews R. T., Lim J. M., Swanier K., Wells L., Pierce J. M. (2012) Developmental expression of the neuron-specific N-acetylglucosaminyltransferase Vb (GnT-Vb/IX) and identification of its in vivo glycan products in comparison with those of its paralog, GnT-V. J. Biol. Chem. 287, 28526–28536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kizuka Y., Kitazume S., Yoshida M., Taniguchi N. (2011) Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J. Biol. Chem. 286, 31875–31884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonasio R., Tu S., Reinberg D. (2010) Molecular signals of epigenetic states. Science 330, 612–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bannister A. J., Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki M. M., Bird A. (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476 [DOI] [PubMed] [Google Scholar]
- 14. Wu H., Zhang Y. (2011) Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 25, 2436–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Williams K., Christensen J., Helin K. (2012) DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 13, 28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koh K. P., Yabuuchi A., Rao S., Huang Y., Cunniff K., Nardone J., Laiho A., Tahiliani M., Sommer C. A., Mostoslavsky G., Lahesmaa R., Orkin S. H., Rodig S. J., Daley G. Q., Rao A. (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8, 200–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y., Xu C., Kato A., Tempel W., Abreu J. G., Bian C., Hu Y., Hu D., Zhao B., Cerovina T., Diao J., Wu F., He H. H., Cui Q., Clark E., Ma C., Barbara A., Veenstra G. J., Xu G., Kaiser U. B., Liu X. S., Sugrue S. P., He X., Min J., Kato Y., Shi Y. G. (2012) Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 151, 1200–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen Q., Chen Y., Bian C., Fujiki R., Yu X. (2013) TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deplus R., Delatte B., Schwinn M. K., Defrance M., Méndez J., Murphy N., Dawson M. A., Volkmar M., Putmans P., Calonne E., Shih A. H., Levine R. L., Bernard O., Mercher T., Solary E., Urh M., Daniels D. L., Fuks F. (2013) TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vella P., Scelfo A., Jammula S., Chiacchiera F., Williams K., Cuomo A., Roberto A., Christensen J., Bonaldi T., Helin K., Pasini D. (2013) Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 49, 645–656 [DOI] [PubMed] [Google Scholar]
- 23. Hanover J. A., Krause M. W., Love D. C. (2012) Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 13, 312–321 [DOI] [PubMed] [Google Scholar]
- 24. Lauc G., Vojta A., Zoldoš V. (2014) Epigenetic regulation of glycosylation is the quantum mechanics of biology. Biochim. Biophys. Acta 1840, 65–70 [DOI] [PubMed] [Google Scholar]
- 25. Suzuki Y., Yanagisawa M., Ariga T., Yu R. K. (2011) Histone acetylation-mediated glycosyltransferase gene regulation in mouse brain during development. J. Neurochem. 116, 874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Horvat T., Deželjin M., Redžić I., Barišić D., Herak Bosnar M., Lauc G., Zoldoš V. (2013) Reversibility of membrane N-glycome of HeLa cells upon treatment with epigenetic inhibitors. PLoS One 8, e54672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsai Y. T., Yu R. K. (2014) Epigenetic activation of mouse ganglioside synthase genes: implications for neurogenesis. J. Neurochem. 128, 101–110 [DOI] [PubMed] [Google Scholar]
- 28. Yoshida M., Kijima M., Akita M., Beppu T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265, 17174–17179 [PubMed] [Google Scholar]
- 29. Miyoshi E., Uozumi N., Noda K., Hayashi N., Hori M., Taniguchi N. (1997) Expression of α1–6 fucosyltransferase in rat tissues and human cancer cell lines. Int. J. Cancer. 72, 1117–1121 [DOI] [PubMed] [Google Scholar]
- 30. Inamori K., Mita S., Gu J., Mizuno-Horikawa Y., Miyoshi E., Dennis J. W., Taniguchi N. (2006) Demonstration of the expression and the enzymatic activity of N-acetylglucosaminyltransferase IX in the mouse brain. Biochim. Biophys. Acta 1760, 678–684 [DOI] [PubMed] [Google Scholar]
- 31. Heller G., Schmidt W. M., Ziegler B., Holzer S., Müllauer L., Bilban M., Zielinski C. C., Drach J., Zöchbauer-Müller S. (2008) Genome-wide transcriptional response to 5-aza-2′-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 68, 44–54 [DOI] [PubMed] [Google Scholar]
- 32. Lopez-Atalaya J. P., Ito S., Valor L. M., Benito E., Barco A. (2013) Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 41, 8072–8084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao L., Cueto M. A., Asselbergs F., Atadja P. (2002) Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 277, 25748–25755 [DOI] [PubMed] [Google Scholar]
- 35. Liu H., Hu Q., D'ercole A. J., Ye P. (2009) Histone deacetylase 11 regulates oligodendrocyte-specific gene expression and cell development in OL-1 oligodendroglia cells. Glia 57, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kreppel L. K., Blomberg M. A., Hart G. W. (1997) Dynamic glycosylation of nuclear and cytosolic proteins: cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 272, 9308–9315 [DOI] [PubMed] [Google Scholar]
- 37. Okuyama R., Marshall S. (2003) UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J. Neurochem. 86, 1271–1280 [DOI] [PubMed] [Google Scholar]
- 38. Szulwach K. E., Li X., Li Y., Song C. X., Wu H., Dai Q., Irier H., Upadhyay A. K., Gearing M., Levey A. I., Vasanthakumar A., Godley L. A., Chang Q., Cheng X., He C., Jin P. (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Globisch D., Münzel M., Müller M., Michalakis S., Wagner M., Koch S., Brückl T., Biel M., Carell T. (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5, e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nestor C. E., Ottaviano R., Reddington J., Sproul D., Reinhardt D., Dunican D., Katz E., Dixon J. M., Harrison D. J., Meehan R. R. (2012) Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 22, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ito R., Katsura S., Shimada H., Tsuchiya H., Hada M., Okumura T., Sugawara A., Yokoyama A. (2014) TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells 19, 52–65 [DOI] [PubMed] [Google Scholar]
- 42. Miyata T., Maeda T., Lee J. E. (1999) NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 13, 1647–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shen S., Li J., Casaccia-Bonnefil P. (2005) Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J. Cell Biol. 169, 577–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujiki R., Hashiba W., Sekine H., Yokoyama A., Chikanishi T., Ito S., Imai Y., Kim J., He H. H., Igarashi K., Kanno J., Ohtake F., Kitagawa H., Roeder R. G., Brown M., Kato S. (2011) GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480, 557–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Daou S., Mashtalir N., Hammond-Martel I., Pak H., Yu H., Sui G., Vogel J. L., Kristie T. M., Affar el B. (2011) Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 2747–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Capotosti F., Guernier S., Lammers F., Waridel P., Cai Y., Jin J., Conaway J. W., Conaway R. C., Herr W. (2011) O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell 144, 376–388 [DOI] [PubMed] [Google Scholar]
- 47. Andrali S. S., Qian Q., Ozcan S. (2007) Glucose mediates the translocation of NeuroD1 by O-linked glycosylation. J. Biol. Chem. 282, 15589–15596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hart G. W., Slawson C., Ramirez-Correa G., Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taniguchi N. (2009) From the γ-glutamyl cycle to the glycan cycle: a road with many turns and pleasant surprises. J. Biol. Chem. 284, 34469–34478 [DOI] [PMC free article] [PubMed] [Google Scholar]