

Background: Mutations in the stomatin family protein podocin are the most common genetic cause of proteinuria.
Results: A conserved proline residue of podocin is essential for its membrane topology.
Conclusion: This study confirms a hairpin-like structure of the membrane-attached PHB domain protein and its significance for cholesterol recruitment.
Significance: PodocinP118L elucidates the pathogenic implication in kidney disease and identifies a novel family of PHB domain proteins.
Keywords: Caenorhabditis elegans, Cholesterol-binding Protein, Glycosylation, Lipid Raft, Protein Conformation, PHB Domain, Membrane Topology, Podocin, Steroid-resistant Nephrotic Syndrome, Stomatin Family Proteins
Abstract
Mutations in the NPHS2 gene are a major cause of steroid-resistant nephrotic syndrome, a severe human kidney disorder. The NPHS2 gene product podocin is a key component of the slit diaphragm cell junction at the kidney filtration barrier and part of a multiprotein-lipid supercomplex. A similar complex with the podocin ortholog MEC-2 is required for touch sensation in Caenorhabditis elegans. Although podocin and MEC-2 are membrane-associated proteins with a predicted hairpin-like structure and amino and carboxyl termini facing the cytoplasm, this membrane topology has not been convincingly confirmed. One particular mutation that causes kidney disease in humans (podocinP118L) has also been identified in C. elegans in genetic screens for touch insensitivity (MEC-2P134S). Here we show that both mutant proteins, in contrast to the wild-type variants, are N-glycosylated because of the fact that the mutant C termini project extracellularly. PodocinP118L and MEC-2P134S did not fractionate in detergent-resistant membrane domains. Moreover, mutant podocin failed to activate the ion channel TRPC6, which is part of the multiprotein-lipid supercomplex, indicative of the fact that cholesterol recruitment to the ion channels, an intrinsic function of both proteins, requires C termini facing the cytoplasmic leaflet of the plasma membrane. Taken together, this study demonstrates that the carboxyl terminus of podocin/MEC-2 has to be placed at the inner leaflet of the plasma membrane to mediate cholesterol binding and contribute to ion channel activity, a prerequisite for mechanosensation and the integrity of the kidney filtration barrier.
Introduction
Nephrotic syndrome (NS)3 is a common kidney disorder characterized by the loss of protein into urine, edema, and decreased levels of albumin in the blood. NS also affects children, about 10% of whom do not respond to standard glucocorticoid therapy and usually progress to end-stage renal disease requiring dialysis or transplantation. An important cause of this disorder is mutation in the NPHS2 gene, which encodes the protein podocin (1). More than 100 different NPHS2 mutations have been identified in both familial and sporadic cases of steroid-resistant nephrotic syndrome (SRNS) (2–5).
Loss of podocin results in failure of the kidney filtration barrier. The human kidneys filter about 180 liters of primary urine per day. This primary filtrate is free of protein. The filter resides in about 1 million of microvascular units, called the glomeruli, which contain a three-layered filtration barrier (6) comprised of podocytes (the visceral epithelial cells), the underlying glomerular basement membrane, and the fenestrated endothelium. Podocytes are highly specialized cells that elaborate primary and secondary foot processes that closely enwrap the glomerular capillaries. Secondary foot processes of adjacent podocytes interdigitate and are connected by the slit diaphragm, a specialized cell-cell contact (7). Glomerular filtration is a tightly regulated process, and the slit diaphragm cell junction has been extensively characterized as a signaling center in podocytes (8). Much effort has been directed toward identifying the protein composition of the slit diaphragm. This cell junctional complex consists of transmembrane proteins such as nephrin and NEPH1, cytoplasmic adaptors including CD2AP and p85, the ion channel TRPC6, and the prohibitin homology (PHB) domain protein podocin, all of which are crucial for downstream signaling and the integrity of the filtration barrier (9–14).
Human podocin is a 383-amino acid membrane protein that belongs to the stomatin protein family. This protein family is characterized by a hairpin-like topology with the amino- and carboxyl termini facing the cytoplasm and a hydrophobic stretch of about 20 amino acids that has been suggested to dip into the inner leaflet of the lipid bilayer and form an intramembrane or membrane-adjacent domain (15, 16). A PHB domain accounts for the major part of the C-terminal tail. In previous studies, we could show that podocin binds cholesterol via a stretch of amino acids at the membrane close and the PHB domain, causing a specialized membrane lipid composition at the site of podocin clusters (14). Through the interaction with podocin, other essential slit diaphragm proteins, such as nephrin, CD2AP, and TRPC6, are targeted to these multiprotein-lipid supercomplexes within the plasma membrane. Formation of these supercomplexes is a prerequisite for proper function of the slit diaphragm signaling platform and the channel function of TRPC6 (8, 14, 17–19).
In this study, we studied a known disease-causing mutation of podocin, P118L, and its ortholog, MEC-2, in the nematode worm Caenorhabditis elegans to assess membrane topology and protein function (4, 20–23). We show that mutating a central proline residue results in a protein with disrupted membrane topology and a carboxyl terminus facing the outside of the cell. We used this information to confirm the membrane topology of the wild-type protein and show that correct membrane topology is essential to protein function. Consistent with our previous findings showing that cholesterol recruitment into ion channel complexes is central to the function of podocin and MEC-2 (14), we now demonstrate that neither mutant MEC-2 nor mutant podocin was associated with detergent-resistant membrane domains (DRMs) and that podocin-mediated TRPC6-channel activity is dramatically diminished in Xenopus oocytes. These experiments highlight the importance of the membrane topology of podocin and MEC-2 and provide a possible mechanism for SRNS in affected humans.
EXPERIMENTAL PROCEDURES
Reagents and Plasmids
Mouse podocin, human nephrin, human TRPC6, human CD2AP, and C. elegans MEC-2 cDNA constructs have been described previously (14, 24). Human podocin was PCR-cloned from a human kidney library. Mutations of podocin and MEC-2 were generated by site-directed mutagenesis. Double-tagged human podocin constructs were generated using standard cloning procedures.
Antibodies were obtained from Sigma (anti-FLAG M2 mAb, catalog no. F3165 and anti-FLAG pAb, catalog no. F7425), Serotec (anti-V5 mAb, catalog no. MCA1360), Millipore (anti-V5 polyclonal antibody, catalog no. AB3792), and Santa Cruz Biotechnology (anti flotillin-2, catalog no. sc-28320; anti-CD71 (Transferrin receptor, catalog no. sc-65882)). The anti MEC-2 antibody (MEC-2C) has been described elsewhere (22). Alexa Fluor 488-conjugated secondary antibodies were from Invitrogen, and Cy3-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories.
Cell Culture and Transfection
HEK 293T cells were cultured in DMEM supplemented with 10% fetal bovine serum under standard conditions (5% CO2, 37 °C). For transfection experiments, cells were grown to 60–80% confluency and transfected with plasmid DNA using the calcium phosphate method for HEK 293T cells.
Immunoblot Analysis and Deglycosylation
HEK 293T cells were transiently transfected with the indicated plasmid DNA by the calcium phosphate method. After incubation for 24 h, cells were rinsed with PBS, lysed on ice in lysis buffer (1% Triton X-100, 20 mm Tris (pH 7.5), 25 mm NaCl, 50 mm NaF, 15 mm Na4P2O7, 1 mm EDTA, 0.25 mm PMSF, and 5 mm Na3VO4) for 15 min and centrifuged (14,000 rpm, 4 °C, 15 min). Supernatants containing equal amounts of total proteins were analyzed by 10% SDS-PAGE.
When indicated, cell lysates were treated with PNGase F (New England Biolabs, catalog no. P0704) according to the protocol of the manufacturer. In brief, 8 μl of cell lysate was incubated with 1 μl of 10× glycoprotein denaturing buffer and 1 μl of double-distilled H2O for 10 min at 95 °C. Then, 2 μl of 10× G7 reaction buffer, 2 μl of 10% Nonidet P-40, and 1 μl of PNGase F were added, filled to 20 μl with double-distilled H2O, and incubated for 1 h at 37 °C. Deglycosylated proteins were analyzed by 10% SDS-PAGE.
Coimmunoprecipitation
Cells were incubated for 24 h after transfection, washed with PBS, lysed in ice-cold modified radioimmune precipitation assay buffer (50 mm Tris (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mm NaF, 1 mm EDTA, 0.25 mm PMSF, and 5 mm Na3VO4) on ice for 20 min and cleared by ultracentrifugation (Beckmann TLA-55, 50,000 rpm, 4 °C, 60 min). Supernatants containing equal amounts of total protein were incubated for 1.5 h at 4 °C with anti-FLAG M2-agarose beads (Sigma-Aldrich). Then the beads were washed three times with radioimmune precipitation assay buffer, and bound proteins were resolved by 10% SDS-PAGE.
Preparation of Lipid Raft Membrane Domains
The preparation of DRMs was performed as described previously (17). Briefly, HEK 293T cells were homogenized in 1 ml of lysis buffer (150 mm NaCl, 20 mm Tris/HCl (pH 7.4), 0.1 mm EDTA, 1% Triton X-100, 5 mm Na3VO4, and 0.25 mm PMSF) by 20 strokes in a Dounce homogenizer. After centrifugation for 10 min at 3.000 rpm at 4 °C (Eppendorf F45-30-10 rotor), supernatants (lysates) containing equal amounts of total proteins were carefully adjusted to 45% sucrose (1.6 ml final volume) and pipetted at the bottom of an ultracentrifuge tube. Samples were then overlaid with 1.6 ml of 30% and 0.8 ml of 5% sucrose (in 150 mm NaCl, 20 mm Tris/HCl (pH 7.4), and 0.1 mm EDTA) to create a sucrose gradient. Samples were centrifuged for 16 h at 41.000 rpm at 4 °C in a swingout rotor (SW60Ti, Beckman Instruments) and seven fractions were collected starting from the top and, together with the non-fractionated lysate, analyzed by SDS-PAGE.
Immunofluorescence
HEK 293T cells were seeded on collagen A-coated coverslips and transfected with double-tagged podocin constructs as indicated using the calcium phosphate method. After 24 h, cells were fixed with 2% paraformaldehyde for 5 min without permeabilizing them, blocked with 5% normal donkey serum, and incubated with anti-FLAG polyclonal antibody. After washing with PBS, the FLAG antibody was fixed at its position by a second fixation step (2% paraformaldehyde for 5 min). Then cells were permeabilized in PBS containing 5% normal donkey serum + 0.1% Triton X-100 and incubated with anti-V5 mouse monoclonal antibody. After extensive washing, cells were incubated with fluorescently labeled secondary antibodies for 45 min. All incubation steps were done for 1 h at room temperature. Between the single incubation steps, cells were rinsed extensively with PBS. Confocal images were taken with a Leica SP8 confocal microscope equipped with a white light laser and a HCX PL APO ×100/1.40 oil objective (Leica Microsystems, Wetzlar, Germany). Images were processed using ImageJ/Fiji software version 1.48 (25).
Mass Spectrometry
Tryptic In-gel Digestion
The lysates of five 10-cm HEK 293T dishes transfected with mouse podocinP120L were pooled and separated using SDS-PAGE. Following electrophoresis, the gels were washed thoroughly in water and stained with Coomassie Blue. Bands of interest were cut out and minced using a scalpel and then transferred to a 1.5-ml reaction tube. After destaining with 100 μl of 50% 10 mm NH4HCO3/50% acetonitrile (ACN) at 55 °C and dehydration in 500 μl of 100% ACN gel, pieces were equilibrated with about 30 μl of 10 mm NH4HCO3 containing porcine trypsin (12.5 ng/μl, Promega, Mannheim, Germany) on ice for 2 h. Excess trypsin solution was removed, and tryptic hydrolysis was performed for 4 h at 37 °C in about 20 μl of 10 mm NH4HCO3.
Enzymatic Deglycosylation
For peptide deglycosylation, tryptic digests of both bands were treated with peptide N-glycosidase F (PNGase F), which hydrolyzes nearly all types of asparagine-linked (N-linked) oligosaccharides from glycopeptides/proteins, leaving the oligosaccharides intact. PNGase F deaminates asparagine to aspartic acid, resulting in a mass difference of +0.984 Da. 5 μl of the in-gel tryptic digests were mixed with 0.5 μl of PNGase F (500,000 units/ml, New England Biolabs, Frankfurt, Germany) or 0.5 μl of PNGase buffer as a control, respectively, and incubated overnight at room temperature.
MALDI Mass Spectrometry
Microcrystalline layers were prepared on MALDI target plates by mixing 0.5 μl of the PNGase F-treated sample and the control, respectively, with 0.5 μl of α-cyano-4-hydroxycinnamic acid (Fluka, 10 mg/ml, 50% ACN/50% 0.1% aqueous TFA) on the target plate. For peptide mass fingerprinting, positively charged ions in the mass-to-charge (m/z) range of 700–4000 were acquired on a 4800 Plus MALDI-TOF/TOF analyzer (AB SCIEX, Darmstadt, Germany) in the reflector mode. Sums of 50 spectra were recorded from 50 different sample spot positions, resulting in 2500 spectra in total using the 4000 Series Explorer operating software (AB SCIEX). The mass spectrometer was calibrated on the basis of near-neighbor calibrant spectra using Calmix solution (Applied Biosystems). Automatic annotation of monoisotopic peptide signals in tryptic digests was performed using internal calibration on trypsin autolysis peaks at 842 and 2211 m/z.
Nano-LC ESI-MS/MS Mass Spectrometry
To determine the amino acid sequence of the tryptic peptides containing amino acid residue 201, nano-LC electrospray ionization (ESI)-MS/MS was performed. The residual PNGase F-treated sample after MALDI MS of the upper band was desalted using STAGE Tipp C18 spin columns (Proxeon) as described elsewhere (26). Tryptic digests of the lower band were acidified with 5 μl of 5% TFA, and the gel pieces were extracted twice with 20 μl of 0.1% TFA and then with 20 μl of 60% ACN/40%H2O/0.1% TFA, followed by a subsequent two-step treatment using 30 μl of 100% ACN. The supernatant and the extractions were combined, concentrated to a volume below 65 μl using a SpeedVac concentrator (Christ, Osterode am Harz, Germany), and desalted using STAGE Tip C18 spin columns (Proxeon). Peptides eluted from STAGE Tips were concentrated to a volume of about 10 μl in vacuo and then diluted with 0.5% acetic acid in water to a final volume of 20 μl.
Experiments were performed on a LTQ Orbitrap Discovery (Thermo Scientific) mass spectrometer that was coupled to a split-less Eksigent nano-LC system (Axel Semrau). Intact peptides were detected in the Orbitrap at 30,000 resolution in the m/z range of 300–2000. Internal calibration was performed using the ion signal of (Si(CH3)2O)6H at m/z 445.120025 as a lock mass. For MS/MS analysis, collision-induced dissociation was used as a peptide fragmentation mode. Up to five collision-induced dissociation spectra were acquired following each full scan. Sample aliquots of 3–8 μl were separated on a 10-cm, 75-μm internal diameter, reversed-phase column (3 μm, 120 Å, ReproSil-Pur C18-AQ, Proxeon). Gradient elution was performed from 2 to 40% acetonitrile within 40 min at a flow rate of 250 nl/min.
Peptide and Protein Identification
For MALDI peptide mass fingerprinting data, a Mascot search (Matrix Science Ltd., London) on a local server (version 2.2) was applied using the Swiss Protein Database of Mus musculus for protein identification (27). A mass tolerance of 20 ppm was allowed for intact peptide masses. For nano-LC ESI-MS/MS data, a Sequest search was applied using the ipi.mouse database version 3.71 (March 2010), including an added entry containing the amino acid sequence of podocin with an aspartic acid instead of asparagine at position 201. A mass tolerance of 10 ppm was allowed for intact peptide masses and 0.8 Da for collision-induced dissociation fragment ions detected in the linear ion trap. Methionine oxidation was allowed as a variable modification.
Oocyte Electrophysiology
cRNA and Double-electrode Voltage Clamp
cRNAs for mouse podocin, mouse podocinP120L, and human TRPC6 were all linearized with Notl or Mlul and in vitro-transcribed using T7, T3, or SP6 promoter and polymerase (Promega). Xenopus laevis oocytes were injected with cRNA (10 ng, 47 nl of double-distilled water). Water-injected oocytes served as controls. Three to four days after injection, oocytes were impaled with two electrodes (Clark Instruments Ltd, Salisbury, UK), which had a resistance of <1 mΩ when filled with 2.7 mol/liter KCI. Using two bath electrodes and a virtual-ground head stage, the voltage drop across Rserial was effectively zero. Membrane currents were measured by voltage clamping (oocyte clamp amplifier, Warner Instruments LLC, Hamden, CT) in intervals from −90 to +30 mV in steps of 10 mV, each 1 s. The bath was perfused continuously (ND96 solution containing 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, and 2.5 mm sodium pyruvate (pH 7.55)) at a rate of 5 ml/min. All experiments were conducted at room temperature (22 °C).
Materials and Statistical Analysis
All compounds used were of highest available grade of purity and were from Sigma or Calbiochem. The compounds used in this study were usually applied at recommended maximal concentrations to achieve full activation or inhibition, respectively. Student's t test was used to compare any two different groups. Differences in multiple groups were analyzed using one-way analysis of variance followed by Tukey's test to compare significant differences between individual groups. p ≤ 0.05 was considered significant.
Database Search and Sequence Alignment
PHB domain proteins of humans and C. elegans were detected using an architecture query for “phb” in the genomic mode of the SMART database (28). After manually removing/replacing outdated records, the 11 human and 13 C. elegans sequences were aligned using the MUSCLE algorithm (29) as provided on the EMBL-EBI web site. The alignment was visualized using Jalview 2.8 (30).
RESULTS
PodocinP120L (Mouse) and PodocinP118L (Human) Are N-Glycosylated
To study the role of membrane topology of podocin and its ortholog in C. elegans for protein function, we studied various disease causing mutations. All members of the family of stomatin-like proteins share a highly conserved central proline residue within the hydrophobic stretch of the protein (31). Mutations of this proline residue are known causes of protein dysfunction throughout species. A P134S mutation in MEC-2 (MEC-2 P134S) causes touch insensitivity in the nematode (22), and the equivalent P118L mutation in humans causes SRNS (3, 4, 20, 21). Because it has been shown that a mutation of this conserved proline in stomatin (P47S) changes the membrane topology of stomatin (31), we concentrated on the molecular mechanisms that cause SRNS in patients bearing the podocin mutation P118L (corresponding to P120L in the mouse protein, collectively named podocinP→L hereafter). Western blot analyses of wild-type podocin, the disease-causing human mutation podocinP118L, or the equivalent mouse protein (podocinP120L) transiently expressed in HEK 293T cells revealed a single protein band of the expected molecular weight for wild-type podocin and one (mouse) or two (human) additional protein bands for the podocinP→L mutants (Fig. 1A). We next tested whether the additional bands in podocinP→L could be attributed to N-glycosylation. HEK 293T cell lysates containing wild-type podocin or podocinP→L were treated with PNGase F, an enzyme that specifically removes N-glycosylations. The additional higher molecular weight protein bands of podocinP→L disappeared with this treatment (Fig. 1B). The number of extra bands in the mutants corresponds with the number of potential N-glycosylation sites (N-X-S/T, where X is any amino acid except for proline). Mouse podocin has only one such site, at amino acid 201, and one additional band, whereas human podocin has two such sites, at amino acids 199 and 355, and two additional bands (Fig. 1C). We used site-directed mutagenesis to mutate the asparagine at the consensus glycosylation site into glutamine or serine (32, 33) both in wild-type podocin and in the P→L mutant. Wild-type podocin was not affected by site-directed mutagenesis, but the additional bands in podocinP→L were eliminated, confirming N-glycosylation of podocinP→L (Fig. 1D).
FIGURE 1.
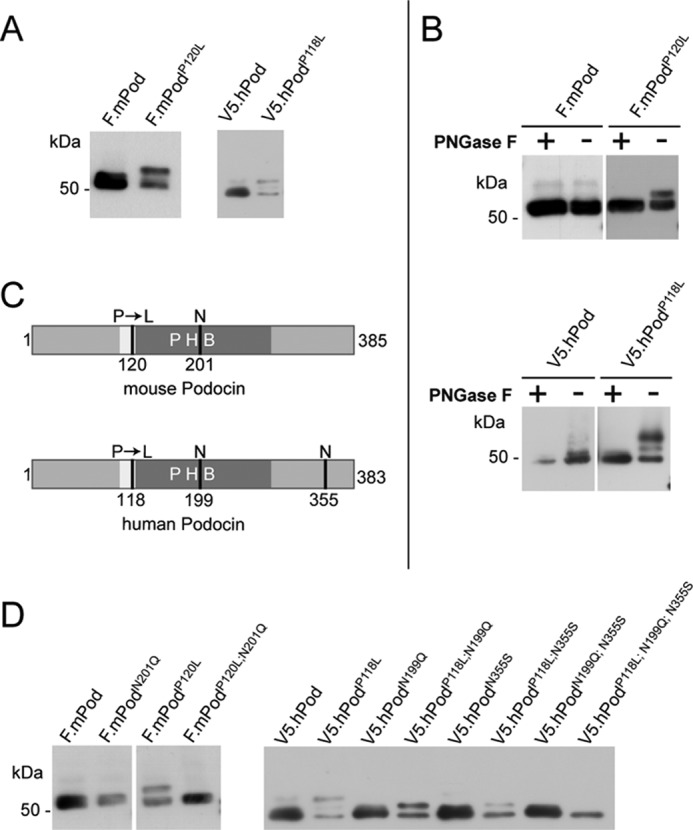
The disease-causing podocin mutation P118L and its mouse equivalent P120L are N-glycosylated. A, B, and D, FLAG- or V5-tagged mouse and human podocin constructs were transiently expressed in HEK 293T cells. Cell lysates were analyzed by Western blot analysis using an antibody against the tag. A, wild-type podocin shows a protein band of the expected molecular weight, whereas podocin with the P→L mutation shows one (mouse) or two (human) additional band(s) of higher molecular weight. B, after PNGase F treatment of cell lysates, the additional bands of podocinP→L disappear. C, Schematic of mouse and human podocin depicting the hydrophobic region (white), the PHB domain (dark gray), the position of the patient mutation (P120L/P118L), and the potential sites of N-glycosylation (N). D, site-directed mutagenesis of the putative N-glycosylation site of mouse podocin Asn-201 into Gln prevents glycosylation. Mutation of one potential N-glycosylation site of human podocin (Asn-199 or Asn-355, respectively) removes the top band of the two additional bands. Mutation of both Asn residues removes both extra bands.
To validate our findings, we next used mass spectrometry to confirm the nature of the posttranslational modification of purified mouse podocinP120L (Fig. 2). The two bands detected in Coomassie-stained gels were digested with trypsin, and one half was treated with PNGase F, whereas the other half was left untreated. If N-glycosylation is present, PNGase F treatment leads to conversion of the asparagine residue carrying the glycosylation to aspartic acid. This results in a mass shift of ∼1 Da. Therefore, non-glycosylated peptides can be clearly distinguished from deglycosylated peptides on the basis of peptide mass as well as peptide sequence in mass spectrometers with high mass accuracy (Fig. 2A). In MALDI mass spectrometry, PNGase F treatment resulted in the detection of two masses matching deglycosylated podocin peptides containing amino acid 201 (Fig. 2B). In the corresponding control, no podocin peptides were detected in this mass region. The lower band only resulted in masses that matched the non-glycosylated form of podocin. To confirm that these detected mass “fingerprints” were, in fact, originating from podocin peptides, aliquots of the remaining samples were subjected to nano LC ESI-MS/MS analyses (Fig. 2C). We found that the masses detected in MALDI MS originated from podocin peptides encompassing amino acid 201. Taken together, the results from MALDI and ESI mass spectrometry suggest that podocin present in the upper band was glycosylated at site 201, whereas podocin in the lower band was not glycosylated at this site.
FIGURE 2.

Mass spectrometric analysis of mouse podocinP120L. A, theoretical peptides and predicted masses containing the potential glycosylation site Asn-201. Deglycosylation by PNGase F treatment results in conversion of asparagine (N) to aspartic acid (D). Because of the presence of a methionine at position 1, these peptides can also be present in oxidized form. B, MALDI MS of the two Coomassie-stained bands containing podocinP120L. Following in-gel tryptic digestion, the samples were treated with PNGase F as indicated. After PNGase F treatment, the upper band contained two peptide masses corresponding to the deglycosylated form, whereas masses corresponding to non-glycosylated peptides were not detected. In the respective control sample (no PNGase F treatment), no podocin peptides were detected in this mass region. Both results suggest glycosylation of the protein at site Asn-201 in the upper band. The lower band contained in the PNGase F-treated sample as well as in the untreated sample only peptide masses that correspond to the non-glycosylated peptide (in oxidized and non-oxidized form), indicating that, in the lower band, the protein was not glycosylated at site Asn-201. C, to analyze the amino acid sequence of the peptide masses detected in MALDI MS, aliquots of the remaining samples were subjected to nano LC ESI-MS/MS analysis. Peptides identified by ESI-MS/MS confirmed that the masses correspond to podocinP120L peptides containing Asn-201 in non- and deglycosylated forms, respectively. Detected ion series in MS2 spectra are indicated in red (y ions) or blue (b ions).
PodocinP→L Shows an Altered Membrane Topology
It has been shown that much of the human podocinP→L is retained in the endoplasmic reticulum (34). We used immunofluorescence to test whether any of the glycosylated mutant podocinP→L was also transferred to the plasma membrane. In this case, the N-glycosylated C terminus should be detectable in the extracellular space. We transiently expressed podocin constructs containing an N-terminal V5 and a C-terminal FLAG tag in HEK 293T cells. In contrast to wild-type podocin, podocinP→L resulted in a positive FLAG staining in non-permeabilizing conditions, indicating that its C terminus was localized extracellularly (Fig. 3). As reported previously (34), the podocin pattern for the P→L mutant shows an accumulation in the endoplasmic reticulum (Fig. 3, A′—D′). However, staining for extracellular, FLAG-tagged C terminus revealed a very clear staining (Fig. 3, A–D), indicating that podocinP→L is at least partially transported to the plasma membrane where it displays a non-conventional membrane topology.
FIGURE 3.
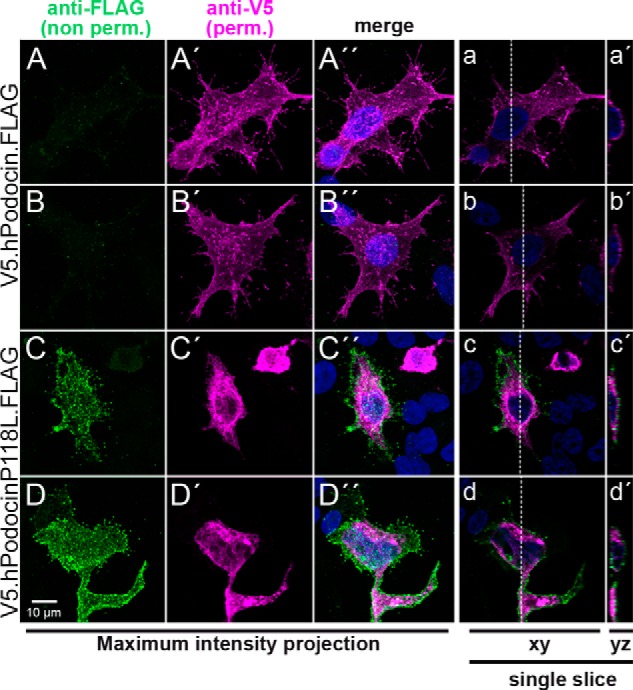
Immunofluorescence analysis confirms the extracellular localization of the podocinP118L C terminus. A, N- and C-terminally double-tagged human podocin wild-type (A and B) or P→L mutant constructs (C and D) were transiently expressed in HEK 293T cells. The cells were fixed and stained with anti-FLAG antibody prior to permeabilization (A–D). Next, cells were permeabilized and stained with anti-V5 antibody (A′—D′). The podocinP118L mutant shows an extracellular FLAG signal. Panels with capital letters show the different channels and a merged image including a DAPI staining of a maximum intensity projection of a confocal stack (28–34 slices each). a—d, a single confocal slice from the corresponding stack. a′—d′, a yz slice of the stack at the position indicated with a dashed line in a–d. The FLAG signal (green) in the podocinP118L-transfected cells is clearly localized at the outer aspect of the cell.
Recruitment of PodocinP→L into Detergent-resistant Membrane Domains Is Impaired
We next tested whether altered membrane topology of mutant podocin interfered with its ability to bind and recruit cholesterol at the membrane. Wild-type podocin partitions into a DRM fraction upon sucrose density gradient centrifugation (17) because podocin binds cholesterol (14). We found that podocinP→L expressed in HEK 293T cells failed to partition into the DRM fraction, in contrast to wild-type podocin and flotillin-2, a marker for DRM fractions (Fig. 4, A and C). PodocinP→L was diminished in the DRM fractions, suggesting that correct membrane topology of podocin with amino and carboxyl termini facing the cytoplasm is required for the interaction with cholesterol (Fig. 4, B and D).
FIGURE 4.
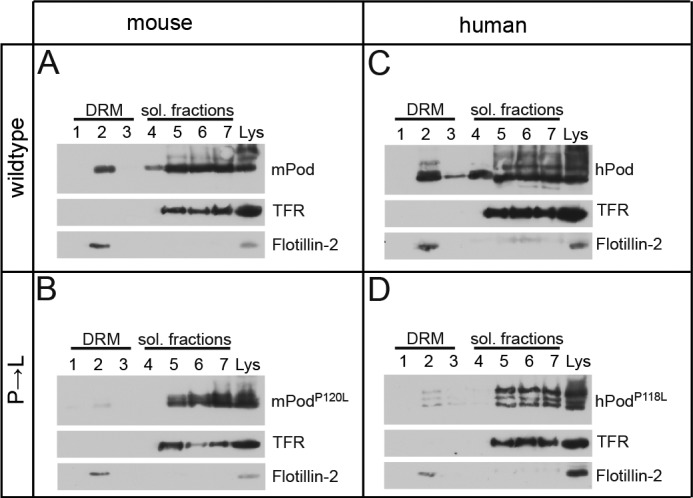
The association of podocinP→L with DRMs is disturbed. A—D, V5-tagged wild-type or mutant podocin was expressed in HEK 293T cells as indicated. The cells were lysed in 1% TX-100 on ice and subjected to sucrose density gradient centrifugation. Seven fractions were collected from the top and were analyzed together with the non-fractionated lysate (Lys) by Western blot analysis. A and C, mouse and human wild-type podocin associates with DRMs. B and D, the DRM association is lost in mouse and human podocinP→L. Antibodies against the transferrin receptor (TFR) and flotillin-2 were used as markers for the Triton-soluble (sol.) and Triton-insoluble fractions, respectively.
PodocinP→L Interacts with Other Slit Diaphragm Proteins but Fails to Activate the TRPC6 Ion Channel
The protein network at the slit diaphragm is essential for its function as a molecular barrier and a signaling platform (18). Podocin is an integral part of this network, interacting with other slit diaphragm proteins such as nephrin, CD2AP, and TRPC6 (10, 13, 24). We therefore tested whether FLAG-tagged podocinP→L would still interact with V5-tagged nephrin, CD2AP, or TRPC6 in HEK 293T cells. Coimmunoprecipitation experiments revealed that all tested proteins interacted with wild-type podocin and with the P→L mutated form of podocin. Because we have shown previously that podocin regulates the activity of the TRPC6 ion channel (14), we next examined the influence of the podocinP→L mutation on TRPC6 activity. TRPC6 currents were measured in X. laevis oocytes in the presence or absence of wild-type mouse podocin or mouse podocinP120L. When wild-type podocin was coexpressed with TRPC6, the inward currents produced by TRPC6 after exposure to 10 μm OAG were augmented, as observed previously (14). However, mouse podocinP→L was unable to augment TRPC6 currents (Fig. 5, B and C). In the absence of OAG, no significant currents were generated by expression of TRPC6 in either the absence or presence of podocin (Fig. 5D).
FIGURE 5.
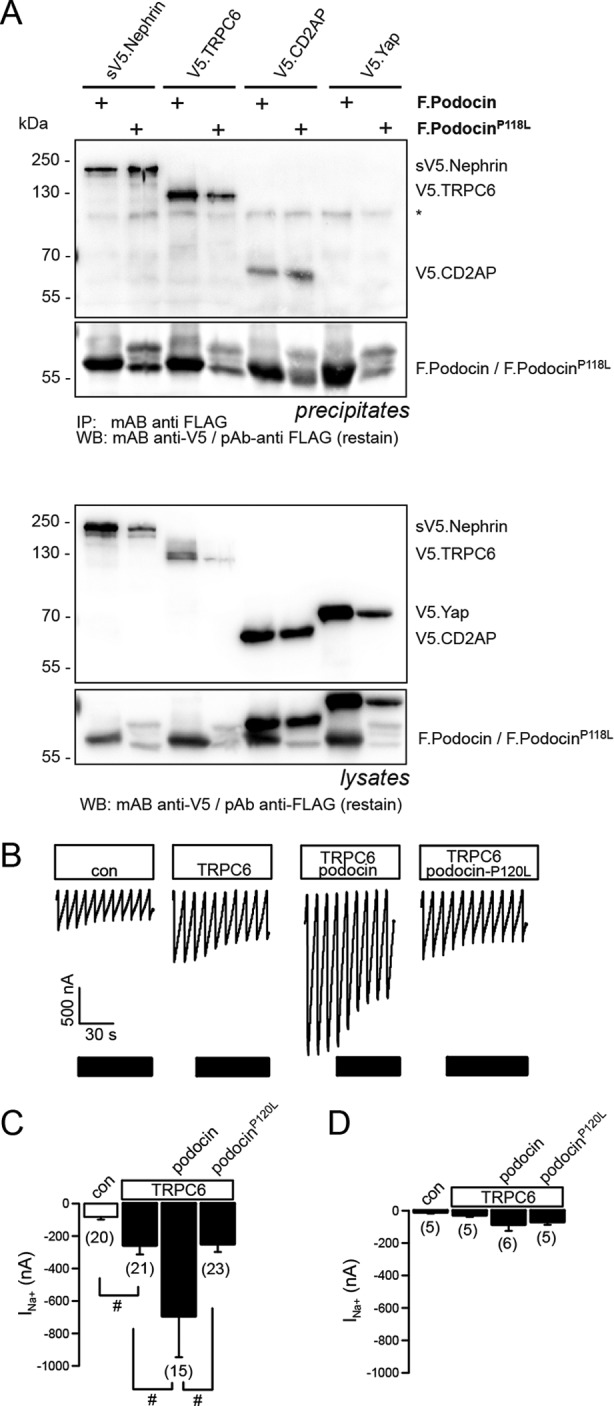
PodocinP118L still interacts with known interaction partners, but the function is impaired. A, V5-tagged nephrin, TRPC6, CD2AP, and a negative control (Yap) were expressed in HEK 293T cells together with podocin or podocinP118L as indicated. Lysates were precipitated (IP) with an anti-FLAG antibody, resolved by SDS-PAGE, and analyzed by Western blot (WB) analysis. PodocinP118L still interacts with all three proteins. Coexpression of the P→S variant has a negative influence on expression levels of the coexpressed proteins, but because this occurs also in the negative control, this is most likely an effect because of overexpression. The asterisk denotes an unspecific band. B—D, the disease-causing mutation P120L eliminates the activating function of podocin. B, whole cell currents measured in Xenopus oocytes at a holding potential of −90 mV. Inward (Na+) currents produced by TRPC6 after exposure to 10 μm OAG were enhanced significantly by wild-type podocin but not by podocinP120L. con, control. C, summary of the inward currents produced by TRPC6 in the absence and presence of coexpressed podocin or podocinP120L after exposure to 10 μm OAG. D, summary of the inward currents produced by TRPC6 in the absence and presence of coexpressed podocin or podocinP120L in the absence of OAG. Data are mean ± S.E. #, indicates significant difference (p ≤ 0.05, unpaired t test). n = number of experiments.
Function of the Central Proline Is Evolutionarily Conserved between Podocin and its C. elegans Ortholog, MEC-2
As with all the members of the stomatin protein family, MEC-2 contains a conserved proline (Pro-134) residue within the hydrophobic stretch that precedes the PHB domain (Fig. 6A). Touch sensitivity, enhancement of DEG/ENaC ion channel activity, and cholesterol binding, all intrinsic properties of wild-type MEC-2, are strongly dependent on this central proline residue; however, multimerization and binding to DEG/ENaC proteins are not (14, 22, 35). To check whether the membrane topology was also altered by the P134S change, we expressed wild-type MEC-2 or MEC-2P134S in HEK 293T cells. The MEC-2P134S band was completely shifted to a higher molecular weight on Western blot analyses compared with wild-type MEC-2. Upon treatment with PNGase F, the size shift of MEC-2P134S disappeared, suggesting that, similarly to podocin, this mutant protein was glycosylated (Fig. 6B). To examine whether a P→S mutation would have the same effect on podocin as a P→L mutation, we repeated the experiment with podocinP→S. Again, the loss of the proline induced an additional band that disappeared upon treatment with PNGase F (Fig. 6C, compare with Fig. 1B). Consistent with our previous studies showing a loss of cholesterol binding (14) and the above studies on podocinP→L, MEC-2P134S was no longer detectable in DRMs (Fig. 6D). Next, we addressed the membrane topology of MEC-2P134S. N-terminally FLAG-tagged, wild-type MEC-2 or MEC-2P134S was expressed in HEK 293T cells. Wild-type MEC-2 and MEC-2P134S protein were detected by both antibodies, but only the MEC-2 P134S C terminus was accessible to antibodies in non-permeabilized cells. These results indicate a change in membrane topology from a hairpin-like structure to a transmembranous form (Fig. 6E) and confirm the evolutionary conservation of membrane topology of podocin, MEC-2, and stomatin. The PHB domain is found in a large number of proteins with different membrane topologies (16). The conservation of the central proline might be an indicator of a hairpin-like topology in a subgroup of PHB family proteins. Of the 11 genes encoding human and mouse PHB domain proteins, five produce proteins containing an equivalent proline within a hydrophobic region N-terminal to the PHB domain (podocin, stomatin, stomatin-like proteins 1–3). In C. elegans, 10 of 13 PHB domain-encoding genes translate into proteins that feature a proline in an equivalent position (MEC-2, UNC-24, UNC-1, STL-1, and STO-1 through STO-6). In addition to this conserved proline, most of these proteins share two conserved cysteine residues that serve as palmitoylation sites. We suggest that these and similar proteins displaying an analogous membrane topography and lipid binding constitute a subclass of PHB domain proteins that we term SL-PHB proteins (SL for stomatin-like, Fig. 7B).
FIGURE 6.

The touch insensitivity causing mutant C. elegans protein MEC-2P134S is glycosylated and has an altered membrane topology. A, schematic of C. elegans MEC-2 depicting the hydrophobic region (white), the PHB domain (dark gray), as well as the position of the P134S mutation and the potential sites of N-glycosylation (N). B and C, FLAG-tagged, wild-type MEC-2 or MEC-2P134S was expressed in HEK 293T cells. Cell lysates were incubated with PNGase F as indicated and analyzed by Western blot analysis using anti-FLAG antibody. Wild-type MEC-2 shows a protein band of the expected molecular weight, whereas the MEC-2P134S band is shifted to a higher molecular weight. A faint band of this size is also visible with wild-type MEC-2. After treatment with PNGase F, the size shift disappears. Mouse podocinP120S shows the same behavior as mouse podocinP120L (cf. Fig. 1B). D, the association of MEC-2P134S with DRMs is disturbed. FLAG-tagged, wild-type or mutant MEC-2 was expressed in HEK 293T cells, lysed in 1% TX-100 on ice, and subjected to sucrose density gradient centrifugation. Fractions were collected from the top and analyzed by Western blot analysis. MEC-2P134S does no longer associate with DRMs. sol., soluble. E, cells were either permeabilized or non-permeabilized and exposed to anti-FLAG and anti-MEC-2 C terminus-specific antibody (anti-MEC-2C), respectively. Immunofluorescence analysis reveals that in non-permeabilized conditions, only the MEC-2P134S C terminus is accessible to antibodies. Scale bar = 20 μm.
FIGURE 7.

The proline preceding the PHB domain is conserved in a subset of PHB proteins. A, schematic of podocin/MEC-2. The conserved proline induces a kink of the C terminus that ensures a membrane proximity of the PHB domain, which is fixed by palmitoylation at two conserved cysteine residues. B, MUSCLE alignment of the 11 human and the 13 C. elegans PHB domain proteins. Podocin and MEC-2 are highlighted with a red box. The conserved proline and cysteine residues are marked (boxes and arrows). We suggest calling these proteins SL-PHB proteins. The UniProt accession numbers are as follows: P27105 STOM_HUMAN, Q9UBI4 STML1_HUMAN, Q9UJZ1 STML2_HUMAN, Q8TAV4 STML3_HUMAN, O75955 FLOT1_HUMAN, Q14254 FLOT2_HUMAN, Q9NP85 PODO_HUMAN, P35232 PHB_HUMAN, Q99623 PHB2_HUMAN, O75477 ERLN1_HUMAN, O94905 ERLN2_HUMAN, Q27433 MEC2_CAEEL, Q19200 STO1_CAEEL, Q19958 STO2_CAEEL, Q20657 STO3_CAEEL, Q22165 STO4_CAEEL, H2L024 STO5_CAEEL, Q9XWC6 STO6_CAEEL, H2FLJ1 STL1_CAEEL, Q21190 UNC1_CAEEL, G5ED76 UNC24_CAEEL, Q9BKU4 PHB1_CAEEL, P50093 PHB2_CAEEL, and A3QMC6 ERL1_CAEEL.
DISCUSSION
Mutation of podocin, an essential component of the slit diaphragm of podocytes, is a major cause of SRNS in children (1). Podocin and MEC-2 are key components of multiprotein-lipid ion channel supercomplexes that are crucial for the maintenance of the kidney filtration barrier and mechanosensation, respectively. Because the cholesterol-binding activity of both proteins is an important but not well understood function of PHB proteins, more structural information on podocin/MEC-2 is essential. Although bioinformatics prediction programs have suggested that podocin, similarly to its C. elegans ortholog, MEC-2, may have a hairpin-like structure with a central hydrophobic membrane-close region and amino and carboxyl termini facing the cytoplasm, the structure has been a matter of debate. Here we take advantage of a disease-causing mutation that alters the structure and that allowed us to clarify the membrane topology of podocin/MEC-2 proteins (4, 20–22). Upon mutation of the central proline, the C termini of podocinP→L/MEC-2P134S were no longer intracellular but flipped through the membrane and became accessible to N-glycosylation. This observation is in agreement with earlier studies on the PHB family member stomatin (31). In contrast to podocinP→L, of which only a fraction is glycosylated, the complete MEC-2P134S pool is glycosylated. The results with podocinP→S rules out that this difference is due to the difference of serine versus leucine because podocinP→S also shows only a partial glycosylation (compare Figs. 1B and 6C). Rather, this observation might be explained by the fact that MEC-2 features three N-glycosylation consensus sites C-terminally of Pro-134, whereas podocin contains only one (mouse) or two (human) such sites. It is conceivable that the transmembranous state is not stable but needs to be stabilized by glycosylations and that more glycosylated asparagine residues may fix the transmembranous state more efficiently. Interestingly, we observed faint bands in Western blot analyses that are susceptible to PNGase F treatment at heights corresponding to the glycosylated forms in wild-type podocin and MEC-2. This may either indicate that the non-mutated wild type proteins can also exist in two topologies or be explained by overexpression artifacts. In any case, mutation of the central proline seems to shift the equilibrium between the two possible topologies toward the transmembranous form, resulting in a severe and progressive kidney disease in the case of podocin and touch insensitivity in the case of MEC-2. Interestingly, we have demonstrated recently that humans also express a shorter isoform of podocin missing most of the PHB domain. This isoform is also partially glycosylated (36).
Unlike the wild type proteins, podocinP→L and MEC-2P134S did not fractionate in detergent-resistant membrane domains, indicative of the fact that they are unable to bind and recruit cholesterol. Consistent with this observation, podocinP→L failed to activate the associated ion channel complexes: TRPC6 in the case of podocin and the MEC-4/MEC-10 DEG/ENaC channel in the case of MEC-2. Our study suggests that the central proline preceding the PHB domain is needed for the formation of the hairpin structure. This structure enables the association of the PHB domain with the inner leaflet of the plasma membrane, which is a prerequisite for cholesterol binding and proper function.
As a result of this study, the membrane topology of podocin and MEC-2 can now be explained. Cholesterol binding is dependent on membrane proximity of the PHB domain, which is achieved through a proline-dependent kink of the C terminus and two cysteine residues (positions 124 and 158 in human podocin) that serve as palmitoylation sites, allowing firm membrane attachment. On the basis of the sequence conservation in many PHB domain proteins, we could identify a subclass of PHB domain proteins that we refer to as SL-PHB proteins (Fig. 7).
Signaling is a major function of the podocin-associated slit diaphragm protein complex (8, 18). PodocinP→L did interact with members of this complex, like nephrin, CD2AP, and TRPC6, indicating that the P→L mutation per se does not interfere with the interaction. The interaction with nephrin, CD2AP, and TRPC6 has been mapped previously to the C terminus of podocin (10, 37). It is likely that the interaction with the glycosylated form is occurring only in the cell lysate. As shown by immunofluorescence microscopy, the C terminus localizes to the extracellular compartment in the glycosylated form, rendering a direct interaction topographically implausible. Podocin has been shown to oligomerize and form high molecular multimers (10). These podocin multimers may also contain the glycosylated form and, thus, mediate the interaction in an indirect way because dimerization or multimerization of the glycosylated and the non-glycosylated form of podocin can still occur via the N terminus (17). Importantly, mixed multimers exhibit a reduced cholesterol affinity and are therefore no longer able to provide the lipid microenvironment needed for a functional slit diaphragm protein complex.
We propose that alterations of the lipid microenvironment are a cause of the pathogenicity of the podocin P118L mutation. However, in addition to the defective lipid assembly, alterations in TRPC6-mediated calcium flux in podocytes bearing the mutant podocinP118L may further compromise the function of the slit diaphragm protein complex and aggravate proteinuria, progressive podocyte loss, and glomerulosclerosis by yet unknown mechanisms.
Taken together, our studies of the patient mutation podocinP118L and the touch-insensitive MEC-2P134S mutant did clarify molecular details of the effect of this mutation. Membrane topology is altered severely, which goes along with a loss of function. In the past, much effort has been put into generating appropriate protein complexes for structural analyses in vitro (38). Our data may be valuable for producing a better picture of cholesterol recruitment through podocin/MEC-2 complexes in vivo.
Acknowledgments
We thank Stefanie Keller, Bettina Maar, Ingo Simons, and Ruth Herzog for technical assistance and members of the laboratories for discussions. Confocal images were acquired at the Imaging Facility of the Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), Cologne, Germany.
This work was supported, in whole or in part, by National Institutes of Health Grant GM30997 (to M. C.). This work was also supported by Deutsche Forschungsgemeinschaft Grants SFB699 (to K. K.) and SFB572, SFB635, and BE2212 (to T. B.).
- NS
- nephrotic syndrome
- SRNS
- steroid-resistant nephrotic syndrome
- PHB
- prohibitin homology
- DRM
- detergent-resistant membrane fraction
- PNGase F
- peptide N-glycosidase F
- SL
- stomatin-like
- ACN
- acetonitrile
- ESI
- electrospray ionization
- OAG
- 1-oleoyl-2-acetyl-sn-glycerol
- DEG/ENaC
- degenerin/epithelial Na+ channel.
REFERENCES
- 1. Hinkes B. G., Mucha B., Vlangos C. N., Gbadegesin R., Liu J., Hasselbacher K., Hangan D., Ozaltin F., Zenker M., Hildebrandt F. (2007) Nephrotic syndrome in the first year of life. Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 119, e907–e919 [DOI] [PubMed] [Google Scholar]
- 2. Boute N., Gribouval O., Roselli S., Benessy F., Lee H., Fuchshuber A., Dahan K., Gubler M.-C., Niaudet P., Antignac C. (2000) NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 24, 349–354 [DOI] [PubMed] [Google Scholar]
- 3. Weber S., Gribouval O., Esquivel E. L., Morinière V., Tête M.-J., Legendre C., Niaudet P., Antignac C. (2004) NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and lowpost-transplant recurrence. Kidney Int. 66, 571–579 [DOI] [PubMed] [Google Scholar]
- 4. Ozer E. A., Aksu N., Erdogan H., Yavascan O., Kara O., Gribouval O., Gubler M.-C., Antignac C. (2004) A novel NPHS2 gene mutation in Turkish children with familial steroid-resistant nephrotic syndrome. Nephrology 9, 310–312 [DOI] [PubMed] [Google Scholar]
- 5. Caridi G., Perfumo F., Ghiggeri G. M. (2005) NPHS2 (Podocin) mutations in nephrotic syndrome. Clinical spectrum and fine mechanisms. Pediatr. Res. 57, 54R–61R [DOI] [PubMed] [Google Scholar]
- 6. Haraldsson B., Nyström J., Deen W. M. (2008) Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 88, 451–487 [DOI] [PubMed] [Google Scholar]
- 7. Pavenstädt H., Kriz W., Kretzler M. (2003) Cell biology of the glomerular podocyte. Physiol. Rev. 83, 253–307 [DOI] [PubMed] [Google Scholar]
- 8. Benzing T. (2004) Signaling at the slit diaphragm. J. Am. Soc. Nephrol. 15, 1382–1391 [DOI] [PubMed] [Google Scholar]
- 9. Kestilä M., Lenkkeri U., Männikkö M., Lamerdin J., McCready P., Putaala H., Ruotsalainen V., Morita T., Nissinen M., Herva R., Kashtan C. E., Peltonen L., Holmberg C., Olsen A., Tryggvason K. (1998) Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol. Cell 1, 575–582 [DOI] [PubMed] [Google Scholar]
- 10. Schwarz K., Simons M., Reiser J., Saleem M. A., Faul C., Kriz W., Shaw A. S., Holzman L. B., Mundel P. (2001) Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Invest. 108, 1621–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roselli S., Gribouval O., Boute N., Sich M., Benessy F., Attié T., Gubler M.-C., Antignac C. (2002) Podocin localizes in the kidney to the slit diaphragm area. Am. J. Pathol. 160, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Winn M. P., Conlon P. J., Lynn K. L., Farrington M. K., Creazzo T., Hawkins A. F., Daskalakis N., Kwan S. Y., Ebersviller S., Burchette J. L., Pericak-Vance M. A., Howell D. N., Vance J. M., Rosenberg P. B. (2005) A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308, 1801–1804 [DOI] [PubMed] [Google Scholar]
- 13. Reiser J., Polu K. R., Möller C. C., Kenlan P., Altintas M. M., Wei C., Faul C., Herbert S., Villegas I., Avila-Casado C., McGee M., Sugimoto H., Brown D., Kalluri R., Mundel P., Smith P. L., Clapham D. E., Pollak M. R. (2005) TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 37, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huber T. B., Schermer B., Müller R. U., Höhne M., Bartram M., Calixto A., Hagmann H., Reinhardt C., Koos F., Kunzelmann K., Shirokova E., Krautwurst D., Harteneck C., Simons M., Pavenstädt H., Kerjaschki D., Thiele C., Walz G., Chalfie M., Benzing T. (2006) Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc. Natl. Acad. Sci. 103, 17079–17086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hiebl-Dirschmied C. M., Entler B., Glotzmann C., Maurer-Fogy I., Stratowa C., Prohaska R. (1991) Cloning and nucleotide sequence of cDNA encoding human erythrocyte band 7 integral membrane protein. Biochim. Biophys. Acta 1090, 123–124 [DOI] [PubMed] [Google Scholar]
- 16. Browman D. T., Hoegg M. B., Robbins S. M. (2007) The SPFH domain-containing proteins. More than lipid raft markers. Trends Cell Biol. 17, 394–402 [DOI] [PubMed] [Google Scholar]
- 17. Huber T. B., Simons M., Hartleben B., Sernetz L., Schmidts M., Gundlach E., Saleem M. A., Walz G., Benzing T. (2003) Molecular basis of the functional podocin-nephrin complex. Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 12, 3397–3405 [DOI] [PubMed] [Google Scholar]
- 18. Huber T. B., Benzing T. (2005) The slit diaphragm. A signaling platform to regulate podocyte function. Curr. Opin. Nephrol. Hypertens. 14, 211–216 [DOI] [PubMed] [Google Scholar]
- 19. Schermer B., Benzing T. (2009) Lipid-protein interactions along the slit diaphragm of podocytes. J. Am. Soc. Nephrol. 20, 473–478 [DOI] [PubMed] [Google Scholar]
- 20. Ekim M., Ozçakar Z. B., Acar B., Yüksel S., Yalçnkaya F., Tulunay O., Ensari A., Erbay B. (2004) Three siblings with steroid-resistant nephrotic syndrome: New NPHS2 mutations in a Turkish family. Am. J. Kidney Dis. 44, e22–e24 [DOI] [PubMed] [Google Scholar]
- 21. Berdeli A., Mir S., Yavascan O., Serdaroglu E., Bak M., Aksu N., Oner A., Anarat A., Donmez O., Yildiz N., Sever L., Tabel Y., Dusunsel R., Sonmez F., Cakar N. (2007) NPHS2 (podicin) mutations in Turkish children with idiopathic nephrotic syndrome. Pediatr. Nephrol. 22, 2031–2040 [DOI] [PubMed] [Google Scholar]
- 22. Zhang S., Arnadottir J., Keller C., Caldwell G. A., Yao C. A., Chalfie M. (2004) MEC-2 is recruited to the putative mechanosensory complex in C. elegans touch receptor neurons through its stomatin-like domain. Curr. Biol. 14, 1888–1896 [DOI] [PubMed] [Google Scholar]
- 23. Bouchireb K., Boyer O., Gribouval O., Nevo F., Huynh-Cong E., Morinière V., Campait R., Ars E., Brackman D., Dantal J., Eckart P., Gigante M., Lipska B. S., Liutkus A., Megarbane A., Mohsin N., Ozaltin F., Saleem M. A., Schaefer F., Soulami K., Torra R., Garcelon N., Mollet G., Dahan K., Antignac C. (2014) NPHS2 mutations in steroid-resistant nephrotic syndrome. A mutation update and the associated phenotypic spectrum. Hum. Mutat. 35, 178–186 [DOI] [PubMed] [Google Scholar]
- 24. Huber T. B., Kottgen M., Schilling B., Walz G., Benzing T. (2001) Interaction with podocin facilitates nephrin signaling. J. Biol. Chem. 276, 41543–41546 [DOI] [PubMed] [Google Scholar]
- 25. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. (2012) Fiji. An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 27. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 28. Letunic I., Doerks T., Bork P. (2012) SMART 7. Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edgar R. C. (2004) MUSCLE. Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Jalview Version 2. A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kadurin I., Huber S., Gründer S. (2009) A single conserved proline residue determines the membrane topology of stomatin. Biochem. J. 418, 587–594 [DOI] [PubMed] [Google Scholar]
- 32. Eckhardt M., Fewou S. N., Ackermann I., Gieselmann V. (2002) N-glycosylation is required for full enzymic activity of the murine galactosylceramide sulphotransferase. Biochem. J. 368, 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rho S., Lee H. M., Lee K., Park C.-S. (2000) Effects of mutation at a conserved N-glycosylation site in the bovine retinal cyclic nucleotide-gated ion channel. FEBS Lett. 478, 246–252 [DOI] [PubMed] [Google Scholar]
- 34. Roselli S., Moutkine I., Gribouval O., Benmerah A., Antignac C. (2004) Plasma membrane targeting of podocin through the classical exocytic pathway. Effect of NPHS2 mutations. Traffic 5, 37–44 [DOI] [PubMed] [Google Scholar]
- 35. Goodman M. B., Ernstrom G. G., Chelur D. S., O'Hagan R., Yao C. A., Chalfie M. (2002) MEC-2 regulates C. elegans DEG/ENaC channels needed for mechanosensation. Nature 415, 1039–1042 [DOI] [PubMed] [Google Scholar]
- 36. Völker L. A., Schurek E.-M., Rinschen M. M., Tax J., Schutte B. A., Lamkemeyer T., Ungrue D., Schermer B., Benzing T., Höhne M. (2013) Characterization of a short isoform of the kidney protein podocin in human kidney. BMC Nephrol. 14, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson M., Kim E. Y., Hagmann H., Benzing T., Dryer S. E. (2013) Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am. J. Physiol. Cell Physiol. 305, C276–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brand J., Smith E. S., Schwefel D., Lapatsina L., Poole K., Omerbašić D., Kozlenkov A., Behlke J., Lewin G. R., Daumke O. (2012) A stomatin dimer modulates the activity of acid-sensing ion channels. EMBO J. 31, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]