

Background: 26S proteasome complex is highly dependent on ATP.
Results: NADH binds the proteasome via the Psmc1 subunit resulting in ATP-independent stabilization of the 26S proteasome complex, in vitro and in cells.
Conclusion: NADH is a novel regulator of the 26S proteasome.
Significance: NADH can maintain proteasomal integrity in the absence of ATP, linking cellular redox state to protein degradation.
Keywords: ATP-dependent Protease, ATPases, Magnesium, NADH, Proteasome, Protein Degradation, Psmc1, Rotenone
Abstract
The 26S proteasome is the end point of the ubiquitin- and ATP-dependent degradation pathway. The 26S proteasome complex (26S PC) integrity and function has been shown to be highly dependent on ATP and its homolog nucleotides. We report here that the redox molecule NADH binds the 26S PC and is sufficient in maintaining 26S PC integrity even in the absence of ATP. Five of the 19S proteasome complex subunits contain a putative NADH binding motif (GxGxxG) including the AAA-ATPase subunit, Psmc1 (Rpt2). We demonstrate that recombinant Psmc1 binds NADH via the GxGxxG motif. Introducing the ΔGxGxxG Psmc1 mutant into cells results in reduced NADH-stabilized 26S proteasomes and decreased viability following redox stress induced by the mitochondrial inhibitor rotenone. The newly identified NADH binding of 26S proteasomes advances our understanding of the molecular mechanisms of protein degradation and highlights a new link between protein homeostasis and the cellular metabolic/redox state.
Introduction
The proteasomes are large proteolytic complexes that can be classified into two main functional particles; the 26S and 20S proteasome complexes (PCs).3 The 26S proteasomes are comprised of the 20S cylindrical catalytic core structure responsible for the peptide bond cleavage and of the regulatory 19S regulatory particle (PA700). The latter is active in the recognition of the polyubiquitinated proteins, deubiquitination, unfolding, and feeding of the substrate into the catalytic chamber where they are degraded (1, 2). Over the years, great insight has been gained on the regulation of proteasome mediated protein degradation, mainly focusing on the ubiquitin pathway and the core catalytic activity of the 20S proteasome. However, the regulated alteration of the 26S/20S proteasomal complex ratio in the cell could also result in a significant protein homeostasis alteration. The 26S/20S ratio in the cell is influenced by the rate of association and dissociation of the 26S proteasome complex. Numerous studies shed light on the chaperone regulation of the 26S proteasome assembly process (3); however, little is known about the cellular factors contributing to 26S proteasome integrity and stability.
In recent years, there has been a growing appreciation of the reciprocal regulation of metabolism and cellular signaling. Several cellular signaling cascades are regulated by the availability of metabolic molecules such as acetyl-CoA (4), UDP-GlcNAc (5) and NAD+ (6). NADH was initially regarded as metabolic cofactor in many key cellular processes such as glycolysis and anaerobic respiration. NADH is consumed when pyruvate is converted to lactate and also in the mitochondria where three of the rate-limiting enzymes in the TCA cycle utilize NADH/NAD+ as cofactors and the oxidation of NADH drives the electron transfer chain leading to ATP synthesis. However, lately, the NADH/NAD+ ratio was shown to function as a metabolic regulatory switch as well. NADH/NAD+ level were shown to be a key component in the epigenetic regulation cascades regulating histone expression (7) and modification (8). Both NAD+ and NADH were shown to directly regulate transcription by binding transcription factors such as p53, p38/GAPDH and Clock:BMAL1 (7, 9, 10), or co-repressor CtBP (11) and affecting their function in involved in regulation of complex biological process such as circadian oscillation (12).
NAD(H) has also been linked to regulating proteasomal degradation as a substrate of NAD(H) utilizing enzymes. NQO1, an NADH-dependent oxidoreductase, has been shown to bind to the 20S proteasome mediating degradation of several client proteins (13). poly-ADP-ribose polymerase has also been shown to associate with the proteasome and directly regulate its function (14, 15). Given the indirect role of NAD(H) in proteasomal function we set out to explore whether NAD(H) can directly bind and regulate proteasomal function.
EXPERIMENTAL PROCEDURES
Animals Cells and Materials
Male ICR mice were used in this study. The cells used in this study include HEK293, HCT116, and NIH3T3 cells that were cultured in DMEM and 10% FBS at 37 °C in a humidified incubator with 5.6% CO2. HEK293(T) cells were transfected by the calcium phosphate method. NAD+, NADH, NADPH, MG132, ATP, and ATPγS were purchased from Sigma.
Construction of the HCT116 and NIH3T3 Stably Overexpressing Psmc1 WT and ΔGxGxxG Mutant Cell Lines
Transducing lentiviral particles were produced in HEK293(T) cells according to the manufacturer's instuctions. HCT116 cells were infected and selected with 1 μg/ml puromycin for a week before experiments were initiated.
Cellular Protein Extraction from Mouse Tissues
Tissue extracts for the proteasomal complex stability assay were taken from tissues of 3-month-old male ICR mice that were homogenized in native buffer without ATP. The extracts were then subjected to centrifugation 13,000 × g for 15 min, and then the supernatant was analyzed for proteasomal composition or subjected for proteasomal complex stability assay. For the analysis of the 26S/20S PC ratio, mouse organs were homogenized in Native buffer and analyzed by native gel.
Proteasomal Complex Stability Assay
Mammalians 26S proteasomes were purified from rabbit muscles as described previously (16), and purified human 26S proteasomes were provided by Dr. Yuval Reiss. Proteasomal Pellets from NIH3T3 cells or purified 26S proteasomes from rabbit muscles were resuspended in Deg buffer (50 mm Tris, pH 7.5, 150 mm NaCl, and 5 mm MgCl2) supplemented with various concentrations of ATP, NAD+ and NAD(P)H in the presence or absence of 8 milliunits/μl apyrase (Sigma). Proteasomal stability in tissue extracts was conducted by incubating tissue extract (3–4 μg/μl concentration) in the presence of 8 milliunits/ml apyrase. In the MgCl2 sensitivity assay, tissues were extracted in minimal buffer (20 mm Tris, pH 7.5, 1 mm DTT, 250 mm sucrose) and MgCl2 was added at the indicated concentrations. In all cases, the mixtures were incubated at 37 °C for the indicated times and then loaded on nondenaturing 4% polyacrylamide gel for detection of proteasomal complex stability (described below).
Nondenaturing PAGE
Proteasomal samples were loaded on a nondenaturing 4% polyacrylamide gel using the protocol described previously (17). Gels were either overlaid with Suc-LLVY-AMC (50 μm) for assessment of proteasomal activity or transferred to nitrocellulose membranes where immunoblotting specific for proteasomal subunits was conducted or stained with Gelcode blue stain reagent (Thermo Fisher Scientific) and dried on a vacuum drier. Proteasomal activity was assessed by measuring the hydrolysis of Suc-LLVY-AMC by substrate overlay assays in native polyacrylamide gels and detected by ImageQuant LAS 4000 (GE Healthcare).
Proteasomal Activity
Proteasomal activity was assessed by measuring the hydrolysis of Suc-LLVY-AMC either in solution where cellular extracts were incubated in 96-well plates, and the kinetics were measured over time as described in manufacturer's instructions (Biomol) or by substrate overlay assays in native polyacrylamide gels and detected by ImageQuant LAS 4000 (GE Healthcare).
Protein Extraction and Immunoblot Analysis
For immunoblotting, protein extraction from different cells was performed by resuspending cells in RIPA buffer as described previously (18). The extract was subjected to ultracentrifugation (13,000 × g for 15 min), and the supernatant was used as the protein extract. Protein concentrations were determined by Bradford assay (Bio-Rad). The protein mix was mixed with Laemmli sample buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, and 0.125 m Tris-HCl), heated at 95 °C for 5 min, and loaded on a 10% polyacrylamide-SDS gel. Following electrophoresis, proteins were transferred to cellulose nitrate 0.45-mm membranes (Schleicher & Schuell). The antibodies used were as follows: rabbit anti Psma4 (Prof. Chaim Kahana), Psmd1 (Acris), and HRP-conjugated His (Promega) and actin (Santa Cruz Biotechnology). Secondary antibodies were HRP-linked goat anti-mouse and rabbit (Jackson ImmunoResearch Laboratories). Signals were detected using the Ez-ECL kit (Biological Industries).
Identification of NADH Motif Search and Its Evolutionary Conservation in Proteasome Subunits
To identify an NADH-binding motif, we used a previously published data set of proteins that were found to bind NADH and are unrelated to the proteasome subunits (19). Using the structures of these proteins in complex with the NADH (after filtering redundant proteins having at least 40% sequence similarity), we identified which residues are in contact with NADH. To reduce the noise and get the most relevant motif, we extracted from each protein windows of residues that contained at least four NAD(H) interacting residues in a consecutive sequence of 6 residues. We extended the window to either 12 or 14 amino acids (centered on the original six-amino acid window) to try and determine a more accurate motif, which may include some of the surrounding residues. We used the program MEME (from the Motif-based sequence analysis suit (20)) with the default parameters, to obtain motifs using our set.
We extracted eukaryotic orthologs of the five proteasome subunits that were found to contain the GxGxxG box using the Inaparanoid program (21) and filtered them to get only highly significant orthologs, using the Inparanoid score. In cases where two or more orthologs were found to have high scores in a given species, we used BLAST (22).
Cloning of the Mutant Psmc1
The WT and ΔGxGxxG Psmc1 mutant were cloned into Pcdna3 vector with a FLAG at the C terminus. The primers used were as follows: for the WT Psmc1, AATTAAGCTTGCCACCATGGGTCAAAGTCAGAGTGGT (forward) AATTTCTAGA-TTACTTGTCATCGTCGTCCTTGTAGTCGAGATACAGCCCCTC (reverse) and for the ΔGxGxxG mutant, ATTAAGCTTGCCACCATGGGTCAAAGTCAGAGTAAGAAGGATGAC (forward) and same as WT (reverse). The template human Psmc1 in a Pcdna3 vector was kindly received from Prof. Shigeo Murata. The PCR product was inserted into pCDNA3 using XbaI and HindIII. The Psmc1 and ΔGxGxxG mutant were then transferred to the gateway® system and introduced into the plenti6 vector (according to manufacturer's instructions) that was used for constructing the HCT116 stably overexpressing cell line.
ATPase Activity Assay
Purified proteasomes were diluted in 50 μl of Deg buffer with indicated concentrations of ATP and NADH and incubated at 37 °C for 1 h. The reaction was stopped by the addition of 50 μl of solution containing 12% SDS. 100 μl of 1:1 mix with 6% ascorbic acid in 1 m HCl and 1% ammonium molybdate were added to the sample and incubated between 3–7 min at room temperature followed by the addition of 150 μl of solution containing 2% sodium citrate, 2% sodium metaarsenite, and 2% acetic acid. Following incubation at 37 °C for 10 min, the amount of inorganic phosphate released was determined by measurement of the absorption at 850 nm.
Psmc1 Subunit Purification
hPsmc1 (and mutant) were overexpressed in Escherichia coli BL21 (DE3) pLysS strain using pRSET B (Invitrogen). Protein was induced at mid-log phase by the addition of 1 mm of isopropyl β-d-1-thiogalactopyranoside and incubating for a further 3 h at 30 °C. Cells were harvested and then lysed by resuspension in extraction buffer (50 mm Tris, pH 8.0, 350 mm NaCl, 20 mm imidazole, 5 mm β-mercaptoethanol, and 1 mm PMSF) incubating (4 °C, 1 h) with 1 mg/ml lysozyme and sonication (X10 10-s pulse, with 40-s intervals). Extract was incubated with 0.01% Triton X-100 (10 min at 4 °C), and insoluble debris was removed by centrifugation. Cell lysate was combined with pre-equilibrated NI-NTA agarose resin, and binding was allowed by gentle shaking overnight at 4 °C. Purification was performed using a chromatography column, and lysate and resin mixture was washed (2×) with 5 bed volumes of wash buffer (50 mm Tris, pH 8, 0.01% Triton X-100, 350 mm NaCl, 20 mm imidazole, 5 mm β-mercaptoethanol, 1 mm PMSF, pH 8) and again (2×) with 5 bed volumes of wash buffer (without Triton X-100 and 40 mm imidazole). Protein was eluted by adding 4 bed volumes of elution buffer (50 mm Tris, pH 8, 350 mm NaCl, 280 mm imidazole) and collecting elution fractions. Elution fractions were analyzed by SDS-PAGE and dialyzed against buffer A (10 mm Tris, pH 7.5, 50 mm NaCl, 2 mm DTT) Protein concentration was estimated by Bradford assay.
NADH/NAD+ Level Quantification
We used the NAD+/NADH quantification kit from BioVision according to the manufacturer's instructions.
NADH Fluorescence Assay
Fluorescence intensity (in arbitrary units) was measured by infinite® 200 multifunctional microplate reader (TECAN) using a NUN96fb LumiNunc/FluoroNunc plate and by a spectrofluorometer (Fluorolog®-3) using a quartz cuvette. Samples were excited at 340 nm and emission spectra between 390 nm to 530 nm in 5-nm steps (1-nm steps when using the Fluorolog®-3) was analyzed. All reactions were performed in 10 mm Tris (pH 8) at room temperature by using indicated concentrations of His6 purified Psmc1 (and ΔGxGxxG mutant) and NADH. Samples were incubated and mixed (5–10′) at room temperature prior to each measurement. Fluorescence readings were corrected for background by subtracting the fluorescence contribution of the buffer and protein alone.
RESULTS
Predicted NADH Binding Motifs Are Found in 19S Proteasome Subunits
Several nucleotides including ATP were reported previously to support 26S PC activity (23). One intriguing overlooked candidate is the nicotinamide adenine dinucleotide (NAD(H)) that is an abundant cellular coenzyme utilized by the cells to transfer electrons. The coenzyme can be found in two forms, the oxidized form (NAD+) that can accept two electrons and be reduced to the second form (NADH) that can later release the two electrons. Given the involvement of NAD(H) in proteasomal degradation (13, 14), we explored the possibility that NAD(H) regulates the proteasomal complex integrity and function.
The NADH binding motif was previously characterized by examination of known NAD(H)-dependent enzymes (24, 25). Given the increase in structural data since the initial NADH motif characterization, we first characterized the determinants of NADH binding regions using motif discovery techniques on an unrelated set of NADH-binding proteins with a structure containing an NAD(H) molecule available in the Protein Data Bank (see “Experimental Procedures” for detailed description). The resulting motif, a GxGxxG box, with some preference to hydrophobic residues prior to the glycines (Fig. 1A), was consistent with previous findings (24). This motif is found in five subunits of the human proteasome: Psmc1 (Rpt2), Psmd9 (p27), Psmd14 (Rpn11), Psmd3 (Rpn3), and Adrm1 (Rpn13), which are all located in the regulatory 19S complex. Interestingly, this motif is highly conserved throughout the vertebrate phylogeny in all subunits except for Psmd3, where the motif appears only in placental mammals (Fig. 1, B–F).
FIGURE 1.

The NADH binding motif in the proteasomal subunits. A, the predicted NADH binding consensus (GxGxxG) as determined by examining a large data set of resolved structures of protein complexes with NADH and by using the Meme motif discovery tool. B–F, the alignment of the NADH binding consensus containing proteasomal subunits Psmc1/Rpt2 (B), Psmd9/p27 (C), Psmd14/Rpn11 (D), Psmd3/Rpn3 (E), and Adrm1/Rpn13 (F) in various vertebrates. H. sapiens, Homo sapiens; F. pygmaeus, Flectonotus pygmaeus; C. porcellus, Cavia porcellus; M. mulatta, Macaca mulatta; C. familiaris, Canis familiaris; M. musculus, Mus musculus; R. norvegicus, Rattus norvegicus; B. taurus, Bos taurus; E. cabballus, Equus ferrus cabballus; M. domestica, Monodelphis domestica; O. anatinus, Ornithorhynchus anatinus; G. gallus, Gallus gallus; X. tropicalis, Xenopus tropicalis; T. rubripes, Takifugu rubripes; G. aculeatus, Gasterosteus aculeatus; T. nigroviridis, Tetraodon nigroviridis; O. latipes, Oryzias latipes; D. rerio, Danio rerio.
NADH Maintains 26S PC Integrity in the Absence of ATP
The 26S proteasomes are very sensitive to ATP depletion (26). Purified 26S proteasomes (Fig. 2A), and proteasomes that were fractionated from cellular extracts (Fig. 2B) exhibited complete dissociation in the absence of ATP in <30 min. As expected the addition of 2 mm ATP prevented the dissociation of the complex (Fig. 2B). Remarkably, NADH was sufficient to maintain the 26S proteasomal integrity and activity whereas, NAD+, was poorly active in this process (Fig. 2, C and D). The stabilization effects of NADH were observed at physiological concentrations of 10–100 μm NADH (Fig. 2E). We further examined the role of NADH, in maintaining the 26S proteasome complex integrity, by incubating purified 26S PC with apyrase to deplete residual ATP and then added either 2 mm NADH, NADPH (a reducing agent similar to NADH), or ATPγS, a non-hydrolysable ATP analog. The latter is known to stabilize the 26S PC (26) and was used as a positive control. NADH-dependent 26S PC was just as stable as the control ATPγS-26S PC (Fig. 2F). NADPH, harboring the same redox potential and structural similarity as NADH, did not stabilize the 26S PC, further indicating the selectivity toward NADH. Our results suggest that in the absence of ATP, NADH can stabilize the 26S PC at physiological concentrations.
FIGURE 2.
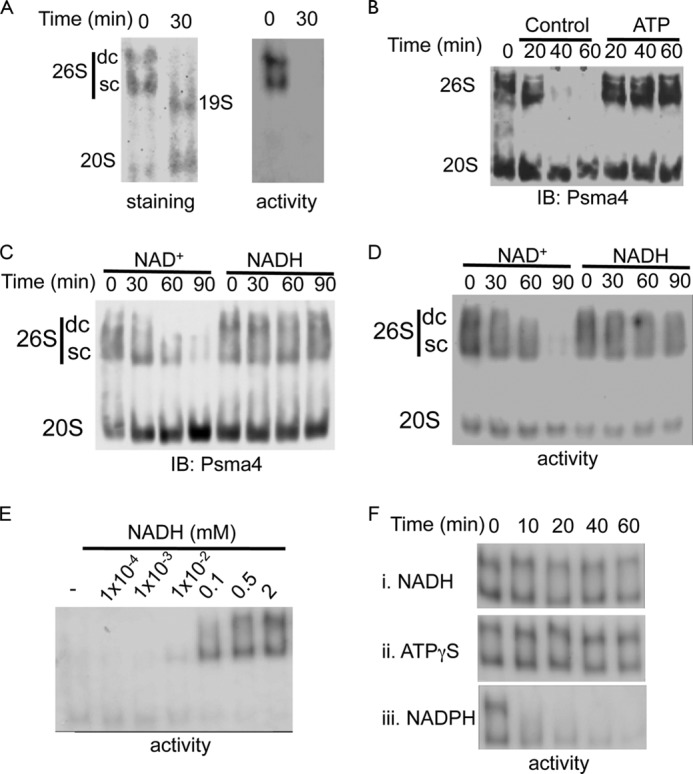
NADH maintains 26S proteasome complex integrity in the absence of ATP. A, purified 26S proteasomes were incubated at 37 °C for 30 min in the absence of ATP, and the 26S PC integrity was analyzed. B, proteasomes fractionated from NIH3T3 cells were incubated at 37 °C for the indicated time points in the absence or presence of 2 mm ATP. C and D, NADH stabilizes the 26S proteasomes. Proteasomes fractionated from NIH3T3 cells were incubated at 37 °C for indicated time in the presence or absence of 2 mm NADH or NAD+. Proteasome complex levels (C) and activity (D) were examined. E. NADH dose dependent stabilization of purified 26S proteasomes. Purified 26S PC were incubated in the presence of 8 milliunits/ml apyrase at 37 °C for 30 min in the presence of increasing concentrations of NADH. Proteasomal activity was analyzed. F, NADH stabilizes the 26S PC in the absence of ATP. In the presence of 8 milliunits/ml apyrase that depleted residual ATP, the purified active 26S proteasomes were stabilized only in the presence of NADH (i) but not NADPH (iii). Incubation with ATPγS (ii) was used as a positive control. SC, single-capped 26S; DC, double-capped 26S.
NADH Binds the 26S Proteasome as Revealed by Partial Trypsin Digest
As shown above, NADH stabilized the 26S PC, we next asked whether NADH physically bound to the 26S PC. To this end, we partially digested 26S PC with trypsin to detect possible conformational changes in the presence of NADH. Both NADH-26S and ATP-26S PCs are more resistant to partial trypsin digestion (Fig. 3A). Trypsin-digested proteasomes samples were separated by SDS-PAGE (Fig. 3B), and the partial resistant bands were analyzed by mass spectrometry. Interestingly, Psmc1 was identified as a protected subunit in both human and rabbit 26S PC. To further validate the interaction of the proteasomal subunits with NADH, we focused on the ATPase subunit Psmc1.
FIGURE 3.

NADH directly binds the Psmc1 subunit of the 26S proteasome. A, purified 26S proteasomes were incubated in the presence or absence of trypsin (5 ng/μl) for 10 min at 37 °C in the presence or absence of either NADH or ATP (2 mm). Levels of 26S proteasomal complexes were analyzed following native gel electrophoresis. B, purified rabbit or human 26S proteasomes were subjected to trypsin digest (5 ng/μl) in the presence or absence of 2 mm NADH. The indicated band was detected by mass spectrometry to be the Psmc1 subunit. C, schematic representation of the wild type and ΔGxGxxG Psmc1 mutant. D and E, the fluorescence of 600 nm NADH was analyzed following excitation at 340 nm in the presence of 1 μm bacterially expressed and purified Psmc1 (C) or in the presence of 200 nm either WT (gray) or ΔNADH mutant psmc1 (dashed line) (D). F and G, ATPase activity of purified 26S proteasomes. F, purified 26S proteasomes were incubated in the presence of either 1 mm ATP, 1 mm NADH, or both. ATPase activity was measured by quantifying the release of Pi following incubation at 37 °C for 1 h. G, purified proteasomes were incubated in Deg buffer in the presence of 100 μm ATP with or without increasing concentrations of NADH (as indicated).
To examine the possibility that free Psmc1 directly binds NADH, we prepared recombinant protein of the WT and a mutant lacking the ΔGxGxxG motif (Fig. 3C) and incubated it with NADH. The binding of NADH to protein was reported to be associated with an enhanced intensity of NADH fluorescence and a blue shift in its peak. The blue shift occurs when fluorescence molecules enter a more hydrophobic environment (27). Unbound NADH excited at 340 nm (or 355 nm) had an emission maximum at the 455 mm. Addition of recombinant Psmc1 caused a marked increase in NADH fluorescence as well as a shift in the emission maximum to 425 nm (Fig. 3D). Interestingly, purified recombinant Psmc1 ΔGxGxxG mutant was capable of neither increasing fluorescence nor inducing NADH blue shift (Fig. 3E). Given the binding of NADH to an ATPase subunit of the proteasome, we examined the effect of NADH on the ATPase activity of purified 26S proteasomes. As expected, NADH did not alter the ATPase activity of the 26S proteasomes (Fig. 3, F and G). Our results indicate that NADH binds the Psmc1 proteasomal subunit at a distinct site from the ATP binding. Although it does not affect the ATPase activity of the proteasome, it could nevertheless alter the nucleotide exchange pattern of ATPases that was suggested to regulate the degradation of structured proteins (28).
The Effect of ATP Depletion on PC Sensitivity in Mouse Tissue Extracts
Different organs exhibit differential protein and metabolic homeostasis. The levels of ATP and the ATP/NADH ratio varies between different tissues, with the muscle exhibiting high ATP levels and the brain exhibiting lower ATP levels and high NADH/NAD+ ratios (29). These differences in the ATP/NADH levels may induce changes in the proteasomal complex ratio. The three major proteasome complexes observed are the 20S, the single-capped, and double-capped 26S proteasome.
To examine whether the proteasomal complexes from the different tissues are differentially sensitive to ATP depletion, we lysed the tissues in buffer devoid of ATP and further treated the tissue extracts with apyrase to rapidly eliminate any residual ATP in the extract. The stability of the PCs was analyzed in these ATP-devoid conditions at 37 °C. The 26S PCs from tissue extracts were more stable in the absence of ATP and the addition of apyrase than the purified or cell fractionated proteasomes (Fig. 4, A–C). Interestingly, the double-capped 26S were much more sensitive to ATP depletion than the single-capped 26S with almost no double-capped 26S observed after 20 min of incubation at 37 °C. We also observed a slight differential decay of the 26S PCs in the different tissues following ATP depletion. Whereas in the liver and muscle, there is a sharp reduction in the level of active 26S PCs after 2-h incubation at 37 °C, the reduction in the brain was, however, more subtle (Fig. 4B). These data suggest that the proteasomal complexes can maintain stability in the absence of ATP in a tissue-specific manner, a process that might be partially mediated by NADH availability.
FIGURE 4.
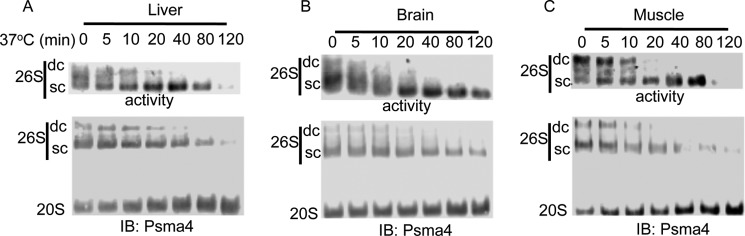
Differential sensitivity of the 26S from different tissues to ATP depletion by apyrase. A–C, equal amounts of protein extracts from different tissues were ATP depleted with the addition of 8 milliunits/μl of apyrase. 26S proteasome activity (upper panel) and 26S/20S PC ratio were analyzed at indicated time points after the addition of apyrase. The organs analyzed by this method were liver (A), brain (B), and muscle (C). IB, immunoblot.
NADH Stabilization of the 26S Proteasome Complex Is Sensitive to High MgCl2 Levels
MgCl2 is required for various ATP-dependent activities of the 26S PC. We examined whether MgCl2 is needed for the NADH stabilization of the 26S PC. Initially, we examined the stability of the 26S PC in the presence of increasing concentrations of ATP and NADH in the presence of standard 10 mm MgCl2. Surprisingly, 2 mm NADH failed to maintain 26S PC (Fig. 5A). In contrast, ATP stabilization of the 26S PC was not altered under this condition (Fig. 5B). Next, we examined the effect of increasing concentrations of MgCl2. MgCl2 at higher concentrations (>10 mm) inhibited the stabilizing effect of NADH (Fig. 5C) but not of ATP (Fig. 5D). However, in the presence of high MgCl2 concentration (20 mm), increasing the concentration of NADH (10–25 mm) can overcome the inhibitory effect of MgCl2 on the NADH-26S PC integrity (Fig. 5E). Thus, the ionic requirement of NADH-26S PC stabilization is different from that of ATP-26S PC. We further followed NADH fluorescence in the presence of the recombinant Psmc1 and MgCl2. The addition of MgCl2 completely eliminated the NADH blue shift that Psmc1 induced (Fig. 5F).
FIGURE 5.

NADH stabilizing effect on the 26S PC is sensitive to MgCl2. A and B, purified 26S proteasomes were incubated in the presence of either increasing concentrations of NADH (A) or increasing concentrations of ATP (B) in the presence of 10 mm MgCl2 for 30 min at 37 °C (input, 0-min incubation). 26S PC levels were determined by on a gel activity assay. C and D, purified 26S proteasomes were incubated in the presence of either 2 mm NADH (C) or 2 mm ATP (D) and increasing concentrations of MgCl2. 26S PC levels and activity were analyzed. E, MgCl2 inhibition of the NADH-26S PC is reversible. Purified 26S PC were incubated in the presence of 20 mm MgCl2 (with the exception of lane 8 that had 2 mm MgCl2) and 8 milliunits/ml of apyrase (except in lane 9) and increasing concentrations of NADH or 2 mm ATP (9) or ATPγS (10) as positive controls. F, MgCl2 inhibits NADH binding to Psmc1. The fluorescence of 600 nm NADH was analyzed in the presence of 200 nm Psmc1 (gray line) with or without 20 mm MgCl2 (black dashed line). G–I, ATP-depleted stable 26S PC is of NADH type. The reaction was conducted under minimal buffer (20 mm Tris, pH 7.5, 250 mm sucrose, and 1 mm DTT) in the presence or absence of MgCl2. Equal amount of protein extract devoid of ATP were incubated at 37 °C for 40 min in the presence or absence of indicated concentrations of MgCl2 (0–20 mm). The organs analyzed were liver (D), brain (E), and muscle (F). IB, immunoblot.
Next, we utilized the selective effect of MgCl2 at high concentrations to discriminate between ATP- and NADH- proteasomal subgroups in different tissues. Muscle, liver, and brain extracts were prepared in the absence of ATP and analyzed following incubation at 37 °C for 40 min in the presence of increasing concentrations of MgCl2 (Fig. 5, G–I). Interestingly, the obtained 26S PC were very sensitive to MgCl2, suggesting NADH might be involved in the stabilization of certain fraction of the 26S PC in these tissues, in agreement with the apyrase experiments described in Fig. 4.
Increasing the NADH/NAD+ Ratio with Mild Mitochondrial Inhibition Results in Elevated 26S Proteasome Levels
We next set out to explore whether alteration of the cellular NADH/NAD+ levels and ratio could directly effect the 26S PC integrity. For this purpose, NIH3T3 cells were initially incubated with either 0.1 or 0.5 mm NAD+ for 16 h. The addition of NAD+ to the media increased the levels and activity of the 26S proteasomes (Fig. 6, A and B). The increase in the 26S proteasome levels was most likely due to altered assembly or stability of the complex as there was no detected change in the subunit expression (Fig. 6C). To further induce an increase in the NADH/NAD+ ratio, we utilized the mitochondrial complex I inhibitor, rotenone. Exposure of NIH3T3 cells to a mild concentration of rotenone (50 nm) resulted in a significant increase in the NADH/NAD+ ratio (Fig. 6D) and to a striking increase in the 26S proteasome levels and activity (Fig. 6E). Under these conditions, no change in the subunit expression was detected (Fig. 6F). These results suggest that NADH might regulate the proteasomal complex dynamics in the cells.
FIGURE 6.

Increasing the NADH/NAD+ levels results in increased 26S proteasome levels in cells. A–C, NIH3T3 cells were treated with 0.1 or 0.5 mm NAD+ for 16 h, and 26S/20S PC levels (A), activity (B), and subunit expression levels (C) were analyzed. D–F, NIH3T3 cells were treated with 50 nm rotenone for 16 h, and the cellular NADH/NAD+ ratio, proteasome levels, and subunit expression were analyzed. Proteasomal complexes were analyzed by both non-denaturing (A and E) and SDS-PAGE (C and F). IB, immunoblot.
The ΔGxGxxG Psmc1 Mutant Incorporates in the 26S PCs
The inability of the ΔGxGxxG Psmc1 mutant to bind NADH in vitro encouraged us to explore whether the ΔGxGxxG Psmc1 mutant incorporates into the 26S PC. To test this possibility, we immunoprecipitated FLAg-tagged wild type and ΔGxGxxG Psmc1 from human cells and examined whether the whole 26S PC co-immunoprecipitated in the presence of either ATP or NADH. In the presence of ATP, both wild type and ΔGxGxxG Psmc1 formed active 26S PCs (ATP-26S PC), indicating their efficient incorporation into the 26S PC (Fig. 7, A and B). In sharp contrast, in the presence of NADH, the ΔGxGxxG Psmc1 mutant did not form stable and active NADH-26PCs. These results suggest that in the transfected cells the ΔGxGxxG Psmc1 mutant is well incorporated into the 26S PC.
FIGURE 7.

Cells expressing the ΔGxGxxG Psmc1 mutant are less viable following rotenone (Rot) treatment. A, FLAG-tagged WT and ΔGxGxxG mutant Psmc1 were overexpressed in HCT116 cells. Proteasomal complexes were immunoprecipitated with FLAG beads in the presence of either 2 mm ATP or NADH. Proteasomal complexes were further eluted with FLAG peptide and analyzed by both non-denaturing and SDS-PAGE. B, proteasomal activity, as determined by the cleavage of the Suc-LLVY-AMC peptide, was analyzed following the immunoprecipitation of the proteasomal complexes with either the WT or ΔGxGxxG Psmc1 mutant in the presence of either ATP or NADH. C and D, NIH3T3 (C) or HCT116 (D) cells stably overexpressing the WT or ΔGxGxxG Psmc1 subunit were analyzed for cell viability 24 h following treatment with indicated concentrations of rotenone (0.1–0.75 μm). IB, immunoblot.
Cells Expressing the ΔGxGxxG Psmc1 Mutant Are Less Viable under Mitochondrial Inhibition
Having demonstrated the presence of ΔGxGxxG Psmc1 mutant 26S PCs in the cells, we next set out to examine the possible physiological role of this 26S-PC mutant. To this end, we stably overexpressed wild type Psmc1 and the ΔGxGxxG Psmc1 mutant in both HCT116 and NIH3T3 cells. The overexpression of the Psmc1 forms did not result in any evident effect on cellular viability under normal conditions (data not shown). However, the ΔGxGxxG Psmc1 mutant cells were significantly less viable following treatment with mild concentrations of rotenone for 24 h in both NIH3T3 (Fig. 7C) and HCT116 cells (Fig. 7D). The observed effect of the Psmc1 ΔGxGxxG mutant, is striking given the fact that the mutant is expressed in the background of the endogenous wild type subunit, and therefore, only a fraction of the 26S PC are expected to carry the mutant. These data suggest that the Psmc1 GxGxxG box involved in NADH binding regulates cell viability.
DISCUSSION
The ability of NADH to maintain 26S proteasome integrity and function raises the mechanistic question of how this could be achieved. NADH was not oxidized during the incubation (data not shown), suggesting that NADH is a 26S PC stabilizing cofactor and not the substrate of an unknown enzyme that facilitates proteasome activity. However, it is not clear whether NADH mediates a distinct function by interaction with a unique domain(s) of the proteasomal complex or is it achieved by nonspecific interacting with the ATP-binding domains. We provide several lines of evidence to rule out the nonspecific possibility. First, we have characterized an NADH binding motif GxGxxG that is present in five subunits of the 19S proteasome complex. Only Psmc1 (Rpt2) binds ATP. Characterization of the NADH interaction with recombinant Psmc1 confirmed that the NADH binding is mediated by the identified putative motif and not by the ATP binding pocket. Second, the four additional proteasomal subunits containing the predicted NADH binding motif (Adrm1/Rpn13, Psmd3/Rpn3, Psmd9/p27, and Psmd14/Rpn11) are likely to bind NADH. Psmc1 and Adrm1 are in the base of the 19S regulatory particle, whereas Psmd3 and Psmd14 are located in the lid. The recently solved structure of the 19S complex suggested that Psmd3 and Psmd14 are extended to the base toward the ATPase. The scaffolding function of the lid, Psmd1, and the ATPase position the Adrm1 (ubiquitin receptor) and Psmd14 (DUB) subunits in the central pore of the AAA+ motor, enabling substrate binding and deubiqutination (30). This “strategic” location of the NADH binding subunits might indicate that NADH interaction can strongly affect the degradation specificity of the proteasomal complex. Third, the vast excess of NADH does not affect 26S PC ATPase activity. This rules out the possibility that NADH binds the ATP pocket of the triple A ATPase (Psmc) subunits.
Psmc1 belongs to the AAA+ superfamily of proteins that consist of an α/β Rossman fold that was shown to mediate nucleotide binding, including NAD(H). Interestingly, the Psmc1 N terminus region containing the NADH binding motif is not part of this fold and is not predicted to have a defined secondary structure. The Psmc1 subunit also harbors the conserved HbYX motif that docks into the 20S proteasome regulating gate opening (31) and was shown to be sufficient to induce the activation of gate opening and degradation by the 20S proteasome (32). Thus, the binding of NADH to the Psmc1 subunit cannot only induce the stabilization of the 26S proteasome but also can potentially result in allosteric regulation of the HbYX-mediated gate opening and substrate specificity of the 26S proteasome. The ATP binding to the ATPase ring is a regulated process whereby the binding of an ATP to the ATPase subunit allosterically alters the ATP affinity of the neighboring subunit. This mechanism was suggested to induce directional cycling of the ATP hydrolysis in the hexameric ATPase ring (28). NADH, by binding to Psmc1, might mediate the initial association of ATP and alter the affinity of ATP to the different ATPase inducing the initial directionality of the ATP hydrolysis cycle.
The stabilization of the 26S proteasome complex by NADH is observed at the 100 μm concentration similar to the ATP concentration required for the same task. The Kd of Psmc1 to NADH as we measured by the extrapolation of the Lineweaver-Burk plot resulted in the 90 nm range (data not shown). This Kd is very close to the Kd of the transcriptional repressor CtBP with NADH that was estimated at 66 nm (33). The higher concentration of NADH that is required for the stabilization of the complex versus the binding to the purified Psmc1 subunit suggests that additional cellular factors might be involved in inhibiting and facilitating this processes such as we show for magnesium as an inhibitory agent.
Protein homeostasis and proteasomal degradation in particular are highly linked to the cellular metabolic/redox state, possibly allowing a more efficient degradation of a subset of proteins. However, to date, little is known on the endogenous metabolic/redox factors that might mediate these processes. The redox state of the cell as induced by oxidative stress has been strongly linked to proteasome complex impairment. Following hydrogen peroxide treatment, the 26S PC integrity is lost, and at acute concentrations, the activity is compromised as well (34, 35). It was suggested that ROS and direct oxidation of the proteasome could be involved in this process. In yeast, it was also shown that the 26S proteasomes integrity is compromised during stationary phase, and the extent of this process dictates the ability to renter cell cycle (36). In neurons, stimulation with NMDA results in a striking reduction only of the 26S proteasome levels (37). Mitochondrial inhibitor rotenone, which we described in our work, was also shown at different concentrations and cells to inactivate the proteasome function by reducing ATP (38). Moreover, the altered cross-talk between mitochondria and proteasome activity has been suggested to be involved in the onset of Parkinson disease (39–41). In all of the cases above, the alteration of the metabolic/redox state in the cell had a profound effect on proteasomal complex integrity and function by a mechanism poorly understood. The possible involvement of NADH in maintaining proteasomal complex integrity could be instrumental to further understand the regulation of protein homeostasis under metabolic/redox stress.
Acknowledgments
We acknowledge Shigeo Murata for human Psmc1 plasmid and Chaim Kahana for the Psma4 antibody and instructive discussions.
This work was supported by Israel Science Foundation Grant 551/11.
- PC
- proteasome complex.
REFERENCES
- 1. Baumeister W., Walz J., Zühl F., Seemüller E. (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92, 367–380 [DOI] [PubMed] [Google Scholar]
- 2. Coux O., Tanaka K., Goldberg A. L. (1996) Structure and functions of the 20 S and 26 S proteasomes. Annu. Rev. Biochem. 65, 801–847 [DOI] [PubMed] [Google Scholar]
- 3. Murata S., Yashiroda H., Tanaka K. (2009) Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 10, 104–115 [DOI] [PubMed] [Google Scholar]
- 4. Yi C. H., Pan H., Seebacher J., Jang I. H., Hyberts S. G., Heffron G. J., Vander Heiden M. G., Yang R., Li F., Locasale J. W., Sharfi H., Zhai B., Rodriguez-Mias R., Luithardt H., Cantley L. C., Daley G. Q., Asara J. M., Gygi S. P., Wagner G., Liu C. F., Yuan J. (2011) Metabolic regulation of protein N-α-acetylation by Bcl-xL promotes cell survival. Cell 146, 607–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fang M., Shen Z., Huang S., Zhao L., Chen S., Mak T. W., Wang X. (2010) The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 143, 711–724 [DOI] [PubMed] [Google Scholar]
- 6. Lin S. J., Defossez P. A., Guarente L. (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128 [DOI] [PubMed] [Google Scholar]
- 7. Zheng L., Roeder R. G., Luo Y. (2003) S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 114, 255–266 [DOI] [PubMed] [Google Scholar]
- 8. Zhang T., Berrocal J. G., Frizzell K. M., Gamble M. J., DuMond M. E., Krishnakumar R., Yang T., Sauve A. A., Kraus W. L. (2009) Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 284, 20408–20417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McLure K. G., Takagi M., Kastan M. B. (2004) NAD+ modulates p53 DNA binding specificity and function. Mol. Cell. Biol. 24, 9958–9967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rutter J., Reick M., Wu L. C., McKnight S. L. (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293, 510–514 [DOI] [PubMed] [Google Scholar]
- 11. Zhang Q., Piston D. W., Goodman R. H. (2002) Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897 [DOI] [PubMed] [Google Scholar]
- 12. Peek C. B., Affinati A. H., Ramsey K. M., Kuo H. Y., Yu W., Sena L. A., Ilkayeva O., Marcheva B., Kobayashi Y., Omura C., Levine D. C., Bacsik D. J., Gius D., Newgard C. B., Goetzman E., Chandel N. S., Denu J. M., Mrksich M., Bass J. (2013) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asher G., Tsvetkov P., Kahana C., Shaul Y. (2005) A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 19, 316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho-Park P. F., Steller H. (2013) Proteasome regulation by ADP-ribosylation. Cell 153, 614–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ullrich O., Reinheckel T., Sitte N., Hass R., Grune T., Davies K. J. (1999) Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc. Natl. Acad. Sci. U.S.A. 96, 6223–6228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berko D., Tabachnick-Cherny S., Shental-Bechor D., Cascio P., Mioletti S., Levy Y., Admon A., Ziv T., Tirosh B., Goldberg A. L., Navon A. (2012) The direction of protein entry into the proteasome determines the variety of products and depends on the force needed to unfold its two termini. Mol. Cell 48, 601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glickman M. H., Rubin D. M., Fried V. A., Finley D. (1998) The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol. 18, 3149–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsvetkov P., Reuven N., Prives C., Shaul Y. (2009) Susceptibility of p53 unstructured N terminus to 20 S proteasomal degradation programs the stress response. J. Biol. Chem. 284, 26234–26242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ansari H. R., Raghava G. P. (2010) Identification of NAD interacting residues in proteins. BMC Bioinformatics 11, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bailey T. L., Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 [PubMed] [Google Scholar]
- 21. Berglund A. C., Sjölund E., Ostlund G., Sonnhammer E. L. (2008) InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 36, D263–D266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hough R., Pratt G., Rechsteiner M. (1987) Purification of two high molecular weight proteases from rabbit reticulocyte lysate. J. Biol. Chem. 262, 8303–8313 [PubMed] [Google Scholar]
- 24. Scrutton N. S., Berry A., Perham R. N. (1990) Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343, 38–43 [DOI] [PubMed] [Google Scholar]
- 25. Bellamacina C. R. (1996) The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins. FASEB. J. 10, 1257–1269 [DOI] [PubMed] [Google Scholar]
- 26. Liu C. W., Li X., Thompson D., Wooding K., Chang T. L., Tang Z., Yu H., Thomas P. J., DeMartino G. N. (2006) ATP binding and ATP hydrolysis play distinct roles in the function of 26 S proteasome. Mol. Cell 24, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolff E. C., Wolff J., Park M. H. (2000) Deoxyhypusine synthase generates and uses bound NADH in a transient hydride transfer mechanism. J. Biol. Chem. 275, 9170–9177 [DOI] [PubMed] [Google Scholar]
- 28. Smith D. M., Fraga H., Reis C., Kafri G., Goldberg A. L. (2011) ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144, 526–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Veech R. L., Lawson J. W., Cornell N. W., Krebs H. A. (1979) Cytosolic phosphorylation potential. J. Biol. Chem. 254, 6538–6547 [PubMed] [Google Scholar]
- 30. Lander G. C., Estrin E., Matyskiela M. E., Bashore C., Nogales E., Martin A. (2012) Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith D. M., Chang S. C., Park S., Finley D., Cheng Y., Goldberg A. L. (2007) Docking of the proteasomal ATPases' carboxyl termini in the 20 S proteasome's α ring opens the gate for substrate entry. Mol. Cell 27, 731–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gillette T. G., Kumar B., Thompson D., Slaughter C. A., DeMartino G. N. (2008) Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J. Biol. Chem. 283, 31813–31822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fjeld C. C., Birdsong W. T., Goodman R. H. (2003) Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. U.S.A. 100, 9202–9207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reinheckel T., Ullrich O., Sitte N., Grune T. (2000) Differential impairment of 20 S and 26 S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch. Biochem. Biophys. 377, 65–68 [DOI] [PubMed] [Google Scholar]
- 35. Wang X., Yen J., Kaiser P., Huang L. (2010) Regulation of the 26 S proteasome complex during oxidative stress. Sci. Signal. 3, ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bajorek M., Finley D., Glickman M. H. (2003) Proteasome disassembly and downregulation is correlated with viability during stationary phase. Curr. Biol. 13, 1140–1144 [DOI] [PubMed] [Google Scholar]
- 37. Tai H. C., Besche H., Goldberg A. L., Schuman E. M. (2010) Characterization of the brain 26 S proteasome and its interacting proteins. Front. Mol. Neurosci. 10.3389/fnmol.2010.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chou A. P., Li S., Fitzmaurice A. G., Bronstein J. M. (2010) Mechanisms of rotenone-induced proteasome inhibition. Neurotoxicology 31, 367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Höglinger G. U., Lannuzel A., Khondiker M. E., Michel P. P., Duyckaerts C., Féger J., Champy P., Prigent A., Medja F., Lombes A., Oertel W. H., Ruberg M., Hirsch E. C. (2005) The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J. Neurochem. 95, 930–939 [DOI] [PubMed] [Google Scholar]
- 40. Um J. W., Im E., Lee H. J., Min B., Yoo L., Yoo J., Lübbert H., Stichel-Gunkel C., Cho H. S., Yoon J. B., Chung K. C. (2010) Parkin directly modulates 26 S proteasome activity. J. Neurosci. 30, 11805–11814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sarraf S. A., Raman M., Guarani-Pereira V., Sowa M. E., Huttlin E. L., Gygi S. P., Harper J. W. (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]