

Background: We studied ribosome and nucleoid distribution in Escherichia coli under growth and quiescence.
Results: Spatially segregated ribosomes and nucleoids show drastically altered distribution in stationary phase or when treated with drugs affecting translation, transcription, nucleoid-topology, or cytoskeleton. Ribosome inheritance in daughter cells is frequently unequal.
Conclusion: Cellular growth processes modulate ribosome and nucleoid distribution.
Significance: This provides insight into subcellular organization of molecular machines.
Keywords: Antibiotics, Cell Division, Cytoskeleton, DNA Replication, Ribosomes, Subcellular Organelles, Transcription, Translation, FtsZ Ring, Nucleoid
Abstract
We have examined the distribution of ribosomes and nucleoids in live Escherichia coli cells under conditions of growth, division, and in quiescence. In exponentially growing cells translating ribosomes are interspersed among and around the nucleoid lobes, appearing as alternative bands under a fluorescence microscope. In contrast, inactive ribosomes either in stationary phase or after treatment with translation inhibitors such as chloramphenicol, tetracycline, and streptomycin gather predominantly at the cell poles and boundaries with concomitant compaction of the nucleoid. However, under all conditions, spatial segregation of the ribosomes and the nucleoids is well maintained. In dividing cells, ribosomes accumulate on both sides of the FtsZ ring at the mid cell. However, the distribution of the ribosomes among the new daughter cells is often unequal. Both the shape of the nucleoid and the pattern of ribosome distribution are also modified when the cells are exposed to rifampicin (transcription inhibitor), nalidixic acid (gyrase inhibitor), or A22 (MreB-cytoskeleton disruptor). Thus we conclude that the intracellular organization of the ribosomes and the nucleoids in bacteria are dynamic and critically dependent on cellular growth processes (replication, transcription, and translation) as well as on the integrity of the MreB cytoskeleton.
Introduction
Recent advances in molecular biology with discovery of fluorescent proteins and their easy introduction into cells as protein tags have provided visual access into the interior of live bacterial cells. The developments in in-cell visualization technology either in live cells by using fluorescent protein tags or in fixed cells by immunofluorescence microscopy, have rendered the earlier image of bacterial cells as semipermeable sacs of chemicals untenable. The notion of structureless, homogeneous bacterial cytoplasm through which macromolecules diffuse freely to interact by random collisions has been replaced by a system of macromolecular machines designed for specific functions assembled at specific locations at appropriate times such that growth, replication, and cell division processes function in coordination to maintain remarkably error-free cycles of growth and reproduction for generations (1–3). This was also suspected earlier with the discovery of plasmids clustered at characteristic intracellular positions (4, 5) and the sequential movement of the bacterial chromosomes during replication (6–9). Now it has been revealed that the bacterial interior possesses a highly ordered subcellular architecture comprising dynamic networks of cytoskeletal fibers (10–12), multiprotein complexes constituting replication, transcription, and translation machineries assembled at characteristic locations (13–19), and oscillatory relocalization of protein complexes in defined trajectories resulting in concentration gradients (20, 21). This paradigm shift in our understanding calls for a re-evaluation of the cellular processes and their regulation in terms of the spatiotemporal organization of macromolecular machines and their interactions inside living cells.
Already in the early 1970s, Miller et al. (22) had demonstrated a close coordination between the transcription and translation machineries in Escherichia coli using electron microscopy. They showed that the mRNAs carrying polyribosomes spread out from the bacterial chromosome with the ribosomes attached to the nascent mRNAs linked to the bacterial chromosome through the transcribing RNA polymerase molecules. This observation lead to the concept of “coupled transcription translation” in bacteria, and a molecular link between these two processes was proposed (23, 24). This model was extended into a “transertion model” according to which a coupled transcription-translation and insertion of the nascent polypeptide chain into the membrane was proposed to regulate nucleoid morphology during its duplication and segregation in coordination with the cell cycle (3, 25). Recent studies in live E. coli and Bacillus subtilis showed RNA polymerase to be co-localized with the bacterial nucleoid in the center of the cell, whereas the ribosomes were traced to occupy the space outside the mass of the nucleoid (15–18). In contrast, in bacteria such as Caulobacter crescentus, Agrobacterium tumefaciens, etc., the ribosomes and the DNA are not segregated in separate chambers (for review, see Ref. 26). In C. crescentus ribosomes are uniformly distributed in the cell cytoplasm irrespective of the location of the cylindrically dispersed DNA (27). Recently, mRNAs were shown as spatially organized in preferred sites in the cell (19, 27, 28), suggesting that protein synthesis may take place on freely diffusing mRNAs (15, 17). These findings put a question mark on the indispensability of the coupled transcription-translation model as the determinant of the organization of the cellular machineries, and the search for alternative principles becomes crucial.
In the last decade our knowledge about the distribution of the ribosomes in bacterial cells has been enriched from high quality microscopic studies performed using gram +ve B. subtilis and gram −ve E. coli as the model systems (15–17, 29–31). Except for some minor differences, the overall picture of ribosome distribution emerging from these studies agrees quite well with each other. Here we have attempted to examine the physiological significance of the observed pattern by comparing the distribution of the fluorescent protein-tagged ribosomes in E. coli during growth and in quiescence. In parallel, the shape and organization of the nucleoids have been followed with DAPI fluorescence. The effect of various drugs known to affect translation, transcription, or the nucleoid topology on the distribution of the ribosomes has been investigated. Moreover, the change in distribution of the ribosomes in growing and dividing cells has been followed in real time using time-lapse imaging. Our results confirm that the relative distribution of the ribosomes and the nucleoid in a bacterial cell is dynamic and highly sensitive to the conditions of growth and its arrest. We demonstrate for the first time to our knowledge that the ribosomes gather in the mid-cell around the septal site determined by the FtsZ ring before cell division, and despite that, the ribosome distribution in the daughter cells is frequently unequal. Furthermore, we show that although the disruption of MreB cytoskeleton severely affects the ribosome distribution, protein synthesis continues uninterrupted at least for two to three generations. Altogether, our results point toward a global interlink between these apparently independent subcellular structures and stages of bacterial growth.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
All bacterial strains used in this work are derivatives of the E. coli K12 MG1655 (WT) (listed in Table 1). Fig. 1A shows the scheme for fusing mCherry tag to the ribosomal protein (r-protein) L9. The termination codon of the gene rplI (encoding L9) on the chromosome of E. coli MG1655 (WT) was replaced by a linear DNA containing the DNA sequence coding for the red fluorescent protein mCherry (32) and kanamycin resistance cassette (KanR) using λ-Red recombineering (33, 34). The recombinants with L9-mCherry fusion were selected against kanamycin and verified by PCR and sequencing. One successful recombinant was named QC101 (MG1655 rplI-mCherryKanR, see Table 1). Colony PCR of QC101 with primers flanking the rplI gene locus produced a band of 2 kb confirming successful fusion of the mCherry-KanR cassette (1.5 kb) to the rplI gene (0.5 kb) (Fig. 1B). A similar fusion of mCherry was also done with tufB gene (encoding elongation factor Tu or EF-Tu) resulting in strain QC702 (MG1655 tufB-mCherry KanR) (Table 1).
TABLE 1.
The description of the strains and plasmids used/generated in this work
The doubling times (rounded to nearest whole number) are estimated from the average of four individual measurements in LB at 37 °C by using Bioscreen-C Automated Growth Curve Analyzer.
| Genetic construction | Strain description | Doubling time | Source | |
|---|---|---|---|---|
| min | ||||
| E. coli strains | ||||
| MG1655 | Wild type E. coli K-12 | WT | 21 ± 0.5 | Laboratory strain |
| QC101 | MG1655 rplI-mCherry Kan | L9-mCherry | 21 ± 0.4 | This work |
| QC901 | MG1655 rpsF-TurboGFP Tet | S6-TurboGFP | 27 ± 1.6 | This work |
| QC501 | MG1655 rplI-TurboGFP Tet | L9-TurboGFP | 21 ± 1.0 | This work |
| QC702 | MG1655 tufB-mCherry Kan | EF-Tu-mCherry | 22 ± 0.5 | This work |
| QC801 | QC501 tufB-mCherry Kan | S6-TurboGFP + EF-Tu-mCherry | 25 ± 0.7 | This work |
| BS433 | QC101 + pEG12 [Plac (ftsZ-GFP)] | L9-mCherry + FtsZ-GFP | 21 ± 3 | This work |
| Plasmids | ||||
| pSIM5 | pSC101repts (λ gam exo bet cat) | (33) | ||
| mCherry | pRSET-B PT7 (mCherry) | (32) | ||
| TurboGFP | pTurboGFP-B PT5 (TurboGFP) | Evrogen, USA | ||
| FtsZ-GFP | pEG12 Plac ftsZ-GFP | (35) | ||
FIGURE 1.
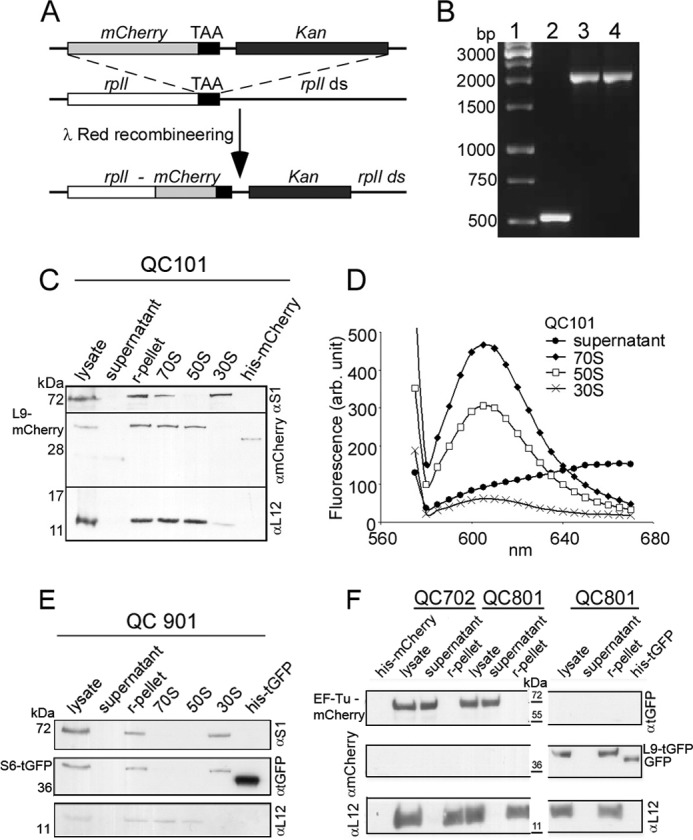
Construction and characterization of E. coli strain QC101 (L9-mCherry), QC901 (S6-TurboGFP), QC702 (EF-Tu-mCherry), and QC801 (EF-Tu-mCherry and L9-TurboGFP). A, scheme showing the strategy for fusion of red fluorescent protein mCherry with ribosomal protein L9. λ-Red recombineering was used to insert a linear DNA containing tandemly arranged genes for mCherry and Kan replacing the stop codon of rplI gene on MG1655 chromosome. The resulting recombinant QC101 produced an in-frame fusion at the 3′-end of rplI gene with the gene for mCherry (see “Experimental Procedures”). B, colony PCR screening for mCherry-Kan (1.5 kb) fusion to rplI (0.5 kb) using primers flanking rplI gene. Lane 1, DNA ladder; lane 2, PCR from colonies of MG1655; lane 3, recombinant QC101 with pSIM5; lane 4, QC101. C, Western blot analysis of cellular fractions from QC101 to trace the L9-mCherry fusion protein. The cell lysate, ribosomal pellet (r-pellet), supernatant (collected after pelleting ribosomes by ultracentrifugation), purified ribosomal particles 70 S, 50 S, and 30 S from QC101, and purified mCherry protein (31.5 kDa) were subjected to Western blot using α-mCherry antibody. Antibodies to the ribosomal proteins S1 and L12 (gift from J. P. Ballesta, CBMSO, Spain) were used to specify 30 S and 50 S subunits. The blot corresponding to L9-mCherry fusion (42.5 kDa) was visible in r-pellet and 70 S and 50 S subunits but not in the supernatant and 30 S subunits. D, fluorescence spectra of 70 S, 50 S, and 30 S ribosomal particles from QC101. Characteristic mCherry fluorescence (excitation at 555 nm and emission λmax 610 nm) was seen in 70 S ribosomes and 50 S subunits but not in the ribosome-free supernatant. Small fluorescence seen in the 30 S subunits could be from minor contamination of 50 S particles. E, Western blot analysis of the cell lysate, ribosomal pellet, ribosome-free supernatant, and the ribosomal particles 70 S, 50 S, and 30 S from QC901 cells separated on a 14% SDS-PAGE using antibodies α-turboGFP, α-S1 (specific to 30 S), and α-L12 (specific to 50 S). Purified TurboGFP (30.5 kDa) was used as a control. Immunoblotting detected S6-turboGFP fusion (40.7 kDa) in the ribosomal pellet and 30 S subunits but neither in the supernatant nor in the 50 S subunits. F, Western blot analysis of cell lysate, ribosomal pellet, and ribosome-free supernatant from QC702 and QC801 cells with α-turboGFP, α-mCherry, and α-L12 antibodies. Purified TurboGFP (∼36 kDa) and mCherry (31.5 kDa) were loaded as positive controls and size markers. In both QC702 and QC801 EF-Tu-mCherry fusion (∼70 kDa) was detected in the lysate and in the supernatant fraction, whereas L9-TurboGFP (∼41 kDa) was present exclusively in the ribosomes of QC801.
For the second line of constructs, a DNA cassette containing genes for TurboGFP and TetR was fused to the rpsF and rplI genes (encoding S6 and L9 proteins) in MG1655 using the same strategy. The resulting strains were QC901 (MG1655 rpsF-TurboGFP TetR) and QC501 (MG1655 rplI-TurboGFP TetR), respectively (Table 1). Furthermore, we moved the EF-Tu-mCherry fusion into QC501 by λ-Red recombineering creating the strain QC801 (QC501 tufB-mCherry KanR) (Table 1). Strain BS433 was constructed by introducing the plasmid pEG12, containing Plac ftsZ-GFP (a gift from Kenn Gerdes) (35) into QC101 (Table 1). All plasmids are listed in Table 1.
Growth Rate Measurement
Growth rates of all the strains used in this work were measured at 37 °C in LB (without any antibiotic) using a Bioscreen C Analyzer (Oy Growth Curves Ab Ltd, Helsinki, Finland). Each well was inoculated with a 1000-fold dilution of an overnight culture, and measurements were made in quadruplicate. The cultures were grown for 16 h with continuous shaking, and A600 values were recorded every 5 min The doubling times were estimated from early log phase (A600 = between 0.02 and 0.1) (Table 1).
Localization of Fluorescent Protein-tagged r-proteins and EF-Tu Using Western Blot and Fluorescence Spectroscopy
The 70 S ribosome and the ribosomal subunits were isolated from QC101, QC901, QC702, and QC801 strains using conventional sucrose gradient ultracentrifugation method. Specific localization of the fluorescence protein-tagged r-proteins and EF-Tu was confirmed by Western blots of total cell lysates, crude ribosome pellets isolated by ultracentrifugation at 200,000 × g at 4 °C for 120 min (marked as r-pellet in Fig. 1), and supernatant (cleared after pelleting the ribosomes by ultracentrifugation) as well as purified 70 S ribosome and its 30 S and 50 S subunits using antibodies raised against mCherry (MBL International Corp.) or TurboGFP (Evrogen) (Fig. 1C). Antibodies specific to the r-proteins S1 and L12 were used to identify ribosomal small and large subunits, respectively. Furthermore, fluorescence spectra of these ribosomal components (15 pmol each) were recorded using a Hitachi F7000 spectrofluorometer (Fig. 1D). For mCherry-tagged components the excitation was done at 555 nm. For Turbo GFP fusions the excitation wavelength was 450 nm.
Fluorescence Microscopy
It has been shown that exponentially growing cultures above a cell density of 5 × 107/ml (A600 = 0.1) contain a subpopulation of stationary phase cells and that the proportion of such quiescent cells increase with cell density (36). Therefore, only bacterial cultures with A600 = 0.1 or less were used for exponential phase studies. For cells in stationary phase, overnight cultures were used. For visualizing DNA, DAPI stain was used. To avoid DNA damage DAPI was added only a few minutes before data collection. Antibiotics were added to early exponential cultures (at final concentrations: 10 μg/ml cephalexin, 200 μg/ml chloramphenicol, 100 μg/ml streptomycin, 50 μg/ml tetracycline, 200 μg/ml rifampicin, 30 μg/ml nalidixic acid, and 10 μg/ml A22). Aliquots withdrawn at different time intervals were spotted directly on thin layers of 1% agarose (AgarGel H/M Mercury) in 0.9% NaCl or nutrient medium for examination under a Carl Zeiss Axioplan 2 Imaging microscope using a 100× objective (NA = 1.40, depth of focus 0.66 μm). The images were captured with a Carl Zeiss Axio-Cam CCD camera. The filter sets for mCherry were HC F 36-504, for TurboGFP were D480/40, and for DAPI were D360/40. The intensity scan for individual cells was performed along the longitudinal axis of the cell using Axiovision digital image analysis software and integrated over the whole cell width. Autofluorescence was measured with MG1655 cells, which varied from 3 to 6%.
Confocal fluorescence images were acquired using an inverted laser scanning confocal microscope (Zeiss LSM 510 Meta) using a 63× oil objective (NA = 1.4) and LSM software. An argon laser (514 nm) was used at 25% intensity, and images were captured using a 512 × 512-pixel frame. Gain settings were between 650 and 800. Scan speed was set to seven and the mean of four lines was detected. Zoom function was set to one, and the pinhole was set to one airy unit. Z-series were used to capture the whole bacterium.
Estimation of Total Protein Synthesis from [3H]Valine Incorporation
QC101 cells were grown in the presence of [3H]valine in M9 culture media without or with drugs chloramphenicol (200 μg/ml) and A22 (10 μg/ml). Aliquots withdrawn at definite time intervals were filtered through glass filters after treating with 5% TCA. The filters were dried after isopropyl alcohol wash, and counts measured in Beckman liquid scintillation counter were plotted against time.
RESULTS
Characterization of the Strains with Fluorescent Protein-tagged Ribosome and EF-Tu
The fluorescent protein-tagged ribosomes were constructed by fusing genes encoding fluorescent proteins mCherry or TurboGFP with two individual r-protein genes from the large and small subunits (rplI and rpsF encoding protein L9 and S6, respectively) at their chromosomal loci. The resulting strains were named QC101 (L9-mCherry), QC501 (L9-TurboGFP), and QC901 (S6-TurboGFP). The L9 fusion strains QC101 and QC501 showed similar doubling time (21 min) in LB at 37 °C as its wild type parent (MG1655). Thus we conclude that the fluorescent protein fusions at this site had no deleterious effect on the growth rate of the bacteria (Table 1). However, QC901 (S6-TurboGFP) showed somewhat impaired growth (doubling time 27 ± 1.5 min) (Table 1). Therefore, this strain was used only for qualitative confirmation of the results obtained with QC101.
The strain QC702 with EF-Tu-mCherry fusion showed a doubling time of 22 min, which is again similar to MG1655. In contrast, the strain with double fusion, QC801 (S6-TurboGFP + EF-Tu-mCherry), showed a longer doubling time (25 ± 0.7 min) (Table 1). We suspect that this slow growth behavior of QC801 is probably due to the S6-TurboGFP fusion as QC901 (S6-TurboGFP) also showed similar growth defect.
Specific Localization of the Fluorescent Protein-tagged Components
The absence of free L9-mCherry protein in the cytosol and its exclusive association with the 50 S ribosomal subunits in the cell were confirmed by Western blots with anti-mCherry antibody. As shown in the Fig. 1C, L9-mcherry could be detected in the cell lysate, ribosomal pellet, and purified 70 S ribosome and 50 S subunit samples. The absence of mCherry in the supernatant fraction after pelleting the ribosomes by ultracentrifugation indicated that there was no extra-ribosomal L9-mCherry protein in the cytosol. Furthermore, the absence of L9-mCherry in the 30 S sample confirmed its exclusive presence in the 50 S subunit (Fig. 1C). This was further assured by demonstrating that the 70 S ribosomes and the 50 S subunits, but not the 30 S subunits, were fluorescent (Fig. 1D). The fluorescence emission from QC101 70 S and 50 S matched the characteristic spectrum of mCherry protein (λmax = 610 nm). The minor fluorescence seen in the 30 S sample was probably due to its incomplete separation from the 50 S subunit in the sucrose gradient step.
Similar to L9-mCherry in QC101, S6-TurboGFP in QC901 also showed exclusive localization in the 30 S subunit of the ribosome when analyzed with Western blot using antibody against TurboGFP (Fig. 1E). In contrast to the r-protein fusions, the EF-Tu-mCherry in QC702 and QC801 was identified solely in the supernatant fractions after pelleting the ribosomes (Fig. 1F). The strain QC801, which carried L9-TurboGFP fusion in addition to EF-Tu-mCherry fusion, showed an exclusive presence of L9-TurboGFP in the ribosome (Fig. 1D). The fusion of the fluorescent proteins and their cellular location were further confirmed by checking mCherry and/or TurboGFP fluorescence of ribosomal pellet or supernatant without ribosomes.
Subcellular Distribution of the Ribosomes and the Nucleoids in Exponential and Stationary Phase
Fig. 2, A–C and E) illustrates the intracellular distribution of the m-Cherry tagged ribosomes and DAPI-stained nucleoids inside live E. coli QC101 (L9-mCherry) cells grown in rich medium. In exponentially growing cells, the translating ribosomes and the nucleoid lobes appeared as alternative bands arranged lengthwise spanning from one pole to the other (Fig. 2A), which is in good agreement with the earlier reports (15–17). The alternative arrangement of the ribosomes and the nucleoids was more evident in the horizontal sectional view of the cells obtained by Z-scan using confocal microscope (Fig. 2B). Both ribosomes and nucleoids could be seen in all Z-sections, peripheral or central, with highest intensity in the middle section of the cell (Fig. 2B, middle row). Furthermore, when cell division was blocked with the drug cephalexin, the cells became filamentous, but the alternative distribution of the ribosomes and the nucleoids was maintained for several generations (Fig. 2C).
FIGURE 2.

Distribution of ribosomes and nucleoids in exponential and stationary phase. Fluorescence images of the ribosomes alone, the nucleoids alone, and both superimposed and digitally colored are as labeled on the top of the figures. Color codes are red for mCherry tagged ribosomes and green for DAPI stained nucleoids. The bar indicates 5 μm. A, images of QC101 cells growing in the exponential phase. The first three panels show a field view with multiple cells; images of mCherry-ribosome alone (1) and (DAPI-stained) nucleoid alone (2), and both superimposed and digitally colored in red and green, respectively (3), as labeled on the top of the images. Panels 4 and 5 show examples of a typical long and a short cell, respectively, following the same color code as in panel 3 together with the intensity scans of the ribosome (red) and the DNA (green) along the longitudinal axis of the cells. a.u., arbitrary units. B, Z-scan images of the exponential phase QC101 cells showing distribution of ribosomes and nucleoids along the longitudinal sections as indicated by the symbols and the labels. C, the distribution of the ribosomes and the nucleoids in a filamentous QC101 cells treated with 10 μg/ml cephalexin, an inhibitor of cell division. D, a field view of exponentially growing QC901 cells; ribosome alone or together with the DNA as indicated in the label. E, a filed view of stationary phase QC101 cells showing ribosomes alone (1), nucleoids alone (2), and both superimposed and colored in red and green, respectively (3). Examples of three enlarged stationary phase cells together with the intensity scans of the ribosomes (red) the DNA (green) are added in the panels 4–6. The bar indicates 5 μm for all panels.
We have examined about 200 log phase QC101 cells, selected randomly from five independent experiments. The average length of the fully grown cells was 6.1 ± 0.2 μm. These cells, classified as “long” cells (example in Fig. 2A, fourth panel) contained four uniformly dispersed nucleoid lobes and five ribosome peaks, usually with the highest intensity in the middle of the cell. The other ribosome peaks were at the two poles and approximately at one-quarter and three-quarter length of the cell, as appeared from the intensity scan along the long axis (Fig. 2A, fourth panel, bottom row). Alternatively, the “short” cells (one typical example shown in Fig. 2A, fifth panel), presumably the newly divided ones, were 2.8 ± 0.2 μm long, contained two distinct nucleoid lobes and three ribosome peaks; two at the poles and one at the center of the cell. Other than these two types, there were cells of all intermediate lengths that showed two to four incompletely divided nucleoid lobes and three to five ribosome peaks. However, irrespective of the length and the shape of the cell, the ribosome peaks always overlapped with nucleoid valleys and vice versa. From the L9-mCherry and DAPI (DNA) intensity scans, the total area covered by the ribosomes was 60 ± 10%, and there was about 27 ± 8% overlap between the two (analysis based on ∼200 log phase cells). However, due to the limitation of the imaging technique, different extents of DAPI staining in different experiments, and varied contribution of the relatively high background fluorescence, the analysis regarding relative proportion of ribosomes and nucleoids and the extent of their overlap should not be taken too literally. The distribution of the ribosome and the nucleoid peaks in relation to the length of the cells was similar to the previous study reported by Bakshi et al. (17) and, therefore, not repeated here.
The strain QC901 (S6-TurboGFP) showed ribosome and nucleoid distribution almost identical to that in QC101 (Fig. 2D), confirming that the ribosome distribution was not influenced by the nature of the fluorescent tag or the site of tag fusion. However, this strain showed some growth defect (Table 1) for which images obtained from QC901 cells were used only for qualitative verification of the QC101 images.
In the stationary phase the cells were more or less uniform in size and much shorter than the long exponential phase cells (average length 2.7 ± 0.2 μm based on the analysis of ∼100 cells). Moreover, in sharp contrast to the exponential phase cells, the nucleoids were compact, devoid of their usual lobed structure, and often placed centrally in the cells, whereas the ribosomes were clumped at the cell poles and the boundaries (Fig. 2E). The stationary phase cells showed more heterogeneity in ribosome and nucleoid distributions than in exponential phase. While some cells showed equal distribution of the ribosomes in two poles, some cells showed unequal and more diffused distribution of the ribosomes. Some cells (presumably dead cells) seemed to have lost their ribosomes (green cells in Fig. 2E, third panel). Fig. 2E also shows some typical stationary phase cells (panels fourth to sixth panels, top row) together with the mCherry-ribosome and DAPI-DNA intensity scans (bottom row). Despite heterogeneity in the relative distribution, the ribosome and the nucleoid peaks were always segregated in the stationary phase as seen in the log phase cells.
Changes in Ribosome Distribution during Growth and Division
Changes in ribosome distribution in individual cells as they grew and underwent cell division were followed further by time-lapse microscopy. QC101 cultures were grown on a thin agarose layer continually soaked with nutrient medium on a glass slide under the fluorescence microscope at room temperature (∼23 °C). Three cells were selected; two new born (#2 and #3 in Fig. 3A) and one grown to an intermediate length (#1) and photographed at 10-min intervals for 2 h tracking the ribosomes with mCherry fluorescence. DAPI staining of the nucleoid was avoided because exposure to UV in presence of DAPI would induce lethal photodynamic damages. Fig. 3A shows a few selected time-lapse images (left column) of three cells and ribosome intensity scans of the cell #1 (right column). Because the valley of the ribosomal intensity scan corresponds to the nucleoid peaks (Fig. 2), the number of the nucleoid peaks was estimated by counting the ribosomal valleys.
FIGURE 3.

Following QC101 cells through growth and division. A, time-lapse images of ribosome distribution in QC101 (L9-mCherry) cells (#1, #2, and #3) and corresponding ribosomal intensity scans of the cell #1 (along the long axis) for each time point (indicated by the number in the images). The fluorescence intensity is integrated over the whole cell-width and normalized to the same level for comparison. The x axis in the intensity scan diagrams indicates the distance relative to cell center (marked with 0) for 0–80-min plots and distance from the new cell pole for 100 and 110-min plots. The gray bars in the 100- and 110-min plots indicate the plane for cell division. B, the change in cell-length (left y axis, the length of a full-grown cell is normalized to 1) and the number of ribosome and nucleoid peaks (right y axis) relative to the generation time. The values are based on the time-lapse images in A, the average from 10 independent cells. The error bars indicate S.D. C, a field view showing mCherry-ribosome (red) distribution in relation to FtsZ ring (yellow) in BS433 (L9-mCherry + FtsZ-GFP) cells. D, zoomed images of three typical fully grown QC101 cells just before dividing or freshly divided showing ribosomes (red) and DNA (green) together with intensity scans of the ribosome along the longitudinal axis of the cells. The black circles highlight the zone for cell division. The gray bars in the intensity scans signify the plane for cell division. The bar in all panels corresponds to 5 μm. a.u., arbitrary units.
The generation time of the QC101 cells at room temperature in the solid media was much longer (120 ± 10 min) than that in LB at 37 °C (∼21 min). Despite that our results showed a definite correspondence between the increase in the length of the cell and the number of the ribosome and the nucleoid peaks per cell (Fig. 3B). The newly divided cells in the log phase showed three ribosome peaks (two at the poles and one in the middle of the cell) (Fig. 3A, 0, 100, and 110 min) and, thus, presumably two nucleoid lobes (Fig. 3B). With the growth of the cell, the middle peak became more and more prominent and stronger than the peaks at the poles (Fig. 3A, 0–80 min). As the cells grew to ∼80% of their final length (before division), at around 65 ± 5% of the generation time, two new ribosome peaks appeared in the intermediate positions (at one quarter and three quarters of the long axis), concomitant with the division of the nucleoids into four lobes (Fig. 3, A, 60 min, and B). The fully grown cells divide by forming the septum through the central ribosome peak (Fig. 3A, 80 and 100 min) thereby reducing the number of the ribosome and nucleoid peaks again to three and two, respectively, in the newborn cells (Fig. 3B). From the change in the shape and size of the ribosome peaks it is clear that the ribosome distribution is highly dynamic in growing E. coli cells.
To further examine the distribution of ribosomes during cell division, we followed the distribution of ribosomes relative to the FtsZ ring using the strain BS433, a derivative of QC101 carrying an FtsZ-GFP expressing plasmid (Table 1). The FtsZ ring indicates the formation of cell septum (35). As shown in Fig. 3C, the FtsZ ring (yellow) was assembled in the mid-cell region overlapping the centrally accumulated ribosomes (red). It was interesting to note that for many cells, the ribosomal fluorescence on both sides of the Fts-Z septum was not equal. This further led to unequal partitioning of the central ribosome peak (Fig. 3, A, 100 and 110 min, and D) in the newly formed poles of the two daughter cells. Three zoomed-in images of dividing or newly divided cells together with the ribosome intensity scan have been added in Fig. 3D for closer inspection. Among all the dividing cells we have analyzed (∼100), the distribution of the central ribosome peak between the newly divided cells varied from almost equal to about 65:35, with the majority showing unequal distribution of the ribosomes (Fig. 3, A 100 min, and D).
EF-Tu Distribution Parallels Ribosome Distribution in the Log Phase
The elongation factor, EF-Tu, is closely associated with the translating ribosomes for continuous loading of charged tRNAs to the ribosome. We have followed EF-Tu distribution in the log and the stationary phase of cell growth, alone in QC702 (EF-Tu-mCherry) and together with ribosomes in QC801 (L9-TurboGFP + EF-Tu-mCherry) cells. Because of the high copy number per cell, EF-Tu showed a more uniform distribution compared with the ribosomes and also a significantly high background fluorescence. However, during exponential growth, the EF-Tu distribution pattern paralleled that of the ribosomes showing a series of bead-like impressions along the length of the cell (Fig. 4A, left; compare with Fig. 2A, first panel). When imaged with QC801 cells carrying fluorescent labels on both EF-Tu (mCherry) and ribosome (TurboGFP), the ribosome and EF-Tu fluorescence showed strong overlap (Fig. 4B), whereas DNA was well separated. Interestingly, the bead-like pattern of EF-Tu disappeared completely in the stationary phase (Fig. 4A, right). Instead, a uniformly diffused EF-Tu fluorescence prevailed throughout the cell without any specific localization. This is in big contrast with the ribosome distribution in the stationary phase, where the ribosomes were localized at the cell poles and boundaries (Fig. 2E). This result suggests that EF-Tu molecules were closely associated with active ribosomes in the log phase but were released and dispersed when translation stopped in the stationary phase.
FIGURE 4.
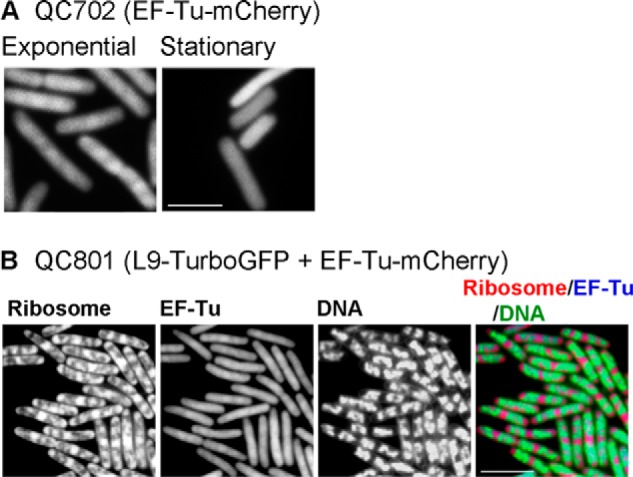
Distribution of EF-Tu in E. coli. A, distribution of EF-Tu-mCherry during exponential and stationary phase of growth. The images are taken using QC702 (EF-Tu-mCherry) cells. B, the distribution of Turbo-GFP tagged ribosomes, EF-Tu-mCherry, DAPI-stained DNA alone or in superposition of all three. The images were taken using QC801 (L9-TurboGFP + EF-Tu-mCherry) cells growing in the exponential phase. The bar indicates 5 μm.
Effect of Exposure to Translation Inhibitors
Fig. 5A shows micrographs of fast-growing QC101 cells after exposure to three different translation-inhibiting antibiotics that stopped bacterial growth almost immediately. These were: chloramphenicol, which binds to the rRNA of the large subunit and inhibits peptide bond formation (Fig. 5A, top panel); tetracycline, which targets the small subunit rRNA and prevents docking of the aminoacyl tRNA to the A-site (middle panel); streptomycin, which binds to the S12 protein of the 30 S subunit and interferes with the binding of initiator tRNA thereby blocking translation initiation (bottom panel) (37). Despite the difference in mechanisms for blocking protein synthesis, all three antibiotics caused collapse of the nucleoid lobes into a compact, central mass and accumulation of ribosomes predominantly at the cell poles as seen in stationary phase cells (Fig. 2E). Thus, it seems that compaction of nucleoids and the polar localization of ribosomes are the common consequences of cessation of active translation in E. coli in stationary phase or from exposure to antibiotics. Because none of these antibiotics interact directly with DNA or DNA-modifying enzymes, a direct effect of the drugs on the nucleoid seemed unlikely, and instead, the change in the nucleoid shape could be attributed to the cessation of global translation activity and disengagement of the ribosomes from translation.
FIGURE 5.

The effect of various antibiotics on ribosome and nucleoid distribution in E. coli. Subcellular distribution of the ribosomes and DNA alone or superimposed and digitally colored in red and green, respectively, in QC101 (L9-mCherry) cells after exposure to three independent translation-inhibiting antibiotics (200 μg/ml chloramphenicol (Chl), 50 μg/ml tetracycline Tet, and 100 μg/ml streptomycin (Strep)) for 10 min (A), transcription inhibitor rifampicin (200 μg/ml) for 5, 30, and 120 min (B), gyrase inhibitor nalidixic acid (30 μg/ml) for 5, 60, and 120 min (arrows highlight the vacuole-like empty spaces (C), both nalidixic acid (30 μg/ml) and chloramphenicol (200 μg/ml) for 120 min (D), and MreB inhibitor drug A22 (10 μg/ml) for 60 min (E). The bar indicates 5 μm for all panels. F, total protein synthesis in QC101 cells were measured by incorporation of [3H]valine without (●) or with drugs chloramphenicol (200 μg/ml) (▴) and A22 (10 μg/ml) (□).
Effect of Transcription Inhibitors
Rifampicin inhibits RNA transcription by binding to RNA polymerase. As shown in Fig. 5B, after treatment of QC101 cultures with rifampicin, the nucleoids were condensed first (within 5 min) but soon (15–30 min) spread out through the length of the cell as uniformly fluorescent cylinders. This was consistent with earlier data on the altered ultrastructure of E. coli nucleoids after exposure to rifampicin (38, 39). The ribosomes too lost their characteristic distribution in exponential phase and were spread uniformly surrounding the nucleoids. Eventually longer exposure (120 min) led to disruption of the cell structure as ribosomes and DNA leaked out from the cell.
Effect of Exposure to Gyrase Inhibitors
Nalidixic acid belongs to a class of broad-spectrum antibacterial agents called quinolones, which blocks DNA replication by specifically targeting DNA gyrases, induces double-strand DNA breaks, leads to elongated, filamentous cells, and kills cells in a protein synthesis-dependent pathway (40, 41). Exposure to nalidixic acid (Fig. 5C) compressed the nucleoid in the mid-cell region within 5 min, whereas ribosomes were seen distributed outside it. Remarkably, some vacuole-like empty spaces were visible among the ribosomes toward the poles (Fig. 5C, marked with an arrowhead). Prolonged exposure (60 min or longer) resulted in significant filamentation of the cells with multiple empty spaces in the cytoplasm. The nucleoids were disrupted into fragments and randomly scattered suggesting topological disruption and possibly double-strand breaks. We have further checked whether the nalidixic acid-mediated damages could be suppressed by blocking protein synthesis as suggested in a previous work (41). When chloramphenicol was added together with nalidixic acid, filamentation was visibly reduced, DNA break was not obvious, and both the nucleoids and the ribosomes acquired typical chloramphenicol phenotype without any empty space in the cell poles (Fig. 5D).
Ribosome-Nucleoid Organization after Disruption of MreB Cytoskeletal Structure
MreB is the prokaryotic actin-homolog contributing toward cell shape and nucleoid segregation (12, 42–44). The dynamics of ribosome distribution during cell growth and division led us to examine whether it was dependent on MreB cytoskeleton or not. For this purpose we treated QC101 cells with A22, a drug known to disrupt MreB polymers (45). When exposed to A22, both ribosomes and the nucleoid lost their distribution pattern. Within 20 min nucleoids in most of the cells collapsed to globular or cylindrical shape and localized randomly in the cell interior surrounded by ribosomes (Fig. 5E). Some cells lost integrity and showed leakage of DNA or ribosomes (Fig. 5E). It is interesting to note that the overall rod shape of the cells was still retained. Also, as shown in Fig. 5F, protein synthesis, monitored by incorporation of radiolabeled amino acid, continued uninhibited for >2 h after A22 treatment similar to the cells without any antibiotics. In comparison, chloramphenicol caused an immediate stop of protein synthesis (Fig. 5F). Thus, MreB-based cytoskeleton seemed not essential for the execution of the translation process.
DISCUSSION
We have studied how the distribution of ribosomes and nucleoids vary in live E. coli cells during different phases of growth and cell division. To gain insight into the factors controlling the distribution, we have also studied the effects of various antibiotics, which block general cellular processes. It is evident from our results that there exists a clear difference in the distribution pattern of the ribosome and the nucleoid between rapidly growing cells (Fig. 2, A–D) and the cells resting in the stationary phase (Fig. 2E). Also, the distribution pattern gets thoroughly disrupted when the cells are treated with drugs affecting translation (Fig. 5A), transcription (Fig. 5B), nucleoid topology (Fig. 5C), and MreB cytoskeleton (Fig. 5D). Despite this extensive variation, the ribosome peaks always remain well segregated from the nucleoid peaks and match with nucleoid valleys and vice versa. This observation is in accordance with earlier reports based on E. coli and B. subtilis cells (15, 17) and, therefore, can be considered as a general feature in most of the bacteria. In the overlay of the intensity scans (Fig. 2A, fourth and fifth panels; also Fig. 2E) some overlap between the ribosome and the nucleoid intensities was observed. We suspect that such overlap originated partly from high background fluorescence from DAPI (DNA) and mCherry-ribosomes and was partly due to diffraction limit and out-of-focus light. The mutual exclusion of the ribosomes and the DNA was more clearly seen in the Z-scan images, especially in the central sectional view where the ribosomes and the nucleoids appeared as disjointed patches (Fig. 2B, middle row). Thus, our results support the notion of functional compartmentalization of the translation apparatus in bacteria (15, 17).
There is biochemical evidence that membrane-bound ribosomes in E. coli are completely devoid of any physical connection with the nucleoids (46). It was also shown by independent studies that freely diffusing mRNAs are localized close to the cell periphery, where they are translated by ribosomes (19, 27, 28). Thus, our results together with other evidence suggest that the transcriptional coupling is not absolutely essential for all translational events. However, the ribosomes close to the nucleoids are probably engaged in coupled-transcription-translation, which might contribute to the ribosome-nucleoid intensity overlap to some extent.
When EF-Tu localization was studied with mCherry fusion, it showed close overlap with the ribosomes in exponential phase but not in stationary phase (Fig. 4). Because EF-Tu is essential in every round of translation elongation, it is expected that EF-Tu will be closely associated with the translating ribosomes. Thus, our EF-Tu data signify that that the log phase ribosomes are translating actively whereas the stationary phase ribosomes are not. Earlier studies have suggested a potential role of EF-Tu in cellular cytoskeleton through their association with MreB (47). Due to high background fluorescence of EF-Tu, we are unable to comment on that aspect.
One interesting observation from our time-lapse study is the asymmetric distribution of the ribosomes in the newly divided cells (Fig. 3). Although unexpected, it signifies that probably the accumulation of ribosomes in the mid-cell region is a rather random process for filling up the nucleoid free space in the cell and, thus, is unrelated to the exact location of septum formation. Because we could not follow the ribosomes under a microscope for more than one generation due to photobleaching of the mCherry fluorescence, it is difficult to ascertain whether the ribosome distribution in the daughter cells follow any definite pattern or not. The unequal inheritance of ribosomes in the daughter cells would also imply different growth rates for these cells. There are attempts to explain heterogeneous distribution of non-genetic components due to partitioning error in cell division using stochastic models (48, 49). It will be interesting to see whether ribosome distribution in the daughter cells can be explained after such models.
Although the governing principle behind the segregated distribution of the ribosomes and the nucleoids are not fully understood, a previous modeling-based study suggested “entropy” and “excluded-volume effect” as two main factors responsible for this segregation (50). According to this model, DNA organized in the bacterial nucleoid localizes in the center of the cell in order to avoid contact with the cell wall, which allows maximum conformational entropy. Alternatively, the ribosomes occupy all available empty spaces (excluded volume) in the cell. Thus, depending on the location, shape, and number of the nucleoids, the ribosome distribution changes in different conditions. This model fits well with the distribution of the ribosome peaks in the exponentially growing cells (Fig. 3B). It can also explain our observation of the dividing cells where the ribosomes overlap the FtsZ ring in the nucleoid-occluded mid-cell (Fig. 3C). However, this statistical model alone cannot explain why the ribosome distribution changes dramatically in stationary phase when translation slows down (Fig. 2E) or when cells are treated with the translation inhibitors, which act specifically on the ribosome (Fig. 5A). In these situations the nucleoid cannot be the primary determinant for the ribosome distribution. Instead, it seems likely that “translation” plays an important role in ribosome and nucleoid distribution as suggested by earlier works (39, 51).
Similar to translation, the contribution of active transcription in maintaining nucleoid conformation and ribosome distribution has long been proposed (25, 38, 39, 44). Our result with rifampicin treatment supports that model (Fig. 5B). The initial compaction of the nucleoids seen immediately after rifampicin treatment was possibly due to fast disruption of the membrane anchorage of the nucleoids mediated by RNA polymerase (38). Although similar in appearance as chloramphenicol treatment, the reason for nucleoid compaction in these two cases can be different. The expansion or diffusion of the nucleoid by prolonged treatment with rifampicin is probably due to the absence of transcription. Thus, our results indicate that transcription and translation, jointly called “transertion,” is not only important to sustain cellular growth processes (25, 39, 52) but is also vital for ribosome and nucleoid distribution in the bacterial cell (39, 51).
The gyrase inhibitor nalidixic acid has been suggested to have multiple effects on the subcellular architecture (40). Our results with nalidixic acid treatment provide visual confirmation that cessation of DNA replication and cell segregation results in filamentous cells with vacuole-like empty areas in the cytoplasm and distorted nucleoids (Fig. 5C). We also confirm that nalidixic acid-mediated cell death is protein synthesis-dependent and thus could be inhibited by translation inhibitors (Fig. 5D) consistent with an earlier suggestion (41). It is interesting to note that the occurrence of similar empty spaces in the cell was reported as a result of osmotic upshift (53). However, it is not obvious that the molecular reasons for such a phenotype are same in these two cases.
Our results with A22 treatment suggest that the intracellular distribution of ribosomes and nucleoids is dependent on the integrity of the MreB cytoskeleton (Fig. 5E). However, protein synthesis could continue for several generations after destruction of MreB polymers (Fig. 5F). Thus, we conclude that the distribution of ribosomes and nucleoids but not protein synthesis is dependent on MreB cytoskeleton. One interesting observation is that the cells retained their cylindrical rod shape even after prolonged treatment with A22 (Fig. 5E). Thus, most likely the cylindrical peptidoglycan sac, once formed, can retain its shape even without the support from MreB cytoskeleton.
In summary, our study has pointed out several cellular factors (DNA replication, transcription, translation, and the MreB cytoskeleton) as responsible for maintenance of ribosome and nucleoid distribution in bacterial cell. We also report time-lapse images of E. coli cells followed over one cycle of growth and cell division, which indicates for the first time unequal inheritance of the ribosomes in the daughter cells. However, understanding of the molecular mechanisms, which govern the ribosome-nucleoid segregation and determine the pattern of ribosome distribution in the newly divided cells, will require further studies with advanced microscopy and other complementary methods, which are the goals of the future work.
Acknowledgments
We thank Roger Tsien, Donald Court and Kenn Gerdes for the gift of plasmid constructs. We also thank J. P. Ballesta for providing antibody against L12 protein.
Footnotes
This work was supported by the research grants from the Swedish Research Council (2010-2619 (M), 2011-6088 (NT), and 2008-6593 (Linnaeus grant to Uppsala RNA Research Center)), the Carl-Tryggers Foundation (CTS 09:341 and 10:330), the Wenner-Gren Foundation, and the Knut and Alice Wallenberg Foundation (KAW 2011.0081 to RiboCORE (to S. S.).
REFERENCES
- 1. Alberts B. (1998) The cell as a collection of protein machines. Preparing the next generation of molecular biologists. Cell 92, 291–294 [DOI] [PubMed] [Google Scholar]
- 2. Laub M. T., McAdams H. H., Feldblyum T., Fraser C. M., Shapiro L. (2000) Global analysis of the genetic network controlling a bacterial cell cycle. Science 290, 2144–2148 [DOI] [PubMed] [Google Scholar]
- 3. Norris V., den Blaauwen T., Doi R. H., Harshey R. M., Janniere L., Jiménez-Sánchez A., Jin D. J., Levin P. A., Mileykovskaya E., Minsky A., Misevic G., Ripoll C., Saier M., Jr., Skarstad K., Thellier M. (2007) Toward a hyperstructure taxonomy. Annu. Rev. Microbiol. 61, 309–329 [DOI] [PubMed] [Google Scholar]
- 4. Weitao T., Dasgupta S., Nordström K. (2000) Plasmid R1 is present as clusters in the cells of Escherichia coli. Plasmid 43, 200–204 [DOI] [PubMed] [Google Scholar]
- 5. Pogliano J., Ho T. Q., Zhong Z., Helinski D. R. (2001) Multicopy plasmids are clustered and localized in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 98, 4486–4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weitao T., Dasgupta S., Nordström K. (2000) Role of the mukB gene in chromosome and plasmid partition in Escherichia coli. Mol. Microbiol. 38, 392–400 [DOI] [PubMed] [Google Scholar]
- 7. Hiraga S. (2000) Dynamic localization of bacterial and plasmid chromosomes. Annu. Rev. Genet. 34, 21–59 [DOI] [PubMed] [Google Scholar]
- 8. Sherratt D. J. (2003) Bacterial chromosome dynamics. Science 301, 780–785 [DOI] [PubMed] [Google Scholar]
- 9. Viollier P. H., Thanbichler M., McGrath P. T., West L., Meewan M., McAdams H. H., Shapiro L. (2004) Rapid and sequential movement of individual chromosomal loci to specific subcellular locations during bacterial DNA replication. Proc. Natl. Acad. Sci. U.S.A. 101, 9257–9262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Erickson H. P., Anderson D. E., Osawa M. (2010) FtsZ in bacterial cytokinesis. Cytoskeleton and force generator all in one. Microbiol. Mol. Biol. Rev. 74, 504–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ausmees N., Kuhn J. R., Jacobs-Wagner C. (2003) The bacterial cytoskeleton. An intermediate filament-like function in cell shape. Cell 115, 705–713 [DOI] [PubMed] [Google Scholar]
- 12. Jones L. J., Carballido-López R., Errington J. (2001) Control of cell shape in bacteria. Helical, actin-like filaments in Bacillus subtilis. Cell 104, 913–922 [DOI] [PubMed] [Google Scholar]
- 13. Lemon K. P., Grossman A. D. (1998) Localization of bacterial DNA polymerase. Evidence for a factory model of replication. Science 282, 1516–1519 [DOI] [PubMed] [Google Scholar]
- 14. Reyes-Lamothe R., Sherratt D. J., Leake M. C. (2010) Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328, 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewis P. J., Thaker S. D., Errington J. (2000) Compartmentalization of transcription and translation in Bacillus subtilis. EMBO J. 19, 710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mascarenhas J., Weber M. H., Graumann P. L. (2001) Specific polar localization of ribosomes in Bacillus subtilis depends on active transcription. EMBO Rep. 2, 685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bakshi S., Siryaporn A., Goulian M., Weisshaar J. C. (2012) Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol. Microbiol. 85, 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cabrera J. E., Jin D. J. (2003) The distribution of RNA polymerase in Escherichia coli is dynamic and sensitive to environmental cues. Mol. Microbiol. 50, 1493–1505 [DOI] [PubMed] [Google Scholar]
- 19. Golding I., Cox E. C. (2004) RNA dynamics in live Escherichia coli cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11310–11315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lutkenhaus J. (2007) Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu. Rev. Biochem. 76, 539–562 [DOI] [PubMed] [Google Scholar]
- 21. Raskin D. M., de Boer P. A. (1999) Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 96, 4971–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller O. L., Jr., Hamkalo B. A., Thomas C. A., Jr. (1970) Visualization of bacterial genes in action. Science 169, 392–395 [DOI] [PubMed] [Google Scholar]
- 23. Burmann B. M., Schweimer K., Luo X., Wahl M. C., Stitt B. L., Gottesman M. E., Rösch P. (2010) A NusE·NusG complex links transcription and translation. Science 328, 501–504 [DOI] [PubMed] [Google Scholar]
- 24. Proshkin S., Rahmouni A. R., Mironov A., Nudler E. (2010) Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328, 504–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woldringh C. L. (2002) The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol. Microbiol. 45, 17–29 [DOI] [PubMed] [Google Scholar]
- 26. Campos M., Jacobs-Wagner C. (2013) Cellular organization of the transfer of genetic information. Curr. Opin. Microbiol. 16, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montero Llopis P., Jackson A. F., Sliusarenko O., Surovtsev I., Heinritz J., Emonet T., Jacobs-Wagner C. (2010) Spatial organization of the flow of genetic information in bacteria. Nature 466, 77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nevo-Dinur K., Nussbaum-Shochat A., Ben-Yehuda S., Amster-Choder O. (2011) Translation-independent localization of mRNA in E. coli. Science 331, 1081–1084 [DOI] [PubMed] [Google Scholar]
- 29. Ortiz J. O., Brandt F., Matias V. R., Sennels L., Rappsilber J., Scheres S. H., Eibauer M., Hartl F. U., Baumeister W. (2010) Structure of hibernating ribosomes studied by cryoelectron tomography in vitro and in situ. J. Cell Biol. 190, 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Azam T. A., Hiraga S., Ishihama A. (2000) Two types of localization of the DNA-binding proteins within the Escherichia coli nucleoid. Genes Cells 5, 613–626 [DOI] [PubMed] [Google Scholar]
- 31. English B. P., Hauryliuk V., Sanamrad A., Tankov S., Dekker N. H., Elf J. (2011) Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc. Natl. Acad. Sci. U.S.A. 108, E365–E373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., Tsien R. Y. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 [DOI] [PubMed] [Google Scholar]
- 33. Datta S., Costantino N., Court D. L. (2006) A set of recombineering plasmids for gram-negative bacteria. Gene 379, 109–115 [DOI] [PubMed] [Google Scholar]
- 34. Ederth J., Mandava C. S., Dasgupta S., Sanyal S. (2009) A single-step method for purification of active His-tagged ribosomes from a genetically engineered Escherichia coli. Nucleic Acids Res. 37, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Galli E., Gerdes K. (2010) Spatial resolution of two bacterial cell division proteins. ZapA recruits ZapB to the inner face of the Z-ring. Mol. Microbiol. 76, 1514–1526 [DOI] [PubMed] [Google Scholar]
- 36. Akerlund T., Nordström K., Bernander R. (1995) Analysis of cell size and DNA content in exponentially growing and stationary-phase batch cultures of Escherichia coli. J. Bacteriol. 177, 6791–6797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson D. N. (2009) The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 44, 393–433 [DOI] [PubMed] [Google Scholar]
- 38. Dworsky P., Schaechter M. (1973) Effect of rifampin on the structure and membrane attachment of the nucleoid of Escherichia coli. J. Bacteriol. 116, 1364–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cabrera J. E., Cagliero C., Quan S., Squires C. L., Jin D. J. (2009) Active transcription of rRNA operons condenses the nucleoid in Escherichia coli. Examining the effect of transcription on nucleoid structure in the absence of transertion. J. Bacteriol. 191, 4180–4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Han X., Geng J., Zhang L., Lu T. (2011) The role of Escherichia coli YrbB in the lethal action of quinolones. J. Antimicrob. Chemother. 66, 323–331 [DOI] [PubMed] [Google Scholar]
- 41. Drlica K., Malik M., Kerns R. J., Zhao X. (2008) Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 52, 385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gitai Z., Dye N. A., Reisenauer A., Wachi M., Shapiro L. (2005) MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 120, 329–341 [DOI] [PubMed] [Google Scholar]
- 43. Osborn M. J., Rothfield L. (2007) Cell shape determination in Escherichia coli. Curr. Opin. Microbiol. 10, 606–610 [DOI] [PubMed] [Google Scholar]
- 44. Kruse T., Møller-Jensen J., Løbner-Olesen A., Gerdes K. (2003) Dysfunctional MreB inhibits chromosome segregation in Escherichia coli. EMBO J. 22, 5283–5292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bean G. J., Flickinger S. T., Westler W. M., McCully M. E., Sept D., Weibel D. B., Amann K. J. (2009) A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry 48, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herskovits A. A., Bibi E. (2000) Association of Escherichia coli ribosomes with the inner membrane requires the signal recognition particle receptor but is independent of the signal recognition particle. Proc. Natl. Acad. Sci. U.S.A. 97, 4621–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Defeu Soufo H. J., Reimold C., Linne U., Knust T., Gescher J., Graumann P. L. (2010) Bacterial translation elongation factor EF-Tu interacts and colocalizes with actin-like MreB protein. Proc. Natl. Acad. Sci. U.S.A. 107, 3163–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huh D., Paulsson J. (2011) Random partitioning of molecules at cell division. Proc. Natl. Acad. Sci. U.S.A. 108, 15004–15009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huh D., Paulsson J. (2011) Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 43, 95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mondal J., Bratton B. P., Li Y., Yethiraj A., Weisshaar J. C. (2011) Entropy-based mechanism of ribosome-nucleoid segregation in E. coli cells. Biophys. J. 100, 2605–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zimmerman S. B. (2006) Shape and compaction of Escherichia coli nucleoids. J. Struct. Biol. 156, 255–261 [DOI] [PubMed] [Google Scholar]
- 52. Fishov I., Norris V. (2012) Membrane heterogeneity created by transertion is a global regulator in bacteria. Curr. Opin. Microbiol. 15, 724–730 [DOI] [PubMed] [Google Scholar]
- 53. Konopka M. C., Shkel I. A., Cayley S., Record M. T., Weisshaar J. C. (2006) Crowding and confinement effects on protein diffusion in vivo. J. Bacteriol. 188, 6115–6123 [DOI] [PMC free article] [PubMed] [Google Scholar]