

Background: The Tim-Tipin complex is a component of the DNA replication machinery that is conserved across eukaryotes.
Results: TIPIN gene knock-out cells showed hypersensitivity to the topoisomerase I inhibitor camptothecin, decreased DNA synthesizing activity, and Top1 degradation.
Conclusion: The Tim-Tipin complex destabilizes the Top1 cleavage complex.
Significance: The Tim-Tipin complex could be a potential drug target in cancer chemotherapy.
Keywords: Anticancer Drug, DNA Repair, DNA Replication, DNA Topoisomerase, Molecular Cell Biology, Camptothecin, DT40, Replication Fork, Tipin, Top1
Abstract
The replication fork temporarily stalls when encountering an obstacle on the DNA, and replication resumes after the barrier is removed. Simultaneously, activation of the replication checkpoint delays the progression of S phase and inhibits late origin firing. Camptothecin (CPT), a topoisomerase I (Top1) inhibitor, acts as a DNA replication barrier by inducing the covalent retention of Top1 on DNA. The Timeless-Tipin complex, a component of the replication fork machinery, plays a role in replication checkpoint activation and stabilization of the replication fork. However, the role of the Timeless-Tipin complex in overcoming the CPT-induced replication block remains elusive. Here, we generated viable TIPIN gene knock-out (KO) DT40 cells showing delayed S phase progression and increased cell death. TIPIN KO cells were hypersensitive to CPT. However, homologous recombination and replication checkpoint were activated normally, whereas DNA synthesis activity was markedly decreased in CPT-treated TIPIN KO cells. Proteasome-dependent degradation of chromatin-bound Top1 was induced in TIPIN KO cells upon CPT treatment, and pretreatment with aphidicolin, a DNA polymerase inhibitor, suppressed both CPT sensitivity and Top1 degradation. Taken together, our data indicate that replication forks formed without Tipin may collide at a high rate with Top1 retained on DNA by CPT treatment, leading to CPT hypersensitivity and Top1 degradation in TIPIN KO cells.
Introduction
In eukaryotic cells, proteins in the DNA replication machinery function in a coordinated manner to ensure accurate DNA replication (1, 2). The MCM2–7 heterohexamer interacts with replication factors such as Cdc45 and GINS (Go, Ichi, Ni, and San) and unwinds double-stranded DNA ahead of the replication fork (3, 4). The leading strand and lagging strand are then synthesized by pol3 ϵ and pol δ/pol α, respectively, and the proliferating cell nuclear antigen clamp promotes the activity of DNA polymerases (5). All of the factors described above are essential for cell survival. Conversely, many factors are not essential for replication but are involved in ensuring the efficient progression of the replication fork. Tof1, Csm3, and Mrc1 in Saccharomyces cerevisiae, which are homologs of Swi1, Swi3, and Mrc1 in Schizosaccharomyces pombe, respectively, are not essential for cell survival; however, they play a role in the stabilization of the replication fork (1, 6). Tof1, Csm3, and Mrc1 translocate with the replication fork and stall, protecting the replication fork when obstacles are encountered (7, 8). In vertebrates, Timeless (Tim), Tipin, and Claspin are orthologs of Tof1, Csm3, and Mrc1. These vertebrate factors contribute to the activation of the replication checkpoint and the establishment of sister chromatid cohesion (9–13). In particular, Tim/Tof1 and Tipin/Csm3 form a tight heterodimer (called a fork protection complex) (6) that is assumed to have a different role than Claspin/Mrc1, although some functions may be partly redundant.
Tof1, a component of the replication fork machinery, was identified as a topoisomerase I (Top1)-binding factor (14). Top1 liberates topological stress generated in front of the replication fork via MCM2–7 helicase. Top1 cleaves the phosphodiester backbone of the DNA strand during the resolution of topological stress and forms a covalent bond with DNA to form the Top1 cleavage complex (Top1-cc) (15–17). Camptothecin (CPT), a Top1 inhibitor, binds to Top1 via hydrogen bonds, stabilizes Top1-cc, and inhibits DNA religation, resulting in cytotoxicity because of the blockage of DNA replication and/or transcription (16, 18). Collision between the replication fork and Top1-cc can produce DNA double strand breaks (DSBs) in the presence of CPT (15, 17). CPT-induced DSBs are mainly repaired by Rad51/BRCA2-mediated homologous recombination (HR) (19). Although DNA repair related to Top1-cc has been analyzed extensively, the interaction between the protein machinery at the replication fork and CPT-induced Top1-cc is poorly understood, especially in vertebrate cells. Because tof1 and csm3 mutant cells in S. cerevisiae and swi1− and swi3− mutant cells in S. pombe are hypersensitive to CPT (20–22), the vertebrate Tim-Tipin complex is expected to be involved in the tolerance to CPT-induced cytotoxicity. However, little is known about the role of the Tim-Tipin complex in overcoming CPT-induced replication barriers.
Here, we generated TIPIN gene knock-out (KO) cells using chicken DT40 cells to elucidate the precise roles of Tipin at the replication fork. TIPIN KO cells were viable but lost their proliferative capacity mainly because of a decrease in DNA replication elongation activity. TIPIN KO cells were hypersensitive to CPT. Characterization of CPT sensitivity in TIPIN KO cells predicts a role for vertebrate Tipin in protecting the replication fork from collapse following CPT treatment.
EXPERIMENTAL PROCEDURES
Plasmid DNA Construction
The targeting vectors for TIPIN gene disruption were designed by inserting a puromycin- or blasticidin-selective marker cassette into exons 3–5 of the TIPIN gene. The pGEM-T Easy vector was used. The expression vector for chicken TIPIN was generated by cloning chicken TIPIN DNA amplified by RT-PCR (SuperScript III, Invitrogen) into the pUHG10-3 vector. A FLAG tag sequence was added to the C-terminal end of the TIPIN coding sequence.
Cell Culture, DNA Transfection, and RT-PCR
The chicken DT40 cell lines used in this study are listed in Table 1. Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% chicken serum, 2 mm l-glutamine, 10 μm 2-mercaptoethanol, and 100 μg/ml kanamycin in 5% CO2 at 39 °C. DNA transfection and RT-PCR were performed as described previously (23). Drug-resistant colonies were selected in 96-well plates in medium containing 0.5 μg/ml puromycin, 30 μg/ml blasticidin, and 2.5 mg/ml hygromycin B. Gene disruption was verified by genomic PCR and RT-PCR. Gene expression was verified by RT-PCR and Western blotting. The primers used in genomic PCR to check marker insertion were 5′-GTGGAGCTCTCCGTCCTCCGAAAGCAGGCG-3′ or 5′-GCACCAGTCAGATCCCGAGCAACTGGGATG-3′ (sense) and 5′-TATTGGTCACCACGGCCGAG-3′ (antisense). The primers used in RT-PCR to amplify TIPIN (KO locus) were 5′-CCCACCTCCTACGTCTCCAGGAAGAGGTGA-3′ (sense) and 5′-AAAGCACCAGTCAGATCCCGAGCAACTGGG-3′ (antisense). The primers used in RT-PCR to amplify TIPIN (full length) were 5′-GAGTGTTGGTGCGGTGCTCGGTATTTTCG-3′ (sense) and 5′-GAAACTCCTGGAGTGAGACTGGAAAGAGC-3′ (antisense), and the primers used to amplify β-actin were 5′-CGTGCTGTGTTCCCATCTATCGTG-3′ (sense) and 5′-TACCTCTTTTGCTCTGGGCTTCATC-3′ (antisense).
TABLE 1.
List of chicken DT40 cell lines used in this study
Puro, puromycin; Bsr, blasticidin; Hyg, hygromycin; Neo, neomycin.
Assessment of Cell Growth and Sensitivity to DNA-damaging Agents
Cell number was determined by flow cytometry using plastic microbeads (07313-5, Polysciences) and propidium iodide (PI). 400 μl of cell solutions were mixed with 100 μl of the plastic microbead suspension, and viable cells were counted when a given number of microbeads were detected by flow cytometry. Cells not stained with PI were regarded as viable cells. To assess drug sensitivity, 1–3 × 104 cells were cultured in 24-well plates containing various concentrations of DNA-damaging agents in 1 ml of medium in duplicate. Cell viability was assessed after 48 h by flow cytometry using plastic microbeads and PI. Percent survival was determined by considering the number of untreated cells as 100%. The final concentration of PI was 1 μg/ml.
Cell Cycle Analysis by Flow Cytometry
To assess cell cycle progression, cells were harvested, fixed in 70% ethanol, stained with PI, and analyzed by flow cytometry. For two-dimensional cell cycle analysis, cells were cultured in medium containing 40 nm CPT and treated with 20 μm bromodeoxyuridine (BrdU; BD Biosciences) for 20 min just before harvesting. Cells were fixed in 70% ethanol, treated with 0.5% (v/v) Triton-X and 2.5 m HCl, and stained with FITC-labeled anti-BrdU antibody (BD Biosciences) and 1 μg/ml PI. Cell cycle distribution was analyzed by flow cytometry.
Cell Death Analysis by Flow Cytometry
Cell death analysis was carried out as described previously (24). To distinguish between viable cells and dead cells, cells were stained with 1 μg/ml PI without fixation and analyzed by flow cytometry.
DNA Fiber Assay
Cells (5 × 105 in 1 ml of medium) were pulse-labeled with 25 μm chlorodeoxyuridine (CldU; Sigma) and then sequentially pulse-labeled with 250 μm iododeoxyuridine (IdU; Sigma). Cells were resuspended in ice-cold PBS and then dropped onto aminopropylsilane-coated glass slides (Matsunami Glass). Cells were lysed with DNA fiber lysis buffer (0.5% SDS, 200 mm Tris-HCl, pH 7.4, 50 mm EDTA), and then glass slides were tilted to extend DNA. For fixation, glass slides were immersed in Carnoy fluid (MeOH:AcOH, 3:1) for 3 min, 70% EtOH for 1 h, and MeOH for 3 min. After washing with PBS, glass slides were immersed in 2.5 mm HCl for 30 min to denature DNA molecules and subsequently in 0.1 m sodium tetraborate for 3 min to neutralize. After washing with PBS, the slides were treated with rat anti-BrdU antibody (1:1000; Abcam) and mouse anti-BrdU antibody (1:500; BD Biosciences), which reacted against CldU and IdU, respectively. Cy3-conjugated anti-rat IgG (1:1000; Jackson ImmunoResearch Laboratories) and Alexa Fluor 488 anti-mouse IgG (1:1000; Invitrogen) were used as the secondary antibodies. The first and second antibodies were incubated for 1 h at room temperature, respectively. Washing of antibodies was performed with 0.05% Tween 20 in PBS. Coverslips were mounted with Fluoromount (Diagnostic BioSystems). Images were captured with a fluorescence microscope (BZ-9000, Keyence). CPT treatment conditions are described in each figure legend. Fiber lengths were measured using ImageJ, and micrometer values were expressed in kilobases using the following conversion factor: 1 μm = 2.59 kb (25). Measurements were recorded from areas of the slides with untangled DNA fibers to prevent the possibility of recording labeled patches from tangled bundles of fibers.
Antibodies
The primary antibodies used were anti-phospho-Histone H2AX-Ser139 (clone JBW301, Millipore), anti-α-tubulin (clone DM1A, Sigma), anti-Rad51 (a kind gift from Dr. Hitoshi Kurumizaka, Waseda University), anti-phospho-Check1 (Chk1)-Ser345 (2341, Cell Signaling Technology), anti-Histone H3 (ab1791, Abcam), anti-Top1 (556597, BD Biosciences), and anti-FLAG (M2, Sigma).
Western Blotting
Western blotting was carried out as described previously (26). Cells were suspended in SDS sample buffer (50 mm Tris-HCl, pH 6.8, 0.1 m DTT, 2% SDS, 10% glycerol, 0.1% bromphenol blue). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG (Cell Signaling Technology) were used as secondary antibodies. Proteins were visualized using ECL Prime Western blotting detection reagents (GE Healthcare). Images were captured with an ImageQuant LAS 4000 Mini imager.
Observation of Subnuclear Focus Formation
After CPT exposure (40 nm for 4 h), cells were harvested and spun onto aminopropylsilane-coated glass slides (Matsunami Glass). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% (v/v) Nonidet P-40 in PBS, and treated with anti-Rad51 antibodies. Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) was used as the secondary antibody, and 0.1 μg/ml DAPI was used for counterstaining. Coverslips were mounted with Fluoromount (Diagnostic BioSystems). Images were captured with a fluorescence microscope (BZ-9000, Keyence).
Subcellular Fractionation
To prepare the subcellular fraction of the nuclear soluble and chromatin-bound fractions, 5 × 106 cells were cultured in 10 ml of medium containing CPT and collected at the indicated time points. A Subcellular Protein Fractionation kit (78840, Thermo Scientific) was used for the fractionation.
RESULTS
Tipin Is Not Essential for Cell Survival but Is Required for the Maintenance of Proliferative Capacity
Little information is available on the chicken TIM gene, whereas the chicken TIPIN gene is located on chromosome 10. In the present study, we generated TIPIN KO DT40 cells. Targeting constructs were designed to delete exons 3–5 of the TIPIN gene, which were then sequentially transfected into DT40 cells (Fig. 1A). Gene disruption was confirmed by genomic PCR and RT-PCR (data not shown and Fig. 1B). Two clones of TIPIN KO cells were obtained, indicating that the TIPIN gene is not essential for vertebrate cell survival. Although TIPIN KO cells were viable, a marked decrease in their proliferative capacity was observed (Fig. 1C). The slow growth of TIPIN KO cells was investigated by analyzing cell cycle progression. Cells were synchronized at the G2/M phase using nocodazole, an inhibitor of microtubule polymerization, and cell cycle progression was monitored by flow cytometry. The results showed a delay in S phase progression in TIPIN KO cells (Fig. 1D). Moreover, cell death was approximately 3 times higher in TIPIN KO cells than in wild-type cells (Fig. 1E). These results suggested that the slow growth of TIPIN KO cells was caused by both a delay in S phase progression and an increase in cell death.
FIGURE 1.

Generation of TIPIN KO cells. A, schematic representation of the targeting construct for TIPIN gene disruption (upper) and the gene targeting procedure (lower). Closed boxes indicate exons. Puro and Bsr indicate the puromycin and blasticidin resistance genes, respectively. B, confirmation of TIPIN gene disruption by RT-PCR. Primer sets a and b were used to detect the KO locus by a targeting construct and full-length coding region in TIPIN mRNA. β-Actin was used as a control. C, growth curve of TIPIN KO cells. The number of viable cells was determined by flow cytometry. D, cell cycle analysis after release from M phase block. Cells were cultured in the presence of nocodazole (500 ng/ml) for 8 h. After release from M phase block, cells were collected at 1-h intervals, fixed, and stained with PI. DNA content was analyzed by flow cytometry. Arrows indicate S phase progression-delayed cells. Asyn, asynchronous. E, detection of dead cells. Cells were stained with PI, analyzed by flow cytometry (left), and quantitated (right). Forward scatter represents the size of cells. The outlined areas, which contain small sized cells heavily stained with PI, represent the population of dead cells. The error bars indicate S.D. from three independent experiments.
Tipin Is Required for Normal DNA Replication Fork Progression
To investigate the alteration of S phase progression in TIPIN KO cells, we examined the effect of TIPIN KO on DNA replication elongation. Cells were pulse-labeled with the thymidine analogs CldU or IdU, and labeled DNA replication tracks were immunostained with specific antibodies to visualize the progression of the replication fork (Fig. 2A). The rate of DNA replication elongation (fork rate) in TIPIN KO cells was almost half of that observed in wild-type cells (Fig. 2, B and C). This result suggests that Tipin is required for the maintenance of normal replication fork progression. The decreased fork rate in TIPIN KO cells partly explains the delayed S phase progression.
FIGURE 2.
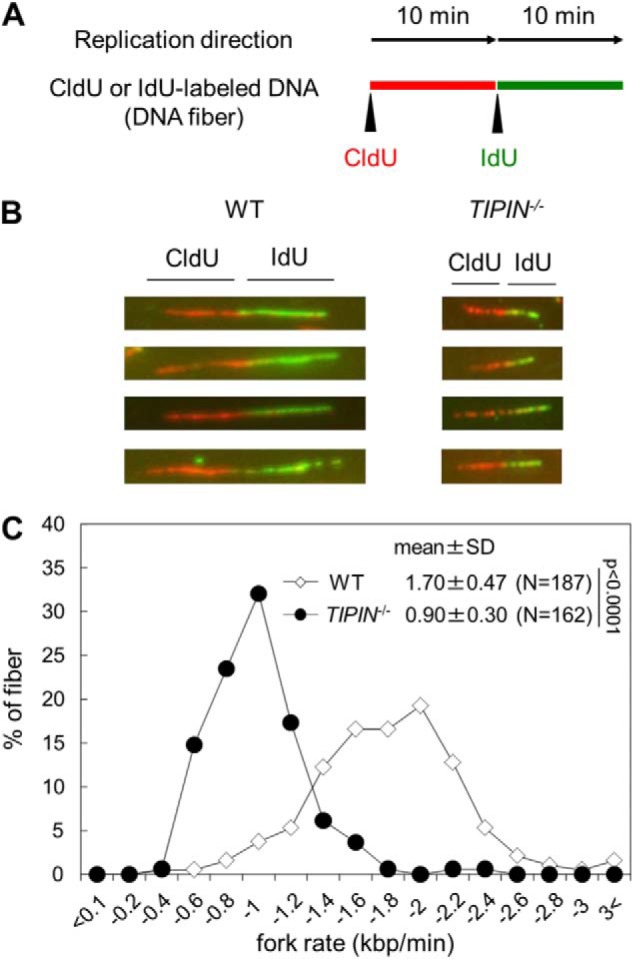
Analysis of DNA replication elongation. A, schematic representation of the DNA fiber assay. Cells were pulse-labeled with CldU (red) and then IdU (green). DNA fibers were stained as described in “Experimental Procedures”. B, image of typical DNA fibers. C, DNA replication elongation rates. The lengths of the DNA fibers (only CldU tracks connected with IdU tracks) prepared as shown in A were measured, and DNA replication elongation rates (fork rates) were calculated as fiber length divided by pulse labeling time. p values were calculated by Student's t test. N, number of measured fibers.
Tipin Is Involved in Tolerance to Topoisomerase I Inhibitor
To determine whether vertebrate Tipin plays a role in the response to CPT-induced replication fork stalling, the sensitivities of TIPIN KO cells to various replication stress-inducing agents including CPT were examined (Fig. 3). Although TIPIN KO cells were hypersensitive to CPT compared with wild-type cells (Fig. 3A), they were not particularly sensitive to etoposide, a topoisomerase II (Top2) inhibitor, or olaparib, a poly(ADP-ribose) polymerase inhibitor (Fig. 3, B and C). TIPIN KO cells were moderately sensitive to hydroxyurea (HU), aphidicolin (APH), and methyl methanesulfonate (Fig. 3, D–F), and their sensitivity to these agents was considerably lower than their sensitivity to CPT.
FIGURE 3.

Assessment of sensitivity to DNA replication stress-inducing agents. A–F, cells were incubated for 48 h in medium containing the indicated agents. Cell viability was assessed after 48 h by flow cytometry using plastic microbeads and PI. Percent survival was determined by considering the number of untreated cells as 100%. The error bars indicate the S.D. of two independent cultures. G, schematic representation of the generation of TIPIN−/− + chTIPIN-FLAG cells. Hyg indicates the hygromycin B resistance gene. H, confirmation of TIPIN gene expression by RT-PCR. Primer set a in Fig. 1B to detect KO locus was used. β-Actin was used as a control. I, confirmation of the expression of chicken Tipin-FLAG protein with anti-FLAG antibody by Western blotting. TIPIN−/− + chTIPIN-FLAG cells were cultured in the presence (+) or absence (−) of 1 μg/ml doxycycline (Dox) for 48 h. The expression of chicken Tipin-FLAG protein was suppressed by doxycycline treatment. α-Tubulin was used as a loading control.
To investigate whether the CPT hypersensitivity of TIPIN KO cells is an intrinsic response, we generated TIPIN KO cells expressing exogenous chicken TIPIN cDNA. Chicken TIPIN cDNA was cloned, and a FLAG tag sequence was added to the C-terminal end. This expression vector was transfected into TIPIN KO cells to generate TIPIN−/− + chTIPIN-FLAG cells (Fig. 3G). Expression of chicken Tipin-FLAG mRNA and protein was assessed by RT-PCR and Western blotting (Fig. 3, H and I). The CPT sensitivity of TIPIN KO cells was reversed by expression of chicken Tipin-FLAG (Fig. 3A). Taken together, these results indicate that Tipin plays a role in resistance to CPT-induced damage.
CPT Treatment Activates Homologous Recombination and Replication Checkpoint in TIPIN KO Cells
Among the DNA-damaging agents tested, we focused on CPT to examine the function of Tipin. γH2AX, the phosphorylated form of H2AX, is a marker of replication stress and/or DSBs (27). High levels of γH2AX were detected in TIPIN KO cells in the absence and presence of CPT (Fig. 4A). Because DNA damage induced by CPT is repaired primarily by Rad51/BRCA2-mediated HR repair (16), we examined the activation of HR in TIPIN KO cells. Rad51 foci, which are indicators of Rad51 filament formation (28), an essential step of the HR pathway, were observed in TIPIN KO cells upon CPT treatment (Fig. 4, B and C). Thus, HR repair appears to operate normally in TIPIN KO cells.
FIGURE 4.

Observation of DNA damage signals in cells. A, H2AX phosphorylation (γH2AX). Cells were treated with 40 nm CPT for 4 or 8 h and analyzed by Western blotting. B and C, Rad51 foci formation. Images of cells treated or not treated with 40 nm CPT for 4 h (B) and the percentage of Rad51 focus-positive cells (C) are shown. The error bars indicate S.D. from two independent experiments. At least 100 nuclei were scored in each case, and nuclei containing more than four bright foci were defined as focus-positive. D, assessment of sensitivity. Cells were incubated for 48 h in medium containing the indicated concentrations of CPT. Cell viability was assessed after 48 h by flow cytometry using plastic microbeads and PI. Percent survival was determined by considering the number of untreated cells as 100%. The error bars indicate S.D. of two independent cultures. E, Chk1 phosphorylation. Cells were treated with 40 nm CPT for 4 or 8 h, harvested, and analyzed by Western blotting. α-Tubulin was used as a loading control. F, Chk1 phosphorylation. Cells were treated with 1 mm HU for 4 or 8 h, harvested, and analyzed by Western blotting. Histone H3 was used as a loading control.
Tipin is required for the activation of replication checkpoint after exposure to HU, APH, or ultraviolet light (11, 12, 29–31). Phosphorylation of Chk1 by the ATR-ATRIP complex activates Chk1, resulting in a delay in cell cycle progression and inhibition of late origin firing (32–34). Rad17 increases the phosphorylation activity of ATR as a 9-1-1 clamp loader (35, 36). To investigate whether the CPT sensitivity of TIPIN KO cells was associated with a defect in the Chk1-mediated replication checkpoint, we compared the sensitivities of RAD17 and CHK1 KO cells to CPT. The sensitivity of these KO cells to CPT was not higher than that of wild-type cells (Fig. 4D). We next examined Chk1 phosphorylation, a marker of replication checkpoint activation. Low levels of Chk1 phosphorylation were detected in wild-type and RAD17 KO cells upon CPT treatment (Fig. 4E). Interestingly, Chk1 phosphorylation upon CPT treatment was increased rather than decreased in TIPIN KO cells, whereas Chk1 phosphorylation following HU treatment was slightly decreased in TIPIN KO cells compared with wild-type cells (Fig. 4, E and F). Taken together, these findings suggest that the CPT sensitivity of TIPIN KO cells is not caused by defects in HR or replication checkpoint activation.
Tipin Promotes Progression of the Replication Fork in the Presence of CPT
We next monitored cell cycle progression upon CPT treatment. Cells were pulse-labeled with BrdU, a thymidine analog, and then stained with a BrdU-specific antibody and PI (Fig. 5A). BrdU-positive cells represent cells in S phase that are actively synthesizing DNA. In the presence of CPT, the number of wild-type cells in S phase decreased, and that in G2/M phase increased, and this was especially evident at 8 h after treatment (Fig. 5, A, left, and B). Conversely, the populations of cells in S and G2/M phases in TIPIN KO cells did not change significantly regardless of the absence or presence of CPT (Fig. 5, A, right, and B). However, two-dimensional analysis showed qualitative changes in the BrdU pattern with a decrease in BrdU incorporation during S phase in TIPIN KO cells compared with wild-type cells (Fig. 5C). These results suggest that DNA synthesis in TIPIN KO cells is defective in the presence of CPT.
FIGURE 5.
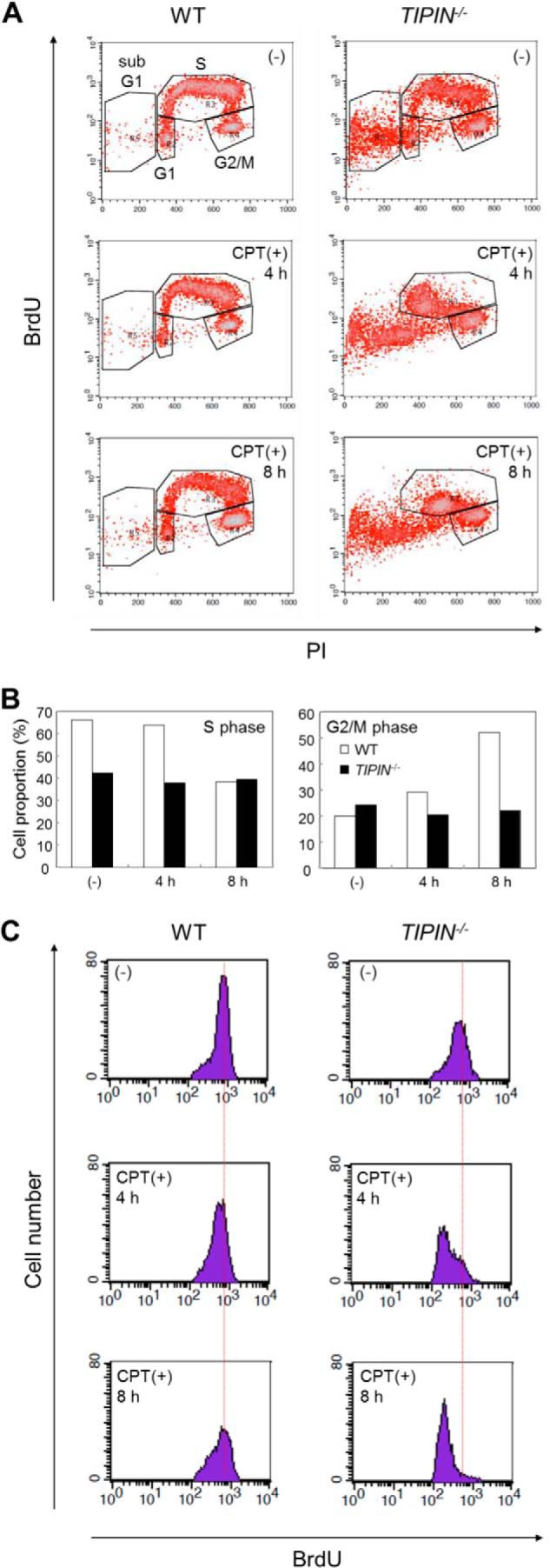
Analysis of cell cycle in the presence of CPT. A, cell cycle analysis with BrdU. Cells were cultured in the presence or absence of 40 nm CPT, pulse-labeled with BrdU for 20 min just before harvesting, collected, and fixed. Cells were stained with FITC-conjugated anti-BrdU antibody to detect BrdU uptake and PI to detect DNA and then analyzed by flow cytometry. B, quantification of the proportion of cells in S or G2/M phase in A. C, histogram of BrdU uptake in cells during S phase. Cell populations contained in the S phase gate in A are plotted by cell number (y axis) and BrdU uptake (x axis).
We used a DNA fiber assay to investigate DNA synthesis in the presence of CPT. Cells were pulse-labeled with CldU in the absence of CPT and then subsequently pulse-labeled with IdU in the presence of CPT (Fig. 6A). DNA replication elongation activity in response to CPT treatment was determined by the CldU/IdU ratio. Attenuation of replication elongation activity by CPT would result in an increase in the CldU/IdU ratio (Fig. 6B). Our results showed that the fork rate in TIPIN KO cells was ∼50% of that in wild-type cells (Fig. 2C). Therefore, if the duration of DNA synthesis is kept constant, the progression distance of the replication fork in TIPIN KO cells should be almost half of that in wild-type cells. Under these conditions, the replication fork would be less likely to encounter Top1-cc in TIPIN KO cells than in wild-type cells. To solve this problem, the incorporation time of CldU/IdU was set as 20 min/20 min or 40 min/40 min for wild-type or TIPIN KO cells, respectively (Fig. 6A). Under these experimental conditions, TIPIN KO cells showed a higher CldU/IdU ratio in the presence of CPT than wild-type cells (Fig. 6C). We then carefully measured the CldU/IdU ratio under the 40-min/40-min condition in wild-type cells upon CPT treatment. The mean CldU/IdU ratio under the 40-min/40-min versus the 20-min/20-min condition in wild-type cells was 1.12 (n = 47) versus 1.24 (n = 158), and the difference was not significant. These results suggested that the progression speed of the DNA replication fork is decreased in TIPIN KO cells treated with CPT.
FIGURE 6.
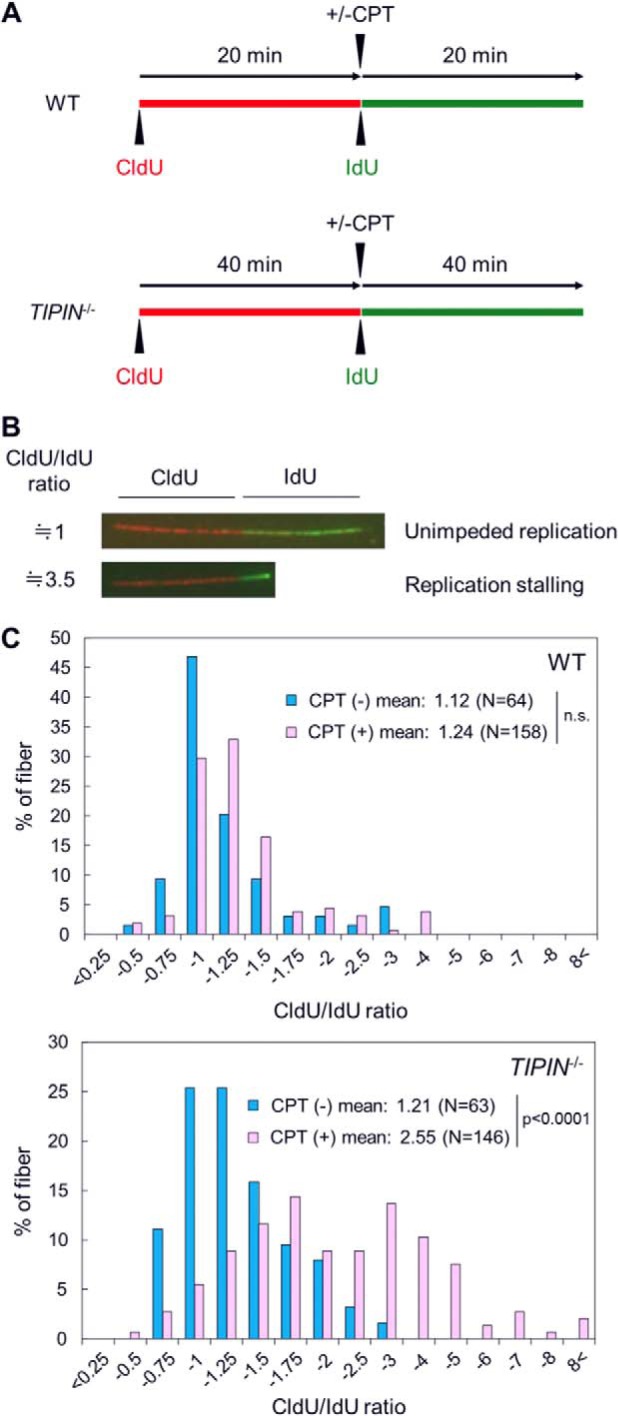
Analysis of DNA replication elongation in the presence of CPT. A, schematic representation of the DNA fiber assay in the presence CPT. Cells were pulse-labeled with CldU (red) and successively pulse-labeled with IdU (green) in the presence of 100 nm CPT. To unify the encounter rate between the replication fork and the Top1 cleavage complex induced by CPT, pulse labeling time with CldU or IdU was doubled in TIPIN KO cells compared with wild-type cells. B, image of typical DNA fibers following CPT treatment. Cells were pulse-labeled first with CldU (red) and then IdU (green) in the presence of CPT. Unimpeded or impeded replication-reflected DNA fibers are shown. The CldU/IdU ratio indicates the value of the CldU-labeled fiber length divided by the IdU-labeled fiber length. C, histograms of the CldU/IdU ratio. The CldU/IdU ratio was calculated as the CldU-labeled fiber length divided by the IdU-labeled fiber length. p values were calculated by Student's t test. N, number of measured fibers. n.s., not significant.
Tipin Prevents CPT-induced Top1 Degradation
CPT traps Top1 on DNA, inhibiting its function, which in turn induces cytotoxicity. To investigate whether the lack of Tipin affects the status of Top1 on chromatin, we performed subcellular fractionation assays. A total fraction and a chromatin fraction were prepared, and Top1 was detected by Western blotting (Fig. 7A). In wild-type cells, Top1 accumulated in the chromatin fraction in response to CPT treatment, suggesting the retention of Top1-cc on chromatin. A new low molecular weight band was detected specifically in the chromatin fraction of TIPIN KO cells that was present even in the absence of CPT and was visible in the total fraction when the film was overexposed. Because Top1 is degraded by the proteasome in response to high concentrations (μm order) of CPT (37–39), we speculated that low concentrations (nm order) of CPT would induce proteasome-dependent degradation of Top1 in TIPIN KO cells but not in wild-type cells. To confirm this, cells were treated with CPT and with a proteasome inhibitor, MG132 or lactacystin. The intensity of the CPT-induced band was attenuated by MG132 or lactacystin in TIPIN KO cells (Fig. 7, B and C), suggesting that this low molecular weight band was generated by proteasome-dependent Top1 degradation. These results indicated that low concentrations of CPT induce rapid degradation of Top1 in TIPIN KO cells.
FIGURE 7.

Detection of Top1 in the presence of CPT. A, Top1 degradation upon CPT treatment. Cells were treated with 40 nm CPT for 4 or 8 h, subjected to subcellular fractionation, and analyzed by Western blotting. The black and white arrowheads indicate the Top1 protein (∼100 kDa) and its degradation products (∼85 kDa), respectively. B, detection of Top1 proteins in the presence of CPT upon MG132 pretreatment. Cells were cultured in the presence or absence of 10 μm MG132 for 1 h, subsequently treated with 40 nm CPT for 4 h, then harvested, and analyzed by Western blotting. Total cell lysate was applied. C, detection of Top1 proteins in the presence of CPT upon lactacystin (LCT) pretreatment. Cells were cultured in the presence or absence of 5 μg/ml lactacystin for 1 h, subsequently treated with 100 nm CPT for 4 h, harvested, and analyzed by Western blotting. Total cell lysate was applied. D, assessment of sensitivity. Cells were cultured in the presence of 1 μm APH for 1 h, then treated with CPT for 4 h, washed, and incubated for 48 h in drug-free medium. Cell viability was assessed after 48 h by flow cytometry using plastic microbeads and PI. Percent survival was determined by considering the number of untreated cells as 100%. The error bars indicate S.D. of two independent cultures. E, detection of Top1 proteins in the presence of CPT upon APH pretreatment. Cells were cultured in the presence or absence of 5 μm APH for 1 h, then treated with 40 nm CPT for 4 h, harvested, and analyzed by Western blotting. Total cell lysate was applied.
In yeast, orthologs of the Tim-Tipin complex play a role in replication fork stabilization and/or protection from replication stress (8). To protect cells from the toxic effects of CPT, collision between the replication fork and Top1-cc should be avoided. Because inhibition of replication fork progression can suppress such collision, we hypothesized that the DNA polymerase inhibitor APH, which induces stalling of the replication fork, would inhibit the effect of CPT in TIPIN KO cells. Our results showed that CPT sensitivity in TIPIN KO cells was suppressed by APH pretreatment (Fig. 7D). Moreover, Top1 degradation in TIPIN KO cells was also suppressed by APH treatment (Fig. 7E). These results suggested that the frequent collisions between the replication fork and Top1-cc are the primary cause of the marked CPT sensitivity of TIPIN KO cells.
DISCUSSION
In the present study, we successfully generated vertebrate TIPIN gene KO cells. Although these cells were viable, they showed decreased proliferative capacity and a reduced rate of DNA replication fork progression. Moreover, TIPIN KO cells were sensitive to replication stress-inducing agents with a marked sensitivity to CPT. CPT delayed S phase progression and replication fork progression in TIPIN KO cells. Top1 degradation in TIPIN KO cells was observed in the absence of CPT and was enhanced by CPT treatment. These findings are indicative of a relationship between Tipin activity and Top1-cc-induced DNA damage.
Tipin Plays a Role in Replication Fork Progression
In previous studies, the function of Tipin in human cells was analyzed using siRNA knockdown (10–12, 29, 40, 41). However, siRNA knockdown did not completely eliminate endogenous Tipin protein expression, whereas the expression of Tipin was completely suppressed in TIPIN KO cells. Therefore, the generation of TIPIN KO cells allowed us to confirm that Tipin is not essential for vertebrate cell survival (Fig. 1). Furthermore, the rate of replication fork progression was obviously decreased in cells lacking Tipin (Fig. 2). However, the mechanism underlying the decreased fork rate in TIPIN KO cells remains unclear. We have formulated three hypotheses to address this question. First, the Tim-Tipin complex may promote DNA polymerase activity during DNA replication elongation and maintain the progression capacity of the replication fork. The Tim-Tipin complex is involved in the association of pol α with And1, a replication factor, in Xenopus egg extracts (42), and it promotes the DNA synthesizing activity of replicative DNA polymerases (43, 44). Second, the Tim-Tipin complex may efficiently load Claspin onto chromatin. Lack of Claspin results in decreased replication fork speed (45, 46), and it has been suggested that the Tim-Tipin complex stably binds Claspin onto chromatin in Xenopus egg extracts (13, 30). Finally, loss of the interaction between the Tim-Tipin complex and Top1 at the replication fork may result in the inability to deal with Top1 properly and thus lead to the degradation of Top1 (Fig. 7A). This possibility will be discussed in detail later. The three possible mechanisms are not mutually exclusive.
Role of Tipin following Top1 Inhibition by CPT
TIPIN KO cells showed hypersensitivity to CPT (Fig. 3A), consistent with a previous report in the fission yeast swi3− mutant (20). Conversely, TIPIN KO cells showed no or little increase of sensitivity to etoposide (Fig. 3B), suggesting an intimate relationship between Tipin and Top1 but not Top2. Tof1, an ortholog of Tim in the budding yeast, interacts with Top1 in vitro (14), and Csm3, an ortholog of Tipin in budding yeast, forms immunoprecipitable replisome progression complexes including Top1 (4). Taken together with these findings, our results suggest that the Tim-Tipin complex is closely associated with Top1 at the replication fork.
CPT-induced damage is repaired by Rad51/BRCA2-mediated HR, and HR-defective cells, such as BRCA2-deficient cells, exhibit extreme sensitivity to olaparib, a poly(ADP-ribose) polymerase inhibitor (47, 48), whereas TIPIN KO cells showed little sensitivity to olaparib (Fig. 3C). In addition, the formation of CPT-induced Rad51 foci was observed in TIPIN KO cells (Fig. 4, B and C). Taken together, these observations indicate that defective HR repair may not be the cause of CPT sensitivity in TIPIN KO cells.
The Tim-Tipin complex is involved in the activation of the replication checkpoint upon HU treatment (11, 12, 29); however, in the present study, the complex was not necessary for activation of the replication checkpoint upon CPT treatment (Fig. 4E). A previous report showed that the phosphorylation of Chk1 is enhanced by TIM knockdown (40). Therefore, the Tim-Tipin complex does not appear to be required for the activation of the replication checkpoint under certain circumstances. One of the replication checkpoint factors, Chk1, reportedly causes inhibition of not only DNA replication initiation but also nascent DNA elongation in the presence of CPT (49). Increased Chk1 phosphorylation in TIPIN KO cells upon CPT treatment could be involved in the decrease of DNA fiber length in the presence of CPT (Fig. 6C).
High concentrations (μm order) of Top1 inhibitors induce the degradation of Top1 (37–39, 50). Collision of CPT-induced Top1-cc with the replication fork leads to Top1 polyubiquitination and degradation by the 26 S proteasome, and the Top1 peptide remaining on the DNA is processed to result in the formation of a DSB (15–17, 37). The effects of low concentrations (nm order) of CPT on TIPIN KO cells, such as replication checkpoint activation, inhibition of DNA fiber elongation, and degradation of Top1, are observed in normal cells treated with high concentrations of CPT (27, 37, 51). This suggests that the DNA replication fork frequently collides with Top1-cc in TIPIN KO cells even in the presence of low concentrations of CPT. In other words, the rate of collision of the replication fork with Top1-cc is decreased in wild-type cells. How the Tim-Tipin complex prevents the collision of the DNA replication fork with Top1-cc remains unclear.
Yeast Tof1 (Tim homolog) interacts with Top1 (14), and vertebrate Tipin interacts with pol α via And1 and has a binding domain for RPA (11, 42). Therefore, it seems possible that the Tim-Tipin complex in association with the DNA replication machinery interacts with Top1-cc, and the interaction may affect Top1 conformation, leading to the dissociation of CPT from Top1-cc because binding of CPT to Top1 is reversible (17). The dissociation of CPT from Top1 destabilizes Top1-cc, resulting in the release of Top1 from DNA after religation of the DNA strand (Fig. 8A). Thus, the replication fork progresses normally in wild-type cells in the presence of low concentrations of CPT. Conversely, the DNA replication machinery collides with Top1-cc in TIPIN KO cells because of the absence of the Tim-Tipin complex, leading to the degradation of Top1 to result in the formation of a DSB (Fig. 8B).
FIGURE 8.

Model of Tipin-mediated protection of the replication fork following CPT treatment. A and B depict models of replication fork progression in the presence of CPT in wild-type and TIPIN KO cells, respectively. The details are provided under “Discussion”.
Another possibility is that the interaction between the Tim-Tipin complex and Top1 directly destabilizes Top1-cc regardless of the presence or absence of CPT. In fact, degradation of Top1 was observed in the absence of CPT in TIPIN KO cells (Fig. 7A). In this context, structurally altered DNAs such as UV adducts or abasic sites also stabilize Top1-cc (15), suggesting that the mechanism leading to the dissociation of Top1-cc even in the absence of CPT is essential for DNA replication elongation. Therefore, one of the physiological functions of the Tim-Tipin complex may be to destabilize the Top1-cc by interacting with Top1.
Concluding Remarks
Our study provides a novel insight into the function of the Tim-Tipin complex in overcoming Top1-cc during the progression of the DNA replication fork. In addition to furthering basic studies, our findings showing that TIPIN KO cells are hypersensitive to CPT may be of value to improve cancer chemotherapy. The stability of Tim and Tipin in their complex is mutually dependent because suppression of either Tim or Tipin expression by siRNA reportedly leads to degradation of both proteins (11, 29, 41). Thus, inhibitors disrupting the interaction between Tim and Tipin will greatly sensitize cells to Top1 inhibitors, which are widely used clinically as anticancer agents.
Acknowledgments
We thank Dr. Hitoshi Kurumizaka for the gift of anti-Rad51 antibodies. We thank Dr. David A. F. Gillespie for the donation of CHK1−/− DT40 cells. We thank Ryosuke Ishihara for technical support.
This work was supported by grants-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (NEXT KAKENHI Grants 22131002 and 24590089).
- pol
- polymerase
- Tim
- Timeless
- Top1
- topoisomerase I
- Top1-cc
- Top1 cleavage complex
- CPT
- camptothecin
- DSB
- double strand break
- HR
- homologous recombination
- PI
- propidium iodide
- CldU
- chlorodeoxyuridine
- IdU
- iododeoxyuridine
- Top2
- topoisomerase II
- HU
- hydroxyurea
- APH
- aphidicolin
- Chk1
- Check1
- ATR
- ataxia-telangiectasia mutated and Rad3-related
- ATRIP
- ATR-interacting protein
- RPA
- replication protein A.
REFERENCES
- 1. Errico A., Costanzo V. (2010) Differences in the DNA replication of unicellular eukaryotes and metazoans: known unknowns. EMBO Rep. 11, 270–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Branzei D., Foiani M. (2010) Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219 [DOI] [PubMed] [Google Scholar]
- 3. Aze A., Zhou J. C., Costa A., Costanzo V. (2013) DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma 122, 401–413 [DOI] [PubMed] [Google Scholar]
- 4. Gambus A., Jones R. C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R. D., Labib K. (2006) GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 8, 358–366 [DOI] [PubMed] [Google Scholar]
- 5. Mailand N., Gibbs-Seymour I., Bekker-Jensen S. (2013) Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 14, 269–282 [DOI] [PubMed] [Google Scholar]
- 6. Leman A. R., Noguchi E. (2012) Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle 11, 3945–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bando M., Katou Y., Komata M., Tanaka H., Itoh T., Sutani T., Shirahige K. (2009) Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J. Biol. Chem. 284, 34355–34365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katou Y., Kanoh Y., Bando M., Noguchi H., Tanaka H., Ashikari T., Sugimoto K., Shirahige K. (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424, 1078–1083 [DOI] [PubMed] [Google Scholar]
- 9. Smith-Roe S. L., Patel S. S., Simpson D. A., Zhou Y. C., Rao S., Ibrahim J. G., Kaiser-Rogers K. A., Cordeiro-Stone M., Kaufmann W. K. (2011) Timeless functions independently of the Tim-Tipin complex to promote sister chromatid cohesion in normal human fibroblasts. Cell Cycle 10, 1618–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leman A. R., Noguchi C., Lee C. Y., Noguchi E. (2010) Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 123, 660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kemp M. G., Akan Z., Yilmaz S., Grillo M., Smith-Roe S. L., Kang T. H., Cordeiro-Stone M., Kaufmann W. K., Abraham R. T., Sancar A., Unsal-Kaçmaz K. (2010) Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem. 285, 16562–16571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshizawa-Sugata N., Masai H. (2007) Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 282, 2729–2740 [DOI] [PubMed] [Google Scholar]
- 13. Tanaka H., Kubota Y., Tsujimura T., Kumano M., Masai H., Takisawa H. (2009) Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells 14, 949–963 [DOI] [PubMed] [Google Scholar]
- 14. Park H., Sternglanz R. (1999) Identification and characterization of the genes for two topoisomerase I-interacting proteins from Saccharomyces cerevisiae. Yeast 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 15. Pommier Y. (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802 [DOI] [PubMed] [Google Scholar]
- 16. Tomicic M. T., Kaina B. (2013) Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim. Biophys. Acta 1835, 11–27 [DOI] [PubMed] [Google Scholar]
- 17. Pommier Y., Barcelo J. M., Rao V. A., Sordet O., Jobson A. G., Thibaut L., Miao Z. H., Seiler J. A., Zhang H., Marchand C., Agama K., Nitiss J. L., Redon C. (2006) Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 81, 179–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pommier Y. (2013) Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 8, 82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arnaudeau C., Tenorio Miranda E., Jenssen D., Helleday T. (2000) Inhibition of DNA synthesis is a potent mechanism by which cytostatic drugs induce homologous recombination in mammalian cells. Mutat. Res. 461, 221–228 [DOI] [PubMed] [Google Scholar]
- 20. Rapp J. B., Noguchi C., Das M. M., Wong L. K., Ansbach A. B., Holmes A. M., Arcangioli B., Noguchi E. (2010) Checkpoint-dependent and -independent roles of Swi3 in replication fork recovery and sister chromatid cohesion in fission yeast. PLoS One 5, e13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roseaulin L. C., Noguchi C., Martinez E., Ziegler M. A., Toda T., Noguchi E. (2013) Coordinated degradation of replisome components ensures genome stability upon replication stress in the absence of the replication fork protection complex. PLoS Genet. 9, e1003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Redon C., Pilch D. R., Bonner W. M. (2006) Genetic analysis of Saccharomyces cerevisiae H2A serine 129 mutant suggests a functional relationship between H2A and the sister-chromatid cohesion partners Csm3-Tof1 for the repair of topoisomerase I-induced DNA damage. Genetics 172, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hosono Y., Abe T., Ishiai M., Takata M., Enomoto T., Seki M. (2011) The role of SNM1 family nucleases in etoposide-induced apoptosis. Biochem. Biophys. Res. Commun. 410, 568–573 [DOI] [PubMed] [Google Scholar]
- 24. Wang W., Seki M., Narita Y., Nakagawa T., Yoshimura A., Otsuki M., Kawabe Y., Tada S., Yagi H., Ishii Y., Enomoto T. (2003) Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol. Cell. Biol. 23, 3527–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jackson D. A., Pombo A. (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 140, 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abe T., Ishiai M., Hosono Y., Yoshimura A., Tada S., Adachi N., Koyama H., Takata M., Takeda S., Enomoto T., Seki M. (2008) KU70/80, DNA-PKcs, and Artemis are essential for the rapid induction of apoptosis after massive DSB formation. Cell. Signal. 20, 1978–1985 [DOI] [PubMed] [Google Scholar]
- 27. Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K. J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M. (2012) Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 19, 417–423 [DOI] [PubMed] [Google Scholar]
- 28. Thompson L. H., Schild D. (2001) Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 477, 131–153 [DOI] [PubMed] [Google Scholar]
- 29. Unsal-Kaçmaz K., Chastain P. D., Qu P. P., Minoo P., Cordeiro-Stone M., Sancar A., Kaufmann W. K. (2007) The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 27, 3131–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Errico A., Costanzo V., Hunt T. (2007) Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc. Natl. Acad. Sci. U.S.A. 104, 14929–14934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gotter A. L., Suppa C., Emanuel B. S. (2007) Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J. Mol. Biol. 366, 36–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Branzei D., Foiani M. (2008) Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 9, 297–308 [DOI] [PubMed] [Google Scholar]
- 33. Finn K., Lowndes N. F., Grenon M. (2012) Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 69, 1447–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zachos G., Rainey M. D., Gillespie D. A. (2003) Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 22, 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delacroix S., Wagner J. M., Kobayashi M., Yamamoto K., Karnitz L. M. (2007) The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21, 1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kobayashi M., Hirano A., Kumano T., Xiang S. L., Mihara K., Haseda Y., Matsui O., Shimizu H., Yamamoto K. (2004) Critical role for chicken Rad17 and Rad9 in the cellular response to DNA damage and stalled DNA replication. Genes Cells 9, 291–303 [DOI] [PubMed] [Google Scholar]
- 37. Lin C. P., Ban Y., Lyu Y. L., Liu L. F. (2009) Proteasome-dependent processing of topoisomerase I-DNA adducts into DNA double strand breaks at arrested replication forks. J. Biol. Chem. 284, 28084–28092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang H. F., Tomida A., Koshimizu R., Ogiso Y., Lei S., Tsuruo T. (2004) Cullin 3 promotes proteasomal degradation of the topoisomerase I-DNA covalent complex. Cancer Res. 64, 1114–1121 [DOI] [PubMed] [Google Scholar]
- 39. Desai S. D., Liu L. F., Vazquez-Abad D., D'Arpa P. (1997) Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J. Biol. Chem. 272, 24159–24164 [DOI] [PubMed] [Google Scholar]
- 40. Smith K. D., Fu M. A., Brown E. J. (2009) Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J. Cell Biol. 187, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chou D. M., Elledge S. J. (2006) Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. U.S.A. 103, 18143–18147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Errico A., Cosentino C., Rivera T., Losada A., Schwob E., Hunt T., Costanzo V. (2009) Tipin/Tim1/And1 protein complex promotes Polα chromatin binding and sister chromatid cohesion. EMBO J. 28, 3681–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aria V., De Felice M., Di Perna R., Uno S., Masai H., Syväoja J. E., van Loon B., Hübscher U., Pisani F. M. (2013) The human Tim-Tipin complex interacts directly with DNA polymerase ϵ and stimulates its synthetic activity. J. Biol. Chem. 288, 12742–12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cho W. H., Kang Y. H., An Y. Y., Tappin I., Hurwitz J., Lee J. K. (2013) Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 110, 2523–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petermann E., Helleday T., Caldecott K. W. (2008) Claspin promotes normal replication fork rates in human cells. Mol. Biol. Cell 19, 2373–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshimura A., Akita M., Hosono Y., Abe T., Kobayashi M., Yamamoto K., Tada S., Seki M., Enomoto T. (2011) Functional relationship between Claspin and Rad17. Biochem. Biophys. Res. Commun. 414, 298–303 [DOI] [PubMed] [Google Scholar]
- 47. Qing Y., Yamazoe M., Hirota K., Dejsuphong D., Sakai W., Yamamoto K. N., Bishop D. K., Wu X., Takeda S. (2011) The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet. 7, e1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maxwell K. N., Domchek S. M. (2012) Cancer treatment according to BRCA1 and BRCA2 mutations. Nat. Rev. Clin. Oncol. 9, 520–528 [DOI] [PubMed] [Google Scholar]
- 49. Seiler J. A., Conti C., Syed A., Aladjem M. I., Pommier Y. (2007) The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 27, 5806–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Desai S. D., Li T. K., Rodriguez-Bauman A., Rubin E. H., Liu L. F. (2001) Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 61, 5926–5932 [PubMed] [Google Scholar]
- 51. Sugimura K., Takebayashi S., Taguchi H., Takeda S., Okumura K. (2008) PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 183, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buerstedde J. M., Takeda S. (1991) Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell 67, 179–188 [DOI] [PubMed] [Google Scholar]