

Background: The mechanisms that control β cell fate following cytokine- and nitric oxide-induced damage remain unknown.
Results: Cytokine-induced nitric oxide activates ATM and ATM-dependent caspase activation in β cells.
Conclusion: ATM regulates the induction of apoptosis in cytokine-treated β cells.
Significance: These studies define a role for DNA damage and ATM activation in nitric oxide-induced β cell apoptosis.
Keywords: Apoptosis, β Cell, Cytokine, DNA Repair, Nitric Oxide, ATM, H2AX
Abstract
In this study, the effects of cytokines on the activation of the DNA double strand break repair factors histone H2AX (H2AX) and ataxia telangiectasia mutated (ATM) were examined in pancreatic β cells. We show that cytokines stimulate H2AX phosphorylation (γH2AX formation) in rat islets and insulinoma cells in a nitric oxide- and ATM-dependent manner. In contrast to the well documented role of ATM in DNA repair, ATM does not appear to participate in the repair of nitric oxide-induced DNA damage. Instead, nitric oxide-induced γH2AX formation correlates temporally with the onset of irreversible DNA damage and the induction of apoptosis. Furthermore, inhibition of ATM attenuates cytokine-induced caspase activation. These findings show that the formation of DNA double strand breaks correlates with ATM activation, irreversible DNA damage, and ATM-dependent induction of apoptosis in cytokine-treated β cells.
Introduction
Type 1 (insulin-dependent) diabetes mellitus is characterized by an inflammatory reaction in and around pancreatic islets that is followed by the selective destruction of insulin-secreting β cells (1). Proinflammatory cytokines such as IL-1, released from infiltrating macrophages, are believed to participate in the development of type 1 diabetes mellitus by directly damaging β cells (2–4). In rat islets, the macrophage-derived cytokine IL-1 is sufficient to induce β cell damage, whereas interferon-γ (IFNγ) potentiates the damaging actions of IL-1 (3, 5, 6). The mechanisms by which cytokines damage β cells include the enhanced expression of inducible nitric oxide synthase (iNOS)3 and the subsequent generation of micromolar levels of nitric oxide within the β cell (7–9). Nitric oxide inhibits mitochondrial aconitase activity and electron transport, leading to decreased levels of cellular ATP (8, 10). Because insulin secretion requires the generation of sufficient levels of ATP to close ATP-sensitive K+ channels, leading to depolarization and Ca2+-dependent exocytosis of insulin, the inhibition of mitochondrial function, and ATP generation is one mechanism by which nitric oxide attenuates glucose-stimulated insulin secretion by β cells (8, 10, 11). In addition, nitric oxide inhibits protein synthesis, induces ER stress responses, and damages β cell DNA (12–17). DNA damage in response to nitric oxide occurs in the form of the oxidation or deamination of base pairs, leading to strand breaks (18, 19). It is likely that the combination of DNA damage, the inhibition of mitochondrial function, and the reduction in cellular levels of ATP contribute to cytokine-mediated β cell death (20).
Although the destructive actions of nitric oxide on β cell function have been described in detail, nitric oxide also activates a number of defense/repair pathways that afford β cells a limited capacity to recover from cytokine-mediated damage (21, 22). The ability to recover from cytokine-induced damage was originally identified in studies where the inhibitory actions of a 15-h incubation with IL-1 on insulin secretion were found to be reversed if the cytokine was removed by washing and the islets were allowed to incubate for an additional 4 days in the absence of the IL-1 (23). We showed that the addition of an iNOS inhibitor (NMMA) following IL-1 treatment and continued culture, without removing the IL-1 from the medium, allows for the time-dependent repair of damaged DNA and the recovery of insulin secretion and oxidative metabolism that is complete after 8 h (13, 21, 22, 24). These studies establish nitric oxide as the primary effector molecule mediating cytokine-induced damage because inhibitors of iNOS prevent cytokine-induced damage and β cells have the capacity to recover from cytokine-induced damage by inhibiting nitric oxide generation in the presence of cytokines. Although β cells have the ability to recover from cytokine-induced damage, this recovery is temporally limited. We have shown that treatment of islets for 36 h or longer with IL-1 results in an irreversible inhibition of mitochondrial oxidation, aconitase activity, protein synthesis, and DNA damage (13, 24). Under conditions in which cytokine-mediated damage is irreversible, the β cell is ultimately committed to death by apoptosis (13, 24).
In this study, the effects of cytokine treatment on activation of the DNA damage response (DDR) and the role of this response in the repair of DNA damage in β cells were examined. Nitric oxide, produced endogenously following iNOS induction or added exogenously using chemical donors induces the phosphorylation of histone variant H2AX on Ser-139. H2AX is a component of the DDR that is phosphorylated (forming γH2AX) in response to DNA double strand breaks (DSBs) (25–27). Consistent with its role as one of the most sensitive markers of DSB lesions (27), γH2AX formation appears to be mediated by the DDR kinase ataxia telangiectasia mutated (ATM) (28). Although these findings implicate DSB formation and the activation of the DDR in response to nitric oxide, ATM does not appear to participate in the repair of nitric oxide-induced DNA damage or the activation of pathways that are required for DNA repair in β cells. In contrast, there is a marked increase in γH2AX formation after 36 h of IL-1 treatment, a time at which nitric oxide-induced damage becomes irreversible and β cells are committed to death by apoptosis (13, 24). Inhibition of ATM under these conditions attenuates IL-1-induced caspase activation, suggesting that ATM may be the kinase responsible for the induction of β cell apoptosis under conditions of irreversible DNA damage in cytokine-treated β cells.
EXPERIMENTAL PROCEDURES
Materials and Animals
Male Sprague-Dawley rats (250–300 g) were purchased from Harlan (Indianapolis, IN). Wild-type and ATM−/− mice (129S6/SvEvTac background) were from The Jackson Laboratory (Bar Harbor, ME). The insulinoma cell line INS 832/13 was obtained from Chris Newgard (Duke University, Durham NC). RPMI 1640 medium, Connaught Medical Research Laboratories (CMRL) 1066 medium, l-glutamine, sodium pyruvate, HEPES, penicillin, streptomycin and β-mercaptoethanol were purchased from Invitrogen. 0.05% trypsin in 0.53 mm EDTA was purchased from Corning (Corning, NY), and human recombinant IL-1β and rat IFNγ were purchased from PeproTech (Rocky Hill, NJ). NG-monomethyl l-arginine (NMMA) was purchased from Axxora (San Diego, CA). (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA/NO) was purchased from Enzo (Farmingdale, NY) and dissolved in 10 mm NaOH prior to use. Hydrogen peroxide, camptothecin, and KU-55933 were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies used and their sources were as follows: mouse anti-phospho-H2AX (Ser-139, γH2AX) (EMD Millipore, Billerica, MA); mouse anti-phospho-ATM (Ser-1981), mouse anti-ATM, and rabbit anti-H2AX (Abcam, Cambridge, MA); mouse anti-GAPDH antibody (Invitrogen); rabbit anti-ACTIVE-JNK (Promega, Madison, WI); rabbit anti-PARP and rabbit anti-cleaved caspase 3 (Cell Signaling Technology, Beverly, MA); rabbit anti-PAR (Trevigen, Gaithersburg, MD); guinea pig anti-insulin (DakoCytomation, Carpinteria, CA); HRP-conjugated donkey anti-rabbit, HRP-conjugated donkey anti-mouse, Alexa Fluor 488-conjugated donkey anti-rabbit, Cy3-conjugated donkey anti-mouse, and DyLight-405 goat anti-guinea pig antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA).
Islet Isolation and Cell Culture
Islets were isolated from male Sprague-Dawley rats by collagenase digestion as described previously (29). Islets were cultured in complete CMRL-1066 medium (CMRL-1066 containing 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin). INS 832/13 cells were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 1 mm sodium pyruvate, 10 mm HEPES, 100 units/ml penicillin, 100 μg/ml streptomycin, and 55 μm β-mercaptoethanol. Cells were cultured at 37 °C at 5% CO2 and 95% air. INS 832/13 cells were detached from culture flasks using 0.05% trypsin and 0.53 mm EDTA and plated at a density of 500,000 cells/ml.
Western Blot Analysis
Cells/islets were washed with PBS and lysed in 150 mm NaCl, 50 mm Tris-HCl, 1 mm EGTA, 1 mm sodium orthovanadate, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm NaF, and Halt protease inhibitor mixture (ThermoFisher Scientific, Waltham, MA). Lysates were incubated on ice for 30 min with occasional vortexing and then sonicated and centrifuged (16,000 × g) at 4 °C for 20 min. Protein concentration was measured using the BCA protein assay (Pierce, Rockford, IL). Lysates were mixed with 6× Laemmli buffer and boiled for 5 min. Proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and blocked in 3% BSA in Tris-buffered saline and Tween 20 (TBST) for 1 h. Membranes were incubated overnight at 4 °C with the following dilutions of primary antibodies: γH2AX (1:2000), H2AX (1:10,000), phospho-JNK (1:2000), PARP (1:1000), PAR (1:1000), cleaved caspase 3 (1:500), ATM (1:1000), and GAPDH (1:10,000). Membranes were then incubated with secondary antibodies for 45 min at the following concentrations: donkey anti-mouse-horseradish peroxidase (1:5000) and donkey anti-rabbit-horseradish peroxidase (1:7000). This was followed by detection using enhanced chemiluminescence.
Nitrite Determination
Nitric oxide generation was assessed by measuring the accumulation of its stable metabolite nitrite in culture supernatants using a Griess assay (30).
Immunofluorescence
Following treatment, islets were dispersed using trypsin (31, 32), washed two times in PBS, and centrifuged onto microscope slides using a Shandon Cytospin II (ThermoFisher Scientific). Cells were fixed using 4% paraformaldehyde for 45 min, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice, and then blocked using 1% BSA in PBS for 30 min. Primary antibodies to γH2AX, phospho-ATM, and insulin were used at 1:500, 1:500, and 1:100, respectively. Alexa Fluor 488-conjugated donkey anti-rabbit, Cy3-conjugated donkey anti-mouse, and DyLight-405 goat anti-guinea pig secondary antibodies were used at 1:400, 1:400, and 1:200, respectively. DAPI was used as a nuclear stain (0.5 ng/ml in PBS for 10 min). Images were captured using a Nikon eclipse 90i confocal microscope. For quantification of γH2AX-positive cells, an intensity threshold was set using ImageJ (National Institutes of Health), and positive cells were counted manually.
Comet Assay
DNA damage was measured using a comet assay (33). Following treatment, cells were detached using PBS, suspended in 0.6% low melting point agarose, and layered onto slides precoated with 1.0% agarose. Samples were lysed (2.5 m NaCl, 100 mm EDTA, 10 mm Tris-base (pH 10.0), 0.1% SDS, and 1.45% Triton X-100) overnight at 4 °C and incubated in an alkaline solution (0.3 m NaOH and 1 mm EDTA (pH >13)) for 45 min. Electrophoresis at 25 V and 300 mA was conducted for 25 min, and the slides were washed three times in 0.4 m Tris base (pH 7.5). Slides were stained with ethidium bromide (2 μg/ml), and images were captured using a Nikon eclipse 80i. DNA damage was quantified as mean tail moment using the Comet Assay Software Project (CASP) program from 30–50 cells/condition.
Quantitative Real-time PCR
Total RNA from whole cell lysates was isolated using an RNeasy kit (Qiagen). First-strand cDNA synthesis was performed using oligo(dTs) and the SuperScript preamplification system according to the instructions of the manufacturer (Invitrogen). Quantitative RT-PCR was performed using SsoFast EvaGreen supermix (Bio-Rad) and a Bio-Rad CFX96 real-time system according to the instructions of the manufacturer. Primers used were as follows: GADD45α, 5′-GTGTGCTGGTGACGAACCCACAT-3′ (forward) and 5′-CCGTTCGGGGAATCACCGTCCG-3′ (reverse). Changes in GADD45α were normalized to GAPDH. The primers were as follows: GAPDH, 5′-TCGGTGTGAACGGATTTGGCCG-3′ (forward) and 5′TGAAGGGGTCGTTGATGGCAACA-3′ (reverse). Primers were purchased from Integrated DNA Technologies (Coralville, IA).
Fast Micromethod
The Fast Micromethod was used to evaluate the extent of DNA damage (34, 35). INS 832/13 cells (400,000/0.8 ml in 12-well plates) were detached using 0.05% trypsin and 0.53 mm EDTA following the indicated treatments. Cells were harvested by centrifugation and resuspended in 2 ml TE buffer (10 mm Tris-HCl and 1 mm EDTA (pH 7.4)), giving an approximate density of 200,000 cells/ml. In a volume of 25 μl (5000 cells), cells were transferred to 96-well plates. 25 μl of TE buffer containing Pico-Green (1:50 Pico-Green/TE buffer, Invitrogen) was added, and the cells were incubated for 20 min in the dark. Following the incubation with Pico-Green, 250 μl of NaOH solution (1 mm NaOH in 20 mm EDTA, made freshly) was added to give a final pH of 12.30 ± 0.02 in each well. The pH of the NaOH solution was attuned beforehand to ensure that the appropriate pH was achieved in the 96-well plate (34). Immediately after addition of the NaOH solution, the loss of Pico-Green binding (extent of DNA damage) was detected by measuring fluorescence at an excitation of 480 nm and emission of 520 nm. DNA damage measured using the Fast Micromethod was expressed as strand scission factor, which is defined as the log (percent of dsDNA in sample/percent of dsDNA in control), calculated following a 20-min incubation with NaOH.
siRNA Transfection
All transfection reagents and siRNA were purchased from Invitrogen. INS 832/13 cells were transfected with 100 nm siRNA targeting ATM or a scramble sequence using Lipofectamine 2000 according to the instructions of the manufacturer. After 24 h of incubation, the transfection medium was removed, and fresh medium was added to the wells. Cells were allowed to incubate in fresh medium for an additional 24 h before treatment.
Statistics
Statistical analyses were performed using one-way analysis of variance with a Tukey-Kramer post hoc test. The minimum level of significance was set at p < 0.05.
RESULTS
Nitric Oxide Induces γH2AX Formation
Histone variant H2AX is one of the core histone proteins that functions to maintain genomic integrity (26). Phosphorylation of H2AX on serine 139 (termed γH2AX) occurs rapidly after induction of DNA DSBs and serves as an initiation site for the assembly of DNA repair complexes (27, 36). The formation of γH2AX is one of the most sensitive markers of DSB damage (27). Because nitric oxide induces DNA damage, the effects of cytokines on γH2AX formation in β cells were examined. Treatment of isolated rat islets with the cytokines IL-1 and IFNγ for 24 h results in a >3.5-fold increase in γH2AX formation (Fig. 1, A and B) that occurs under conditions in which there is a 5-fold increase in nitric oxide production (Fig. 1C). Nitric oxide is required for γH2AX formation in cytokine-treated islets because iNOS inhibition prevents its formation (Fig. 1, A and C). In agreement with β cells being the cellular source of iNOS (37), cytokine-induced γH2AX formation occurs in insulin-containing β cells (Fig. 1D). The formation of cytokine-induced γH2AX is attenuated by the NOS inhibitor NMMA. Following a 24-h incubation with IL-1 + IFNγ, 26 ± 10% of β cells contain detectable levels of γH2AX. This represents a 2.5-fold increase above the levels identified in untreated islets (Fig. 1, D and E). Consistent with nitric oxide being the mediator of γH2AX formation following cytokine treatment, nitric oxide provided exogenously using the donor DEA/NO stimulates formation of γH2AX following a 120-min incubation in both rat islets and INS 832/13 insulinoma cells (Fig. 2). Together, these findings demonstrate that nitric oxide produced endogenously or supplied exogenously induces γH2AX formation in pancreatic β cells.
FIGURE 1.

Cytokines stimulate γH2AX formation in a nitric oxide-dependent manner. Rat islets (150 islets/400 μl of complete CMRL-1066 tissue culture medium) were treated with cytokines (10 units/ml IL-1 and 150 units/ml IFNγ) ± NMMA (2 mm) for the indicated times. γH2AX levels were determined by Western blot analysis (A) and quantified by densitometry (B). Total H2AX was used as a loading control. C, nitrite accumulation was measured in the culture supernatants. D, rat islets were dispersed and centrifuged onto microscope slides following treatment. Immunofluorescence was carried out using antibodies to insulin (green) and γH2AX (red), and the nuclei were stained with DAPI (blue). E, insulin-containing cells positive for γH2AX were quantified. Results are the mean ± S.E. of three independent experiments with statistically significant differences versus control. *, p < 0.05.
FIGURE 2.

DEA/NO stimulates γH2AX formation in islets and INS 832/13 cells. Rat islets (150 islets/400 μl of complete CMRL-1066 tissue culture medium (A) or INS 832/13 cells (200,000 cells/400 μl) (B) were treated with 1 mm DEA/NO for the indicated times. γH2AX was determined by Western blot analysis and quantified by densitometry for islets (C) and INS 832/13 cells (D). GAPDH was used to confirm equal protein loading. Results are the mean ± S.E. of three independent experiments with statistically significant differences versus control. *, p < 0.05.
ATM Inhibition Prevents Nitric Oxide-induced γH2AX Formation
Although H2AX can be phosphorylated by any of the phosphatidylinositol 3-kinase-related kinases, including ATM, ataxia telangiectasia and rad3-related protein, and DNA-dependent protein kinase (26), ATM is the principal kinase responsible for γH2AX formation following DNA damage induced by agents that induce genotoxic stress (28, 38, 39). Using the ATM-selective inhibitor KU-55933 (40), the role of this phosphatidylinositol 3-kinase-related kinase in nitric oxide-induced γH2AX formation was examined. KU-55933 attenuates γH2AX formation in INS 832/13 cells treated for 3 h with DEA/NO (Fig. 3A) and in rat islets following a 24-h cytokine treatment (Fig. 3B). Consistent with a role for ATM in γH2AX formation, treatment of INS 832/13 cells for 2 h with DEA/NO results in ATM activation, as determined using antibodies selective for phosphorylated ATM (immunocytochemistry and green fluorescence) in cells containing γH2AX formation (red fluorescence, Fig. 3C). Although consistent with a role for ATM in stimulating γH2AX formation, antibodies that recognize phospho-ATM have been reported to identify activated ATM substrates as well, so an increase in signal may reflect an increase in ATM activity in addition to the phosphorylation of ATM itself.
FIGURE 3.

ATM inhibition attenuates nitric oxide-induced γH2AX formation. A, INS 832/13 cells were pretreated for 30 min with the ATM inhibitor KU-55933 (5 μm) and then incubated for 3 h with DEA/NO (500 μm). γH2AX formation was determined by Western blot analysis (top panel) and quantified by densitometry (bottom panel). B, rat islets (200 islets/400 μl medium) were treated with cytokines (10 units/ml IL-1 and 150 units/ml IFNγ) ± KU-55933 (5 μm) for 24 h. γH2AX formation was determined by Western blot analysis (top panel) and quantified by densitometry (bottom panel). GAPDH was used to confirm equal protein loading. C, INS 832/13 cells were treated with DEA/NO (500 μm) for 2 h, and ATM phosphorylation (p-ATM, green) and γH2AX formation (red) were identified by immunofluorescence. Nuclei were identified by DAPI (blue). D, siRNA (100 nm) was used to knock down ATM in INS 832/13 cells (inset). Following siRNA depletion (48 h), the cells were exposed to DEA/NO (500 μm) for 90 min, harvested, and then γH2AX formation and GAPDH levels were determined by Western blot analysis. γH2AX formation was quantified by densitometry and normalized to GAPDH levels (bottom panel). E, islets isolated from wild-type and ATM−/− mice were treated with or without DEA/NO (500 μm) for 2 h. The islets were harvested by centrifugation, dispersed into individual cells, and centrifuged onto microscope slides. γH2AX formation (red) and insulin (green) were identified by immunofluorescence, and DAPI was used to identify nuclei (blue). Results are representative of three experiments or the mean ± S.E. of three (A–D) or one (E) independent experiments with statistically significant differences versus control. *, p < 0.05.
Molecular approaches have been used to confirm that ATM is responsible for nitric oxide-induced γH2AX formation in β cells. Treatment of INS 832/13 cells with siRNA targeting ATM results in a greater than 80% reduction in ATM levels compared with cells transfected with nonspecific scrambled siRNA. In the absence of ATM, there is an almost complete inhibition of nitric oxide-induced γH2AX formation (Fig. 3D).
Consistent with the effects of siRNA-mediated ATM knockdown, nitric oxide fails to stimulate γH2AX formation in islets isolated from mice deficient in ATM (Fig. 3E). In contrast, DEA/NO stimulates γH2AX in islet cells isolated from wild-type littermate controls (Fig. 3E). These findings support ATM as the kinase responsible for mediating β cell γH2AX formation in response to nitric oxide.
The Role of ATM in the Repair of Nitric Oxide-induced DNA Damage
In addition to inducing DNA damage, nitric oxide stimulates the activation of pathways that promote DNA repair in β cells (41–43). We have shown that nitric oxide activates the JNK-dependent expression of growth arrest and DNA damage (GADD) 45α and that both JNK and GADD45α are necessary for repair of DNA damage (41). ATM has been reported to initiate signaling pathways leading to JNK and GADD45α activation during genotoxic stress (44–47). Therefore, the role of ATM in the activation of JNK and the expression of GADD45α were examined. Treatment of INS 832/13 cells for 3 h with DEA/NO results in a >8-fold increase in GADD45α mRNA accumulation and a 2-fold increase in JNK phosphorylation (Fig. 4). The ATM inhibitor KU-55933 does not modify either nitric oxide-stimulated GADD45α expression (Fig. 4A) or the phosphorylation of JNK (Fig. 4, B and C). These findings suggest that ATM does not participate in the regulation of pathways known to be required for the repair of nitric oxide-induced DNA damage in β cells.
FIGURE 4.

The role of ATM in nitric oxide-induced GADD45α expression and JNK activation. A, INS 832/13 cells were treated with 500 μm DEA/NO for 3 h ± KU-55933 (5 μm). Total RNA was isolated, and GADD45α mRNA accumulation was determined by quantitative RT-PCR and normalized to GAPDH. B, INS 832/13 cells were treated with 500 μm DEA/NO for the indicated times ± 5 μm KU-55933. JNK phosphorylation (p-JNK) and GAPDH levels were determined by Western blot analysis, and phospho-JNK was quantified by densitometry and normalized to the protein loading control, GAPDH (C). Results are mean ± S.E. of three independent experiments (B and C) or four independent experiments (A) with statistically significant differences versus control. *, p < 0.05.
To determine whether ATM is required for DNA repair following nitric oxide-induced damage, isolated islets were treated for 18 h with IL-1 or treated with IL-1, followed by the addition of the NOS inhibitor NMMA and continued incubation for another 8 h in the presence of both NMMA and IL-1 (repair conditions). IL-1 induces a >2-fold increase in DNA damage, as measured by comet assay, and this damage is repaired completely during the recovery period that is stimulated by the inhibition of NOS (Fig. 5, A and B). Inhibition of ATM using KU-55933 has no effect on the level of DNA damage induced by IL-1 or the repair of this damage (Fig. 5, A and B). These findings suggest that, although nitric oxide induces γH2AX formation in an ATM-dependent manner, ATM does not participate in the repair of damaged DNA.
FIGURE 5.

The role of ATM in the repair of nitric oxide-induced DNA damage. A, rat islets (150 islets/condition) were left untreated, treated for 18 h with 10 units/ml IL-1 ± 5 μm KU-55933, or treated with IL-1 for 18 h ± KU-55933 and then cultured in the presence of NMMA for an additional 8 h (recovery conditions). Islets were then dispersed, and DNA damage was determined by the comet assay. Representative comets are shown. B, damage was quantified from imaged cells and expressed as mean tail moment. Results are mean ± S.E. of three independent experiments. Differences between groups were found to be statistically significant using analysis of variance.
Differential γH2AX Formation and DNA Damage Induction in Response to Nitric Oxide and Hydrogen Peroxide (H2O2)
Although γH2AX formation is a rapid event that occurs within minutes of DSB formation, nitric oxide-induced γH2AX formation is a late event, first apparent 2 h post-treatment (Fig. 6A, top panel). Conversely, the induction of γH2AX following nitric oxide treatment does not correlate with the induction of DNA damage because a >3.5-fold increase in DNA damage is detectable as early as 15 min after the addition of the nitric oxide donor (Fig. 6B). H2O2, a reactive oxygen species known to induce DSBs, induces the rapid formation of γH2AX (Fig. 6A, bottom panel) and DNA damage (Fig. 6C) as early as 15 min after treatment of INS 832/13 cells. Because there is a temporal discordance between the induction of DNA damage and the formation of γH2AX in response to nitric oxide and H2O2, additional methodologies were used to gain information on the extent of DNA damage in β cells in response to both oxidants. The Fast Micromethod relies on the ability of the fluorescent dye Pico-Green to exclusively bind to dsDNA but not to single-stranded DNA or protein. The assay measures the loss of Pico-Green fluorescence under denaturing conditions (basic conditions), where an increased number of DNA strand breaks results in accelerated DNA unwinding and the loss of Pico-Green fluorescence (34). The strand scission factor is used to quantify the levels of DNA strand breaks (34). As shown in Fig. 6B, there is a >5-fold increase in the level of DNA strand breaks in INS 832/13 cells treated with 100 μm H2O2 for 30 min, whereas levels of DNA damage induced by 1 mm DEA/NO were not significantly different from the untreated control (Fig. 6D). These findings indicate that the extent of dsDNA damage in response to nitric oxide is minimal when compared with the damage induced by H2O2 even though it is possible to detect single-stranded DNA strand breaks in β cells treated with cytokines or nitric oxide donors by TUNEL (48) and comet assay (Fig. 6B) (49). Furthermore, the marked DNA damage induced by H2O2 treatment of INS 832/13 cells results in the rapid activation of poly(ADP-ribose) polymerase (PARP) that is evident by ADP-ribosylation (PAR formation) (Fig. 6, E and F). In contrast, nitric oxide does not stimulate PAR formation (Fig. 6, E and F), a finding consistent with the lower levels of DNA strand breaks quantified by the Fast Micromethod (Fig. 6D).
FIGURE 6.

γH2AX formation and DNA damage induced by nitric oxide and hydrogen peroxide (H2O2). INS 832/13 cells were treated for the indicated times with 1 mm DEA/NO or 100 μm H2O2. A, formation of γH2AX was determined by Western blot analysis with GAPDH levels shown to control for protein loading. B, DNA damage (mean tail moment) was evaluated using the comet assay. C and D, DNA damage was evaluated using the Fast Micromethod (see “Experimental Procedures”) and quantified as a strand scission factor (SSF). ADP-ribosylation of proteins (PAR formation) was evaluated by Western blot analysis (E) and quantified by densitometry (F). Results are mean ± S.E. of three independent experiments (A,C, E, and F) or five independent experiments (D) with statistically significant differences versus control. *, p < 0.05.
The Role of Caspase Activation in Nitric Oxide-induced γH2AX Formation
Following formation of a DSB, γH2AX accumulates in the vicinity of the lesion to aid in the recruitment and retention of DNA repair factors (27, 36). When visualized by immunofluorescence, DSB-induced γH2AX formation occurs in punctate foci flanking the region of the DSB lesion (50). To determine whether cytokine-induced γH2AX formation occurs in a similar pattern in β cells, isolated rat islets were treated for 24 h with cytokines (IL-1 and IFNγ) or with camptothecin, a topoisomerase inhibitor that induces DSBs (51). Cytokine-induced γH2AX forms in a pan-nuclear pattern in β cells (Fig. 7A, top row), whereas camptothecin-induced γH2AX formation occurs in punctate foci (Fig. 7A, bottom row). The presence of pan-nuclear γH2AX formation has been implicated in apoptosis as a preapoptotic signal (52) and as a consequence of caspase activation and DNA fragmentation during apoptosis (53–55). To explore these possibilities, the temporal relationship between DEA/NO-induced γH2AX formation and caspase 3 cleavage was examined. As shown in Fig. 7B, γH2AX formation precedes DEA/NO-induced caspase 3 cleavage, indicating that γH2AX occurs as a preapoptotic signal and not a consequence of caspase activation. Consistent with this interpretation, the inhibition of caspases (caspase inhibitor 1) does not affect DEA/NO-induced γH2AX formation in INS 832/13 cells (>65-fold increase in γH2AX with both DEA/NO and DEA/NO + caspase inhibitor 1) but is effective at inhibiting camptothecin-induced caspase 3 cleavage (positive control to show caspase inhibitor efficacy, Fig. 7C). These findings show that the pattern of γH2AX formation induced by nitric oxide is suggestive of an apoptotic event that occurs before caspase activation.
FIGURE 7.
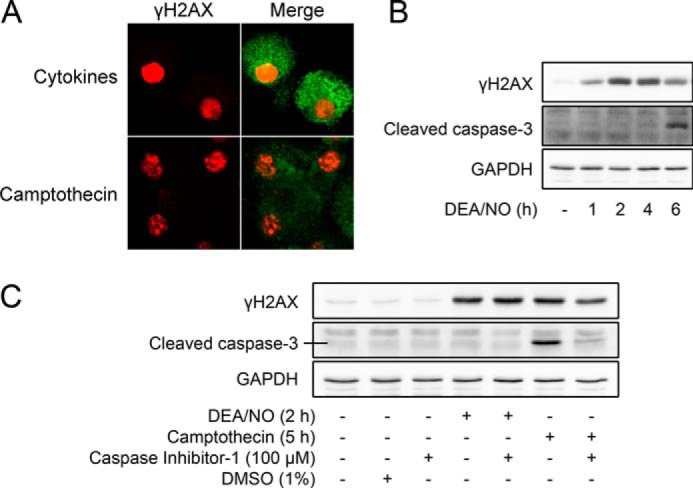
The role of caspase activation in nitric oxide-induced γH2AX formation. A, isolated rat islets (150 islets/condition) were treated for 24 h with cytokines (10 units/ml IL-1 and 150 units/ml IFN-γ) or for 2 h with camptothecin (25 μm). Islets were dispersed and centrifuged onto slides, and the localization of γH2AX (red) formation in insulin-containing cells (green) was determined by immunofluorescent microscopy. Images were captured using a ×60 objective and ×2 field zoom. B, INS 832/13 cells were treated for the indicated times with 1 mm DEA/NO and harvested, and cleaved caspase-3, γH2AX, and GAPDH were detected by Western blot analysis. C, INS 832/13 cells were pretreated for 1 h with or without caspase inhibitor 1 (10 μm). DEA/NO (1 mm) or camptothecin (25 μm) were added, and the cells were cultured for the indicated times. Dimethyl sulfoxide (DMSO) was used as a vehicle control. Cells were harvested, and cleaved caspase 3, γH2AX, and GAPDH levels were determined by Western blot analysis, with GAPDH serving as a protein loading control. Results are representative of three independent experiments.
IL-1-induced γH2AX Formation Correlates with an Increase in β Cell Apoptosis
We have shown that treatment of islets or β cells for 36 h with IL-1 results in an irreversible inhibition of insulin secretion, DNA damage, and commitment of β cells to death in a nitric oxide-dependent manner (13, 24). Associated with this irreversible damage is the induction of apoptosis, as evidenced by a 3-fold increase in the accumulation of mRNA for the proapoptotic factor p53-up-regulated mediator of apoptosis and detectable caspase 3 cleavage (24). To determine whether γH2AX formation temporally correlates with the induction of apoptosis, INS 832/13 cells were treated for 24 or 36 h with IL-1, and γH2AX formation was assessed by Western blot analysis. Following 24 h of incubation with IL-1, there is a small (1.7-fold) increase in γH2AX formation. However, following a 36-h incubation, γH2AX formation is increased 4-fold (Fig. 8, A and C). Associated with γH2AX formation following 36 h of exposure to IL-1 is a 3.2-fold increase in the cleavage of the caspase substrate PARP (56). However, this increase did not achieve statistical significance (p = 0.06; Fig. 8, A and C). Similar to INS 832/13 cells, treatment of rat islets with IL-1 for 36 h results in high levels of γH2AX formation that occurs under conditions associated with the cleavage of caspase 3 to its mature and active form (Fig. 8, B and D). In response to a 24-h incubation, IL-1 does not stimulate caspase cleavage.
FIGURE 8.

IL-1-induced γH2AX formation correlates with an increase in apoptosis. A, INS 832/13 cells were treated for the indicated times with IL-1 (10 units/ml), and caspase activation (evaluated by the cleavage of PARP) and γH2AX formation were determined by Western blot analysis and quantified by densitometry (C). GAPDH levels were determined to control for protein loading. B, rat islets (150–200 islets/condition/experiment) were treated with IL-1 (10 units/ml) for the indicated times, and the cleavage of caspase 3 and γH2AX formation were determined by Western blot analysis and quantified by densitometry (D). Results are representative (Western blots) or the mean ± S.E. of three independent experiments with statistically significant differences versus control. *, p < 0.05.
Because ATM mediates γH2AX formation in cytokine- and nitric oxide-treated β cells and high levels of γH2AX formation occur under conditions associated with the induction of β-cell apoptosis, the role of ATM in cytokine-induced caspase activation was examined. Consistent with a role in the induction of apoptosis, ATM inhibition using KU-55933 prevents cytokine-induced caspase 3 cleavage in rat islets treated for 36 h with IL-1 (Fig. 9). These data support a role for ATM in the initiation of apoptosis under conditions in which cytokine-induced β cell damage becomes irreversible.
FIGURE 9.

ATM is required for IL-1-induced caspase cleavage. A, rat islets (150 islets/condition) were treated for 36 h with IL-1 (10 units/ml) ± KU-55933 (5 μm). The islets were harvested, and cleavage of caspase 3 was examined by Western blot analysis and quantified by densitometry (B). GAPDH was used to control for protein loading. Results are representative or the mean ± S.E. of three independent experiments with statistically significant differences versus control. *, p < 0.05.
DISCUSSION
Nitric oxide, produced in micromolar amounts in response to proinflammatory cytokines, inhibits secretory function and oxidative metabolism, induces DNA damage, and, ultimately, causes β cell death (57). Although damaging, nitric oxide also stimulates repair pathways that allow the β cell to regain metabolic and secretory function and to repair damaged DNA (57). The recovery pathway was uncovered using a temporal experimental design in which a NOS inhibitor was added to islets following treatment with IL-1, and the islets were then incubated with both the NOS inhibitor and cytokine (22). Under these conditions, the inhibition of iNOS stimulates a time-dependent recovery of metabolic and secretory function and the repair of damaged DNA that is complete in 8 h (13, 21, 22, 24). Further, this ability to recover from cytokine-induced damage occurs in the presence of the cytokine, indicating that nitric oxide is the mediator of cytokine-induced damage (22). Although β cells have the ability to recover from cytokine-induced damage, it is temporally limited. Treatment of β cells for 36 h and longer with cytokines results in the irreversible inhibition of metabolic and secretory function and irreversible DNA damage that ultimately leads to β cell death (13, 24). In this work, the signaling mechanisms that initiate DNA repair pathways and the mechanisms controlling the induction of irreversible DNA damage were investigated.
To explore the mechanisms of DNA repair, the potential activation of the DDR was examined by evaluating the effects of cytokines and nitric oxide on the phosphorylation of H2AX. Treatment of INS 832/13 cells and rat islets with cytokines results in the phosphorylation of γH2AX on Ser-139 (γH2AX). Cytokine-induced γH2AX is dependent on the production of nitric oxide (Fig. 1) and can be induced by treatment with nitric oxide donors (Fig. 2). γH2AX is a sensitive and selective marker of DNA DSBs that can be detected within minutes of DSB formation (27). It functions in the recruitment and retention of repair factors at the site of the DNA damage (26, 27, 36). Multiple kinases have been reported to phosphorylate H2AX, primarily members of the PI3K-like kinases, including ATM, ataxia telangiectasia and rad3-related protein, and DNA-dependent protein kinase (26). ATM and DNA-dependent protein kinase are activated in response to DSBs that arise from genotoxic stressors, whereas ataxia telangiectasia and rad3-related protein is activated by replication-induced DSBs (25). Using molecular and pharmacological approaches, we show that the cytokine- and nitric oxide-induced γH2AX formation in β cells is dependent on the activation ATM (Fig. 3). These findings are consistent with two recent reports demonstrating that nitric oxide can stimulate γH2AX formation in RAW 264.7 macrophages and esophageal adenocarcinoma cells (58, 59).
Detailed information concerning the mechanisms of DNA repair in β cells is lacking. We have shown that the repair of nitric oxide-induced DNA damage in β cells does not require the transcription factor p53 but does require one of its targets, GADD45α (41). The expression of GADD45α is controlled by JNK and the forkhead transcription factor FOXO1 (41). siRNA knockdown of GADD45α attenuates the repair of cytokine-induced DNA damage, and nitric oxide stimulates GADD45α mRNA accumulation in a FOXO1- and JNK-dependent manner in β cells (41). We now show that nitric oxide stimulates γH2AX formation in an ATM-dependent manner, suggesting that nitric oxide activates the DNA damage response in β cells. Interestingly, ATM inhibition does not modify DNA repair following IL-1-induced damage (Fig. 5). Furthermore, although ATM can regulate JNK and GADD45α through intermediate factors such as BRCA1 and ATF-2 following DNA damage in U2OS and Hep3B cells (44–47), ATM is not required for JNK activation or GADD45α expression in β cells exposed to nitric oxide (Fig. 4). Collectively, these findings suggest that ATM and γH2AX do not participate in the repair of damaged DNA in β cells exposed to nitric oxide.
Consistent with recent findings showing that β cells respond differently to nitric oxide and H2O2 (48), temporal differences occur between nitric oxide- and H2O2-induced γH2AX formation. Interestingly, there is a temporal dissociation between nitric oxide-induced γH2AX formation and nitric oxide-induced DNA damage (Fig. 6, A and B). In response to nitric oxide donor treatment, a >3.5-fold increase in DNA damage can be detected using a comet assay within 15 min of exposure (Fig. 6B), yet γH2AX formation is much later, not detectable until 2 h post-treatment (Fig. 6A). In contrast, H2O2 causes the rapid formation of γH2AX (detectable as early as 15 min after treatment, Fig. 6A) that correlates with the induction of DNA damage (Fig. 6C) (48). Nitric oxide damages DNA by base oxidation or deamination, leading to single strand breaks (18, 19). The delay in γH2AX formation in nitric oxide-treated β cells is consistent with low levels of single strand breaks in DNA that may eventually give rise to DSBs. In support of this hypothesis, an increase in the formation of dsDNA breaks as determined using the Fast Micromethod and quantified as strand scission factor occurs after 2-h treatment with DEA/NO, although, in these experiments, statistical significance was not reached (Fig. 6D). In contrast to nitric oxide, H2O2 induces extensive single strand breaks, leading to DSB formation, the rapid induction of γH2AX, and the activation of PARP (Fig. 6).
Even though β cells can recover from short exposures (0–24 h) to IL-1, long exposures (36 h and longer) cause irreversible damage to DNA and inhibition of β-cell function, and, under this prolonged exposure, β cells are committed to death (13, 24). Associated with this commitment to death is a 3-fold increase in the expression of the proapoptotic factor p53-up-regulated mediator of apoptosis and caspase 3 cleavage, suggesting that β cell death under these conditions is apoptotic. In this study, γH2AX formation is shown to coincide with the induction of apoptosis, as measured by cleavage of caspase 3 and its substrate, PARP, following 36 h IL-1 treatment of rat islets and insulinoma cells. Importantly, high levels of γH2AX are not observed under conditions in which DNA damage is reversible (1 h of DEA/NO or 24 h of IL-1 treatment) (24, 41). It is only when DNA damage becomes irreversible (2 h of DEA/NO or 36 h of IL-1 treatment) that large increases in γH2AX are detected (Figs. 2 and 8) (24). We hypothesize that the formation of γH2AX does not cause β cell apoptosis but identifies cells progressing to apoptosis. The pan-nuclear localization pattern of γH2AX in cytokine treated β cells is consistent with this hypothesis because previous reports have shown that pan-nuclear localization of γH2AX functions as a preapoptotic signal (52). In addition, we show that DEA/NO-induced γH2AX formation precedes caspase 3 cleavage in INS 832/13 cells (Fig. 7B) and that caspase inhibition does not modify γH2AX formation in β cells exposed to nitric oxide (Fig. 7C).
Although there is a correlation between γH2AX formation and apoptosis, our findings point to ATM as a key component that regulates the induction of apoptosis in cytokine-treated β cells. We hypothesize that the formation of DSBs (evident by γH2AX) leads to irreversible DNA damage and apoptotic induction in cytokine-treated β cells. ATM, which is activated by DSBs, is responsible for γH2AX formation in cytokine-treated islets and appears to promote apoptosis following irreversible cytokine-induced damage (Fig. 9). Consistent with these findings, ATM has been shown to promote apoptosis following extensive DNA damage, primarily through p53-dependent mechanisms (60). The dissociation of ATM activation and γH2AX formation from the induction of DNA damage suggests that activation of this pathway is not a consequence of the DDR but may represent a proapoptotic signaling event that occurs only when β cells can no longer recover from cytokine-mediated damage.
Acknowledgments
We thank Jennifer A. McGraw and Aaron Naatz for technical assistance.
This work was supported by National Institutes of Health Grants DK52194 and AI-44458 (to J. A. C.).
- iNOS
- inducible nitric oxide synthase
- NMMA
- NG-monomethyl l-arginine
- DDR
- DNA damage response
- DSB
- double-strand break
- ATM
- ataxia telangiectasia mutated
- DEA/NO
- (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- PARP
- poly(ADP-ribose) polymerase.
REFERENCES
- 1. Gepts W. (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14, 619–633 [DOI] [PubMed] [Google Scholar]
- 2. Mandrup-Poulsen T., Bendtzen K., Nielsen J. H., Bendixen G., Nerup J. (1985) Cytokines cause functional and structural damage to isolated islets of Langerhans. Allergy 40, 424–429 [DOI] [PubMed] [Google Scholar]
- 3. Mandrup-Poulsen T., Bendtzen K., Nerup J., Dinarello C. A., Svenson M., Nielsen J. H. (1986) Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia 29, 63–67 [DOI] [PubMed] [Google Scholar]
- 4. Lacy P. E. (1994) The intraislet macrophage and type I diabetes. Mt. Sinai J. Med. 61, 170–174 [PubMed] [Google Scholar]
- 5. Cetkovic-Cvrlje M., Eizirik D. L. (1994) TNF-α and IFN-γ potentiate the deleterious effects of IL-1 β on mouse pancreatic islets mainly via generation of nitric oxide. Cytokine 6, 399–406 [DOI] [PubMed] [Google Scholar]
- 6. Heitmeier M. R., Scarim A. L., Corbett J. A. (1997) Interferon-γ increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J. Biol. Chem. 272, 13697–13704 [DOI] [PubMed] [Google Scholar]
- 7. Corbett J. A., Lancaster J. R., Jr., Sweetland M. A., McDaniel M. L. (1991) Interleukin-1 β-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 β-induced inhibition of insulin secretion. J. Biol. Chem. 266, 21351–21354 [PubMed] [Google Scholar]
- 8. Welsh N., Eizirik D. L., Bendtzen K., Sandler S. (1991) Interleukin-1 β-induced nitric oxide production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology 129, 3167–3173 [DOI] [PubMed] [Google Scholar]
- 9. Southern C., Schulster D., Green I. C. (1990) Inhibition of insulin secretion by interleukin-1 β and tumour necrosis factor-α via an l-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 276, 42–44 [DOI] [PubMed] [Google Scholar]
- 10. Corbett J. A., Wang J. L., Hughes J. H., Wolf B. A., Sweetland M. A., Lancaster J. R., Jr., McDaniel M. L. (1992) Nitric oxide and cyclic GMP formation induced by interleukin 1 β in islets of Langerhans. Evidence for an effector role of nitric oxide in islet dysfunction. Biochem. J. 287, 229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wollheim C. B., Maechler P. (2002) β-Cell mitochondria and insulin secretion: messenger role of nucleotides and metabolites. Diabetes 51, S37–42 [DOI] [PubMed] [Google Scholar]
- 12. Hughes J. H., Colca J. R., Easom R. A., Turk J., McDaniel M. L. (1990) Interleukin 1 inhibits insulin secretion from isolated rat pancreatic islets by a process that requires gene transcription and mRNA translation. J. Clin. Invest. 86, 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scarim A. L., Heitmeier M. R., Corbett J. A. (1997) Irreversible inhibition of metabolic function and islet destruction after a 36-hour exposure to interleukin-1β. Endocrinology 138, 5301–5307 [DOI] [PubMed] [Google Scholar]
- 14. Oyadomari S., Takeda K., Takiguchi M., Gotoh T., Matsumoto M., Wada I., Akira S., Araki E., Mori M. (2001) Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. U.S.A. 98, 10845–10850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chambers K. T., Unverferth J. A., Weber S. M., Wek R. C., Urano F., Corbett J. A. (2008) The role of nitric oxide and the unfolded protein response in cytokine-induced β-cell death. Diabetes 57, 124–132 [DOI] [PubMed] [Google Scholar]
- 16. Delaney C. A., Green M. H., Lowe J. E., Green I. C. (1993) Endogenous nitric oxide induced by interleukin-1 β in rat islets of Langerhans and HIT-T15 cells causes significant DNA damage as measured by the “comet” assay. FEBS Lett. 333, 291–295 [DOI] [PubMed] [Google Scholar]
- 17. Fehsel K., Jalowy A., Qi S., Burkart V., Hartmann B., Kolb H. (1993) Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes 42, 496–500 [DOI] [PubMed] [Google Scholar]
- 18. Wink D. A., Kasprzak K. S., Maragos C. M., Elespuru R. K., Misra M., Dunams T. M., Cebula T. A., Koch W. H., Andrews A. W., Allen J. S. (1991) DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 254, 1001–1003 [DOI] [PubMed] [Google Scholar]
- 19. Burney S., Caulfield J. L., Niles J. C., Wishnok J. S., Tannenbaum S. R. (1999) The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 424, 37–49 [DOI] [PubMed] [Google Scholar]
- 20. McDaniel M. L., Kwon G., Hill J. R., Marshall C. A., Corbett J. A. (1996) Cytokines and nitric oxide in islet inflammation and diabetes. Proc. Soc. Exp. Biol. Med. 211, 24–32 [DOI] [PubMed] [Google Scholar]
- 21. Rosales A. L., Cunningham J. M., Bone A. J., Green I. C., Green M. H. (2004) Repair of cytokine-induced DNA damage in cultured rat islets of Langerhans. Free Radic. Res. 38, 665–674 [DOI] [PubMed] [Google Scholar]
- 22. Corbett J. A., McDaniel M. L. (1994) Reversibility of interleukin-1 β-induced islet destruction and dysfunction by the inhibition of nitric oxide synthase. Biochem. J. 299, 719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Comens P. G., Wolf B. A., Unanue E. R., Lacy P. E., McDaniel M. L. (1987) Interleukin 1 is potent modulator of insulin secretion from isolated rat islets of Langerhans. Diabetes 36, 963–970 [DOI] [PubMed] [Google Scholar]
- 24. Hughes K. J., Chambers K. T., Meares G. P., Corbett J. A. (2009) Nitric oxides mediates a shift from early necrosis to late apoptosis in cytokine-treated β-cells that is associated with irreversible DNA damage. Am. J. Physiol. Endocrinol. Metab. 297, E1187–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ciccia A., Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pinto D. M., Flaus A. (2010) Structure and function of histone H2AX. Subcell. Biochem. 50, 55–78 [DOI] [PubMed] [Google Scholar]
- 27. Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 28. Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J. (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467 [DOI] [PubMed] [Google Scholar]
- 29. Kelly C. B., Blair L. A., Corbett J. A., Scarim A. L. (2003) Isolation of islets of Langerhans from rodent pancreas. Methods Mol. Med. 83, 3–14 [DOI] [PubMed] [Google Scholar]
- 30. Green L. C., Wagner D. A., Glogowski J., Skipper P. L., Wishnok J. S., Tannenbaum S. R. (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 126, 131–138 [DOI] [PubMed] [Google Scholar]
- 31. McDaniel M. L., Colca J. R., Kotagal N., Lacy P. E. (1983) A subcellular fractionation approach for studying insulin release mechanisms and calcium metabolism in islets of Langerhans. Methods Enzymol. 98, 182–200 [DOI] [PubMed] [Google Scholar]
- 32. Scarim A. L., Arnush M., Blair L. A., Concepcion J., Heitmeier M. R., Scheuner D., Kaufman R. J., Ryerse J., Buller R. M., Corbett J. A. (2001) Mechanisms of β-cell death in response to double-stranded (ds) RNA and interferon-γ: dsRNA-dependent protein kinase apoptosis and nitric oxide-dependent necrosis. Am. J. Pathol. 159, 273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tice R. R., Andrews P. W., Hirai O., Singh N. P. (1991) The single cell gel (SCG) assay: an electrophoretic technique for the detection of DNA damage in individual cells. Adv. Exp. Med. Biol. 283, 157–164 [DOI] [PubMed] [Google Scholar]
- 34. Schröder H. C., Batel R., Schwertner H., Boreiko O., Müller W. E. (2006) Fast micromethod DNA single-strand-break assay. Methods Mol. Biol. 314, 287–305 [DOI] [PubMed] [Google Scholar]
- 35. Ullmann K., Müller C., Steinberg P. (2008) Two essential modifications strongly improve the performance of the Fast Micromethod to identify DNA single- and double-strand breaks. Arch. Toxicol. 82, 861–867 [DOI] [PubMed] [Google Scholar]
- 36. Paull T. T., Rogakou E. P., Yamazaki V., Kirchgessner C. U., Gellert M., Bonner W. M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 [DOI] [PubMed] [Google Scholar]
- 37. Corbett J. A., Wang J. L., Sweetland M. A., Lancaster J. R., Jr., McDaniel M. L. (1992) Interleukin 1 β induces the formation of nitric oxide by β-cells purified from rodent islets of Langerhans. Evidence for the β-cell as a source and site of action of nitric oxide. J. Clin. Invest. 90, 2384–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhatti S., Kozlov S., Farooqi A. A., Naqi A., Lavin M., Khanna K. K. (2011) ATM protein kinase: the linchpin of cellular defenses to stress. Cell. Mol. Life Sci. 68, 2977–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shiloh Y., Ziv Y. (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 [PubMed] [Google Scholar]
- 40. Hickson I., Zhao Y., Richardson C. J., Green S. J., Martin N. M., Orr A. I., Reaper P. M., Jackson S. P., Curtin N. J., Smith G. C. (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64, 9152–9159 [DOI] [PubMed] [Google Scholar]
- 41. Hughes K. J., Meares G. P., Chambers K. T., Corbett J. A. (2009) Repair of nitric oxide-damaged DNA in β-cells requires JNK-dependent GADD45α expression. J. Biol. Chem. 284, 27402–27408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hughes K. J., Meares G. P., Hansen P. A., Corbett J. A. (2011) FoxO1 and SIRT1 regulate beta-cell responses to nitric oxide. J. Biol. Chem. 286, 8338–8348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scarim A. L., Nishimoto S. Y., Weber S. M., Corbett J. A. (2003) Role for c-Jun N-terminal kinase in β-cell recovery from nitric oxide-mediated damage. Endocrinology 144, 3415–3422 [DOI] [PubMed] [Google Scholar]
- 44. Jang E. R., Choi J. D., Park M. A., Jeong G., Cho H., Lee J. S. (2010) ATM modulates transcription in response to histone deacetylase inhibition as part of its DNA damage response. Exp. Mol. Med. 42, 195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maekawa T., Sano Y., Shinagawa T., Rahman Z., Sakuma T., Nomura S., Licht J. D., Ishii S. (2008) ATF-2 controls transcription of Maspin and GADD45 α genes independently from p53 to suppress mammary tumors. Oncogene 27, 1045–1054 [DOI] [PubMed] [Google Scholar]
- 46. Li S., Ting N. S., Zheng L., Chen P. L., Ziv Y., Shiloh Y., Lee E. Y., Lee W. H. (2000) Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 406, 210–215 [DOI] [PubMed] [Google Scholar]
- 47. Wang Z., Wang M., Kar S., Carr B. I. (2009) Involvement of ATM-mediated Chk1/2 and JNK kinase signaling activation in HKH40A-induced cell growth inhibition. J. Cell. Physiol. 221, 213–220 [DOI] [PubMed] [Google Scholar]
- 48. Meares G. P., Fontanilla D., Broniowska K. A., Andreone T., Lancaster J. R., Jr., Corbett J. A. (2013) Differential responses of pancreatic β-cells to ROS and RNS. Am. J. Physiol. Endocrinol. Metab. 304, E614–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meares G. P., Hughes K. J., Jaimes K. F., Salvatori A. S., Rhodes C. J., Corbett J. A. (2010) AMP-activated protein kinase attenuates nitric oxide-induced β-cell death. J. Biol. Chem. 285, 3191–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rogakou E. P., Boon C., Redon C., Bonner W. M. (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pommier Y., Redon C., Rao V. A., Seiler J. A., Sordet O., Takemura H., Antony S., Meng L., Liao Z., Kohlhagen G., Zhang H., Kohn K. W. (2003) Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. 532, 173–203 [DOI] [PubMed] [Google Scholar]
- 52. de Feraudy S., Revet I., Bezrookove V., Feeney L., Cleaver J. E. (2010) A minority of foci or pan-nuclear apoptotic staining of γH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 107, 6870–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu C., Zhu F., Cho Y. Y., Tang F., Zykova T., Ma W. Y., Bode A. M., Dong Z. (2006) Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 23, 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mukherjee B., Kessinger C., Kobayashi J., Chen B. P., Chen D. J., Chatterjee A., Burma S. (2006) DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair 5, 575–590 [DOI] [PubMed] [Google Scholar]
- 55. Solier S., Sordet O., Kohn K. W., Pommier Y. (2009) Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol. Cell. Biol. 29, 68–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lazebnik Y. A., Kaufmann S. H., Desnoyers S., Poirier G. G., Earnshaw W. C. (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371, 346–347 [DOI] [PubMed] [Google Scholar]
- 57. Padgett L. E., Broniowska K. A., Hansen P. A., Corbett J. A., Tse H. M. (2013) The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann. N.Y. Acad. Sci. 1281, 16–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang Y. C., Chou H. Y., Shen T. L., Chang W. J., Tai P. H., Li T. K. (2013) Topoisomerase II-mediated DNA cleavage and mutagenesis activated by nitric oxide underlie the inflammation-associated tumorigenesis. Antioxid. Redox Signal. 18, 1129–1140 [DOI] [PubMed] [Google Scholar]
- 59. Murata M., Thanan R., Ma N., Kawanishi S. (2012) Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J. Biomed. Biotechnol. 18, 623019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roos W. P., Kaina B. (2013) DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 332, 237–248 [DOI] [PubMed] [Google Scholar]