

Background: Although Cdc42 signaling is a known requirement for insulin secretion to occur, how it is initiated remains unknown.
Results: YES kinase is required for Cdc42 activation in human islets and β cells.
Conclusion: YES is a proximal, glucose-specific activator of Cdc42 and glucose-simulated insulin secretion.
Significance: YES may provide a novel target for improvement of functional β cell mass.
Keywords: Actin, Cdc42, Insulin Secretion, Pancreatic Islets, Src
Abstract
Second-phase insulin secretion sustains insulin release in the face of hyperglycemia associated with insulin resistance, requiring the continued mobilization of insulin secretory granules to the plasma membrane. Cdc42, the small Rho family GTPase recognized as the proximal glucose-specific trigger to elicit second-phase insulin secretion, signals downstream to activate the p21-activated kinase (PAK1), which then signals to Raf-1/MEK/ERK to induce filamentous actin (F-actin) remodeling, to ultimately mobilize insulin granules to the plasma membrane. However, the steps required to initiate Cdc42 activation in a glucose-specific manner in β cells have remained elusive. Toward this, we identified the involvement of the Src family kinases (SFKs), based upon the ability of SFK inhibitors to block glucose-stimulated Cdc42 and PAK1 activation events as well as the amplifying pathway of glucose-stimulated insulin release, in MIN6 β cells. Indeed, subsequent studies performed in human islets revealed that SFK phosphorylation was induced only by glucose and within 1 min of stimulation before the activation of Cdc42 at 3 min. Furthermore, pervanadate treatment validated the phosphorylation event to be tyrosine-specific. Although RT-PCR showed β cells to express five different SFK proteins, only two of these, YES and Fyn kinases, were found localized to the plasma membrane, and of these two, only YES kinase underwent glucose-stimulated tyrosine phosphorylation. Immunodetection and RNAi analyses further established YES kinase as a proximal glucose-specific signal in the Cdc42-signaling cascade. Identification of YES kinase provides new insight into the mechanisms underlying the sustainment of insulin secretion via granule mobilization/replenishment and F-actin remodeling.
Introduction
After glucose stimulation, insulin secretion from the pancreatic β cell occurs in two distinct phases. First-phase insulin secretion is initiated via glucose uptake through GLUT2 glucose transporters and subsequent glucose metabolism, leading to an elevated ATP:ADP ratio and closure of ATP-sensitive potassium (KATP) channels (1), and the ensuing cellular depolarization results in an influx of calcium through voltage-gated calcium channels to trigger the fusion of 50–100 insulin granules (2–6). First-phase secretion occurs within ∼10 min of glucose stimulation as a kinetically defined and transient event, which is then followed by a sustained second-phase during which insulin is secreted for a longer duration but at a reduced rate. Granules released during the second-phase of insulin secretion require recruitment from storage/reserve pools deep within the cell (7, 8). It has become apparent that the mobilization of insulin secretory granules from the storage pools to the cell surface requires the dynamic reorganization of the actin cytoskeleton, although the precise molecular events underpinning glucose-induced actin remodeling remain unclear (9–11).
Cdc42, a small Rho family GTPase, has been recognized as the most proximal event in the course of second-phase insulin release and is proposed to sit at the head of a signaling node central to filamentous actin remodeling. In human islets and MIN6 β cells, Cdc42 activation occurs within 3 min of glucose stimulation, during the time of first-phase insulin release; however, Cdc42 is not actually required for first-phase secretion (12, 13). Cdc42 activation is glucose-specific and requires glucose metabolism beyond fructose 6-phosphate production (14–16). Cdc42 signals downstream to activate PAK12 and N-WASP, each of which are involved in actin remodeling and second-phase glucose-stimulated insulin secretion (GSIS) in human and mouse islets (15, 16). PAK1 activation signals downstream to MEK/ERK, leading to changes in actin remodeling and promoting GSIS (17). PAK1 is also a downstream signaling target for SAD-A kinase (18). Pertinent to humans, islets from type 2 diabetic individuals present with an 80% loss of total PAK1 protein; this dramatic loss of PAK1 in diabetic patients may explain the loss of second-phase insulin secretion (13). However, how Cdc42 gets activated by glucose to begin this signaling cascade through PAK1 remains unknown.
Within the past decade the pertinent guanine nucleotide dissociation inhibitor (GDI) and guanine nucleotide exchange factor (GEF) that play integral roles in the activation of Cdc42 in the β cell were identified as Caveolin-1 (Cav-1) and βPix, respectively (19, 20). The dissociation of Cav-1-Cdc42 complexes by βPix is dependent on the tyrosine phosphorylation of Cav-1 (20). In other cell types Cav-1-Cdc42 and βPix-Cdc42 interactions are dependent on the phosphorylation statuses of both the GDI and GEF. Members of the Src family kinases (SFKs) have been implicated in these crucial phosphorylation events (21–25). Furthermore, SFKs have been shown to play a role in Cdc42 activation as well as stimulus-induced filamentous actin reorganization in other non-β-cell types (26, 27). SFKs are a family of protein-tyrosine kinases containing nine members: Src, YES, Fyn, Fgr, Lck, Hck, Blk, Lyn, and Frk. A multitude of cellular functions, including cellular motility, proliferation and survival, and cell-cell adhesion, are dependent on SFK activity. Importantly, many of these SFK-requiring processes also involve dynamic cytoskeleton remodeling (28, 29). Given the known role of SFKs in actin dynamics and in the phosphorylation of both Cav-1 and βPix, we investigated the contribution of SFKs in the glucose-specific activation of Cdc42 and PAK1 and sustained insulin secretion in the β cell.
In this study we show that glucose-stimulated Cdc42 activation, PAK1 phosphorylation, and GSIS are all dependent on SFK activity and identify YES kinase as the particular SFK involved in these processes. YES kinase is specifically activated by glucose within 1 min at the cell surface, occurring before Cdc42 activation. YES activation of Cdc42 represents one of the first-recognized proximal steps in glucose-specific release of insulin to date.
EXPERIMENTAL PROCEDURES
Human Islet Culture
Pancreatic human islets were obtained through the Integrated Islet Distribution Program. Criteria for human donor islet acceptance: receipt within 36 h of isolation and of at least 80% purity and 75% viability. Upon receipt, human islets were first allowed to recover in CMRL (Invitrogen) medium for 2 h at 37 °C and then handpicked using a green gelatin filter to eliminate residual non-islet material. Islets were incubated for 2 h at 37 °C in Krebs-Ringer bicarbonate buffer (KRBH) (10 mm HEPES (pH 7.4), 134 mm NaCl, 5 mm NaHCO3, 4.8 mm KCl, 1 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, 2.8 mm glucose, and 0.5 mg/ml RIA-grade BSA; Sigma) followed by a 1-min stimulation with 16.7 mm glucose or 5-min treatment with 0.1 or 1 mm pervanadate. Islet proteins were resolved by 10% SDS-PAGE for immunoblotting.
MIN6 Cell Culture and Insulin Secretion Assays
MIN6 β cells were cultured in DMEM (25 mm glucose) supplemented with 15% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml l-glutamate, and 50 μm β-mercaptoethanol as described previously (30). MIN6 cells were washed twice and incubated for 2 h at 37 °C in modified Krebs-Ringer bicarbonate buffer (MKRBB) (15 mm HEPES (pH 7.4), 120 mm NaCl, 24 mm NaHCO3, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, and 1 mg/ml RIA-grade BSA; Sigma). SFK inhibitors (PP2 or SU-6656, 20 μm (EMD Millipore Chemicals)) were included during MKRBB incubations and treatments/stimulations with 1 mm pervanadate or 20 mm glucose, as indicated in the figure legends. After stimulation, cells were harvested in 1% Nonidet P-40 lysis buffer (1% Nonidet P-40, 25 mm HEPES (pH 7.4), 137 mm NaCl, 10% glycerol, 50 mm sodium fluoride, 10 mm sodium pyrophosphate, 1 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 μg/ml pepstatin) and cleared of insoluble material by centrifugation for 10 min at 4 °C. For GSIS using the diazoxide paradigm, MIN6 cells were preincubated with MKRBB for 2 h, treated with 250 μm diazoxide (Sigma), and stimulated with 40 mm KCl or 40 mm KCl plus 20 mm glucose as described (17). Insulin secreted was quantified using a rat insulin radioimmunoassay (Millipore).
siRNA Transfection
Two siRNA oligonucleotides for YES (J-040156-06 and J-040156-07) were purchased from Thermo (Dharmacon ON-TARGET plus siRNAs) along with the control non-targeting siRNA oligonucleotide (D-001810-01). YES siRNA (J-040156-07) was employed for the PAK1 and SFK activation assays. siRNA oligonucleotides were transfected into MIN6 cells using Lipofectamine 2000 (Invitrogen) at 100 nm. Forty-eight hours after transfection, MIN6 cells were incubated in MKRBB, stimulated with 20 mm glucose, and harvested in 1% Nonidet P-40 lysis buffer for detection of SFK and PAK1 phosphorylation.
Immunoblotting
Proteins were resolved by 10–12% SDS-PAGE and transferred to PVDF membranes for immunoblotting. Non-stimulated A431 Cell Lysate (Millipore) was used as a positive control where indicated in the figure legends. Mouse anti-VAMP2 (Synaptic Systems) and rabbit anti-Syntaxin 4 (produced in-house, as described previously (31)) were used at 1:5000. Rabbit anti-GAPDH (Abcam), mouse anti-clathrin (BD Biosciences), rabbit anti-Src, rabbit anti-phospho-SrcTyr-416, rabbit anti-phospho-SrcTyr-527, rabbit anti-PAK1, and rabbit anti-phospho-PAK1Thr-423 (Cell Signaling Technology), and rabbit anti-RhoGDI (Santa Cruz Biotechnology) were used at 1:1000. Src and phospho-Src antibodies from Cell Signaling Technologies cross-react with other SFKs and are thus referred to as SFK or phospho-SFK. Mouse anti-YES (BD Biosciences) was used at 1:500, and mouse anti-Cdc42 (Thermo Fisher Scientific) was used at 1:250. Rabbit anti-Fyn, rabbit anti-Hck, and rabbit anti-c-Src (Santa Cruz Biotechnology) were all used at 1:200, whereas mouse anti-Rak/Frk (Santa Cruz Biotechnology) was used at 1:100. Goat anti-mouse and anti-rabbit horseradish peroxidase secondary antibodies were obtained from Bio-Rad and used at a concentration of 1:5000. Immunoreactive bands were visualized with ECL or ECL Prime (GE Healthcare) reagents and imaged using a Chemi-Doc gel documentation system (Bio-Rad). Bløk-PO Reagent (Millipore) was utilized to reduce noise in the phospho-specific blots.
Cdc42 Activation Assays
The Active Cdc42 Pull-Down and Detection Kit from Thermo was used to detect GTP-loaded Cdc42. Freshly prepared whole cell lysates (600 μg) were combined with 20 μg of GST-PAK1-PBD protein and rotated with the provided glutathione resin for 1 h at 4 °C. Proteins were then eluted from the resin after 3 washes with the kit lysis/binding/wash buffer and subjected to 12% SDS-PAGE followed by transfer to PVDF membrane for immunoblotting with mouse anti-Cdc42. Data were normalized to GST-PAK1-PBD detected by Ponceau S staining in each matched lane to control for lane loading differences.
Subcellular Fractionation
All fractionation steps were completed at 4 °C using an established protocol (32). In brief, after glucose stimulation MIN6 cells were washed twice with cold phosphate-buffered saline (PBS) and harvested with 500 μl of homogenization buffer (20 mm Tris-HCl (pH 7.4), 0.5 mm EDTA, 0.5 mm EGTA, 250 mm sucrose, 1 mm DTT, 100 μm PMSF, 10 μg/ml leupeptin, 4 μg/ml aprotinin, and 2 μg/ml pepstatin). Cell lysates were disrupted by 10 strokes through a 27-gauge needle and centrifuged at 0.9 × g for 10 min. Post-nuclear supernatants were centrifuged at 5500 × g for 15 min, and the subsequent supernatant was centrifuged at 25,000 × g for 20 min to obtain the secretory granule fraction in the pellet. The supernatant was further centrifuged at 100,000 × g for 1 h to obtain the cytosolic fraction. To isolate the plasma membrane fraction, the post-nuclear pellet was combined with 1 ml of Buffer A (10 mm Tris-HCl (pH 7.4), 0.25 m sucrose, 1 mm MgCl2) and 2 ml of Buffer B (10 mm Tris-HCl (pH 7.4), 2 m sucrose, 1 mm MgCl2). The resulting mixture was overlaid with Buffer A and centrifuged at 113,000 × g for 1 h. The sucrose gradient achieved with Buffers A and B yielded an interface containing the plasma membrane fraction. This interface was collected, washed twice with homogenization buffer, and collected via centrifugation at 6000 × g for 10 min. Both the plasma membrane and secretory granule pellets were resuspended in 1% Nonidet P-40 lysis buffer and stored at −80 °C until further analysis.
Immunofluorescence and Confocal Microscopy
MIN6 cells plated onto glass coverslips were incubated in MKRBB with or without 20 μm SU-6656 for 2 h and treated for 5 min with 1 mm pervanadate where indicated in the figure legends. Cells were immediately fixed and permeabilized in 4% paraformaldehyde plus 0.1% Triton X-100 for 10 min at room temperature. Fixed and permeabilized cells were blocked in a 1% BSA, 5% donkey serum solution for 1 h at room temperature followed by incubation with primary antibody overnight at 4 °C. Rabbit anti-Src and rabbit anti-phospho-SrcTyr-416 (Cell Signaling Technology) were used at 1:100. Cells were then washed 3 times with PBS and incubated with Alexa Fluor 488 secondary antibody (Invitrogen) at 1:100 for 1 h at room temperature. Cells were washed 3 times with PBS; during the second wash, 4′,6-diamidino-2-phenylindole (DAPI) was added to stain nuclei. Using Vectashield, coverslips were mounted onto slides for confocal fluorescence microscopy. Fluorescent images from at least five fields of each condition were captured using an Olympus FV1000-MPE confocal microscope (single-channel scanning, 60× objective, 2× zoom). ImageJ (National Institutes of Health) was used to prepare final images.
RNA Isolation and RT-PCR
Total RNA from MIN6 cells and mouse spleen was isolated using the RNeasy Mini Kit (Qiagen). RNA (1 μg) was reverse-transcribed with SuperScript reverse transcriptase (Invitrogen), and 5% of the product was used for subsequent PCR. PCR was performed with BioMix Red (Bioline) for 30 cycles: 94 °C for 1 min, 58 °C for 1 min, and 72 °C for 1 min with a final 10-min elongation step at 71 °C. PCR products were visualized on 2% agarose gels with GAPDH used as a positive reaction control. Using PrimerBank, specific primers (Table 1) for each of the nine Src kinase family members were designed to be unique to that SFK with no cross-reactivity to the other eight family members (optimal Tm = 60–63 °C).
TABLE 1.
Primer sequences for Src family kinase members
| Family member | 5′ Primer | 3′ Primer |
|---|---|---|
| Fyn | ACCTCCATCCCGAACTACAAC | CATAAAGCGCCACAAACAGTG |
| Lck | CCTGCACAGTTATGAGCCCTC | CGAAGTTGAAGGGAATGAAGCC |
| Lyn | AGTGCAGGAGCTTTCCTTATCA | CGAGGAGAGATGTAATAGCCACC |
| Hck | TCGTTGTCTGTTCGAGACTTTG | TCTTGTAGTGGAGCACGAGTT |
| Src | CAATGCCAAGGGCCTAAATGT | TGTTTGGAGTAGTAAGCCACGA |
| Yes | ATGCTACAGTTGCCCCGAC | TCCAAAAGGAGTCACCCCTGA |
| Blk | TTGTGGCCCCAGTAGAGACTC | GGGGAGATGTAATAGCCTCCA |
| Fgr | CTCAAGGCCGGACTTCGTC | CCCACCAGTCATACTCCGTATTG |
| Frk | GAAGGGTGACTTTTCCCTCTCA | GTTGTGGTGTAGTAGTTCACGAA |
Co-immunoprecipitation and Immunoblotting
Cleared MIN6 detergent lysates (2 mg) were combined with 2 μg of mouse anti-phosphotyrosine (Clone 4G10, Millipore) for 2 h at 4 °C followed by the addition of Protein G PLUS-agarose beads (Santa Cruz Biotechnology) for an additional 2 h. Normal mouse IgG (Santa Cruz Biotechnology) was used as a negative control. After 3 washes with 1% Nonidet P-40 lysis buffer, the resulting immunoprecipitates were subjected to 10% SDS-PAGE followed by transfer to PVDF membranes for immunoblotting mouse anti-YES as described earlier. Secondary antibody incubation and immunoreactive band visualization was completed as stated previously.
Adenoviral Transduction of MIN6 Cells and Mouse Islets
Adenoviral delivery plasmids were constructed by insertion of the annealed complementary oligonucleotide sequence of siCtrl or siYES (Dharmacon: J-040156–07) into the 5′-XhoI and 3′-SpeI sites of the pSilencer Adeno-CMV vector (Ambion) and verified by DNA sequencing analysis. High purity cesium chloride viral particles were obtained from Viraquest Inc. (North Liberty, IA), and viruses were packaged with GFP to identify transduced cells. shCtrl-Ad or shYES-Ad viral particles were used at a multiplicity of infection = 100. Pancreatic mouse islets were isolated as described previously (33, 34) from 10–16-week-old C57Bl/6J mice and immediately transduced for 1 h at 37 °C, washed, and cultured for 48 h in complete CMRL-1066 culture medium (CMRL-1066 medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin). MIN6 cells were transduced for 2 h. GFP fluorescence was visualized in >95% of MIN6 cells and ∼70% of cells of the islets (including those at the islet core) in all experiments. GFP-positive islets were handpicked and washed twice in KRBH. Groups of 10 islets were preincubated in KRBH for 2 h at 37 °C followed by a 5-min treatment with 250 μm diazoxide (Sigma). Islets were stimulated with 35 mm KCl or 35 mm KCl plus 16.7 mm glucose for 30 min. Supernatants were collected to measure insulin secretion, and islets were harvested in 1% Nonidet P-40 lysis buffer to determine islet insulin content.
Statistical Analysis
Data were evaluated for statistical significance using a two-tailed Student's t test or analysis of variance (with Tukey's or Dunnett's test as a post hoc analysis). Differences were considered significant if p < 0.05. Data are expressed as the average ± S.E.
RESULTS
SFK Protein Activity Is Detectable in Human Islets and MIN6 Cells
To determine the potential for SFKs to undergo activation in human islets, three independent batches of cadaveric islets were treated for 5 min with increasing doses of freshly made protein-tyrosine phosphatase inhibitor pervanadate (pV) and assessed for relative levels of phosphorylated SFKTyr-416 via immunoblotting with a pan-phosphospecific antibody that recognizes phosphorylation of all SFKs (Fig. 1A). Phosphorylation of tyrosine 416, located within the active site of the kinase domain, is required for SFK activity. SFKs became increasingly tyrosine-phosphorylated as the concentration of pervanadate increased (p-SFKTyr-416/GAPDH for 0.1 mm pV = 16.2 ± 0.9 and for 1 mm pV = 33.7 ± 2.8-fold over basal = 1.0 ± 0.3). Because standard stripping protocols were insufficient to accurately and reliably remove phospho-SFKTyr-416 from membranes containing human islet proteins, p-SFK was normalized to GAPDH rather than total SFK levels. Similar to human islets, MIN6 β cells treated with 1 mm pV for 5 min showed an ∼24-fold increase in SFKTyr-416 phosphorylation (Fig. 1B). Inhibitors of SFK activity, either PP2 or SU-6656 (20 μm), significantly reduced SFK activation (p-SFKTyr-416/SFK after pV stimulation for DMSO = 24.3 ± 3.1-fold, for PP2 = 11.0 ± 0.8-fold, and for SU-6656 = 12.3 ± 1.5-fold). SU-6656 was used for subsequent studies as PP2 treatment consistently elevated basal levels of phosphorylated SFKTyr-416 (basal p-SFKTyr-416/SFK for PP2 = 1.5 ± 0.2 over DMSO-treated = 1.0 ± 0.2 basal cells), suggesting potential spurious, off-target effects.
FIGURE 1.
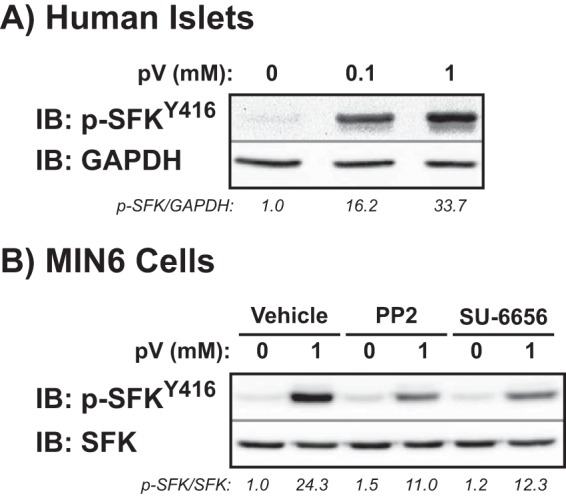
SFK protein abundance and activity in healthy human islets. A, human islets were preincubated in KRBH plus 2.8 mm glucose and then treated for 5 min with 0.1 or 1 mm freshly made pV. Islets were lysed and resolved by 10% SDS-PAGE for immunoblotting (IB) with phospho-SFKTyr-416 and GAPDH antibodies. Total phosphorylation was adjusted for loading and normalized to basal equal to 1.0 in each of three independent experiments, shown as the average ratio below each gel lane. B, MIN6 β cells preincubated in modified MKRBB with the SFK inhibitors PP2 or SU-6656 (20 μm) or vehicle (DMSO) were treated with 1 mm pV for 5 min. Lysates were resolved by 10% SDS-PAGE for immunodetection of phospho-SFKTyr-416. Phosphorylated levels were adjusted to total SFK abundance and normalized to basal equal to 1.0 in each of four independent experiments, shown as the average ratio below each gel lane.
Glucose-stimulated Cdc42 and PAK1 Activation Is Dependent on SFK Function
To evaluate the requirement for SFK activity in the glucose-induced activation of Cdc42, MIN6 β cells were pretreated with either 20 μm SU-6656 or vehicle (DMSO) for 2 h followed by glucose stimulation for 3 min as previously described (12, 15, 20). Cdc42 activation significantly increased after glucose stimulation in vehicle-treated cells (Fig. 2A); however, SU-6656 treatment ablated this glucose-induced activation event. A similar loss of glucose-induced PAK1 phosphorylation after SU-6656 treatment was observed (Fig. 2B). These data implicated the SFKs in the activation of Cdc42 and its downstream effector PAK1 after glucose stimulation.
FIGURE 2.
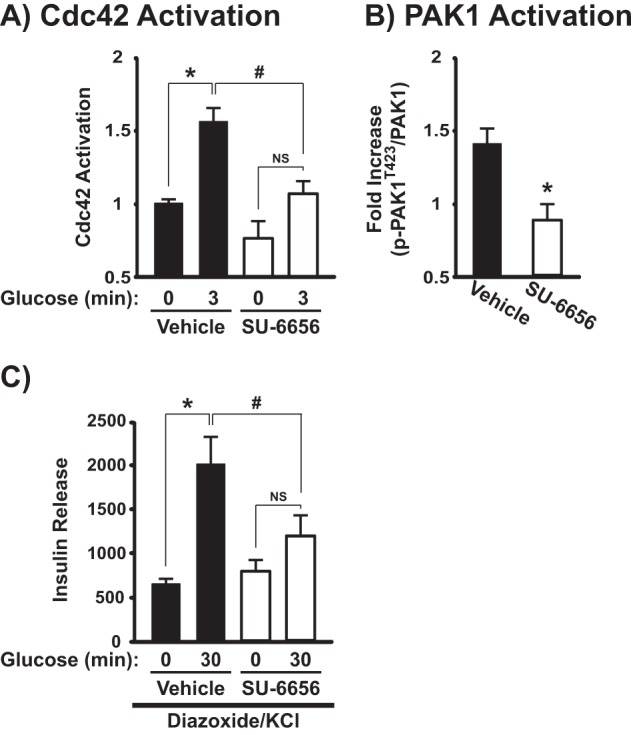
SFK activity is required for glucose-induced Cdc42 and PAK1 activation and the amplifying pathway of GSIS. MIN6 cells were preincubated in MKRBB with the SFK inhibitor SU-6656 (20 μm) or vehicle (DMSO) before stimulation with 20 mm glucose for 3 min for assessment of Cdc42-GTP (A) or for 5 min for assessment of PAK1 phosphorylation (B). A, Cdc42-GTP was captured on GST-PAK1-PBD beads and resolved by 12% SDS-PAGE for detection by anti-Cdc42 immunoblotting. Activation levels, shown as the average ± S.E., were adjusted for GST loading and normalized to untreated basal activation in each of three independent experiments: *, p < 0.05 versus basal; #, p < 0.05 versus respective condition in SU-6656-treated cells; NS = not statistically significant. B, glucose-induced PAK1Thr-423 phosphorylation was detected by resolving lysate protein on 10% SDS-PAGE for immunodetection; gels were stripped and reprobed for comparison against total PAK1 abundance and normalized to basal levels within each treatment group. Data are shown as the -fold increase in glucose-induced phosphorylation compared with basal and represent the average ± S.E. of five independent experiments: *, p < 0.05 versus vehicle-treated control. C, after pretreatment with the SFK inhibitor SU-6656 (20 μm) or vehicle (DMSO), MIN6 cells were treated with 250 μm diazoxide for 5 min and then stimulated with 40 mm KCl ± 20 mm glucose for 30 min. Insulin secretion was normalized to corresponding cellular protein content. Data are displayed as insulin release (ng/mg of protein) and represent the average ± S.E. of eight independent experiments: *, p < 0.05 versus KCl alone; #, p < 0.05 versus respective condition in SU-6656-treated cells.
The Amplifying Pathway of GSIS Requires SFK Activity
The diazoxide paradigm was utilized to assess the requirement of the SFKs during GSIS, specifically their role in either the triggering or amplifying the pathway of GSIS. Subjecting β cells to depolarizing concentrations of KCl while in the presence of the KATP channel opener diazoxide results in low concentrations of glucose stimulating the triggering pathway of insulin secretion, with high concentrations of glucose stimulating the amplifying pathway of insulin secretion (35–39). Previous work has demonstrated that PAK1 is required for both second-phase GSIS and the amplification pathway during the diazoxide paradigm (13). Although vehicle-treated cells exhibited a >3-fold increase in insulin secretion after the addition of KCl plus glucose (as compared with KCl alone), this increase in the amplifying pathway was significantly reduced in the SU-6656-treated cells (Fig. 2C). Moreover, SU-6656 had no effect on the triggering pathway. These data further implicate the SFKs in the sustainment of GSIS through the glucose-specific activation of Cdc42 and PAK1.
SFKs Are Specifically Activated by Glucose at the Cell Surface before the Activation of Cdc42
Because Cdc42 activation occurs within 3 min of glucose stimulation, we predicted that glucose would induce activation of the SFKs before 3 min. Indeed, maximal SFKTyr-416 phosphorylation occurred within 1 min of glucose stimulation, rapidly returning to basal levels after 2 min (Fig. 3A). Human islets showed a similar level of SFKTyr-416 phosphorylation/activation after a 1-min glucose stimulation (Fig. 3B), establishing the continuity of function between human islets and MIN6 β cells (p-SFKTyr-416/GAPDH for 1-min glucose = 1.5 ± 0.1 versus basal, p < 0.05). In contrast, MIN6 cells treated with 50 mm KCl for 0.5 to 2 min showed no significant increase in phospho-SFKTyr-416 levels above basal (Fig. 3C). Tyrosine 527 is positioned in the N-terminal negative regulatory tail of SFKs, which when phosphorylated, participates in an intramolecular interaction with the SH2 domain to keep the kinase in a closed, inactive conformation (28). Compared with basal levels, there was no change in the glucose-induced phosphorylation of SFKs at tyrosine 527 (data not shown).
FIGURE 3.
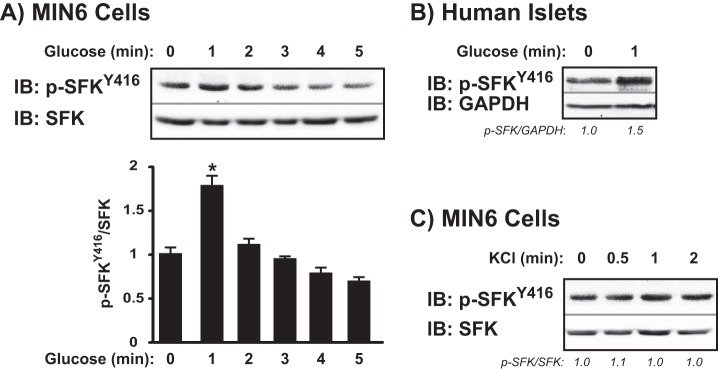
SFKs are activated by glucose but not KCl. MIN6 cells were preincubated in MKRBB and then stimulated with 20 mm glucose (A) or 50 mm KCl (C) for the times indicated. Human islets were preincubated in KRBH containing 2.8 mm glucose and then stimulated with 16.7 mm glucose (B) for the times indicated. Proteins were resolved on 10% SDS-PAGE for immunodetection of phospho-SFKTyr-416. Phosphorylated protein was adjusted for total SFK content or GAPDH and normalized to basal levels as described in the legend to Fig. 1. Data are representative of three independent experiments each. *, p < 0.05 versus basal. IB, immunoblot.
Cdc42 activation is known to occur at the plasma membrane of the β cell. To determine the predominant subcellular localization of SFK activation in response to glucose, we used both subcellular fractionation and immunofluorescence as complementary methods. In non-stimulated MIN6 cells, both methods revealed strong SFK labeling in the plasma membrane (PM) compartment (Fig. 4, A and B). Subcellular fraction integrity was verified by the localization of the known PM t-SNARE protein Syntaxin 4, the cytosolic factor RhoGDI, and the insulin granule v-SNARE protein VAMP2. The membranous localization of SFK is consistent with the known lipid modifications within the SH4 domain (40). Although Fig. 4C shows trace amounts of SFK staining detected in the storage granule fraction (SG), the rapid glucose-induced phospho-SFKTyr-416 activation was found only in the PM fraction; pV treatment yielded the same result (Fig. 4D, panels 1 versus 2). Acute pretreatment with SU-6656 blocked pV-induced phospho-SFKTyr-416 activation, confirming the specificity of SFK activation at the PM (Fig. 4D, panels 2 versus 4) and not under basal conditions (Fig. 4D, panels 1 and 3).
FIGURE 4.

SFK activation occurs at the plasma membrane. A, MIN6 β cells subfractionated into PM or soluble (Cyto) fractions were assessed for total SFK content by immunoblot (IB). Syntaxin 4, RhoGDI, and VAMP2 were used to validate the integrity of the fractions. Data are representative of three independent experiments. B, MIN6 cells were fixed, permeabilized, and immunostained for total SFK (panel 1); DAPI labeling was used to identify individual nuclei in merged images (panel 2). Images were taken at the midplane of the cell cluster by single channel scanning confocal microscopy. Data are representative of five fields, each from two independent experiments; scale bar = 0.2 μm. C, subcellular (PM and SG) fractions prepared from MIN6 cells under basal or 1-min glucose-stimulated (20 mm) conditions were subjected to phospho-SFKTyr-416 immunoblotting relative to total SFK amounts. Optical density scanning quantitation of PM changes is shown in the bar graph format to the right of a representative of three independent experiments: *, p < 0.05 versus basal PM. D, MIN6 cells preincubated in MKRBB with vehicle (DMSO, panels 1 and 2) or SU-6656 (20 μm, panels 3 and 4) followed by treatment with 1 mm pV (panels 2 and 4) for 5 min were immediately fixed and permeabilized for immunofluorescent confocal imaging detection of phospho-SFKTyr-416. Data are representative of five fields, each from two independent experiments; scale bar = 0.2 μm.
YES and Fyn, but Not Src, Are Exclusively Present at the β Cell Plasma Membrane
To determine which of the nine possible SFK members were upstream activator candidates of Cdc42 in the β cell, a candidate PCR screen was initiated. Using specific primer sets for each SFK member (Table 1), mRNA expression for five of the nine known family members (Src, Hck, YES, Fyn, and Frk) was detected (Fig. 5A). Mouse spleen was employed as a positive control for all primer sets but Frk; however, MIN6 cells did show a strong Frk band of the predicted size. Positive hits from the PCR screen were secondarily assessed via immunoblotting using antibodies specific to each protein (Fig. 5B). Although Hck and Frk mRNA was expressed in MIN6 cells, only Src, Fyn, and YES kinases were detected at the protein level. For both Frk and Src, commercially available A431 cell lysate (human epithelial carcinoma cell line) was used a positive control, whereas mouse spleen lysate sufficed for the remaining three targets; clathrin was used as a loading control. We further verified the presence of Src, Fyn, and YES proteins in human islets (Fig. 5C).
FIGURE 5.
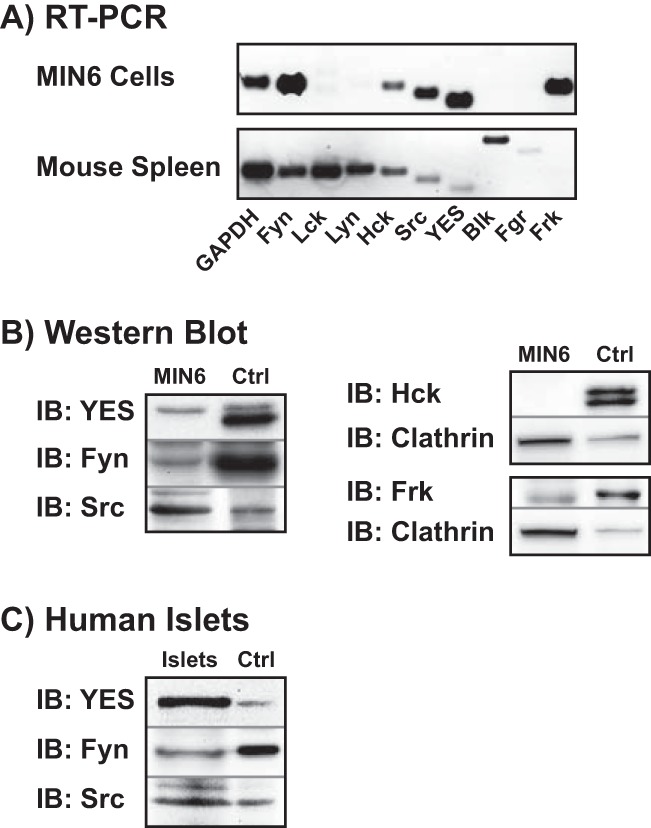
MIN6 β cells and human islets express the SFK family members YES, Fyn, and Src. A, RT-PCR was used to detect which of the nine SFK members is expressed in MIN6 cells. Mouse spleen, which contains all the family members except Frk, was used as a positive control for the primers, and GAPDH was a positive control for sample integrity. Data are representative of two independent experiments. B, MIN6 cell lysates were immunoblotted for Fyn, Hck, Src, YES, and Frk (positive hits from A). Mouse spleen was used as a positive control (Ctrl) for Fyn, Hck, and YES, whereas A431 lysate was used as a control for Src and Frk. Clathrin served as a loading control for SFKs less clearly detected in MIN6 cells. Data are representative of four independent experiments. IB, immunoblot. C, Specific antibodies were used to confirm positive hits from B in human islets. Mouse spleen was used as a positive control for YES and Fyn, whereas A431 lysate was used as a control for Src. Data are representative of three independent experiments.
We next analyzed the subcellular localization of Src, YES, and Fyn as a means to identify which of the SFKs exhibited PM localization and activation in response to glucose. Similar to the pan-SFK blot in Fig. 4A, YES and Fyn were found principally at the PM, with lesser abundance in the SG fraction (Fig. 6A). By contrast, Src was almost exclusively confined to the SG fraction. To determine if Src might translocate into the PM fraction from the SG fraction upon glucose stimulation and thus might have been missed in assessments of unstimulated cells, 1-min glucose-stimulated cells were fractionated and assessed for Src translocation. Despite glucose stimulation, Src remained in the SG fraction (Fig. 6B). These data indicated that of the nine known SFK members, two (YES and Fyn) were viable candidates for the activation of Cdc42.
FIGURE 6.
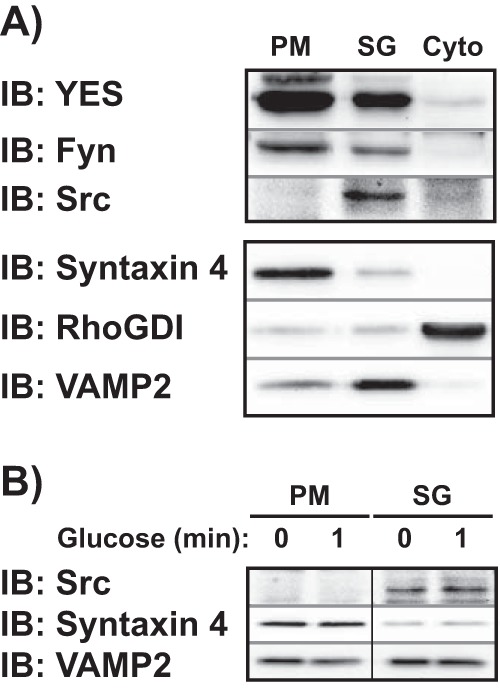
YES and Fyn are the exclusive SFKs present at the PM in MIN6 cells. A, MIN6 cells were fractionated into PM, SG, and remaining soluble (Cyto) fractions. The subcellular localization of YES, Fyn, and Src in each fraction was assessed by immunoblots (IB). Syntaxin 4, RhoGDI, and VAMP2 were used to validate the integrity of the fractions. Data are representative of three independent experiments. B, subcellular fractions (PM and SG) were prepared from MIN6 cells stimulated with 20 mm glucose for 1 min after a 2-h incubation in MKRBB to assess for potential translocation of Src from the SG pool to the PM upon glucose stimulation. Data are representative of three independent experiments. Vertical lines denote splicing of lanes from within the same gel exposure.
YES Is the SFK Member Activated in Response to Glucose
To determine which of the two PM-localized SFK members were accountable for the SFKTyr-416 phosphorylation in response to glucose, MIN6 cell lysates were subjected to anti-phosphotyrosine immunoprecipitation for immunoblotting with either YES or Fyn antibodies (no specific phospho-YES or phospho-Fyn antibodies have been reported). Phospho-reactive bands were normalized against the IgG heavy chain to control for protein loading. Glucose stimulation resulted in a significant increase in YES phosphorylation within 1 min (Fig. 7A); no Fyn phosphorylation was detected within the same time frame (Fig. 7B). Via the comparative analyses completed in Figs. 5, 6, and 7, we systematically identified YES kinase as the putative SFK member involved in the glucose-induced activation of Cdc42 and PAK1.
FIGURE 7.
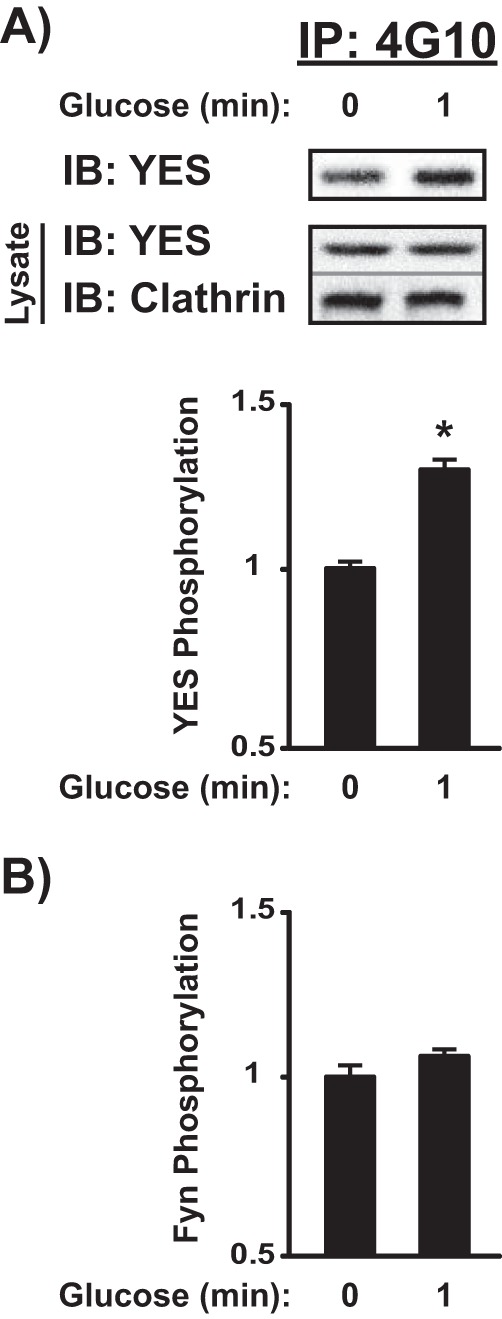
YES, but not Fyn, is phosphorylated after 1-min glucose stimulation in MIN6 cells. MIN6 cells preincubated in MKRBB were left unstimulated or were stimulated with 20 mm glucose for 1 min, and detergent cell lysates were prepared for immunoprecipitation (IP) reactions with anti-4G10 (anti-phosphotyrosine) antibody. Co-immunoprecipitation of YES (A) or Fyn kinase (B) was quantified by immunodetection and optical density scanning. Control IgG was used to control for non-specificity within the immunoprecipitation. YES and Fyn levels were adjusted for loading using the nonspecific heavy chain band detected in the immunoblot (IB) and then normalized to unstimulated levels in each of three independent experiments. Bar graph quantifications of immunodetections are shown as the average ± S.E. *, p < 0.05 versus basal. A representative image for YES phosphorylation is displayed in A.
Depletion of YES Kinase Impairs Cdc42 and PAK1 Activation
To determine the requirement for YES as the upstream mediator of Cdc42 activation and subsequent signaling events, RNAi-mediated depletion of endogenous YES was employed. MIN6 cells were transiently transfected with one of two commercially available siRNA nucleotides; both oligos reduced endogenous YES expression by ∼50% compared with cells transfected with the non-targeting siRNA control oligonucleotides (Fig. 8A). Using the siYES oligo #2, we examined the effects of YES knockdown upon glucose-stimulated PAK1 activation (Fig. 8B). Although 5-min glucose stimulation significantly elevated phospho-PAK1Thr-423 in siCtrl-transfected cells by nearly 2-fold, there was a significant ∼50% reduction in the level of glucose-stimulated PAK1 activation in the siYES-transfected cells (a substantial decrease in light of the efficiency of knockdown at ∼50%). Moreover, siYES-transfected cell lysates showed a significant ablation of the pSFK signal (Fig. 8C), altogether revealing YES kinase to be the operative SFK in the initiation of the Cdc42-PAK1 signaling pathway.
FIGURE 8.
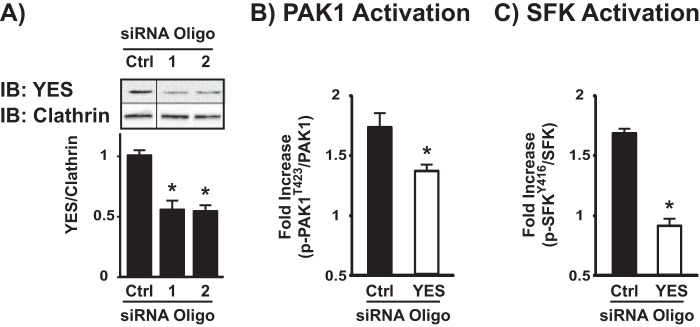
Depletion of YES via siRNA impairs glucose-stimulated total SFK and PAK1 phosphorylation in MIN6 β cells. A, MIN6 cells were transfected with non-targeting commercially available control (Ctrl) or YES siRNA oligonucleotides. After a 48-h incubation, detergent cell lysates were prepared, and proteins were resolved by 10% SDS-PAGE for immunoblotting (IB) with YES and clathrin (for loading) antibodies. Vertical lines denote splicing of lanes from within the same gel exposure. Data represent the average ± S.E. of four independent experiments: *, p < 0.05 versus siCtrl. MIN6 cells transfected with Ctrl or YES siRNA oligonucleotides were preincubated in MKRBB and then subsequently stimulated with 20 mm glucose for 5 min for immunodetection of PAK1Thr-423 phosphorylation (B) or 1 min for immunodetection of SFKTyr-416 phosphorylation (C). Cell lysates were prepared, and proteins were resolved by 10% SDS-PAGE. The data are reported as the -fold increase in glucose-induced phosphorylation compared with siCtrl-transfected cells. Data represent the average ± S.E. of three independent experiments. *, p < 0.05 versus siCtrl-transfected cells.
YES Kinase Depletion Inhibits the Amplifying Pathway of GSIS in MIN6 β Cells and Mouse Islets
To assess the specific effect of YES knockdown on the amplifying pathway of insulin secretion in a pure β cell environment, MIN6 cells were transduced with either an adenovirus encoding the siYES sequence used earlier (shYES-Ad) or a non-targeting control sequence (shCtrl-Ad) and subjected to the diazoxide paradigm. Despite only ∼30% knockdown in YES protein in MIN6 cells (Fig. 9A), the glucose-induced amplification pathway was significantly impaired (Fig. 9B) but was without effect on the triggering pathway. In mouse islets the shYES-Ad elicited a ∼50% reduction in YES protein (Fig. 9C), and again only impaired the amplifying pathway of insulin secretion (Fig. 9D). These data demonstrate that glucose-induced YES activation is required for the amplifying pathway of insulin release.
FIGURE 9.
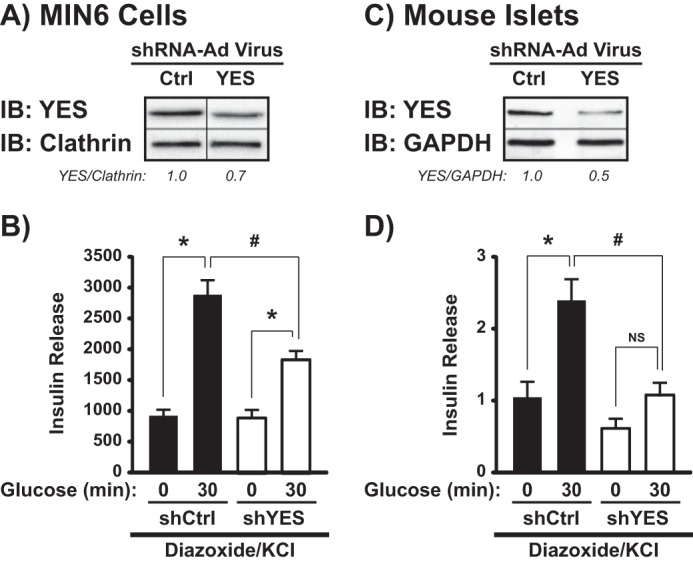
RNAi-mediated depletion of YES from MIN6 cells and mouse islets inhibits the amplifying pathway of GSIS. A, MIN6 cells were transduced with shYES-Ad or shCtrl-Ad viruses, and after 48-h cell lysates were prepared and proteins were resolved by 10% SDS-PAGE for immunoblotting (IB) for YES. A representative image of five independent experiments is provided; vertical lines denote splicing of lanes from within the same gel exposure. B, transduced MIN6 cells were incubated for 5 min with 250 μm diazoxide followed by stimulation for 30 min with KCl ± glucose. Insulin secreted was normalized to corresponding cellular protein content. Data are displayed as insulin release (ng/mg of protein) and represent the average ± S.E. of five independent experiments: *, p < 0.05 versus KCl alone; #, p < 0.05 versus respective condition in shYES-transduced cells. shCtrl- and shYES-transduced mouse islets were validated for YES depletion (C) and insulin secretion (D) as described above. Insulin secretion from four independent experiments was quantified and normalized to islet insulin content. Data are shown as insulin release (-fold above diazoxide alone) for each independent experiment: *, p < 0.05 versus KCl alone; #, p < 0.05 versus respective condition in shYES-transduced islets; NS = not statistically significant.
DISCUSSION
In the current study we have identified YES kinase, an SFK family member, as a proximal factor in the glucose-specific activation of Cdc42 in islet β cells. Of the nine known family members, five transcripts were expressed in β cells, and three of those were confirmed by immunoblotting, and yet only YES kinase fit the criteria expected of a Cdc42 activator in β cells. YES undergoes phosphorylation within 1 min in both human islets and MIN6 β cells, preceding the time of activation of Cdc42 (3 min) and of PAK1 (5 min), and YES is required for these subsequent signaling events to occur. As did the p-SFKTyr-416 signal, YES kinase was localized to the plasma membrane, as is consistent with its role in Cdc42 activation. Additionally, YES kinase was required for GSIS from both isolated mouse islets and MIN6 β cells. Taken together, we propose the following testable model: upon entry and rapid metabolism in the β cell, glucose activates YES kinase, which in turn activates Cdc42 via phosphorylation of its GDI on the insulin granule (Cav-1) to trigger dissociation from Cdc42. Activated Cdc42 can then proceed to directly activate its downstream effector, PAK1, which subsequently activates Raf-1, MEK1/2, and ERK1/2, culminating in F-actin remodeling to mobilize insulin granules to the PM, supporting the amplification pathway of GSIS (Fig. 10).
FIGURE 10.
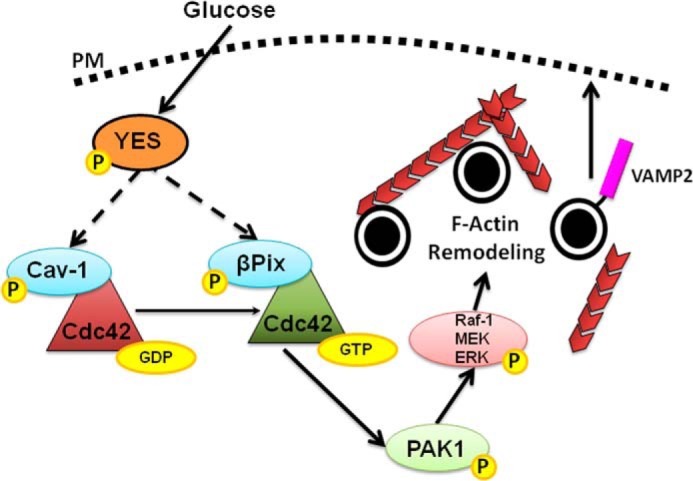
A proposed model for the proximal role of YES in regulating glucose-specific Cdc42-PAK1 signaling. Glucose enters the β cell, activating YES then Cdc42 and PAK1, possible via phosphorylating Cav-1 and βPix (designated by dashed arrows). The subsequent signals of PAK1 to Raf-1, MEK1/2, and ERK1/2 induce F-actin remodeling and insulin granule recruitment at the PM, events required for the sustainability of GSIS.
Having identified YES kinase as a proximal, glucose-specific activator of Cdc42, the next steps toward testing this model include determination of the substrate(s) for YES kinase in β cells. Based upon the literature, βPix and Cav-1 are both candidates (21, 23) and in β cells serve as the requisite GDI and GEF, respectively, for glucose-induced Cdc42 activation and GSIS (19, 20). For example, the Tyr-14 SFK phosphorylation site in Cav-1 is required for Cdc42-Cav-1 interactions, and the Cav-1Y14F mutant is unable to compete with βPix for Cdc42 association in β cells (20). Moreover, in response to growth factors in other cell types, tyrosine phosphorylation of βPix at tyrosine 442 by SFKs is required for its GEF abilities and subsequent activation of Cdc42 (23, 24). Furthermore, Cdc42 itself has been suggested to undergo tyrosine phosphorylation in COS-7 cells (41). In this manner it is conceivable that YES kinase activation could ignite additional activation events centered at the Cdc42 signaling node. Despite implicating YES kinase in the Cdc42-PAK1 signaling pathway, YES depletion only partially reduced glucose-induced PAK1 activation, suggesting that another non-SFK factor may participate. Indeed, occurring concurrent with YES kinase activation and related to Cdc42 activation is the activation of the small GTPase Arf6 (42). On the other hand, how Arf6 fits in with YES action is unclear. Although Arf6 inhibition can block GSIS and Cdc42 activation, Arf6 inhibition-impaired Cdc42 activation was measured after 30 min of glucose stimulation and not the initial activation event known to occur at 3 min, past the time noted for Cdc42 to have already down-regulated in its cycling (12). Another caveat is that Arf6 inhibition also impairs KCl- and arginine-stimulated insulin secretion, which places it outside of the strictly glucose-sensitive Cdc42 pathway and implicates Arf6 and ARNO in KATP channel-dependent pathways, as suggested by others (43). In pursuing efforts to determine linkage between these events in Cdc42 signaling, there is precedence also pointing to a Src-Pix mechanism involving post-translational modifications of Rac1 (44). Like Rac1, Cdc42 undergoes substantial modification and requires both geranylgeranylation and carboxylmethylation as well as glucosylation to facilitate its cycling and GSIS (12, 14, 45–48).
The identification of factors/steps upstream of Cdc42 has been challenging due perhaps in large part to the reliance upon biochemical Cdc42 activation assays as the output signal, assays that are notoriously difficult in β cells, particularly at time points earlier than 2 min; real-time imaging biosensors have also proven challenging given the abundance of Cdc42 in all major subcompartments of the β cell. Our identification herein of YES phosphorylation permits a more approachable assay of the early events upstream of Cdc42 activation given the ease of use of the high quality p-SFKTyr-416 antibodies that might be suitable for high throughput screening. Such a screening platform would be advantageous for identification of the metabolic amplifying factor(s) responsible for activating the pathway for granule recruitment to sustain insulin release. Although insulin release has been used previously as an output measure of SFK involvement using SFK inhibitors (49), reliable GSIS readings require at a minimum of 5–10 min of incubation time with glucose, and hence existing reports could actually represent a net average of positive and negative concurrent effects by one or more SFKs acting upon a multitude of factors impinging upon GSIS, thus requiring more than insulin secretion alone as a readout for SFK function. Moreover, pharmacologic inhibitors PP2 and SU-6656 are active against all SFKs (50, 51), and our results here show the β cell to express three or more different SFK proteins. As such, our data pinpointing the p-SFKTyr-416 activation event to YES kinase, occurring in response only to glucose and at 1 min in the context of GSIS, now provide the ability to use this as a single event output measure for future identification of the upstream glucose metabolite/amplifying factor required for inducing insulin release via Cdc42.
How glucose stimulation might trigger the activation of YES kinase in β cells is an intriguing question. Activation of SFKs requires the autophosphorylation of tyrosine 416 within the kinase domain. Commonly, activation of SFKs at Tyr-416 is induced through a receptor-induced signal transduction pathway, such as immune recognition receptors on T and B cells (28). Given that Cdc42 activation requires glucose metabolism beyond fructose 6-phosphate production, it seems unlikely that YES phosphorylation would be linked to a receptor-mediated signal. However, three possibilities exist: 1) phosphorylation through an integrin such as β1 integrin to focal adhesion kinase (26, 27, 52–54), 2) phosphorylation through the insulin receptor, a receptor protein-tyrosine kinase, or 3) that YES phosphorylation occurs in response to reactive oxygen species (ROS) generated via rapid glucose metabolism. Supporting the first possibility, Rondas et al. (55, 56) have demonstrated a role for focal adhesion kinase, paxillin, and ERK1/2 in focal adhesion/cytoskeleton remodeling and subsequent GSIS in β cells through β1 integrin. If YES were to connect β1 integrin signaling to Cdc42 and PAK1, this could explain the dynamic cytoskeletal rearrangement compulsory for sustained insulin release. Further work is needed to elucidate how glucose mediates the activation of an integrin receptor. In support of the second possibility, the insulin receptor substrate (IRS) proteins associated with activated IR contain several SH2-binding sites and have been shown to interact with SFKs in other cell types (57). In this scenario, after glucose simulation the rapid release of insulin in an auto- or paracrine fashion may activate the insulin receptor, YES phosphorylation, and the Cdc42 signaling pathway. Disputing this, however, are new proteomic data questioning the validity of interactions between SFKs with IRS/IR (58) and whether or not sufficient insulin is released within 1 min of glucose stimulation to activate the insulin receptor and, even if so, whether the insulin receptor could bind to the immediately released insulin, which is secreted in a hexameric form (59). In the third possibility, that YES phosphorylation may result from ROS produced during rapid glucose metabolism, ROS such as H2O2, are known to activate SFKs (60–65). In the β cell, the generation of ROS can be an important positive mediator of insulin secretion (66–69). Moreover, ROS generation occurs half-maximally within 22 s of glucose stimulation (70, 71), placing this intracellular signaling message temporally upstream of YES activation. Thus, it is tempting to speculate that upon stimulation, glucose metabolism increases superoxide (O2⨪) generation, which once converted to H2O2 by superoxide dismutase activates the YES → Cdc42 → PAK1 signaling cascade. Future work will be required to substantiate the validity of this hypothesis.
In summary, we demonstrate here for the first time that YES kinase, one of five SFKs expressed in β cells, accounts for the glucose-induced increase in phospho-SFK and is required for the activation of the Cdc42-PAK1 signaling cascade. Given the recent interest in targeting PAK1 signaling as a means to improve functional β cell mass (13, 72, 73), our elucidation of this upstream signal is a step forward. Because Cdc42-PAK1 signaling also oversees GLP-1 secretion from the intestinal L cells (74) and GLP-1 is a potentiating factor of functional β cell mass and important for mediating glucose homeostasis, there could be reverberating beneficial effects if YES kinase were to also be implicated in activating Cdc42-PAK1 signaling in L cells.
Acknowledgments
We thank Michael Kalwat, Latha Ramalingam, Eunjin Oh, Ragadeepthi Tunduguru, and Dean Wiseman for many helpful discussions during the progress of this manuscript. We also thank Kara Benninger, Emily Mulpuri, and Carrie Sedam for technical assistance. Human pancreatic islets were obtained through the Integrated Islet Distribution Program.
This work was supported, in whole or in part, by National Institutes of Health Grants F32 DK094488 (to S. M. Y.), CTSI-KL2 RR025760 (to Z. W.), and DK067912 and DK076614 (to D. C. T).
- PAK1
- p21-activated kinase
- Cav-1
- caveolin-1
- GDI
- guanine nucleotide dissociation inhibitor
- GEF
- guanine nucleotide exchange factor
- GSIS
- glucose-stimulated insulin secretion
- KRBH
- Krebs-Ringer bicarbonate buffer
- MKRBB
- modified KRBB
- pV
- pervanadate
- SFK
- Src family kinase
- SG
- storage granule
- ROS
- reactive oxygen species
- PM
- plasma membrane.
REFERENCES
- 1. Ashcroft F. M., Harrison D. E., Ashcroft S. J. (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312, 446–448 [DOI] [PubMed] [Google Scholar]
- 2. Cook D. L., Hales C. N. (1984) Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 311, 271–273 [DOI] [PubMed] [Google Scholar]
- 3. Rorsman P., Ashcroft F. M., Trube G. (1988) Single calcium channel currents in mouse pancreatic B-cells. Pflugers Arch. 412, 597–603 [DOI] [PubMed] [Google Scholar]
- 4. Satin L. S., Cook D. L. (1985) Voltage-gated Ca2+ current in pancreatic B-cells. Pflugers Arch. 404, 385–387 [DOI] [PubMed] [Google Scholar]
- 5. Daniel S., Noda M., Straub S. G., Sharp G. W. (1999) Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes 48, 1686–1690 [DOI] [PubMed] [Google Scholar]
- 6. Shibasaki T., Takahashi H., Miki T., Sunaga Y., Matsumura K., Yamanaka M., Zhang C., Tamamoto A., Satoh T., Miyazaki J., Seino S. (2007) Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Natl. Acad. Sci. U.S.A. 104, 19333–19338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rorsman P., Eliasson L., Renström E., Gromada J., Barg S., Göpel S. (2000) The cell physiology of biphasic insulin secretion. News Physiol. Sci. 15, 72–77 [DOI] [PubMed] [Google Scholar]
- 8. Rorsman P., Braun M. (2013) Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 75, 155–179 [DOI] [PubMed] [Google Scholar]
- 9. Orci L., Gabbay K. H., Malaisse W. J. (1972) Pancreatic beta-cell web. Its possible role in insulin secretion. Science 175, 1128–1130 [DOI] [PubMed] [Google Scholar]
- 10. Thurmond D. C., Gonelle-Gispert C., Furukawa M., Halban P. A., Pessin J. E. (2003) Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 17, 732–742 [DOI] [PubMed] [Google Scholar]
- 11. Mourad N. I., Nenquin M., Henquin J. C. (2010) Metabolic amplifying pathway increases both phases of insulin secretion independently of beta-cell actin microfilaments. Am. J. Physiol. Cell Physiol. 299, C389–C398 [DOI] [PubMed] [Google Scholar]
- 12. Nevins A. K., Thurmond D. C. (2003) Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am. J. Physiol. Cell Physiol. 285, C698–C710 [DOI] [PubMed] [Google Scholar]
- 13. Wang Z., Oh E., Clapp D. W., Chernoff J., Thurmond D. C. (2011) Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 286, 41359–41367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kowluru A., Seavey S. E., Li G., Sorenson R. L., Weinhaus A. J., Nesher R., Rabaglia M. E., Vadakekalam J., Metz S. A. (1996) Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure beta cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J. Clin. Invest. 98, 540–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Z., Oh E., Thurmond D. C. (2007) Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J. Biol. Chem. 282, 9536–9546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Uenishi E., Shibasaki T., Takahashi H., Seki C., Hamaguchi H., Yasuda T., Tatebe M., Oiso Y., Takenawa T., Seino S. (2013) Actin dynamics regulated by the balance of neuronal Wiskott-Aldrich syndrome protein (N-WASP) and cofilin activities determines the biphasic response of glucose-induced insulin secretion. J. Biol. Chem. 288, 25851–25864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalwat M. A., Yoder S. M., Wang Z., Thurmond D. C. (2013) A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic beta cells. Biochem. Pharmacol. 85, 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nie J., Sun C., Faruque O., Ye G., Li J., Liang Q., Chang Z., Yang W., Han X., Shi Y. (2012) Synapses of amphids defective (SAD-A) kinase promotes glucose-stimulated insulin secretion through activation of p21-activated kinase (PAK1) in pancreatic beta-cells. J. Biol. Chem. 287, 26435–26444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nevins A. K., Thurmond D. C. (2006) Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J. Biol. Chem. 281, 18961–18972 [DOI] [PubMed] [Google Scholar]
- 20. Kepner E. M., Yoder S. M., Oh E., Kalwat M. A., Wang Z., Quilliam L. A., Thurmond D. C. (2011) Cool-1/betaPIX functions as a guanine nucleotide exchange factor in the cycling of Cdc42 to regulate insulin secretion. Am. J. Physiol. Endocrinol. Metab. 301, E1072–E1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li S., Seitz R., Lisanti M. P. (1996) Phosphorylation of caveolin by src tyrosine kinases. The α-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J. Biol. Chem. 271, 3863–3868 [PubMed] [Google Scholar]
- 22. Nomura R., Fujimoto T. (1999) Tyrosine-phosphorylated caveolin-1. Immunolocalization and molecular characterization. Mol. Biol. Cell 10, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng Q., Baird D., Peng X., Wang J., Ly T., Guan J. L., Cerione R. A. (2006) Cool-1 functions as an essential regulatory node for EGF receptor- and Src-mediated cell growth. Nat. Cell Biol. 8, 945–956 [DOI] [PubMed] [Google Scholar]
- 24. Feng Q., Baird D., Yoo S., Antonyak M., Cerione R. A. (2010) Phosphorylation of the cool-1/β-Pix protein serves as a regulatory signal for the migration and invasive activity of Src-transformed cells. J. Biol. Chem. 285, 18806–18816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuroiwa M., Oneyama C., Nada S., Okada M. (2011) The guanine nucleotide exchange factor Arhgef5 plays crucial roles in Src-induced podosome formation. J. Cell Sci. 124, 1726–1738 [DOI] [PubMed] [Google Scholar]
- 26. ten Klooster J. P., Jaffer Z. M., Chernoff J., Hordijk P. L. (2006) Targeting and activation of Rac1 are mediated by the exchange factor β-Pix. J. Cell Biol. 172, 759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huveneers S., Danen E. H. (2009) Adhesion signaling-crosstalk between integrins, Src and Rho. J. Cell Sci. 122, 1059–1069 [DOI] [PubMed] [Google Scholar]
- 28. Thomas S. M., Brugge J. S. (1997) Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13, 513–609 [DOI] [PubMed] [Google Scholar]
- 29. Martin G. S. (2001) The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2, 467–475 [DOI] [PubMed] [Google Scholar]
- 30. Wasmeier C., Hutton J. C. (1999) Secretagogue-dependent phosphorylation of phogrin, an insulin granule membrane protein tyrosine phosphatase homologue. Biochem. J. 341, 563–569 [PMC free article] [PubMed] [Google Scholar]
- 31. Wiseman D. A., Kalwat M. A., Thurmond D. C. (2011) Stimulus-induced S-nitrosylation of syntaxin 4 impacts insulin granule exocytosis. J. Biol. Chem. 286, 16344–16354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kowluru A., Seavey S. E., Rhodes C. J., Metz S. A. (1996) A novel regulatory mechanism for trimeric GTP-binding proteins in the membrane and secretory granule fractions of human and rodent beta cells. Biochem. J. 313, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lacy P. E., Kostianovsky M. (1967) Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes 16, 35–39 [DOI] [PubMed] [Google Scholar]
- 34. Spurlin B. A., Thomas R. M., Nevins A. K., Kim H. J., Kim Y. J., Noh H. L., Shulman G. I., Kim J. K., Thurmond D. C. (2003) Insulin resistance in tetracycline-repressible Munc18c transgenic mice. Diabetes 52, 1910–1917 [DOI] [PubMed] [Google Scholar]
- 35. Henquin J. C. (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760 [DOI] [PubMed] [Google Scholar]
- 36. Bratanova-Tochkova T. K., Cheng H., Daniel S., Gunawardana S., Liu Y. J., Mulvaney-Musa J., Schermerhorn T., Straub S. G., Yajima H., Sharp G. W. (2002) Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 51, S83–S90 [DOI] [PubMed] [Google Scholar]
- 37. Straub S. G., Sharp G. W. (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab. Res. Rev. 18, 451–463 [DOI] [PubMed] [Google Scholar]
- 38. Ohara-Imaizumi M., Nakamichi Y., Tanaka T., Ishida H., Nagamatsu S. (2002) Imaging exocytosis of single insulin secretory granules with evanescent wave microscopy. Distinct behavior of granule motion in biphasic insulin release. J. Biol. Chem. 277, 3805–3808 [DOI] [PubMed] [Google Scholar]
- 39. Henquin J. C. (2009) Regulation of insulin secretion. A matter of phase control and amplitude modulation. Diabetologia 52, 739–751 [DOI] [PubMed] [Google Scholar]
- 40. Alland L., Peseckis S. M., Atherton R. E., Berthiaume L., Resh M. D. (1994) Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J. Biol. Chem. 269, 16701–16705 [PubMed] [Google Scholar]
- 41. Tu S., Wu W. J., Wang J., Cerione R. A. (2003) Epidermal growth factor-dependent regulation of Cdc42 is mediated by the Src tyrosine kinase. J. Biol. Chem. 278, 49293–49300 [DOI] [PubMed] [Google Scholar]
- 42. Jayaram B., Syed I., Kyathanahalli C. N., Rhodes C. J., Kowluru A. (2011) Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42, and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem. Pharmacol. 81, 1016–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lawrence J. T., Birnbaum M. J. (2003) ADP-ribosylation factor 6 regulates insulin secretion through plasma membrane phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. U.S.A. 100, 13320–13325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Singh N. K., Kundumani-Sridharan V., Rao G. N. (2011) 12/15-Lipoxygenase gene knockout severely impairs ischemia-induced angiogenesis due to lack of Rac1 farnesylation. Blood 118, 5701–5712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li G., Regazzi R., Roche E., Wollheim C. B. (1993) Blockade of mevalonate production by lovastatin attenuates bombesin and vasopressin potentiation of nutrient-induced insulin secretion in HIT-T15 cells. Probable involvement of small GTP-binding proteins. Biochem. J. 289, 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Metz S. A., Rabaglia M. E., Stock J. B., Kowluru A. (1993) Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem. J. 295, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amin R., Chen H. Q., Tannous M., Gibbs R., Kowluru A. (2002) Inhibition of glucose- and calcium-induced insulin secretion from betaTC3 cells by novel inhibitors of protein isoprenylation. J. Pharmacol. Exp. Ther. 303, 82–88 [DOI] [PubMed] [Google Scholar]
- 48. Veluthakal R., Kaur H., Goalstone M., Kowluru A. (2007) Dominant-negative α-subunit of farnesyl- and geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes 56, 204–210 [DOI] [PubMed] [Google Scholar]
- 49. Cheng H., Straub S. G., Sharp G. W. (2007) Inhibitory role of Src family tyrosine kinases on Ca2+-dependent insulin release. Am. J. Physiol. Endocrinol. Metab. 292, E845–E852 [DOI] [PubMed] [Google Scholar]
- 50. Blake R. A., Broome M. A., Liu X., Wu J., Gishizky M., Sun L., Courtneidge S. A. (2000) SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 20, 9018–9027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., Cohen P. (2007) The selectivity of protein kinase inhibitors. A further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wary K. K., Mariotti A., Zurzolo C., Giancotti F. G. (1998) A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 94, 625–634 [DOI] [PubMed] [Google Scholar]
- 53. Mitra S. K., Schlaepfer D. D. (2006) Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 [DOI] [PubMed] [Google Scholar]
- 54. Huveneers S., van den Bout I., Sonneveld P., Sancho A., Sonnenberg A., Danen E. H. (2007) Integrin αvβ3 controls activity and oncogenic potential of primed c-Src. Cancer Res. 67, 2693–2700 [DOI] [PubMed] [Google Scholar]
- 55. Rondas D., Tomas A., Halban P. A. (2011) Focal adhesion remodeling is crucial for glucose-stimulated insulin secretion and involves activation of focal adhesion kinase and paxillin. Diabetes 60, 1146–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rondas D., Tomas A., Soto-Ribeiro M., Wehrle-Haller B., Halban P. A. (2012) Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem. 287, 2423–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun X. J., Pons S., Asano T., Myers M. G., Jr., Glasheen E., White M. F. (1996) The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J. Biol. Chem. 271, 10583–10587 [DOI] [PubMed] [Google Scholar]
- 58. Liu B. A., Engelmann B. W., Jablonowski K., Higginbotham K., Stergachis A. B., Nash P. D. (2012) SRC homology 2 domain binding sites in insulin, IGF-1, and FGF receptor mediated signaling networks reveal an extensive potential interactome. Cell. Commun. Signal. 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., Ward C. W., Lawrence M. C. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cao H., Sanguinetti A. R., Mastick C. C. (2004) Oxidative stress activates both Src kinases and their negative regulator Csk and induces phosphorylation of two targeting proteins for Csk. Caveolin-1 and paxillin. Exp. Cell Res. 294, 159–171 [DOI] [PubMed] [Google Scholar]
- 61. Nakamura K., Hori T., Sato N., Sugie K., Kawakami T., Yodoi J. (1993) Redox regulation of a src family protein-tyrosine kinase p56lck in T cells. Oncogene 8, 3133–3139 [PubMed] [Google Scholar]
- 62. Hardwick J. S., Sefton B. M. (1995) Activation of the Lck tyrosine protein kinase by hydrogen peroxide requires the phosphorylation of Tyr-394. Proc. Natl. Acad. Sci. U.S.A. 92, 4527–4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Giannoni E., Buricchi F., Raugei G., Ramponi G., Chiarugi P. (2005) Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 25, 6391–6403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xi G., Shen X., Maile L. A., Wai C., Gollahon K., Clemmons D. R. (2012) Hyperglycemia enhances IGF-I-stimulated Src activation via increasing Nox4-derived reactive oxygen species in a PKCζ-dependent manner in vascular smooth muscle cells. Diabetes 61, 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Weaver J. R., Taylor-Fishwick D. A. (2013) Regulation of NOX-1 expression in beta cells. A positive feedback loop involving the Src-kinase signaling pathway. Mol. Cell. Endocrinol. 369, 35–41 [DOI] [PubMed] [Google Scholar]
- 66. Leloup C., Tourrel-Cuzin C., Magnan C., Karaca M., Castel J., Carneiro L., Colombani A. L., Ktorza A., Casteilla L., Pénicaud L. (2009) Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 58, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pi J., Bai Y., Zhang Q., Wong V., Floering L. M., Daniel K., Reece J. M., Deeney J. T., Andersen M. E., Corkey B. E., Collins S. (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56, 1783–1791 [DOI] [PubMed] [Google Scholar]
- 68. Newsholme P., Morgan D., Rebelato E., Oliveira-Emilio H. C., Procopio J., Curi R., Carpinelli A. (2009) Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 52, 2489–2498 [DOI] [PubMed] [Google Scholar]
- 69. Rebelato E., Abdulkader F., Curi R., Carpinelli A. R. (2011) Control of the intracellular redox state by glucose participates in the insulin secretion mechanism. PLoS ONE 6, e24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Civelek V. N., Deeney J. T., Kubik K., Schultz V., Tornheim K., Corkey B. E. (1996) Temporal sequence of metabolic and ionic events in glucose-stimulated clonal pancreatic beta-cells (HIT). Biochem. J. 315, 1015–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Corkey B. E. (2012) Banting lecture 2011. Hyperinsulinemia. Cause or consequence? Diabetes 61, 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang Z., Thurmond D. C. (2012) PAK1 limits the expression of the pro-apoptotic protein Bad in pancreatic islet β-cells. FEBS Open Bio. 2, 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen Y. C., Fueger P. T., Wang Z. (2013) Depletion of PAK1 enhances ubiquitin-mediated Survivin degradation in pancreatic beta-cells. Islets 5, 22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lim G. E., Xu M., Sun J., Jin T., Brubaker P. L. (2009) The Rho GTPase, Cdc42, is required for insulin-induced actin remodeling and glucagon-like peptide-1 secretion in the intestinal endocrine L cell. Endocrinology 150, 5249–5261 [DOI] [PubMed] [Google Scholar]