

Background: Calmodulin binding to KCNQ subunit is required for maintaining the M-current.
Results: Protein kinase CK2-mediated phosphorylation of CaM enhances KCNQ2 current. KCNQ2 subunit tethers protein kinase CK2 and protein phosphatase 1.
Conclusion: Phosphorylation status of CaM regulates the M-current.
Significance: Phosphorylation status of calmodulin regulates neuronal excitability via M-current modulation.
Keywords: Calmodulin, Potassium Channels, PP1, Protein Kinases, Protein Phosphorylation, CK2, KCNQ2
Abstract
M-type potassium channels, encoded by the KCNQ family genes (KCNQ2–5), require calmodulin as an essential co-factor. Calmodulin bound to the KCNQ2 subunit regulates channel trafficking and stabilizes channel activity. We demonstrate that phosphorylation of calmodulin by protein kinase CK2 (casein kinase 2) rapidly and reversibly modulated KCNQ2 current. CK2-mediated phosphorylation of calmodulin strengthened its binding to KCNQ2 channel, caused resistance to phosphatidylinositol 4,5-bisphosphate depletion, and increased KCNQ2 current amplitude. Accordingly, application of CK2-selective inhibitors suppressed KCNQ2 current. This suppression was prevented by co-expression of CK2 phosphomimetic calmodulin mutants or pretreatment with a protein phosphatase inhibitor, calyculin A. We also demonstrated that functional CK2 and protein phosphatase 1 (PP1) were selectively tethered to the KCNQ2 subunit. We identified a functional KVXF consensus site for PP1 binding in the N-terminal tail of KCNQ2 subunit: mutation of this site augmented current density. CK2 inhibitor treatment suppressed M-current in rat superior cervical ganglion neurons, an effect negated by overexpression of phosphomimetic calmodulin or pretreatment with calyculin A Furthermore, CK2 inhibition diminished the medium after hyperpolarization by suppressing the M-current. These findings suggest that CK2-mediated phosphorylation of calmodulin regulates the M-current, which is tonically regulated by CK2 and PP1 anchored to the KCNQ2 channel complex.
Introduction
The M-current is generated by voltage-gated potassium channels composed of a combination of Kv7/KCNQ subunits (KCNQ2, 3, 4, and 5), which require calmodulin (CaM)2 as an essential co-factor (1–3). The interaction between CaM and KCNQ2 subunit plays a role not only in calcium-dependent regulation (4, 5), but also in channel trafficking (6) and stabilization of the binding of another essential co-factor, phosphatidylinositol 4,5-bisphosphate (PIP2) (5, 7).
We have demonstrated previously that PKC-mediated phosphorylation of KCNQ2 subunit regulates KCNQ2-CaM binding (7). Interestingly, 15–40% of endogenous CaM is known to be phosphorylated (8, 9), although the functional role of phosphorylated CaM on KCNQ2 channel remains unknown. Protein kinase CK2 (casein kinase 2) has been identified as key kinase for phosphorylating CaM (10, 11). Various functional consequences of phosphorylated CaM have been demonstrated in a number of CaM-dependent proteins (10–12) including the small conductance calcium-activated potassium channel 2, SK2 channel (13). However, because protein kinase CK2 is constitutively active and phosphorylates a wide range of proteins (14, 15), it has long been considered to be engaged in housekeeping functions (14). This traditional view has been revised by recent studies, where CK2-mediated phosphorylation of CaM was shown to play an active role in muscarinic facilitation of long term potentiation in the hippocampus by modulating SK2 channel activity (16, 17).
In this report, we demonstrate that CK2-mediated phosphorylation of CaM modulated KCNQ2 channel activity and facilitated CaM-KCNQ2 binding. We found that CK2 and protein phosphatase 1 (PP1) were selectively tethered to the KCNQ2 subunit. Accordingly, we identified the PP1 binding KVXF motif in the N-terminal of KCNQ2 subunit, which is not present in other KCNQ subunits. We demonstrate that pharmacological inhibition of CK2 attenuated KCNQ2 current, whereas overexpression of phosphomimetic CaM enhanced KCNQ2 current. In superior cervical ganglion neurons, pharmacological inhibition of CK2 reduced the M-current corresponding to a decrease in apamin-resistant medium afterhyperpolarization (mAHP). These findings suggest that modulation of the phosphorylation status of CaM by CK2 is an important regulatory mechanism for the M-current.
MATERIALS AND METHODS
Expression Constructs
The wild-type FLAG-tagged rat KCNQ2 construct (7, 18), V5 epitope-tagged calmodulin (7), and PP1 construct (9) have been described. HA-tagged CK2 construct is a gift from Dr. Litchfield (Western University, London, ON, Canada) via Addgene. Calmodulin mutations were generated by QuikChange II XL site-directed mutagenesis (Agilent Technologies, San Diego, CA). All PCR-derived constructs were verified by sequencing.
Reagents
Tetrabromobenzotriazole (TBB), tetrabromocinnamic acid (TBCA), and apamin were purchased from EMD Millipore. XE991, calyculin A, and anti-FLAG M2 antibody-conjugated beads were purchased from Sigma-Aldrich. Purified protein kinase CK2, λ protein phosphatase (λPP), and PP1 were purchased from New England Biolabs. Anti-PP1 monoclonal antibodies were purchased from Santa Cruz Biotechnology. Anti-V5 antibody was purchased from Invitrogen.
Cell Culture and Transfection
Human embryonic kidney (HEK) 293A cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. Chinese hamster ovary (CHO) hm1 cells were grown in α-modified Eagle's medium with 5% fetal bovine serum and 500 μg/ml G418 sulfate. HEK293A and CHO hm1 cells were transiently transfected with DNA plasmids using Trans-IT1 reagent (Mirus Bio, Madison, WI). Sympathetic cervical ganglion (SCG) neurons were isolated from 14–19-day-old Sprague-Dawley rats and cultured according to the procedure described previously (18). Acquisition of primary cultures was done under the regulation of the Institutional Animal Care and Use Committee at University of California, Irvine. For SCG neuron transfection, mammalian expression plasmids were microinjected into the nucleus as described (18).
Immunoprecipitation
FLAG-tagged KCNQ2 channel complexes were immunoprecipitated from HEK293A cells that had been transiently transfected with combinations of relevant expression constructs as described (5, 7). Briefly, cells were harvested 30–48 h after transfection and lysed in HSE buffer 150 mm NaCl, 5 mm EDTA, 5 mm EGTA, 20 mm HEPES (pH 7.4), 1% Triton X-100, and Complete protease inhibitor mixture (Roche Applied Science). After centrifugation at 22,000 × g for 15 min, supernatants were incubated with anti-FLAG antibody-conjugated beads. After washing with HSE buffer, bound proteins were eluted in SDS sample buffer and analyzed by SDS-PAGE and immunoblotting.
CaM Purification from HEK293A Cells and in Vitro Dephosphorylation
His6/V5-tagged CaM, CaM-V5, was purified from transiently transfected HEK293A cells using phenyl-Sepharose 6 Fast Flow (GE Healthcare) (19) followed by Ni-NTA beads. Phosphate buffers (8) and phosphatase inhibitors were used to protect phosphorylated CaM. Cells were collected in a buffer containing 50 mm NaF, 1 mm Na3VO4, 1 mm DTT, 0.5 mm EDTA, 50 mm phosphate buffer (pH 7.2) and Complete protease inhibitor mixture. After freeze-thaw followed by Dounce homogenization, lysates were centrifuged at 22,000 × g for 15 min. Supernatant was collected and adjusted to 1 mm CaCl2 and then applied to a phenyl-Sepharose column. The column was washed with buffer A (50 mm phosphate buffer (pH 7.2) and 1 mm CaCl2) followed by buffer B (500 mm NaCl, 1 mm CaCl2, 50 mm phosphate buffer (pH 7.2)) and then with buffer C (0.1 mm CaCl2 and 50 mm phosphate buffer (pH 7.2)). CaM was eluted in buffer D (1 mm EGTA and 50 mm phosphate buffer (pH 7.2)). His6-tagged CaM was further purified by incubating recovered CaM in buffer D with Ni-NTA beads followed by a wash with 50 mm phosphate buffer (pH 7.2) and then with T-HBS (0.2% Tween 20, 150 mm NaCl, 10 mm HEPES (pH 7.4)). For dephosphorylation, His6-CaM-bound beads were briefly washed with λPP buffer (100 mm NaCl, 1 mm MnCl2, 20 mm β-mercaptoethanol, 10 mm HEPES (pH 7.4) and incubated for 30 min at 30 °C with λPP. λPP was removed by washes with T-HBS. For rephosphorylation, beads were further incubated with purified CK2 for 60 min at 30 °C in a CK2 buffer containing 10 mm MgCl2, 50 mm KCl, 10 mm ATP, 0.1 mg/ml polylysine, 50 mm NaF, 1 mm Na3VO4, and 10 mm HEPES (pH 7.4). In some experiments, [γ-32P]ATP was included to monitor phosphorylation of CaM. After a wash with T-HBS, CaM-V5 was eluted from Ni-NTA beads by an elution buffer containing 250 mm imidazole (pH 6.0), 50 mm NaF, 1 mm Na3VO4 and then pH was adjusted to 7.0 by adding Tris. These procedures provided a single band CaM-V5/His6 in Coomassie Brilliant Blue staining of SDS-PAGE (>95% purity).
CaM Purification from Escherichia coli
E. coli BL21 carrying pET-30a(+)/CaM-V5 was sonicated in a lysis buffer (0.2% Tween 20, 150 mm NaCl, 10 mm HEPES (pH 7.4), 10 mm imidazole, 50 mm NaF, 1 mm Na3VO4, and Complete mini protease inhibitor mixture). After centrifugation at 22,000 × g for 15 min, lysates were incubated with Ni-NTA resin, followed by washes with the lysis buffer containing 30 mm imidazole. After a brief wash with T-HBS, CaM-bound Ni-NTA beads were incubated with CK2 and ATP for 60 min at 30 °C in the CK2 buffer described above followed by washes with T-HBS. For dephosphorylation, CK2-treated CaM samples were further treated with λPP and washed with T-HBS. [γ-32P]ATP was included in some experiments to monitor phosphorylation status of CaM. CaM was eluted from Ni-NTA beads with the elution buffer described above. Because of an additional S-tag, CaM generated by E. coli migrates slower (∼30 kDa) in SDS-PAGE than CaM-V5 purified from HEK293A cells (∼23 kDa). These procedures provided CaM-His6/V5 with >95% purity.
In Vitro KCNQ2-CaM Binding Assay
FLAG-tagged full-length KCNQ2 protein was purified from transiently transfected HEK293A cells as described above in the immunoprecipitation section except with an additional calcium wash (1 mm CaCl2) to eliminate KCNQ2-bound CaM as we described previously (5). KCNQ2 beads and purified CaMs were incubated in HSE buffer containing 50 mm NaF and 1 mm Na3VO4 for 1 h at 4 °C. KCNQ2 beads were then washed with HSE, and bound CaM was assessed by immunoblotting.
GST Pulldown Assay
GST-KCNQ2 fusion proteins were purified from E. coli BL21 by glutathione-Sepharose beads (GE Healthcare) according to a standard procedure. After washes with PBS, purified KCNQ2(321–569) beads in HSE buffer were mixed with purified CaM and incubated for 1 h at 4 °C. After washes with HSE buffer, bound CaM was analyzed by SDS-PAGE and immunoblotting.
GST-KCNQ2(1–59) or GST control proteins prebound to glutathione-Sepharose beads were mixed with 100 ng of pure PP1 protein, incubated for 90 min, washed with 0.1% Triton X-100 in phosphate-buffered Ca2+/Mg2+-free saline, and then eluted by heating in SDS sample buffer. HEK293 cell lysates were obtained by washing near confluent 10-cm plates with PBS and lysing in the buffer (1% Triton X-100, 1× Complete protease inhibitor, 50 mm NaF) on ice, followed by incubation on ice with mixing for 30 min. After centrifugation (16,000 × g) for 20 min, supernatant was added to glutathione beads precoated with wild-type and F24A mutant GST-KCNQ2(1–59). After a 3-h incubation with mixing at 4 °C, beads were centrifuged and washed with lysis buffer, and bound proteins were eluted in SDS sample buffer.
Kinase Assay
CK2-specific kinase activities of KCNQ2-FLAG immunoprecipitate from one 10-cm dish/sample were measured using a CK2 substrate peptide (RRRDDDSDDD) (Millipore) and [γ-32P]ATP (PerkinElmer Life Sciences) in a buffer containing 166 μm CK2 substrate peptide, 100 μm ATP, 15 mm MgCl2, 5 mm EGTA, 25 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm DTT, and 20 mm MOPS (pH 7.2). To evaluate the effect of calyculin A on KCNQ2 immunoprecipitate, CK2 activities were measured in 166 μm CK2 substrate peptide, 100 μm ATP, 15 mm MgCl2, 1 mm MnCl2, 20 mm MOPS (pH 7.2), and 500 nm FK506 with or without 500 nm calyculin A. After a 10-min incubation at 30 °C, reactions were terminated by spotting aliquots on Whatman P81 phosphocellulose paper and washing with phosphoric acid. Blanks in the absence of substrate peptide were subtracted from all samples.
Electrophysiology
Patch clamp recordings were performed at room temperature on isolated cells using an Axon multiclamp 700B patch clamp amplifier (Molecular Devices). Data acquisition was performed using pClamp software (version 10; Molecular Devices). Signals were sampled at 2 kHz and low pass-filtered at 1 kHz. Whole cell clamp recordings in CHO hm1 cells were performed as described (7). Briefly, cells were perfused with a solution containing 144 mm NaCl, 5 mm KCl, 2 mm CaCl2, 0.5 mm MgCl2, 10 mm glucose, 10 mm HEPES (pH 7.4). Patch pipettes (2–4 megohms) were filled with intracellular solution containing 135 mm potassium aspartate, 2 mm MgCl2, 3 mm EGTA, 1 mm CaCl2, 4 mm ATP, 0.1 mm GTP, 10 mm HEPES (pH 7.2). Perforated patch clamp in SCG neurons has been described (18). Briefly, SCG neurons were perfused with a solution containing 120 mm NaCl, 6 mm KCl, 1.5 mm MgCl2, 2.5 mm CaCl2, 11 mm glucose, and 10 mm HEPES. Patch pipettes (1–2 megohms) were filled with an intracellular solution containing 130 mm potassium acetate, 15 mm KCl, 3 mm MgCl2, 6 mm NaCl, 10 mm HEPES, and amphotericin B (0.1–0.2 mg/ml). After amphotericin B permeabilization, access resistances ranged from 5 to 20 megohms. Current clamp recordings were used to study afterhyperpolarization. mAHP was evoked by an injection of depolarizing current pulse (100 ms) into a neuron, which was adjusted to evoke three action potentials/pulse. Data analysis and statistical comparisons were performed using Clampfit (Molecular Devices) and Prism (GraphPad, La Jolla, CA). All quantitative data are expressed as mean ± S.E.
RESULTS
Our previous study indicates that the phosphorylation status of KCNQ2 protein regulates CaM binding (7). In this study, we investigated the role of CK2-mediated phosphorylation of CaM on KCNQ2 binding and its functional consequences. Because previous studies indicate that endogenous CaM can be phosphorylated by CK2 (8, 10), we examined whether dephosphorylation of CaM would change KCNQ2 binding properties. To this end, we purified His6/V5-tagged CaM from HEK293A cells by phenyl-Sepharose followed by Ni-NTA resin. Purified CaM was then dephosphorylated by λPP and tested for its binding ability to purified FLAG-tagged KCNQ2 protein in vitro. Dephosphorylation of CaM diminished KCNQ2 binding (Fig. 1A). This reduced KCNQ2 binding was restored by rephosphorylation of CaM by purified CK2 (Fig. 1A).
FIGURE 1.

CK2 phosphorylation of calmodulin facilitates binding of calmodulin to the KCNQ2 channel. A, in vitro binding assay showing that dephosphorylation of CaM purified from HEK293A cells by λPP diminished the ability to bind KCNQ2, which was restored by rephosphorylation by CK2. Coomassie Blue staining of input CaM and its autoradiography are shown to demonstrate that CK2 phosphorylated purified CaM. Bottom histogram shows summary of quantification from independent experiments. Corresponding lane numbers are indicated. IP, immunoprecipitation. B, in vitro binding assay showing that CaM purified from E. coli, which lacks endogenous CK2, showed increased binding after CK2 phosphorylation, which was diminished by dephosphorylation by λPP treatment. Protein staining and 32P uptake into CaM are also shown. C, in vitro binding assay showing GST fusion protein containing KCNQ2(321–569) and E. coli generated CaM with indicated CaM treatment and mutation. CK2 phosphorylation and untreated CaM(3D) mutant showed facilitated KCNQ2 binding compared with untreated wild-type CaM. D, immunoprecipitation of KCNQ2-FLAG co-expressed with aspartate substitutions of CK2 phosphorylation sites of CaM. E, immunoprecipitation of KCNQ2-FLAG co-expressed with alanine substitution of CK2 phosphorylation sites. *, p < 0.05; **, p < 0.01; ***, p < 0.001 by nonparametric ANOVA followed by Dunn's multiple comparisons test. Error bars show S.E.
To confirm the roles of CK2-mediated phosphorylation in CaM-KCNQ2 binding, we performed similar in vitro binding experiments using CaM produced in E. coli, which lacks endogenous CK2 (20). His6/V5-tagged CaM generated in E. coli was purified using Ni-NTA beads followed by CK2 treatment and then tested for its binding to full-length KCNQ2-FLAG protein purified from HEK293A cells. CK2 phosphorylation of CaM increased its binding to KCNQ2 protein, which was abolished by further dephosphorylation of CaM by λPP (Fig. 1B). To confirm that these changes were not intervened by other mammalian proteins co-purified with the KCNQ2 channel complex, we examined CaM binding with E. coli-generated GST fusion protein containing the C-terminal tail of KCNQ2, GST-Q2(321–569). CK2-phosphorylated CaM-His6/V5 purified from E. coli showed increased binding to GST-Q2(321–569) protein (Fig. 1C). These results suggest that CK2-mediated phosphorylation of CaM enhances its binding to KCNQ2 protein.
Three residues in CaM have been identified as physiological CK2 phosphorylation acceptor sites (9, 13, 21): threonine 80, serine 82, and serine 102. We generated a phosphomimetic mutation of CaM in which all three phosphorylation sites were substituted with aspartate, CaM(3D). As expected, CaM(3D) purified from E. coli without any treatment showed enhanced binding to KCNQ2, similar to that of CK2-phosphorylated CaM (Fig. 1C).
To identify the critical residue within these three CK2 phosphorylation sites, we co-expressed single residue aspartate or alanine mutants of CaM with KCNQ2 channel in HEK293A cells and tested their binding by co-immunoprecipitation (Fig. 1D). Any single aspartate mutations increased KCNQ2 binding to an extent similar to that of CaM(3D). This observation is consistent with a previous report that single aspartate mutation facilitates phosphorylation of other sites in mammalian cells (11). However, co-immunoprecipitation assays using alanine-substituted mutants identified Thr-80 as the critical residue (Fig. 1E), which showed reduced binding, similar to that of the three alanine-substituted mutant, CaM(3A). These results suggest that phosphorylation of CaM(Thr-80) is key for CaM binding to KCNQ2.
Because CK2 is constitutively active, some CK2-mediated regulation is known to be controlled through microdomains generated by channel complexes (13). To test whether KCNQ2 channel uses this mechanism, we co-expressed HA-tagged CK2α subunit with KCNQ2-FLAG and examined whether it was co-precipitated with the KCNQ2-FLAG protein. As expected, CK2α protein was detected in KCNQ2 immunoprecipitates (Fig. 2A). In contrast, neither Kv1.2-FLAG nor KCNQ3-FLAG co-immunoprecipitated CK2α (Fig. 2A). Consistent with these results, CK2 kinase activity was detected selectively in KCNQ2 immunoprecipitates (Fig. 2B). This CK2 kinase activity was completely abolished in the presence of a CK2-selective inhibitor, 50 μm TBCA, confirming that the detected kinase activity was derived from CK2 (Fig. 2B). These findings indicate that the KCNQ2 channel complex contains functional CK2.
FIGURE 2.

KCNQ2 channel tethers functional CK2. A, immunoprecipitation (IP) showing selective binding of HA-tagged CK2α to KCNQ2-FLAG, but not to Kv1.2-FLAG or KCNQ3-FLAG. *, p < 0.05 by nonparametric ANOVA followed by Dunn's multiple comparisons test. IB, immunoblotting. Error bars show S.E. B, CK2 assay of immunoprecipitates from Kv1.2-FLAG or KCNQ2-FLAG. TBCA completely eliminated CK2 activity. **, p < 0.01 by Mann-Whitney test.
Because CK2 is constitutively active, dephosphorylation is an important regulatory mechanism for CK2-mediated phosphorylation. Interestingly, we found the consensus PP1 binding R/K-V/I-X-F motif (22) at residues 21–24 (KVGF) located in the intracellular N-terminal region of the KCNQ2 channel (Fig. 3A). This putative KCNQ2 PP1 binding motif is conserved in fish, mouse, and human KCNQ2, consistent with the fact that the KCNQ2 gene arose in a common ancestor of jawed vertebrates (23). However, the motif is absent in all other KCNQ subtypes (Fig. 3A). Pulldown experiments using an N-terminal fragment of KCNQ2 fused to GST, GST-KCNQ2(1–59), showed dose-dependent PP1 binding to GST-KCNQ2(1–59) but not to GST alone (Fig. 3B). This GST-KCNQ2(1–59) binding of PP1 was abolished by F24A mutation within the RVXF motif (Fig. 3C). Consistent with these observations, full-length KCNQ2 protein, but not Kv1.2 protein, co-immunoprecipitated PP1 (Fig. 3D). However, we detected trace amounts of PP1 in KCNQ3-FLAG and KCNQ2(F24A)-FLAG (Fig. 3D), which could be attributed to binding through endogenous AKAP79 (24) that associates with KCNQ channels (18).
FIGURE 3.

KCNQ2 channel tethers functional PP1. A, alignment of N terminus of KCNQ channel showing conserved the KVXF motif in KCNQ2 subtype in vertebrates but not in other KCNQ subtypes. The KVXF motif is shaded. B, in vitro binding assay showing selective PP1 binding to KCNQ2 N terminus in a dose-dependent manner. C, pulldown experiments of GST-KCNQ2 N-terminal fusion proteins, GST-Q2(1–59)(WT) and GST-Q2(1–59)(F24A), showing binding of PP1 and its disruption by F24A mutation. The bottom panel shows protein staining of input proteins. D, immunoprecipitation (IP) of FLAG-tagged channels with PP1. Wild-type KCNQ2-FLAG co-precipitated a significant amount of PP1 compared with that by Kv1.2-FLAG. *, p < 0.05 by nonparametric ANOVA followed by Dunn's multiple comparisons test. IB, immunoblotting. E, CK2 assay with or without a phosphatase-specific inhibitor, calyculin A (CalyA). *, p < 0.05 by Mann-Whitney test. Error bars, S.E.
Because KCNQ2-anchored PP1 is the best candidate to counteract CK2-mediated phosphorylation in the KCNQ2 channel complex, we examined the effects of KCNQ2-anchored PP1 on CK2 activity. To test this, we measured CK2 activity in KCNQ2 immunoprecipitates in a PP1 permissive condition and examined whether a PP1 inhibitor, calyculin A, could increase kinase activity. As expected, CK2 activity was increased to 162.4 ± 25% (n = 4) by adding calyculin A (Fig. 3E). These results indicate that KCNQ2-anchored PP1 counteracts phosphorylation mediated by KCNQ2-anchored CK2.
We next investigated functional roles of CK2-mediated CaM regulation on KCNQ2 channel. Because TBB has been widely used in cellular experiments (14, 16, 17), we tested the effect of TBB on KCNQ2 current. TBB rapidly suppressed KCNQ2 current (Fig. 4, A and B). This suppression was attenuated by calyculin A (Fig. 4, A and B). In addition, overexpression of CaM(3D) attenuated TBB response to a degree similar to that of calyculin A-treated cells (Fig. 4, C and E), which suggests that the TBB effect occurred through phosphorylated CaM. Similar results were obtained with TBCA (Fig. 4, D and E), which supports the idea that this KCNQ2 current suppression is a result of CK2 inhibition rather than off-target effects of these compounds. Concomitantly, immunoprecipitation showed a reduction in KCNQ2-bound CaM in TBB- and TBCA-treated cells (Fig. 4F). Furthermore, KCNQ2-bound CaM(3D) was not affected by TBB or TBCA treatment (Fig. 4G), which supports the idea that these changes are because of the phosphorylation status of CaM. Collectively, these results suggest that CK2 inhibitors promote dephosphorylation of KCNQ2-bound CaM, which destabilizes CaM binding to KCNQ2 subunit and causes current suppression.
FIGURE 4.

Pharmacological inhibition of CK2 reduced KCNQ2 current and CaM-KCNQ2 binding. A and B, current traces (A) and pooled results (B) showing KCNQ2 current suppression by CK2 inhibitor, TBB (10 μm, green), and its attenuation by pretreatment with phosphatase inhibitor, calyculin A (10 nm, blue). KCNQ2 currents were activated from a holding potential of −80 mV by two-step test pulses to −10 mV for 500 ms followed by −60 mV for 300 ms. Amplitudes of KCNQ2 currents were normalized to those at t = 0. The black box indicates the presence of TBB. C, KCNQ2 current traces compared before (black) and after (red) TBB treatment in the presence of CaM(3D). D, KCNQ2 current traces showing TBCA (5 μm) effect on the KCNQ2 currents (blue) and KCNQ2 co-expressed with CaM(3D) (red). E, summary of pooled data showing that treatment with CK2 inhibitor (10 μm TBB, 5 μm TBCA) suppressed KCNQ2 currents (green), and overexpression of CaM(3D) attenuated the effects of CK2 inhibitors (red). Relative KCNQ2 currents at t = 5 min as shown in B were normalized to current recorded at t = 0. *, p < 0.05; **, p < 0.01 by nonparametric ANOVA followed by Dunn's multiple comparisons test. F, co-immunoprecipitation (IP) showing that CK2 inhibitor treatments reduced the binding between KCNQ2 protein and CaM. Quantification from four independent experiments is shown. IB, immunoblotting. G, co-immunoprecipitation showing that overexpression of CaM(3D) maintained KCNQ2 channel binding in the presence of TBB or TBCA. Error bars show S.E.
To further characterize how CK2-phosphorylated CaM regulates KCNQ2 current, we measured current densities of KCNQ2 co-expressed with CaM mutants or KCNQ2(F24A) + CaM(WT) (Fig. 5A). Co-expression of KCNQ2(WT) + CaM(3D) or KCNQ2(F24A) + CaM(WT) showed higher current density (Fig. 5, A and B), which accords well with our CK2 inhibitor study.
FIGURE 5.

CK2 phosphorylation mutations of CaM modified KCNQ2 current and PIP2 susceptibility. A, representative current traces of KCNQ2 currents expressed in CHO cells in the presence of CaM(WT), CaM(3D), or CaM(3A) as well as KCNQ2(F24A) + CaM(WT). At a holding potential of −80 mV, KCNQ2 currents were activated by 500-ms step depolarizations up to +30 mV, followed by −60 mV. B, KCNQ2 current density at −10 mV. ***, p < 0.001 by nonparametric ANOVA followed by Dunn's multiple comparisons test. C, voltage clamp current traces showing Ci-VSP-induced rundown of KCNQ2 current. Step depolarizations (10 s), which activated noninactivating current when KCNQ2 channel was expressed alone (upper traces), showed depolarization-dependent run-down (lower traces) when co-expressed with Ci-VSP. D, average current traces ± S.E. (shaded) of scaled currents showing that the Ci-VSP-induced rundown of the KCNQ2 current was reduced by co-expression of CaM(3D) or in KCNQ2(F24A) channel. Shaded area surrounding each trace shows S.E. The data were collected from 10-s depolarization steps to +30 mV. E, histogram of relative KCNQ2 currents at t = 10 s shown in D. ***, p < 0.001 by nonparametric ANOVA followed by Dunn's multiple comparisons test. Error bars show S.E.
CaM is known to engage in KCNQ2 channel trafficking (6). Therefore, the observed enhanced KCNQ2 currents may be attributed to a change in membrane trafficking of the KCNQ2 subunit. However, our previous studies (5, 7) suggest that increased CaM-KCNQ2 binding enhanced KCNQ2 currents by stabilizing PIP2 affinity of KCNQ2 subunit. Because CK2 inhibitors suppressed KCNQ2 current within 10 s (Fig. 4B), we reasoned that it would be too rapid to explain the current suppression exclusively by KCNQ2 channel trafficking. Thus, we examined whether phospho-CaMs modify susceptibility of the channel to PIP2 depletion to maintain KCNQ2 current. Voltage-sensitive phosphatase (VSP) is a unique phosphatase that degrades PIP2 upon membrane depolarization. It has been widely used to investigate PIP2 sensitivity of ion channels including KCNQ2 channel (5, 25–28). In addition, KCNQ2 is optimal for this assay because KCNQ2 current per se does not inactivate during prolonged depolarization steps (Fig. 5C). Co-expression of VSP from Cinona intestinalis, Ci-VSP, with KCNQ2(WT) channel induced voltage-dependent current decay of KCNQ2 current because of PIP2 depletion as reported previously (5, 29). When we compared KCNQ2 current decays at +30 mV, co-expression of CaM(3D) protected KCNQ2 current from VSP-induced current decay (Fig. 5, D and E). In addition, KCNQ2(F24A) + CaM(WT) current showed slower current decay compared with that of KCNQ2(WT) (Fig. 5, D and E). These results suggest that KCNQ2 channel with CK2-phosphorylated CaM is more resistant to PIP2 depletion.
This resistance to PIP2 depletion observed in KCNQ2 currents co-expressed with phosphomutants of CaM or KCNQ2(F24A) (Fig. 5E) were closely correlated with their current densities (r2 = 0.902, p = 0.036) (Fig. 5B), as well as relative CaM binding to KCNQ2 channel (r2 = 0.829, p = 0.042) (Fig. 1, D and E). Such intercorrelation among KCNQ2 current density, CaM-KCNQ2 binding, and sensitivity to PIP2 depletion is consistent with our previous observations that KCNQ2-CaM association facilitates PIP2 efficacy for maintaining functional KCNQ2 channels (7).
It has been shown that CK2 phosphorylation changes calcium sensitivity of the SK2 channel (13). To evaluate the effect of phosphorylated CaM on calcium sensitivity of KCNQ2 current, we co-expressed CaM(3D) with KCNQ2 channel and measured calcium responses induced by 1 μm ionomycin as we described previously (5). Co-expression of CaM(3D) did not alter ionomycin induced responses (Fig. 6).
FIGURE 6.
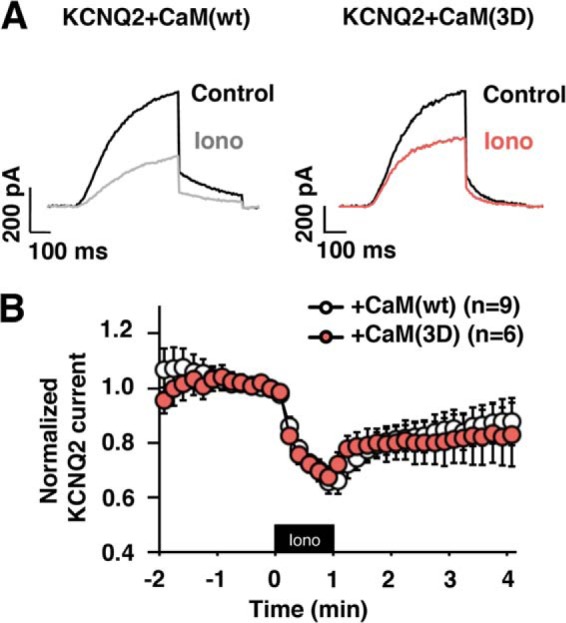
Ionomycin-induced KCNQ2 current suppression is not affected by co-expression of CaM(3D). A, representative current traces of ionomycin responses of KCNQ2 channel co-expressed with CaM(WT) or CaM(3D). B, pooled results showing equivalent ionomycin responses for KCNQ2 channel co-expressed either with CaM(WT) or CaM(3D). Black box indicates presence of 1 μm ionomycin. Error bars show S.E.
Finally, we evaluated the role of CK2 on M-current regulation in rat SCG sympathetic neurons. Application of TBB onto SCG neurons reduced the M-current (Fig. 7, A and B). Similarly to what we observed in CHO cells, suppression of the M-current was attenuated by calyculin A pretreatment. When CaM(3D) was overexpressed, SCG neurons showed attenuated M-current response to TBB as we observed in CHO cells (Fig. 7C). Because the M-current contributes to the mAHP, we examined how CK2 inhibition affected mAHP (Fig. 7, D–F). The mAHP was partially suppressed by apamin, indicating the contribution of SK channels as expected. TBB reduced apamin-resistant mAHP. Subsequent application of XE991, an M-current blocker, completely eliminated residual mAHP, indicating that TBB-induced reduction of mAHP was exclusively generated by XE991-sensitive current (Fig. 7, D–F). These results suggest that CK2-mediated phosphorylation of CaM regulates mAHP in SCG neurons.
FIGURE 7.

CK2 mediates modulation of the M-current and mAHP in SCG neurons. A, current traces of perforated patch clamp recordings from rat SCG neurons showing that the M-current was partially inhibited by 10 μm TBB and completely inhibited by a combined application of TBB and 10 μm XE991. B, pooled results showing M-current inhibition by TBB and XE991. Responses from untreated control (open symbols) or pretreatment with 10 nm calyculin A (blue symbols) are shown. The gray bar and the black bar indicate the presence of TBB and XE991, respectively. C, summary histogram of current at t = 5 in C showing that calyculin A attenuated TBB-induced suppression. Overexpression of CaM(3D) induced similar attenuated response to TBB (red). *, p < 0.05 by Mann-Whitney test. D, current clamp recordings showing the mAHP in SCG neurons before (black) and after (red) application of 100 nm apamin, subsequent addition of 10 μm TBB (blue), and 10 μm XE991 (green). The resting membrane potential of the cell was kept at −60 mV. mAHP was induced by 100-ms current injection. E, magnified views of the dotted gray box shown in D (top) and dissected components of mAHP in the traces shown. F, summary of incremental decrease of peak mAHP under indicated pharmacological treatments showing that TBB suppressed XE991-sensitive mAHP. Error bars show S.E.
DISCUSSION
The present study demonstrates that CK2-mediated phosphorylation of CaM facilitated its binding to KCNQ2 and enhanced channel activities. We also demonstrate that CK2 and PP1 were anchored to the KCNQ2 subunit, providing tonic regulation for protein phosphorylation. Accordingly, application of CK2 inhibitors decreased KCNQ2-bound CaM and suppressed KCNQ2 current. Consistent with our hypothesis that KCNQ2-CaM interaction regulates PIP2 sensitivity of KCNQ2 channel (5, 7), CK2 inhibitor decreased the amount of KCNQ2-anchored CaM and increased channel susceptibility to PIP2 depletion. Thus, CK2-mediated phosphorylation of CaM is a novel mechanism that regulates PIP2 susceptibility of the M-channel.
CK2-mediated CaM phosphorylation has been shown to modify CaM-dependent processes with variable effects depending on the enzymes affected (10–12). For instance, phosphodiesterase exhibits lower binding affinity to phosphorylated CaM without affecting its Vmax (10). In contrast, the endothelial NO synthetase shows a reduced Vmax in the presence of phosphorylated CaM without changing its affinity for phosphorylated CaM (11). Regarding the effect of CaM phosphorylation on ion channels, detailed analysis has been described for the SK2 small conductance calcium-activated potassium channels (13, 16, 17). CK2 phosphomimetic CaM induces rightward shift of calcium-dependent activation curve for SK2 channel, which leads to a reduced SK2 channel activity (13). This change in calcium sensitivity by phosphomimetic CaM makes a clear contrast to the KCNQ2 channel, in which co-expression of phosphomimetic CaM does not change calcium sensitivity. This difference is interesting because phosphorylation of CaM(Thr-80) plays a key role for both channels (13). Because CaM(Thr-80) locates outside the EF hand motifs (10) and phosphorylation of Thr-80 per se does not change calcium sensitivity of CaM (11), the observed difference may be derived from a channel-specific role for CaM. In fact, the SK2 channel is activated by calcium-bound CaM, whereas calcium-free CaM has been identified as the preferred binding form for the KCNQ2 channel (1, 5, 30). Because CK2-phosphorylated CaM modulates KCNQ2 current independent of cellular calcium, CaM can be considered to be a phosphorylation sensor for the KCNQ2 channel.
CK2 is enriched in the axon initial segment, where KCNQ2 subunits are clustered (31, 32). Thus, subcellular compartments could be a potential mechanism for the facilitation of CK2 activity surrounding the M-channel. However, we found that KCNQ2 subunit selectively tethers CK2 and PP1. These anchored enzymes were active in KCNQ2 immunoprecipitates. Thus, it is likely that the balance between anchored CK2 and PP1 tonically regulates the phosphorylation status of CaM in the vicinity of KCNQ2 subunits. Similarly, SK2 channels anchor CK2 and protein phosphatase 2B (13). Thus, co-existence of CK2 and protein phosphatase in target protein complexes is a fundamental mechanism to regulate CK2-mediated phosphorylation. However, it has been shown that norepinephrine facilitates phosphorylation of SK2-anchored CaM but not KCNQ2-anchored CaM (17), indicating distinct regulations of CaM phosphorylation at these channels.
We identified the KVXF motif that is responsible for anchoring PP1 to the KCNQ2 subunit. However, a KCNQ2-associated protein, AKAP79/150, also binds PP1 (24). Interestingly, AKAP79/150 interaction has been demonstrated to suppress PP1 activity (24), whereas PP1 bound to the KVXF motif maintains its phosphatase activity (33). These different modes of PP1 association should provide additional regulation for PP1 activity at the KCNQ channel complex, substrates of which may not be limited to CaM.
Functionally, application of CK2 inhibitors suppressed KCNQ2 current as well as the M-current, which led to a reduction in mAHP. These effects of CK2 inhibitors were negated by a pretreatment with PP1 inhibitors as expected. Furthermore, a PP1-deficient mutant, KCNQ2(F24A) or overexpression of CaM(3D) with KCNQ2 channel were resistant to CK2 inhibitors. These pieces of evidence suggest that CK2 inhibitors modulate KCNQ2 and M-currents through changes in the phosphorylation status of CaM.
In conclusion, we demonstrated that CK2-mediated phosphorylation of CaM regulates the M-current by facilitating its binding to KCNQ2, hence modulating mAHP. This pathway may present a novel important regulatory mechanism to control neuronal excitability.
Acknowledgments
We thank Tingching Kao for excellent technical assistance and Yahsushi Okamura for Ci-VSP cDNA (Integrative Physiology, Osaka University, Osaka, Japan).
This work was supported, in whole or in part, by National Institutes of Health Grants R01 NS067288 (to N. H.) and R01 NS49119 (to E. C. C.).
- CaM
- calmodulin
- ANOVA
- analysis of variance
- Ci-VSP
- VSP from Cinona intestinalis
- CK2
- casein kinase 2
- mAHP
- medium afterhyperpolarization
- Ni-NTA
- nickel-nitrilotriacetic acid
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- λPP
- λ protein phosphatase
- PP1
- protein phosphatase 1
- SCG
- sympathetic cervical ganglion
- TBB
- tetrabromobenzotriazole
- TBCA
- tetrabromocinnamic acid
- VSP
- voltage-sensitive phosphatase.
REFERENCES
- 1. Yus-Najera E., Santana-Castro I., Villarroel A. (2002) The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 277, 28545–28553 [DOI] [PubMed] [Google Scholar]
- 2. Shahidullah M., Santarelli L. C., Wen H., Levitan I. B. (2005) Expression of a calmodulin-binding KCNQ2 potassium channel fragment modulates neuronal M-current and membrane excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16454–16459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Delmas P., Brown D. A. (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862 [DOI] [PubMed] [Google Scholar]
- 4. Gamper N., Shapiro M. S. (2003) Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J. Gen. Physiol. 122, 17–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kosenko A., Hoshi N. (2013) A change in configuration of the calmodulin-KCNQ channel complex underlies Ca2+-dependent modulation of KCNQ channel activity. PLoS One 8, e82290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Etxeberria A., Aivar P., Rodriguez-Alfaro J. A., Alaimo A., Villacé P., Gómez-Posada J. C., Areso P., Villarroel A. (2008) Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB J. 22, 1135–1143 [DOI] [PubMed] [Google Scholar]
- 7. Kosenko A., Kang S., Smith I. M., Greene D. L., Langeberg L. K., Scott J. D., Hoshi N. (2012) Coordinated signal integration at the M-type potassium channel upon muscarinic stimulation. EMBO J. 31, 3147–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plancke Y. D., Lazarides E. (1983) Evidence for a phosphorylated form of calmodulin in chicken brain and muscle. Mol. Cell. Biol. 3, 1412–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quadroni M., James P., Carafoli E. (1994) Isolation of phosphorylated calmodulin from rat liver and identification of the in vivo phosphorylation sites. J. Biol. Chem. 269, 16116–16122 [PubMed] [Google Scholar]
- 10. Benaim G., Villalobo A. (2002) Phosphorylation of calmodulin: functional implications. Eur. J. Biochem. 269, 3619–3631 [DOI] [PubMed] [Google Scholar]
- 11. Greif D. M., Sacks D. B., Michel T. (2004) Calmodulin phosphorylation and modulation of endothelial nitric oxide synthase catalysis. Proc. Natl. Acad. Sci. U.S.A. 101, 1165–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quadroni M., L'Hostis E. L., Corti C., Myagkikh I., Durussel I., Cox J., James P., Carafoli E. (1998) Phosphorylation of calmodulin alters its potency as an activator of target enzymes. Biochemistry 37, 6523–6532 [DOI] [PubMed] [Google Scholar]
- 13. Bildl W., Strassmaier T., Thurm H., Andersen J., Eble S., Oliver D., Knipper M., Mann M., Schulte U., Adelman J. P., Fakler B. (2004) Protein kinase CK2 is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron 43, 847–858 [DOI] [PubMed] [Google Scholar]
- 14. Litchfield D. W. (2003) Protein kinase CK2: structure, regulation, and role in cellular decisions of life and death. Biochem. J. 369, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meggio F., Pinna L. A. (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–368 [DOI] [PubMed] [Google Scholar]
- 16. Buchanan K. A., Petrovic M. M., Chamberlain S. E., Marrion N. V., Mellor J. R. (2010) Facilitation of long-term potentiation by muscarinic M1 receptors is mediated by inhibition of SK channels. Neuron 68, 948–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maingret F., Coste B., Hao J., Giamarchi A., Allen D., Crest M., Litchfield D. W., Adelman J. P., Delmas P. (2008) Neurotransmitter modulation of small-conductance Ca2+-activated K+ channels by regulation of Ca2+ gating. Neuron 59, 439–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoshi N., Zhang J. S., Omaki M., Takeuchi T., Yokoyama S., Wanaverbecq N., Langeberg L. K., Yoneda Y., Scott J. D., Brown D. A., Higashida H. (2003) AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 6, 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gopalakrishna R., Anderson W. B. (1982) Ca2+-induced hydrophobic site on calmodulin: application for purification of calmodulin by phenyl-Sepharose affinity chromatography. Biochem. Biophys. Res. Commun. 104, 830–836 [DOI] [PubMed] [Google Scholar]
- 20. Olsten M. E., Litchfield D. W. (2004) Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem. Cell Biol. 82, 681–693 [DOI] [PubMed] [Google Scholar]
- 21. Arrigoni G., Marin O., Pagano M. A., Settimo L., Paolin B., Meggio F., Pinna L. A. (2004) Phosphorylation of calmodulin fragments by protein kinase CK2: mechanistic aspects and structural consequences. Biochemistry 43, 12788–12798 [DOI] [PubMed] [Google Scholar]
- 22. Egloff M. P., Johnson D. F., Moorhead G., Cohen P. T., Cohen P., Barford D. (1997) Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 16, 1876–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hill A. S., Nishino A., Nakajo K., Zhang G., Fineman J. R., Selzer M. E., Okamura Y., Cooper E. C. (2008) Ion channel clustering at the axon initial segment and node of Ranvier evolved sequentially in early chordates. PLoS Genet. 4, e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Le A. V., Tavalin S. J., Dodge-Kafka K. L. (2011) Identification of AKAP79 as a protein phosphatase 1 catalytic binding protein. Biochemistry 50, 5279–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murata Y., Okamura Y. (2007) Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J. Physiol. 583, 875–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Villalba-Galea C. A., Miceli F., Taglialatela M., Bezanilla F. (2009) Coupling between the voltage-sensing and phosphatase domains of Ci-VSP. J. Gen. Physiol. 134, 5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Falkenburger B. H., Jensen J. B., Hille B. (2010) Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 135, 99–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suh B. C., Kim D. I., Falkenburger B. H., Hille B. (2012) Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 109, 3161–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okamura Y., Murata Y., Iwasaki H. (2009) Voltage-sensing phosphatase: actions and potentials. J. Physiol. 587, 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alaimo A., Alberdi A., Gomis-Perez C., Fernández-Orth J., Bernardo-Seisdedos G., Malo C., Millet O., Areso P., Villarroel A. (2014) Pivoting between calmodulin lobes triggered by calcium in the Kv7.2/calmodulin complex. PLoS One 9, e86711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bréchet A., Fache M. P., Brachet A., Ferracci G., Baude A., Irondelle M., Pereira S., Leterrier C., Dargent B. (2008) Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J. Cell Biol. 183, 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pan Z., Kao T., Horvath Z., Lemos J., Sul J. Y., Cranstoun S. D., Bennett V., Scherer S. S., Cooper E. C. (2006) A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cohen P. T. (2002) Protein phosphatase 1: targeted in many directions. J. Cell Sci. 115, 241–256 [DOI] [PubMed] [Google Scholar]