

Background: Certain hereditary spastic paraplegia (HSP)-related proteins possess hairpin domains and regulate the morphology of the endoplasmic reticulum (ER) network.
Results: Protrudin possesses a hairpin domain and interacts with HSP-related proteins.
Conclusion: Protrudin regulates ER morphology and function.
Significance: Mutant protrudin produced in certain individuals with HSP is prone to form microaggregates that induce ER stress.
Keywords: Endoplasmic Reticulum Stress, Endoplasmic Reticulum (ER), Membrane Structure, Neurodegenerative Diseases, Proteomics
Abstract
Protrudin is a membrane protein that regulates polarized vesicular trafficking in neurons. The protrudin gene (ZFYVE27) is mutated in a subset of individuals with hereditary spastic paraplegia (HSP), and protrudin is therefore also referred to as spastic paraplegia (SPG) 33. We have now generated mice that express a transgene for dual epitope-tagged protrudin under control of a neuron-specific promoter, and we have subjected highly purified protrudin-containing complexes isolated from the brain of these mice to proteomics analysis to identify proteins that associate with protrudin. Protrudin was found to interact with other HSP-related proteins including myelin proteolipid protein 1 (SPG2), atlastin-1 (SPG3A), REEP1 (SPG31), REEP5 (similar to REEP1), Kif5A (SPG10), Kif5B, Kif5C, and reticulon 1, 3, and 4 (similar to reticulon 2, SPG12). Membrane topology analysis indicated that one of three hydrophobic segments of protrudin forms a hydrophobic hairpin domain similar to those of other SPG proteins. Protrudin was found to localize predominantly to the tubular endoplasmic reticulum (ER), and forced expression of protrudin promoted the formation and stabilization of the tubular ER network. The protrudin(G191V) mutant, which has been identified in a subset of HSP patients, manifested an increased intracellular stability, and cells expressing this mutant showed an increased susceptibility to ER stress. Our results thus suggest that protrudin contributes to the regulation of ER morphology and function, and that its deregulation by mutation is a causative defect in HSP.
Introduction
The endoplasmic reticulum (ER)3 is a continuous membrane system that comprises the nuclear envelope, ribosome-studded peripheral sheets, and an interconnected network of smooth tubules that extend throughout the cell. The ER is a multifunctional organelle that plays a key role in the synthesis, modification, quality control, and trafficking of integral membrane and secreted proteins (1, 2). It also contributes to Ca2+ sequestration and release, cellular signaling, sterol synthesis, as well as lipid synthesis and distribution. In neurons, the ER participates in the massive polarized membrane expansion that occurs during axon and dendrite formation as well as serves as an intracellular Ca2+ store that is integrated into pre- and postsynaptic signaling pathways (3).
Hereditary spastic paraplegia (HSP) is an inherited neurological disorder characterized by spastic weakness of the lower extremities (4–6). More than half of HSP cases result from autosomal dominant mutations in the genes for atlastin-1 (also known as spastic paraplegia 3A, or SPG3A), receptor expression enhancing protein (REEP) 1 (SPG31), and spastin (SPG4). Atlastin-1, spastin, and REEP1 interact with each other in the tubular ER membrane of neurons to coordinate ER morphogenesis and microtubule dynamics (7). These three proteins as well as reticulon 2 (SPG12) possess hydrophobic hairpin domains that shape high-curvature ER tubules and mediate intramembrane protein interactions (8). Members of the atlastin/RHD3/Sey1 family of dynamin-related GTPases mediate the formation of three-way junctions that characterize the tubular ER network (9–11), and additional classes of hydrophobic hairpin-containing ER proteins interact with and remodel the microtubule cytoskeleton. Indeed, overexpression or depletion of these various proteins affects ER morphology (10, 12, 13), and abnormal ER morphology is thought to be a major causative defect in HSP pathogenesis.
Protrudin (ZFYVE27 or SPG33) was also identified as a protein whose gene is mutated in a subset of HSP patients (14). Protrudin contains a Rab11 binding domain and a FYVE domain, and promotes neurite formation through activation of polarized vesicular trafficking (15). Protrudin also contains a short sequence motif designated FFAT (two phenylalanines in an acidic tract) between the Rab11 binding domain and the FYVE domain, and this motif mediates the interaction of protrudin with vesicle-associated membrane protein-associated protein (VAP) (16) as well as with Kif5A (SPG10), a motor protein that mediates anterograde vesicular transport in neurons. VAP is an important determinant of the subcellular localization of protrudin at the ER. Protrudin facilitates the interaction of Kif5 with Rab11, VAP family proteins, Surf4, and reticulon proteins, suggesting that it serves as an adaptor protein and that the protrudin-Kif5 complex contributes to the transport of these proteins in neurons (17). Given that mutations in the genes for protrudin, Kif5A, and reticulons give rise to HSP, protrudin-containing complexes appear to be fundamental to neuronal function associated with HSP pathogenesis.
To identify proteins that engage in physiologically relevant interactions with protrudin in the central nervous system, we generated mice that express a protrudin transgene under control of a neuron-specific prion promoter. We isolated protrudin complexes from the brain of these animals and identified component proteins by proteomics analysis. We now show that protrudin is associated with major HSP-related proteins including myelin proteolipid protein 1 (PLP1, SPG2); reticulon 1, 3, and 4; atlastin-1; REEP1 and REEP5; and Kif5A, -B, and -C. Protrudin was found to colocalize with atlastin-1, REEP1, and REEP5 at the tubular ER network, and overexpression of protrudin promoted the formation and stabilization of this network. Topological analysis revealed that protrudin possesses a hydrophobic hairpin domain similar to those of other HSP-related ER-shaping proteins. A mutant (G191V) of protrudin that is associated with HSP in a subset of patients was found to have an increased intracellular stability and to increase the sensitivity of cells to ER stress. Our results thus suggest that protrudin contributes to formation of the ER network and that altered protrudin function contributes to the pathology of HSP.
EXPERIMENTAL PROCEDURES
Generation of Protrudin Transgenic Mice
Animals were handled in accordance with the guidelines of Kyushu University. A cDNA for mouse protrudin with the His6 and FLAG epitopes at its NH2 terminus was subcloned into the pPrPpE1/E2,3sal plasmid, which contains a prion promoter. The resulting vector was linearized and injected into fertilized mouse eggs of the (C57BL/6NCrSlc × DBA/2CrSlc)F1 (BDF1) background. Primary genotyping was performed by PCR and Southern blot analysis and was followed by immunoblot analysis with antibodies to (anti-) FLAG. WT littermates of the transgenic mice were studied as controls.
Construction of Expression Plasmids
Construction of vectors encoding human protrudin (15) and VAP-A(ΔTM) (16), as well as mouse protrudin (17), was described previously. Mouse cDNAs encoding atlastin-1, REEP5, REEP1, and VAP-A were generated by PCR with PrimeSTAR HS DNA polymerase (Takara, Shiga, Japan) from cDNA prepared from Neuro2A cells. Human cDNAs encoding Sec61β and Climp63 were generated by PCR from cDNA prepared from HeLa cells. The cDNAs encoding mouse atlastin-1, REEP5, REEP1, and VAP-A were subcloned into pEFBOS-HHg (kindly provided by S. Nagata, Kyoto University, Japan, and H. Sumimoto, Kyushu University, Japan). Complementary DNAs encoding deletion mutants of human protrudin were generated by PCR and subcloned into p3xFLAG-CMV-7.1 (Sigma), and those encoding deletion mutants of mouse protrudin tagged with the HA and Myc epitopes at their NH2 and COOH termini, respectively, were generated by PCR and subcloned into the pEFBOS vector. The cDNA encoding human Sec61β was subcloned into pEFBOS-HHg or ptdTomato (Clontech, Palo Alto, CA), and that encoding human Climp63 was subcloned into pEGFP (Clontech). The cDNAs encoding WT or G191V mutant forms of human protrudin tagged with the HA epitope were subcloned into pMX-puro (kindly provided by T. Kitamura, Tokyo University, Japan). The pcDNA3-NHK-FLAG vector was kindly provided by H. Ichijo, Tokyo University, Japan.
Antibodies
Antibodies to protrudin and FKBP38 were generated as described previously (15, 18). Antibodies to GM130 and heat shock protein 90 (HSP90) were from BD Biosciences; FLAG (mouse monoclonal M2 and rabbit polyclonal) and the Myc epitope (9E10) were from Sigma; HA epitope (HA.11) were from Covance (Princeton, NJ); α-tubulin (TU-01) from Zymed Laboratories Inc. (South San Francisco, CA); calreticulin from Thermo Scientific (Rockford, IL); Climp63 (clone G1/296) from Enzo Life Science (Farmingdale, NY); calnexin (N and C epitopes) from Stressgen (Victoria, British Columbia, Canada); protrudin from Proteintech (Chicago, IL); and HA epitope (Y-11) and cyclin D1-(72–13G) as well as normal mouse IgG (sc-2025) from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa Fluor 488- or Alexa Fluor 546-conjugated goat antibodies to mouse or rabbit IgG were obtained from Molecular Probes (Eugene, OR).
Cell Culture, Transfection, and Retroviral Infection
Neuro2A, HEK293T, HeLa, COS-7, and Plat-E cells were cultured under a humidified atmosphere of 5% CO2 at 37 °C in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen). The culture medium for Plat-E cells was also supplemented with blasticidin (10 μg/ml). Cells were transfected with the use of the FuGENE HD (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) reagents. For retroviral infection, Plat-E cells were transiently transfected with pMX-puro-based vectors and then cultured for 48 h. The retroviruses in the resulting culture supernatants were used to infect Neuro2A cells, and the cells were then subjected to selection with puromycin (1 μg/ml). The infected cells were incubated with tunicamycin (Sigma), thapsigargin (Sigma), or DTT (Sigma) to induce the unfolded protein response (UPR), and exposed to cycloheximide (Sigma) to inhibit protein synthesis or MG132 (Sigma) to inhibit proteasome activity. Nocodazole was obtained from Sigma.
Isolation of Protrudin Complexes by Dual Affinity Purification
The brains of protrudin transgenic mice were disrupted with a Potter homogenizer, the homogenate was centrifuged at 1000 × g for 5 min at 4 °C to remove nuclei and nondisrupted cells, and the resulting supernatant was centrifuged at 100,000 × g for 30 min at 4 °C to isolate a membrane pellet. The pellet was solubilized with lysis buffer (40 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 10% glycerol, 0.5% Triton X-100, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF). The protein concentration of the resulting supernatant was determined with the Bradford assay (Bio-Rad), and this soluble membrane fraction was then incubated with rotation for 60 min at 4 °C with anti-FLAG (M2)-agarose affinity gel (Sigma). The beads were washed three times with lysis buffer, after which protein complexes were eluted by incubation for several minutes at 4 °C with lysis buffer containing the FLAG peptide (Sigma). For the second affinity purification step, nickel-nitrilotriacetic acid (Ni-NTA)-agarose (ProBond resin, Invitrogen) was added to the eluate, and the mixture was incubated with rotation for 90 min at 4 °C. The beads were washed three times with lysis buffer, and protein complexes were eluted by incubation for several minutes at 4 °C with lysis buffer containing 300 mm imidazole.
Identification of Protrudin-associated Proteins by LC-MS/MS Analysis
LC-MS/MS analysis was performed as described previously (17). Proteins reproducibly detected in both of two independent experiments with brain extract from protrudin transgenic mice, but not with that from nontransgenic mice, were considered protrudin-associated proteins (17). Subcellular categorization was based on gene ontology annotation with the Mouse Genome Informatics (MGI) GO Term Finder. Genes responsible for HSP or other neurodegenerative diseases were based on Online Mendelian Inheritance in Man (OMIM) data. Semiquantitative estimation of protein abundance was based on identification frequency (19), which was normalized by the sum of the identification frequencies for all identified proteins in each experiment and then multiplied by 100 (17).
Immunoprecipitation and Immunoblot Analysis
HEK293T cells cultured for 1 day after transfection were lysed by incubation for 10 min at 4 °C with a lysis buffer (40 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 10% glycerol, 0.5% CHAPS, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF). The lysates were centrifuged at 20,400 × g for 10 min at 4 °C, and equal amounts of protein from the resulting supernatants were subjected to immunoblot analysis directly or to immunoprecipitation for 30 min at 4 °C with anti-FLAG and protein G-Sepharose 4 Fast Flow (Amersham Biosciences). The immunoprecipitates were washed three times with lysis buffer and then subjected to immunoblot analysis as described previously (15). The blot images were scanned with a LAS-4000 instrument (GE Healthcare), and the intensity of immunoblot bands was quantified with the use of ImageJ software.
Immunostaining and Image Analysis
HeLa or COS-7 cells were fixed for 10 min at room temperature with 4% paraformaldehyde in PBS before consecutive incubation with primary antibodies and Alexa Fluor 488- or Alexa Fluor 546-labeled goat secondary antibodies in PBS containing 1% Triton X-100. They were also stained with Hoechst 33258 (Wako, Tokyo, Japan) in some experiments. Cells were covered with a drop of GEL/MOUNT (Biomeda, Hayward, CA) and examined with a fluorescence microscope (Olympus BX51) or a laser-scanning confocal microscope (LSM510; Carl Zeiss, Oberkochen, Germany). The PSC colocalization plug-in for ImageJ was used to quantify Pearson's correlation coefficient for signals in two channels in the selected region. The default threshold value of 40 for 8-bit images, which was above the background fluorescence, was used for quantification in both channels. The density of three-way junctions of ER was quantified on the basis of the number of empty spaces surrounded by ER in a selected square area of 200 μm2. The empty spaces were counted manually.
Subcellular Fractionation
The brains of WT or protrudin-deficient mice were homogenized in a solution containing 20 mm HEPES-NaOH (pH 7.4), 0.32 m sucrose, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 10 μm MG132, 1 mm PMSF, and 1 mm EDTA. The homogenate was centrifuged at 500 × g for 5 min at 4 °C to remove debris, and the resulting supernatant was then centrifuged at 100,000 × g for 1 h at 4 °C to obtain cytosolic and membrane fractions.
Sodium Carbonate Extraction
HEK293T cells transfected with a vector for HA-protrudin-Myc were cultured for 1 day, washed with PBS, and homogenized in an alkaline solution (0.25 M sucrose, 0.1 m sodium carbonate (pH 11.4), 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 10 μm MG132, 1 mm PMSF). The homogenate was centrifuged at 100,000 × g for 1 h at 4 °C, and the resulting pellet was resuspended in a lysis buffer (40 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 10% glycerol, 0.5% Triton X-100, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF).
Protease Protection Assay
HEK293T cells transfected with WT or mutant forms of HA-protrudin-Myc were cultured for 1 day, washed with PBS, and homogenized in a solution identical to that used for subcellular fractionation with the exception that the sucrose concentration was 0.25 m. The homogenate was centrifuged at 500 × g for 5 min at 4 °C to remove debris, and the resulting supernatant was centrifuged at 100,000 × g for 1 h at 4 °C. The new pellet was suspended in 0.25 m sucrose buffer and incubated for 20 min on ice with proteinase K (40 μg/ml, Roche Applied Science) in the absence or presence of 1% Triton X-100. Proteolysis was terminated by the addition of PMSF to a final concentration of 1 mm.
Chemical Modification with mPEG
HeLa cells expressing mutant forms of protrudin tagged at the NH2 terminus with the HA epitope were harvested by exposure to trypsin and then washed twice with DMEM and once with ice-cold HCN buffer (50 mm HEPES-NaOH (pH 7.5), 2 mm CaCl2, 150 mm NaCl). They were then incubated for 20 min at 4 °C in HCN buffer containing 0.04% digitonin (to permeabilize the plasma membrane) or 0.1% Triton X-100 (to permeabilize all cell membranes), washed twice with ice-cold HNE buffer (50 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 1 mm EGTA), and transferred to a new tube. They were then incubated first for 10 min at 37 °C in HNE buffer and then for 30 min at 30 °C with 1.5 mm methoxypolyethylene glycol maleimide (mPEG, Sigma) in 20 mm Tris-HCl (pH 7.5). The reaction was terminated by the addition of DTT to a final concentration of 10 mm and incubation for 10 min at 4 °C. The cells were lysed by incubation for 20 min at 4 °C with lysis buffer (50 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 2 mm CaCl2, 1% Triton X-100, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF), after which the lysates were centrifuged at 20,400 × g for 10 min at 4 °C and the resulting supernatant was subjected to immunoblot analysis.
RNAi
Stealth siRNAs designed for human protrudin (ZFYVE27, 5′-AGGAUGCAGGUGAUGGUGUUCGAUA-3′ and 5′-AAGAAGAGGCGGAGCUGCAGUAAUU-3′; numbers 1 and 2, respectively) or negative control duplexes (Invitrogen) were introduced into HeLa cells by transfection with Lipofectamine RNAiMax (Invitrogen). For rescue experiments, plasmids were introduced into the cells by transfection 2 days after the siRNAs.
RT and Real-time PCR Analysis
Total RNA was isolated from Neuro2A cells and purified with the use of an RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Portions of the RNA were subjected to RT with the use of a QuantiTect RT kit (Qiagen), and the resulting cDNA was subjected to real-time PCR analysis with a StepOne real-time PCR system (Applied Biosystems, Foster City, CA) and SYBR Premix Ex Taq (Takara). The amplification protocol comprised the initial incubation at 60 °C for 30 s and 95 °C for 3 s, followed by 40 cycles of denaturation, annealing, and extension. Data were analyzed according to the 2−ΔΔCT method and were normalized relative to the amount of GAPDH mRNA. PCR was performed with primers (forward and reverse, respectively) for mouse Gapdh (5′-CATGGCCTTCCGTGTTCCTA-3′ and 5′-GCGGCACGTCAGATCCA-3′) and mouse Bip (5′-TCTCACTAAAATGAAGGAGA-3′ and 5′-TTGTCGCTGGGCATCATTGA-3′).
Luciferase Assay
Neuro2A cells were transfected with pGL3-GRP78P(−132)-luc (kindly provided by K. Mori, Kyoto University) as an ER stress response element reporter together with pRL-TK (Promega, Madison, WI). The cells were collected 24 h after transfection, lysed, and assayed for luciferase activity with a dual-luciferase reporter assay system (Promega).
XBP1 mRNA Splicing Assay
Total RNA was isolated from Neuro2A cells and purified with the use of ISOGEN (Nippon Gene, Tokyo, Japan). Portions of the RNA were subjected to RT with the use of a QuantiTect RT kit (Qiagen), and the resulting cDNA was subjected to PCR with primers (forward and reverse, respectively) for mouse Hprt (hypoxanthine-guanine phosphoribosyltransferase) (Hprt, 5′-GCCTAAGATGAGCGCAAGTTG-3′ and 5′-TACTAGGCAGATGGCCACAGG-3′) and mouse X-box binding protein 1 (Xbp1, 5′-ACACGCTTGGGAATGGACAC-3′ and 5′-CCATGGGAAGATGTTCTGGG-3′) followed by agarose gel electrophoresis and staining with ethidium bromide. The gels were scanned with a LAS-4000 instrument, and band intensity was quantified with the use of ImageJ software.
Gel Filtration Chromatography
Gel filtration chromatography was performed with an ÄKTAexplorer 10S system (GE Healthcare) fitted with a Superose 6 10/300 GL column (GE Healthcare). Cells were lysed by incubation for 10 min at 4 °C with a lysis buffer (50 mm sodium phosphate (pH 7.5), 150 mm NaCl, 10% glycerol, 1% CHAPS, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF). The lysates were centrifuged at 20,400 × g for 10 min at 4 °C, and equal amounts of protein from the resulting supernatants were injected into the column equilibrated with running buffer (50 mm sodium phosphate (pH 7.5), 150 mm NaCl, 10% glycerol, 1% CHAPS). Elution was performed at a flow rate of 0.4 ml/min, and 0.48 ml fractions were collected.
Statistical Analysis
Where indicated, quantitative data are presented as mean ± S.D. and analyzed by Student's t test or by one-way analysis of variance followed by Tukey's test. A p value of <0.05 was considered statistically significant.
RESULTS
Identification of Protrudin-interacting Proteins in Mouse Brain
To provide further insight into the physiological role of protrudin, we generated transgenic mice that express a form of protrudin tagged with both His6 and FLAG epitopes under control of a neuron-specific prion promoter (Fig. 1A). Protrudin-containing complexes were purified from the brain of these transgenic mice by tandem affinity chromatography with anti-FLAG and Ni-NTA resin (which binds to the His6 epitope). Proteins in the final eluate were digested with trypsin, and the generated peptides were subjected to LC-MS/MS. We identified 141 proteins that were present in the complexes isolated from the transgenic mice but were not recovered from control mice (Fig. 1A). Assignment of the protrudin-associated proteins to subcellular compartments on the basis of gene ontology annotation with the use of the Mouse Genome Informatics (MGI) GO Term Finder revealed that more than half of these proteins were localized at the plasma membrane or ER (Fig. 1B, Table 1), consistent with the notion that protrudin plays an important role in the endomembrane system. Given that protrudin is implicated in the pathogenesis of HSP, protrudin-associated proteins were categorized on the basis of OMIM annotation. A substantial proportion (11.1%) of the proteins implicated in HSP pathology were found in protrudin complexes, whereas <5% of those related to other neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), Charcot-Marie-Tooth disease, Parkinsons disease, and spinocerebellar ataxia, were detected in these complexes (Fig. 1C, Table 2). All protrudin-associated proteins directly implicated in or potentially related to HSP were subjected to semiquantitative analysis on the basis of the normalized identification frequency in each LC-MS/MS experiment (Fig. 1D). PLP1 and several reticulon family members ranked high in the list, and altastin-1, REEP5, and Kif5 isoforms were also identified. In addition, REEP1 was identified as a protein that interacts with protrudin by proteomics analysis of immunoprecipitates prepared from the brain of C57BL/6 mice with anti-protrudin (data not shown; see also Fig. 2, C and F). These results suggested that protrudin forms complexes with many HSP-related proteins.
FIGURE 1.

Identification of protrudin-associated proteins with a proteomics approach. A, schematic workflow for identification of protrudin-associated proteins by proteomics analysis. An extract prepared from the brain of protrudin transgenic mice was subjected to dual affinity purification with anti-FLAG and Ni-NTA-agarose. The isolated protrudin complexes were subjected to SDS-PAGE, slices of the resulting gel were exposed to trypsin, and the generated peptides were analyzed by LC-MS/MS. The mass and partial amino acid sequence data were simultaneously compared with protein and nucleotide sequence databases for protein identification. B, subcellular localization of protrudin-associated proteins identified by LC-MS/MS analysis. C, proportion of proteins encoded by genes responsible for the indicated diseases in OMIM that were identified in protrudin complexes. D, proteins directly implicated in or potentially related to HSP identified in A. The amount of each protein was estimated semiquantitatively on the basis of the normalized identification frequency (IF). The proteins were ranked according to the average of the scores from two independent experiments (Exp. 1 and Exp. 2).
TABLE 1.
Protein symbols of protrudin-associated proteins categorized in terms of subcellular localization
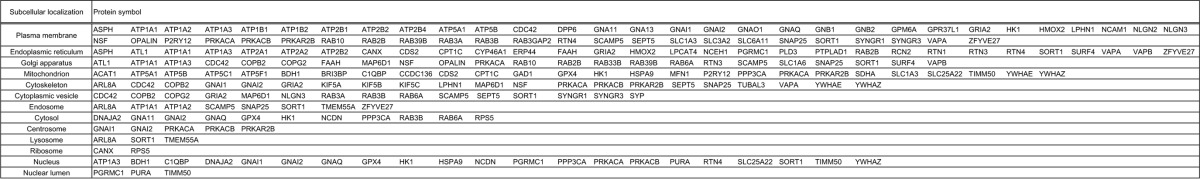
TABLE 2.
Responsible genes for HSP, ALS, Charcot-Marie-Tooth disease (CMT), Parkinson disease (PD), and spinocerebellar ataxia (SCA)
Gene symbols of protrudin-associated proteins identified by proteomics analysis were represented in bold.
| HSP | ALS | CMT | PD | SCA |
|---|---|---|---|---|
| AP4B1 | ALS2 | AARS | ADH1C | AFG3L2 |
| AP4E1 | ANG | AIFM1 | ATP13A2 | ATXN1 |
| AP4M1 | ATXN2 | ARHGEF10 | EIF4G1 | ATXN10 |
| AP4S1 | C9ORF72 | BSCL2 | FBXO7 | ATXN2 |
| AP5Z1 | CHMP2B | DNM2 | GBA | ATXN3 |
| ATL1 | DAO | DYNC1H1 | GIGYF2 | ATXN7 |
| BSCL2 | DCTN1 | EGR2 | HTRA2 | CACNA1A |
| C12orf65 | FIG4 | FGD4 | LRRK2 | FGF14 |
| CYP2U1 | FUS | FIG4 | MAPT | ITPR1 |
| CYP7B1 | NEFH | GARS | PARK2 | KCNC3 |
| DDHD1 | OPTN | GDAP1 | PARK7 | KCND3 |
| DDHD2 | PRPH | GJB1 | PINK1 | KLHL1 |
| ERLIN2 | SETX | HK1 | PLA2G6 | NOP56 |
| FA2H | SIGMAR1 | HSPB1 | SNCA | PDYN |
| GJC2 | SOD1 | HSPB8 | TBP | PPP2R2B |
| HSPD1 | SPG11 | KARS | UCHL1 | PRKCG |
| KIAA0196 | TAF15 | KIF1B | VPS35 | SPTBN2 |
| KIF1A | TARDBP | LITAF | TBP | |
| KIF5A | UBQLN2 | LMNA | TGM6 | |
| L1CAM | VAPB | LRSAM1 | TK2 | |
| NIPA1 | VCP | MED25 | TTBK2 | |
| PGN | MFN2 | |||
| PLP1 | MPZ | |||
| PNPLA6 | MTMR2 | |||
| REEP1 | NAMSD | |||
| RTN2 | NDRG1 | |||
| SLC16A2 | NEFL | |||
| SLC33A1 | PMP22 | |||
| SPAST | PRPS1 | |||
| SPG11 | PRX | |||
| SPG20 | RAB7A | |||
| SPG21 | SBF2 | |||
| TECPR2 | SH3TC2 | |||
| VPS37A | TRPV4 | |||
| ZFYVE26 | YARS | |||
| ZFYVE27 |
FIGURE 2.

Protrudin interacts with atlastin-1 and REEP family members. A–C, extracts of HEK293T cells transiently transfected with expression vectors for FLAG-tagged protrudin and HA epitope-tagged forms of atlastin-1, REEP5, or REEP1 were subjected to immunoprecipitation (IP) with anti-FLAG. VAP-A and VAP-A(ΔTM) were studied as positive and negative controls, respectively, for interaction with protrudin. The resulting precipitates, as well as a portion (1% of the input for immunoprecipitation) of the cell extracts, were subjected to immunoblot (IB) analysis with anti-HA, anti-FLAG, and anti-HSP90 (loading control). D–F, HeLa cells expressing FLAG-tagged protrudin and HA epitope-tagged forms of atlastin-1, REEP5, or REEP1 were fixed and processed for immunofluorescence analysis with anti-FLAG (red) and anti-HA (green). Merged images in which nuclei are stained with Hoechst 33258 (blue) are also shown. The boxed areas in the upper panels are shown at higher magnification in the lower panels. Scale bars, 50 μm. G, domain organization of human protrudin and structure of deletion mutants thereof. RBD, Rab binding domain; HP1 to HP3, hydrophobic domains; CC, coiled-coil domain. H and I, extracts of HEK293T cells expressing full-length (FL) protrudin or its mutants shown in G (fused at their NH2 termini to the FLAG tag) together with HA-atlastin-1 (H) or HA-REEP5 (I) were subjected to immunoprecipitation with anti-HA, and the resulting precipitates, as well as a portion (1% of the input for immunoprecipitation) of the cell extracts, were subjected to immunoblot analysis with anti-FLAG, anti-HA, and anti-HSP90.
Protrudin Associates with Atlastin-1, REEP5, and REEP1 at the ER Network
To confirm the interaction of protrudin with atlastin-1, REEP5, and REEP1, we performed co-immunoprecipitation analysis. Lysates of HEK293T cells transfected with expression vectors for both FLAG-tagged mouse protrudin and HA epitope-tagged forms of mouse atlastin-1, REEP5, REEP1, VAP-A (positive control), or a VAP-A mutant (ΔTM) lacking the transmembrane domain (negative control) were subjected to immunoprecipitation with anti-FLAG. The resulting precipitates were then subjected to immunoblot analysis with anti-HA and anti-FLAG. Atlastin-1 (Fig. 2A), REEP5 (Fig. 2B), and REEP1 (Fig. 2C) were detected in the FLAG-protrudin immunoprecipitates. We next examined the subcellular localization of these proteins in HeLa cells. FLAG-tagged protrudin was colocalized with HA-tagged atlastin-1 (Fig. 2D), HA-REEP5 (Fig. 2E), and HA-REEP1 (Fig. 2F) at tubular and reticular structures, suggesting that protrudin forms complexes with atlastin-1, REEP5, and REEP1 in the tubular ER network.
We next investigated which region of protrudin is required for binding to atlastin-1 or REEP5 by generating FLAG-tagged deletion mutants of protrudin (Fig. 2G) and examining their ability to associate with HA-tagged atlastin-1 (Fig. 2H) or HA-REEP5 (Fig. 2I) in a co-immunoprecipitation assay with HEK293T cells. Whereas full-length protrudin and a mutant that included the NH2-terminal region of protrudin (amino acids 1 to 206) interacted with atlastin-1 and REEP5, a mutant that lacked this region failed to do so, suggesting that the NH2-terminal half of protrudin including three putative hydrophobic domains is necessary and sufficient for the interaction of protrudin with either atlastin-1 or REEP5.
Protrudin Is an Integral Membrane Protein
In silico analysis with SMART (Simple Modular Architecture Research Tool) suggested that protrudin contains three hydrophobic or transmembrane domains. Cytosolic and membrane fractions prepared from the brain of WT or protrudin knock-out mice were subjected to immunoblot analysis with anti-protrudin. Protrudin was detected in the membrane fraction but not in the cytosolic fraction of WT mice (Fig. 3A). We next investigated whether protrudin is substantially integrated in or simply attached to cellular membranes. HEK293T cells were thus transfected with an expression vector encoding mouse protrudin tagged with the HA epitope at its NH2 terminus and with the Myc epitope at its COOH terminus (HA-protrudin-Myc), and cell lysates were subjected to alkaline extraction with sodium carbonate. Immunoblot analysis revealed that protrudin was resistant to such extraction, as was the integral membrane protein calnexin, whereas the membrane-attached protein GM130 was extracted into the supernatant (Fig. 3B), suggesting that protrudin is an authentic integral membrane protein.
FIGURE 3.
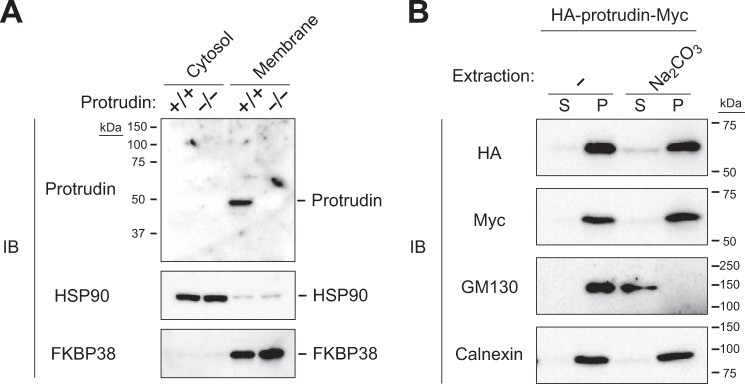
Protrudin is an integral membrane protein. A, cytosolic and membrane fractions were isolated from the brain of WT (+/+) or protrudin-deficient (−/−) mice, and equal amounts of protein from each fraction were subjected to immunoblot analysis with anti-protrudin, anti-HSP90, and anti-FKBP38. HSP90 and FKBP38 were examined as controls for cytosolic and membrane proteins, respectively. B, homogenates of HEK293T cells expressing protrudin tagged at its NH2 and COOH termini with HA and Myc epitopes, respectively, were subjected to extraction with or without Na2CO3 followed by centrifugation to isolate supernatant (S) and pellet (P) fractions. Equal amounts of protein from each fraction were subjected to immunoblot analysis with anti-HA, anti-Myc, anti-GM130, and anti-calnexin. GM130 and calnexin were examined as controls for peripheral and integral membrane proteins, respectively.
Protrudin Contains a Hydrophobic Hairpin Domain
Other HSP-related proteins found to associate with protrudin, including reticulon family members, atlastin-1, REEP5, and REEP1, contain hydrophobic hairpin domains. Given that these domains affect ER morphology by shaping the lipid bilayer into high-curvature tubules, we investigated whether protrudin also contains such sequences. We examined the membrane topology of protrudin with two approaches. First, we subjected a membrane fraction prepared from cells expressing HA-protrudin-Myc to protease treatment. As a control, we confirmed that the NH2 terminus of calnexin, which faces the luminal side of the ER, was protected from proteolytic attack, whereas the COOH terminus of this protein, which is exposed to the cytosolic side, was cleaved (Fig. 4, A and B). In contrast, both the NH2 and COOH termini of HA-protrudin-Myc were completely processed by treatment with proteinase K (Fig. 4B), suggesting that both termini of the protein are exposed to the cytosol. These results thus raised the question of whether the three hydrophobic regions (HP1 to HP3) present in protrudin span the entire membrane or are buried within the phospholipid bilayer. To determine the membrane topology around the hydrophobic regions of protrudin, we subjected a deletion mutant of HA-protrudin-Myc to the same analysis. The region between HP2 and HP3 (examined with the mutant 1–188) was cleaved by the protease, suggesting that this region is exposed to the cytosol (Fig. 4C).
FIGURE 4.

Analysis of the membrane topology of protrudin. A, schematic representation of calnexin as well as full-length (FL) and mutant forms of mouse protrudin. The transmembrane (TM) domain of calnexin as well as the hydrophobic regions (HP1 to HP3) of protrudin are indicated. NH2- or COOH-terminal epitopes recognized by anti-calnexin (calnexin-N and calnexin-C, respectively) are denoted by black bars. Full-length and mutant forms of protrudin were tagged at their NH2 and COOH termini with HA and Myc epitopes, respectively, as indicated. B, microsomes prepared from HEK293T cells expressing full-length HA-protrudin-Myc were incubated in the absence or presence of proteinase K and Triton X-100 and then subjected to immunoblot analysis with anti-HA, anti-Myc, anti-calnexin-N, or anti-calnexin-C, as indicated. C, microsomes prepared from HEK293T cells expressing HA-protrudin-(1–188)-Myc were analyzed as in B. D, schematic representation of the topology of HA epitope-tagged protrudin mutants. E, HeLa cells were incubated with digitonin to permeabilize the plasma membrane or with Triton X-100 to permeabilize all cell membranes. They were then treated with or without mPEG and subjected to immunoblot analysis with anti-calnexin. F, cysteine residues of protrudin-(1–228) were mutated to alanine, or sequences GGCGG or GGECEGG were inserted before residue 88, as indicated in D. HeLa cells expressing the various HA epitope-tagged protrudin mutants were then analyzed as in E with the exception that immunoblot analysis was performed with anti-HA and anti-calnexin.
As a second approach, we performed a PEGylation assay, which is based on the premise that cysteine residues in the cytosolic region of an ER protein, but not those in the luminal region or within the phospholipid bilayer, are accessible to mPEG. Cysteine residues are present at positions 72, 77, 113, 169, 172, 199, and 207 in the region of mouse protrudin spanning HP1 to HP3 (Fig. 4D). Calnexin, which contains one cysteine residue in the cytosolic portion and five cysteines in the luminal portion of the protein, showed one and three band shifts after PEGylation in the absence or presence of ER membrane permeabilization, respectively (Fig. 4E). To identify the accessible cysteine residues around the hydrophobic regions of protrudin, we generated a series of mutant proteins by inserting a cysteine-containing sequence (GGCGG or GGECEGG) between residues 87 and 88 in the region between HP1 and HP2, or by replacing the cysteines at positions 113, 169, and 172 in the region between HP2 and HP3 with alanine, in combination with replacement of the cysteine at position 25 (Fig. 4, A and D). The PEGylation analysis revealed that the region between HP1 and HP2 was not accessible to mPEG, whereas that between HP2 and HP3 was accessible (Fig. 4F), suggesting that the region between HP1 and HP2 faces the ER lumen, although this sequence is short and might be present to a variable extent within the membrane. Although we are unable to exclude the possibility that the overexpressed tagged proteins do not necessarily reflect the topology of endogenous protrudin, the results of our two approaches (protease protection assay and mPEG modification assay) together suggested that HP1 and HP2 domains span the membrane fully, with the loop between HP1 and HP2 residing in the ER lumen, and that the HP3 domain folds into a hairpin.
Forced Expression of Protrudin Promotes ER Network Formation
Given that our results indicated that protrudin contains a hydrophobic hairpin domain and that many proteins that possess such domains are localized to the tubular ER network, we examined in more detail whether protrudin might also reside in tubular ER as opposed to sheetlike ER. To this end, we compared the intracellular localization of protrudin with those of Climp63 and REEP5, which largely reside in sheetlike and tubular ER, respectively (20). Immunofluorescence analysis revealed that FLAG-tagged protrudin did not colocalize with enhanced green fluorescent protein (EGFP)-tagged Climp63 in COS-7 cells, whereas the distribution of FLAG-protrudin was almost identical to that of HA-REEP5 (Fig. 5, A–C). We also examined the potential colocalization of FLAG-protrudin with endogenous Climp63 (Fig. 5D) and calreticulin (a marker for all of the ER) (Fig. 5E). The extent of colocalization between FLAG-protrudin and endogenous Climp63 was markedly lower than that between FLAG-protrudin and endogenous calreticulin (Fig. 5F). These results suggested that protrudin is predominantly localized to the tubular ER.
FIGURE 5.

Forced expression of protrudin promotes ER network formation. A, COS-7 cells expressing EGFP-tagged Climp63 and FLAG-tagged human protrudin were fixed and processed for immunofluorescence analysis with anti-FLAG (red) and confocal microscopy. The fluorescence of EGFP was monitored directly. Merged images are also shown. The boxed regions of the upper panels are shown at higher magnification in the lower panels. Scale bar, 10 μm. B, COS-7 cells expressing HA epitope-tagged REEP5 and FLAG-tagged human protrudin were fixed and processed for confocal immunofluorescence analysis with anti-HA (green) and anti-FLAG (red). Scale bar, 50 μm. C, percentage colocalization of FLAG-protrudin with EGFP-Climp63 or HA-REEP5 as determined in A and B and as measured with Pearson's correlation coefficient in a square area of 2000 μm2. Data are mean ± S.D. from four or five cells. D, COS-7 cells expressing FLAG-tagged human protrudin were fixed and processed for confocal immunofluorescence analysis with anti-Climp63 (green) and anti-FLAG (red). Scale bar, 10 μm. E, COS-7 cells transfected with an expression vector for FLAG-tagged human protrudin (or with the corresponding empty vector, Mock) were fixed and processed for confocal immunofluorescence analysis with anti-calreticulin (green) and anti-FLAG (red). Scale bars, 10 μm. F, percentage colocalization of FLAG-protrudin with Climp63 or calreticulin as determined in D and E and as measured with Pearson's correlation coefficient in a square area of 2000 μm2. Data are mean ± S.D. for three cells. G, density of three-way junctions of the ER for cells examined as in E. Data are mean ± S.D. for five cells. A.U., arbitrary units. *, p < 0.05 (Student's t test). H, HeLa cells transfected with protrudin or control siRNAs were subjected to immunoblot analysis with anti-protrudin and anti-HSP90. The arrowhead and asterisk indicate specific and nonspecific bands, respectively. I, HeLa cells transfected with protrudin or control siRNAs were subsequently transfected with an expression vector for FLAG-tagged human protrudin (or with the corresponding empty vector, Mock) before fixation and processing for confocal immunofluorescence analysis with anti-calreticulin (green) and anti-FLAG (red). Left panels are higher magnification views of the boxed areas. Scale bar, 10 μm. J, density of three-way junctions of the ER for cells examined as in I. Data are mean ± S.D. for 7 to 10 cells. *, p < 0.05 (one-way analysis of variance followed by Tukey's test). K, COS-7 cells transfected with expression vectors for tdTomato-tagged Sec61β and FLAG-tagged human protrudin (or with the corresponding empty vector, Mock) were treated (or not) with 100 μm nocodazole for 60 min to induce microtubule depolymerization. The cells were then fixed and processed for immunofluorescence analysis with anti-α-tubulin (green). The fluorescence of tdTomato was monitored directly. Merged images are also shown. The boxed areas in the upper panels are shown at higher magnification in the lower panels. Scale bars, 50 μm. The density of three-way junctions of the ER was also measured. Data are mean ± S.D. for three to seven cells. *, p < 0.05 (Student's t test).
Given that we found that protrudin interacts with atlastin-1, a dynamin-like GTPase that promotes ER network formation through homotypic membrane fusion of ER tubules (9, 10, 21, 22), we investigated whether protrudin also promotes ER network formation. The reticular mesh of the ER network (as revealed by immunofluorescence analysis of calreticulin) became finer and more complex, as reflected by an increase in the density of three-way junctions especially at the cell periphery, as a result of overexpression of protrudin (Fig. 5, E and G). These results indicated that protrudin may promote the fusion of ER tubules and ER network formation. We also found that depletion of protrudin by RNAi in HeLa cells (Fig. 5H) rendered the sheetlike structure of the ER more prominent (Fig. 5, I and J). Expression of FLAG-protrudin in such protrudin-depleted HeLa cells resulted in reversion of the morphology of the ER to a pattern similar to that observed in control cells, suggesting that protrudin indeed contributes to regulation of the sheet versus tubule structure of this organelle.
Given that formation of the ER network is dependent on microtubules, we investigated the effects of nocodazole, which induces microtubule depolymerization, on the ER network in cells transfected with a vector for FLAG-protrudin or with the corresponding empty vector. Whereas the ER network (as revealed by the fluorescence of a tdTomato-tagged form of the ER protein Sec61β) disappeared in response to nocodazole treatment in control cells, it was resistant to this agent in those overexpressing protrudin (Fig. 5K). These results thus suggested that protrudin contributes to both the formation and stabilization of the tubular ER network.
Cells Expressing Mutant Protrudin Are Susceptible to ER Stress
We next examined whether protrudin(G191V), which has been identified in a subset of HSP patients, also interacts with atlastin-1, REEP5, and REEP1. The interaction between protrudin and each of these three proteins was not affected by the G191V mutation (Fig. 6, A–C). We also examined the localization of FLAG-protrudin(G191V) in COS-7 cells by immunofluorescence analysis. The distribution of the mutant protein did not differ substantially from that of WT protrudin (Fig. 6D), suggesting that the pathogenesis of HSP associated with this mutation of protrudin is not attributable to a change in the subcellular localization of the protein. Aggregate formation by the mutant protein was also not apparent, at least at the level of resolution achieved with the light microscope used for immunofluorescence analysis. Furthermore, forced expression of protrudin(G191V) induced a change in ER morphology similar to that induced by overexpression of the WT protein in COS-7 cells, and it resulted in stabilization of the ER network in a manner similar to that apparent with WT protrudin in cells treated with nocodazole (Fig. 6E). We therefore conclude that the HSP-associated G191V mutation of protrudin affects neither the localization of the protein nor its function in the regulation of ER morphology and stability.
FIGURE 6.

An HSP-associated mutant of protrudin associates with atlastin-1 and REEP family members. A–C, extracts of HEK293T cells transiently transfected with expression vectors for FLAG-tagged WT or G191V mutant forms of human protrudin as well as for HA epitope-tagged forms of atlastin-1, REEP5, or REEP1 were subjected to immunoprecipitation with anti-FLAG. VAP-A(ΔTM) was studied as a negative control for interaction with protrudin. The resulting precipitates, as well as a portion (1% of the input for immunoprecipitation) of the cell extracts, were subjected to immunoblot analysis with anti-HA, anti-FLAG, and anti-HSP90 (loading control). D, COS-7 cells expressing FLAG-tagged WT or G191V mutant forms of protrudin were fixed and processed for confocal immunofluorescence analysis with anti-calreticulin (green) and anti-FLAG (red). The boxed regions of the upper panels are shown at higher magnification in the lower panels. Scale bars, 10 μm. The percentage colocalization of FLAG-protrudin(WT) or FLAG-protrudin(G191V) with calreticulin was measured with Pearson's correlation coefficient in a square area of 2000 μm2. Data are mean ± S.D. for four cells. E, COS-7 cells expressing tdTomato-tagged Sec61β and either FLAG-tagged protrudin(WT) or FLAG-protrudin(G191V) were treated (or not) with 100 μm nocodazole for 60 min. The cells were then fixed and processed for immunofluorescence analysis with anti-α-tubulin (green). The fluorescence of tdTomato was monitored directly. Scale bars, 50 μm. The density of three-way junctions of the ER was also measured. Data are mean ± S.D. for four or five cells.
Impairment of ER function may result in abnormal accumulation of unfolded or misfolded proteins, a condition referred to as ER stress. Mutations in some SPG genes, such as SPG17 (23), have been shown to result in misfolding and aggregation of the encoded proteins, which triggers ER stress and may play a role in HSP pathogenesis. We therefore examined the effect of protrudin mutation on the ER stress response. Forced expression of WT human protrudin did not affect the increase in the amount of mRNA for the ER stress-inducible gene Bip (also known as Grp78) elicited by exposure of Neuro2A cells to ER stress-inducing agents such as tunicamycin, thapsigargin, and DTT. However, expression of the HSP-associated protrudin(G191V) mutant enhanced the Bip gene response to all three agents (Fig. 7, A–C). We also examined whether cells expressing protrudin(G191V) manifest ER stress with the use of an ER stress response element reporter assay and by monitoring the splicing of Xbp1 mRNA. Neuro2A cells infected with retroviruses encoding WT or G191V mutant forms of protrudin were transfected with a luciferase reporter vector under the control of the BIP/GRP78 gene promoter. Expression of protrudin(G191V), but not that of the WT protein, resulted in a moderate but significant increase in luciferase activity compared with that of control cells (Fig. 7D), suggestive of the presence of ER stress triggered by expression of the mutant protein even in the absence of an ER stress-inducing agent. We also examined the splicing of Xbp1 mRNA in cells expressing WT or mutant protrudin in the absence or presence of tunicamycin treatment. Whereas the spliced form of Xbp1 mRNA was essentially undetectable in cells not exposed to tunicamycin, this drug increased the amount of the spliced mRNA to a greater extent in cells expressing the G191V mutant than in those expressing WT protrudin (Fig. 7E). On the basis of these results, we speculate that the relatively modest effect of protrudin(G191V) on ER stress might accumulate over long periods of time (decades) before the onset of symptoms in individuals with HSP, similar to the long time courses for the development of other human neurodegenerative diseases.
FIGURE 7.

Expression of an HSP-associated mutant of protrudin induces ER stress. A–C, RT and real-time PCR analysis of BiP mRNA in Neuro2A cells infected with retroviruses encoding WT or G191V mutant forms of human protrudin and exposed to 5 μg/ml of tunicamycin (A), 1 μm thapsigargin (B), or 5 mm DTT (C) for 8 h. Data are expressed relative to the corresponding normalized value for cells infected with the empty retrovirus (Mock) and exposed to vehicle, and are mean ± S.D. from four to six independent experiments. *, p < 0.05 (one-way analysis of variance followed by Tukey's test). D, activity of an ER stress response element reporter plasmid relative to that of the reference plasmid pRL-TK in Neuro2A cells expressing WT or G191V mutant forms of protrudin. Data are mean ± S.D. from six independent experiments. *, p < 0.05 (one-way analysis of variance followed by Tukey's test). E, Neuro2A cells infected with retroviruses for HA epitope-tagged WT or G191V mutant forms of protrudin were incubated in the absence or presence of tunicamycin (2 μg/ml) for 7 h and then subjected to RT-PCR analysis of XBP1 mRNA and hypoxanthine-guanine phosphoribosyltransferase (HPRT) mRNA (loading control). Open and filled arrowheads indicate bands corresponding to unspliced (uXBP1) and spliced (sXBP1) forms of XBP1 mRNA, respectively. The asterisk indicates a nonspecific band. The sXBP1/uXBP1 band intensity ratio was measured. F, Neuro2A cells infected with retroviruses for HA epitope-tagged WT or G191V mutant forms of protrudin were incubated with cycloheximide (CHX, 10 μg/ml) for the indicated times, lysed, and subjected to immunoblot analysis with anti-HA, anti-cyclin D1 (positive control), and anti-HSP90 (loading control). The intensity of the HA-protrudin and cyclin D1 bands was measured. Data are mean ± S.D. for four independent experiments. G, Neuro2A cells infected with retroviruses for HA epitope-tagged WT or G191V mutant forms of protrudin were transfected for 48 h with a vector for FLAG-tagged NHK, incubated with cycloheximide (10 μg/ml) for the indicated times, lysed, and subjected to immunoblot analysis with anti-FLAG, anti-HA, anti-cyclin D1, and anti-HSP90. The intensity of the NHK-FLAG and cyclin D1 bands was measured. H, Neuro2A cells infected with retroviruses for HA epitope-tagged WT or G191V mutant forms of protrudin were incubated with cycloheximide (10 μg/ml) in the absence or presence of MG132 (10 μm) for the indicated times, lysed, and subjected to immunoblot analysis with anti-HA, anti-cyclin D1, and anti-HSP90. I, Neuro2A cells infected with retroviruses for HA epitope-tagged forms of WT or G191V mutant forms of protrudin were exposed to 1 μm thapsigargin for 8 h, lysed, and subjected to gel filtration chromatography with a running buffer containing 1% CHAPS. The resulting fractions were subjected to immunoblot analysis with anti-HA and anti-calnexin (negative control), and the intensity of the HA-protrudin and calnexin bands was measured. Similar results were obtained in two independent experiments.
Some SPG proteins have been functionally linked to ER-associated degradation (ERAD), a multistep pathway encompassing the degradation of ER proteins by the ubiquitin-proteasome system. We therefore examined the relationship of ERAD to the mutation of protrudin. The half-life of protrudin(G191V) in Neuro2A cells was markedly longer than that of protrudin(WT) (Fig. 7F), suggesting that the HSP-associated mutation of protrudin may result in a defect in the ERAD system. Consistent with this observation, the degradation of NHK, a typical substrate of the ERAD system, was delayed in cells expressing protrudin(G191V) compared with that in cells expressing the WT protein (Fig. 7G). Furthermore, exposure of cells to the proteasome inhibitor MG132 resulted in partial attenuation of the degradation of WT protrudin to an extent similar to that apparent for cyclin D1 (a soluble proteasome substrate), whereas MG132 almost completely blocked the degradation of protrudin(G191V) (Fig. 7H), suggesting that degradation of the mutant protein is highly sensitive to proteasome inhibition. The subcellular localization of the WT and G191V mutant forms of protrudin was not affected by MG132 treatment (data not shown). Given that excessive accumulation of unfolded protein in the ER leads to the UPR or ER overload response, the protrudin(G191V) mutant might be misfolded in the ER, leading to a defect in the ERAD system that enhances the ER stress response. Consistent with this notion, gel filtration analysis revealed that the apparent molecular size of protrudin(G191V) in Neuro2A cells was larger than that of the WT protein (Fig. 7I), suggesting that the mutant protein was part of a larger complex. Collectively, these results indicated that mutant protrudin produced in certain individuals with HSP may be prone to aggregation and tend to increase ER stress, which may account for the pathogenesis of HSP.
DISCUSSION
Protrudin is categorized as an HSP-associated protein (SPG33). The relationship between protrudin mutation and HSP pathogenesis has remained largely unclear, however, mainly because (i) the number of HSP patients harboring protrudin mutations is much smaller than that of those with mutations in other SPG genes, such as those for atlastin-1 (SPG3A), spastin (SPG4), and REEP1 (SPG31); (ii) the molecular function of protrudin has not been fully elucidated; and (iii) the mechanism by which the mutation of protrudin affects cell function associated with HSP etiology has been unknown. We recently applied a proteomics approach to Neuro2A cells and found that protrudin associates with Kif5A (SPG10), -B, and -C, and that this interaction is required for the function of protrudin in neurite extension (17). In the present study, we sought to identify molecules that interact with protrudin in mouse brain, a more physiologically relevant system than Neuro2A cells. With this and other approaches, we have now obtained several lines of evidence for a causative relationship between mutation of protrudin and HSP.
First, we found that protrudin interacts with other HSP-related proteins including PLP1 (SPG2), atlastin-1 (SPG3A), REEP1 (SPG31), REEP5 (similar to REEP1), Kif5A (SPG10), Kif5B, Kif5C, and reticulon 1, 3, and 4 (similar to reticulon 2, SPG12). Although we did not detect spastin (SPG4) in the protrudin complexes isolated from mouse brain, others have demonstrated an interaction between protrudin and spastin (14, 24, 25).
Second, many of the HSP-related proteins found to interact with protrudin are thought to contribute to regulation of the morphology of the ER, a heterogeneous organelle with distinct morphologies of sheets and an interconnected network of tubules that share a common lumen. These proteins generate membrane curvature through scaffolding and hydrophobic insertion mechanisms and thereby shape the lipid bilayer of the ER into tubules, resulting in the formation of the tubular ER network (11). The HSP-related proteins possess long hydrophobic stretches of amino acids that form intramembrane hairpin domains and are thought to partially span the lipid bilayer, inducing or stabilizing the high curvature of ER tubules via hydrophobic wedging (12, 26). Depletion of atlastin-1 in cultured cortical neurons was found to inhibit axon elongation (27), and proper ER morphology is thought to be essential for maintenance of long cellular processes such as axons (3). Defects in tubular ER shaping and in interactions of the ER network with the microtubule cytoskeleton thus appear to be largely responsible for the pathogenesis of HSP. Mutation of seipin (SPG17) has been shown to result in misfolding of the protein, aggregate formation, and ER stress, with these events likely playing a role in HSP pathogenesis (23, 28). Furthermore, the SPG18 protein Erlin2 has been functionally linked to ERAD (29). We have now shown that protrudin is localized predominantly to the tubular ER, and that expression of protrudin promotes formation of the tubular ER network.
Third, expression of the G191V mutant of protrudin rendered cells more vulnerable to ER stress, probably as a result of abnormal stability of the mutant protein. Glycine 191 is positioned in the hydrophobic hairpin domain of protrudin, and topology prediction in silico with the use of SOSUI software suggested that this mutation might result in a conformational change in the three-dimensional structure of protrudin, particularly in that of the FYVE domain, leading to misfolding of the COOH-terminal region (14). These results suggest that the G191V mutant of protrudin may be misfolded in the ER and therefore might stimulate the UPR.
We therefore conclude that protrudin shares common characteristics with other members of the SPG family: it interacts with other SPG proteins, harbors a hydrophobic hairpin domain, and regulates ER morphology. These characteristics suggest that, like other SPG proteins, protrudin contributes to ER network formation by regulating membrane curvature, and that mutation of protrudin is a causative defect in HSP. It is also possible that protrudin functions as a tethering factor through its FYVE domain, which is a lipid binding domain. EEA1, a typical FYVE-domain protein localized to early endosomes, possesses a structure similar to that of protrudin and serves as a tethering factor during fusion of early endosomes. Protrudin might thus function in cooperation with atlastin-1 to tether ER membranes for fusion during the formation of three-way junctions.
It remains unclear how protrudin links ER morphology and neuronal function. We previously showed that protrudin binds to Kif5 through its FFAT motif and coiled-coil domain, and that it serves as an adaptor protein to link the motor protein Kif5 and its cargo molecules including Rab11, VAP family members, and Surf4 (17). The protrudin-Kif5 complex contributes to the transport of these proteins in neurons and is essential for neurite elongation. The identification of mutations in the KIF5A gene in families with the SPG10 subtype of HSP has provided direct evidence for impairment of motor-based transport as an underlying cause of HSP (30). In mammals, the Kif5A motor protein mediates the anterograde transport of cargo such as vesicles along axons. Kif5 also regulates transport of cargo in dendrites and functions in several different membrane trafficking pathways. A VAP mutant that gives rise to familial ALS (ALS8) has been found to induce ER restructuring, providing further support for a role of aberrant ER morphogenesis in neurological disorders (31, 32). In addition, VAP contributes to tethering between the ER and the plasma membrane (33–35). Given that the UPR is activated in the ER of cells deficient in proteins that tether the ER to the plasma membrane, our results suggest that the protrudin-VAP interaction may play an integral role in regulation of ER morphology, function, and maintenance. The fact that several other HSP proteins also localize to the ER suggests that a fuller understanding of the function of these proteins may clarify the contribution of the ER to HSP pathogenesis. Further study of the pathological mechanisms of mutant forms of protrudin may lead to important new insights into motor neuron diseases, including other spastic paraplegias and ALS.
During preparation of the present manuscript, Chang et al. (36) described the interaction of protrudin with other HSP-related proteins in HEK293 cells (a human embryonic kidney cancer cell line) and a role for protrudin in the regulation of ER morphology. Although the results of the two independent studies overlap in part, we adopted a more physiological and comprehensive approach by applying proteomics analysis to neuron-specific protrudin transgenic mice. We thereby identified many HSP-related proteins as protrudin-interacting proteins in an unbiased manner. The detection of such interactions in mouse brain supports their physiological relevance. Furthermore, and most importantly, only our study includes characterization of a pathological mutant of protrudin(G191V) and provides insight into the pathogenesis of HSP caused by this mutation. The two studies performed independently and in parallel, however, reinforce and complement each other.
Acknowledgments
We thank S. Nagata and H. Sumimoto for providing the pEFBOS-HHg vector; T. Kitamura for providing pMX-puro; H. Ichijo for providing pcDNA3-NHK-FLAG; K. Mori for providing pGL3-GRP78P(−132)-luc and p5×ATF6GL3; and A. Hamasaki and other laboratory members for technical assistance.
This work was supported in part by a Japan Society for the Promotion of Science KAKENHI Grant-in-Aid for Young Scientists (S) (to M. S.).
- ER
- endoplasmic reticulum
- HSP
- hereditary spastic paraplegia
- SPG
- spastic paraplegia
- REEP
- receptor expression enhancing protein
- VAP
- vesicle-associated membrane protein-associated protein
- PLP1
- myelin proteolipid protein 1
- UPR
- unfolded protein response
- Ni-NTA
- nickel-nitrilotriacetic acid
- OMIM
- Online Mendelian Inheritance in Man
- mPEG
- methoxypolyethylene glycol maleimide
- XBP1
- X-box binding protein 1
- ALS
- amyotrophic lateral sclerosis
- EGFP
- enhanced green fluorescent protein
- ERAD
- ER-associated degradation.
REFERENCES
- 1. Baumann O., Walz B. (2001) Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 205, 149–214 [DOI] [PubMed] [Google Scholar]
- 2. Levine T., Rabouille C. (2005) Endoplasmic reticulum: one continuous network compartmentalized by extrinsic cues. Curr. Opin. Cell Biol. 17, 362–368 [DOI] [PubMed] [Google Scholar]
- 3. Ramírez O. A., Couve A. (2011) The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol. 21, 219–227 [DOI] [PubMed] [Google Scholar]
- 4. Harding A. E. (1983) Classification of the hereditary ataxias and paraplegias. Lancet 1, 1151–1155 [DOI] [PubMed] [Google Scholar]
- 5. Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. (2012) Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 318, 1–18 [DOI] [PubMed] [Google Scholar]
- 6. Blackstone C. (2012) Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 35, 25–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park S. H., Blackstone C. (2010) Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 11, 515–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park S. H., Zhu P. P., Parker R. L., Blackstone C. (2010) Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 120, 1097–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Orso G., Pendin D., Liu S., Tosetto J., Moss T. J., Faust J. E., Micaroni M., Egorova A., Martinuzzi A., McNew J. A., Daga A. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983 [DOI] [PubMed] [Google Scholar]
- 10. Hu J., Shibata Y., Zhu P. P., Voss C., Rismanchi N., Prinz W. A., Rapoport T. A., Blackstone C. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu J., Prinz W. A., Rapoport T. A. (2011) Weaving the web of ER tubules. Cell 147, 1226–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 [DOI] [PubMed] [Google Scholar]
- 13. Anderson D. J., Hetzer M. W. (2008) Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J. Cell Biol. 182, 911–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mannan A. U., Krawen P., Sauter S. M., Boehm J., Chronowska A., Paulus W., Neesen J., Engel W. (2006) ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 79, 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shirane M., Nakayama K. I. (2006) Protrudin induces neurite formation by directional membrane trafficking. Science 314, 818–821 [DOI] [PubMed] [Google Scholar]
- 16. Saita S., Shirane M., Natume T., Iemura S., Nakayama K. I. (2009) Promotion of neurite extension by protrudin requires its interaction with vesicle-associated membrane protein-associated protein. J. Biol. Chem. 284, 13766–13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsuzaki F., Shirane M., Matsumoto M., Nakayama K. I. (2011) Protrudin serves as an adaptor molecule that connects KIF5 and its cargoes in vesicular transport during process formation. Mol. Biol. Cell 22, 4602–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shirane M., Nakayama K. I. (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 5, 28–37 [DOI] [PubMed] [Google Scholar]
- 19. Matsumoto M., Oyamada K., Takahashi H., Sato T., Hatakeyama S., Nakayama K. I. (2009) Large-scale proteomic analysis of tyrosine-phosphorylation induced by T-cell receptor or B-cell receptor activation reveals new signaling pathways. Proteomics 9, 3549–3563 [DOI] [PubMed] [Google Scholar]
- 20. Shibata Y., Hu J., Kozlov M. M., Rapoport T. A. (2009) Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 25, 329–354 [DOI] [PubMed] [Google Scholar]
- 21. Bian X., Klemm R. W., Liu T. Y., Zhang M., Sun S., Sui X., Liu X., Rapoport T. A., Hu J. (2011) Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. U.S.A. 108, 3976–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anwar K., Klemm R. W., Condon A., Severin K. N., Zhang M., Ghirlando R., Hu J., Rapoport T. A., Prinz W. A. (2012) The dynamin-like GTPase Sey1p mediates homotypic ER fusion in S. cerevisiae. J. Cell Biol. 197, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yagi T., Ito D., Nihei Y., Ishihara T., Suzuki N. (2011) N88S seipin mutant transgenic mice develop features of seipinopathy/BSCL2-related motor neuron disease via endoplasmic reticulum stress. Hum. Mol. Genet. 20, 3831–3840 [DOI] [PubMed] [Google Scholar]
- 24. Martignoni M., Riano E., Rugarli E. I. (2008) The role of ZFYVE27/protrudin in hereditary spastic paraplegia. Am. J. Hum. Genet. 83, 127–128; author reply 128–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang C., Li D., Ma Y., Yan J., Yang B., Li P., Yu A., Lu C., Ma X. (2012) Role of spastin and protrudin in neurite outgrowth. J. Cell Biochem. 113, 2296–2307 [DOI] [PubMed] [Google Scholar]
- 26. Shibata Y., Voss C., Rist J. M., Hu J., Rapoport T. A., Prinz W. A., Voeltz G. K. (2008) The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 283, 18892–18904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu P. P., Soderblom C., Tao-Cheng J. H., Stadler J., Blackstone C. (2006) SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 15, 1343–1353 [DOI] [PubMed] [Google Scholar]
- 28. Windpassinger C., Auer-Grumbach M., Irobi J., Patel H., Petek E., Hörl G., Malli R., Reed J. A., Dierick I., Verpoorten N., Warner T. T., Proukakis C., Van den Bergh P., Verellen C., Van Maldergem L., Merlini L., De Jonghe P., Timmerman V., Crosby A. H., Wagner K. (2004) Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 36, 271–276 [DOI] [PubMed] [Google Scholar]
- 29. Alazami A. M., Adly N., Al Dhalaan H., Alkuraya F. S. (2011) A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18). Neurogenetics 12, 333–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reid E., Kloos M., Ashley-Koch A., Hughes L., Bevan S., Svenson I. K., Graham F. L., Gaskell P. C., Dearlove A., Pericak-Vance M. A., Rubinsztein D. C., Marchuk D. A. (2002) A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 71, 1189–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishimura A. L., Mitne-Neto M., Silva H. C., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J. R., Gillingwater T., Webb J., Skehel P., Zatz M. (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marques V. D., Barreira A. A., Davis M. B., Abou-Sleiman P. M., Silva W. A., Jr., Zago M. A., Sobreira C., Fazan V., Marques W., Jr. (2006) Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve 34, 731–739 [DOI] [PubMed] [Google Scholar]
- 33. Loewen C. J., Young B. P., Tavassoli S., Levine T. P. (2007) Inheritance of cortical ER in yeast is required for normal septin organization. J. Cell Biol. 179, 467–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stefan C. J., Manford A. G., Baird D., Yamada-Hanff J., Mao Y., Emr S. D. (2011) Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 144, 389–401 [DOI] [PubMed] [Google Scholar]
- 35. Manford A. G., Stefan C. J., Yuan H. L., Macgurn J. A., Emr S. D. (2012) ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell 23, 1129–1140 [DOI] [PubMed] [Google Scholar]
- 36. Chang J., Lee S., Blackstone C. (2013) Protrudin binds atlastins and endoplasmic reticulum-shaping proteins and regulates network formation. Proc. Natl. Acad. Sci. U.S.A. 110, 14954–14959 [DOI] [PMC free article] [PubMed] [Google Scholar]