

Background: PLCG1 plays an important role in calcium signaling.
Results: PLCG1 up-regulates 20E-induced calcium signaling and regulates USP1 PKC phosphorylation in the lepidopteran insect Helicoverpa armigera.
Conclusion: 20E activates PLCG1 to induce calcium influx to regulate USP1 PKC phosphorylation for gene expression.
Significance: Our study establishes a link between the nongenomic pathway and genomic pathway in steroid hormone 20E signaling.
Keywords: Calcium Signaling, Gene Transcription, Phosphotyrosine, Protein Phosphorylation, Steroid Hormone Receptor, 20-Hydroxyecdysone, Calcium Influx, Phospholipase Cγ1
Abstract
In addition to the classical nuclear receptor pathway, there is a nongenomic pathway in the cell membrane that regulates gene expression in animal steroid hormone signaling; however, this mechanism is unclear. Here, we report that the insect steroid hormone 20-hydroxyecdysone (20E) regulates calcium influx via phospholipase Cγ1 (PLCG1) to modulate the protein kinase C phosphorylation of the transcription factor ultraspiracle (USP1) in the lepidopteran insect Helicoverpa armigera. The PLCG1 mRNA levels are increased during the molting and metamorphic stages. The depletion of PLCG1 by RNA interference can block 20E-enhanced pupation, cause larvae death and pupation defects, and repress 20E-induced gene expression. 20E may induce the tyrosine phosphorylation of PLCG1 at the cytosolic tyrosine kinase (Src) homology 2 domains and then determine the migration of PLCG1 toward the plasma membrane. The G-protein-coupled receptor (GPCR) inhibitor suramin, Src family kinase inhibitor PP2, and the depletions of ecdysone-responsible GPCR (ErGPCR) and Gαq restrain the 20E-induced tyrosine phosphorylation of PLCG1. PLCG1 participates in the 20E-induced Ca2+ influx. The inhibition of GPCR, PLC, inositol 1,4,5-trisphosphate receptor, and calcium channels represses the 20E-induced Ca2+ influx. Through calcium signaling, PLCG1 mediates the transcriptional activation driven by the ecdysone-response element. Through PLCG1 and calcium signaling, 20E regulates PKC phosphorylation of USP1 at Ser-21 to determine its ecdysone-response element binding activity. These results suggest that 20E activates PLCG1 via the ErGPCR and Src family kinases to regulate Ca2+ influx and PKC phosphorylation of USP1 to subsequently modulate gene transcription for metamorphosis.
Introduction
Phosphoinositide metabolism is a vital intracellular signaling system participating in a variety of cellular functions, including the transduction of hormones and neurotransmitters, growth factor-mediated signaling, cell morphology, and cell division (1). Phospholipase C (PLC)2 is a key enzyme in this system that hydrolyzes phosphatidylinositol 4,5-bisphosphate to generate inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) in response to the cellular setting of ligand-mediated signal transduction by hormones, neurotransmitters, growth factors, and other molecules (2). The binding of IP3 to its receptor in the endoplasmic reticulum membrane drives the release of calcium ions from intracellular stores, and DAG and Ca2+ bind to the protein kinase C (PKC) conserved region domains (C1 and C2) to activate PKC (3). Because of the presence of the cytosolic tyrosine kinase (Src) homology (SH) domain, PLCγ (PLCG) is distinct from other PLC isozymes and is activated by receptor tyrosine kinases (RTKs) (4). Studies have shown that GPCRs can activate PLCG1 through RTK or Src (5, 6), suggesting the participation of PLCG1 in GPCR- and Src-regulated signaling.
Previous studies have indicated that the mammalian steroid hormone estrogen triggers gene expression via the nuclear receptor genomic pathway and GPCR-regulated nongenomic pathway (7). In insects, steroid hormone 20-hydroxyecdysone (20E) is also known to transmit a signal via the nuclear receptor genomic pathway and the GPCR-regulated nongenomic pathway. In the nuclear receptor genomic pathway, 20E binds to the ecdysone receptor (EcR) and forms a heterodimeric transcription complex with ultraspiracle (USP) to bind to the ecdysone-response element (EcRE) for gene transcription. Drosophila USP can also directly bind to EcRE (8). The 20E-induced genes, including hormone receptor 3 (HR3), and the metamorphosis initiation factor Broad (Br) subsequently mediate insect molting and metamorphosis (9, 10).
In the GPCR-regulated nongenomic pathway, 20E directly binds to a GPCR (dopamine/ecdysteroid receptor, DopEcR) to regulate development and signaling in the mature adult nervous system in Drosophila melanogaster (11). The programmed cell death in the silkworm anterior silk glands is triggered by 20E-induced GPCR-PLC-IP3-Ca2+-PKC signaling (12, 13). In Helicoverpa armigera, through GPCR-PLC-Ca2+ signaling, 20E induces the rapid phosphorylation of cyclin-dependent kinase 10 (CDK10) to promote gene transcription (14). On the H. armigera plasma membrane, an ecdysone-responsible GPCR (ErGPCR) regulates the nongenomic pathway in 20E signaling, but it does not bind to the ecdysone analog [3H]ponasterone A (15). 20E induces USP phosphorylation in Chironomus tentans and Tenebrio molitor (16, 17). In D. melanogaster, the PKC-mediated phosphorylation of USP at Ser-35 is essential for 20E-induced transcriptional activation (18). However, the connection between the upstream cell membrane signaling and the downstream nuclear receptor signaling has not been demonstrated.
In this study, we found that 20E increases the PLCG1 expression levels during the molting and metamorphic stages in H. armigera, which is one of the most serious insect pests in cotton, vegetables, corn, and other crops (19). PLCG1 is essential for larva development and pupation. Through ErGPCR, Gαq, and Src family kinases, 20E rapidly induces the tyrosine phosphorylation at the SH2 domains in PLCG1 and the migration of PLCG1 toward the plasma membrane. PLCG1 participates in the 20E-induced Ca2+ influx depending on its tyrosine phosphorylation status. Through PLCG1 and Ca2+ signaling, 20E activates EcRE transcriptional activity by regulating USP1 PKC phosphorylation at Ser-21, which determines its binding activity to EcRE. These results suggest that ErGPCR transducts the 20E signal to Src family kinases to activate PLCG1 and that this activation then triggers calcium signaling to induce PKC-mediated USP1 phosphorylation for transcriptional activation.
EXPERIMENTAL PROCEDURES
Chemicals
Chemicals were purchased commercially as follows: restriction enzymes and ExTaq polymerase (Fermentas International Inc., Thermo Fisher Scientific Inc., Waltham, MA); TRIzol reagent kit and genomic DNA extraction kit (BioTek, Beijing, China); mouse monoclonal antibodies against RFP and His tag (CWbio, Beijing, China); anti-phosphotyrosine mouse monoclonal antibody (Tyr(P)-102) (Cell Signaling Technology Inc., Beverly, MA); first strand cDNA synthesis kit (Sangon, Shanghai, China); 20E (Sigma); inhibitors (suramin sodium salt, U73122, pyrazole compound, flunarizine dihydrochloride, chelerythrine chloride, and xestospongin C) (Sigma); Src inhibitor PP2 and RTK inhibitor SU6668 (Selleckchem, Houston, TX); phorbol 12-myristate 13-acetate (PMA) and ionomycin (Beyotime, Shanghai, China). All other reagents used were of analytical grade.
Animals
H. armigera larvae were raised on an artificial diet at 28 °C with 60–70% relative humidity and were maintained under 10-h dark/14-h light cycles in an insectarium (20). The molting stage from larvae to larvae is distinguished by the head capsule slippage, and the metamorphically commitment stage from the final instar to pupae is discriminated by the wandering behavior and stopping feeding.
Cloning of the cDNA and Sequence Analysis
Full-length cDNA sequence was obtained by transcriptome sequencing H. armigera epidermis cells. BLASTX (www.ncbi.nlm.nih.gov) analysis showed the gene is homologous to PLCG1 from other animals. The open reading frame was identified using the Expert Protein Analysis System (ExPASy). The domain predictions were undertaken with SMART (Simple Modular Architecture Research Toll). Sequence alignments and phylogenetic trees were performed with the GENEDOC computer program and MEGA 3.1 software.
Cell Culture
The epidermal cell line HaEpi of H. armigera (21) was used in all of the related experiments. HaEpi cells were cultured as a loosely attached monolayer and were maintained at 26 °C in 25-cm2 tissue culture flasks with 4 ml of antibiotic-free Grace's medium supplemented with 10% heat-inactivated fetal bovine serum. The cell density was estimated by counting the cells in a suspension aliquot using a hemocytometer under a microscope. All of the experiments were initiated by seeding the flasks with 5 × 105 cells and cultured under the above-mentioned normal growth conditions for 96 h.
Western Blot
Protein concentration was determined using the Bradford method (22). Equal amounts of protein (50 μg) were subjected to 12.5% SDS-PAGE and then electrotransferred onto nitrocellulose membranes. The resulting membranes were incubated for 1 h in a blocking buffer (10 mm Tris-buffered saline solution) containing 3% fat-free milk powder at room temperature and then with the primary anti-RFP polyclonal antibody (1:1000 dilution in the blocking buffer) at 4 °C overnight. Goat anti-rabbit IgG conjugated with alkaline phosphatase diluted 1:10,000 in the blocking buffer was adopted as a secondary antibody. The signal of immunoblotting was visualized by 10 ml of Tris-buffered saline, 45 μl of p-nitro blue tetrazolium chloride, 5%, and 35 μl of 5-bromo-4-chloro-3-indolyl phosphate, 5%, in the dark for 10 min.
Quantitative Real Time PCR (qRT-PCR)
The total RNA was extracted from the treated cells or staged insects using the TRIzol reagent according to the manufacturer's instructions (CWbio, Beijing, China). Four micrograms of RNA were reversely transcribed into cDNA after the determination of the RNA quality through electrophoresis on an agarose gel (1%). qRT-PCR was performed using SsoFastTM EvaGreen Supermix (Bio-Rad) according to the manufacturer's instructions and a real time thermal cycler (Bio-Rad). β-Actin was amplified for internal standardization. Primers for H. armigera PLCG1, EcRB1 (GenBankTM accession number EU526831), USP1 (EU526832), HR3 (AF337637), BrZ2 (not released), ErGPCR (JQ809653), Gαq (AAX56092.1), DopEcR (not released), and β-actin (EU527017) were used in the qRT-PCR assay (Table 1). The data from three independent experiments were statistically analyzed by Student's t test. The relative expression data were statistically analyzed using the 2−ΔΔCT method (23).
TABLE 1.
Primer sequences for RT-PCR, RNAi, and overexpression
| Primer name | (5′ →3′) nucleotide sequence |
|---|---|
| β-actin F | Agtagccgccctggttgtagac |
| β-actin R | Ttctccatgtcgtcccagt |
| PLCG1-qRTF | Ccagtgctcgctccctacaac |
| PLCG1-qRTR | Ggcttatcaggagtctggtagttca |
| PLCG1-OEF1 | tactcacaattggatgaatccggtctgctataat |
| PLCG1-OER1 | tactcaggcgcgccgatttgctcgtgatgaaggatat |
| dsGFPT7F | gcgtaatacgactcactataggtggtcccaattctcgtggaac |
| dsGFPT7R | Gcgtaatacgactcactataggcttgaagttgaccttgatgcc |
| dsPLCG1T7F | gcgtaatacgactcactataggggttccgcacatttctgaggt |
| dsPLCG1T7R | gcgtaatacgactcactatagggaacgggattgtagttcgacg |
| dsErGPCRT7F | Gcgtaatacgactcactatagggttcatccttctaacggtggc |
| dsErGPCRT7R | Gcgtaatacgactcactatagggtcgcttcatcttcgctatct |
| ErGPCR-qRTF1 | Aaacggttcacctactacgc |
| ErGPCR-qRTR1 | Cgcttcatcttcgctatct |
| dsDopEcRT7F | Gcgtaatacgactcactataggtgaccaacgatgctggttac |
| dsDopEcRT7R | Gcgtaatacgactcactataggctgctctgatgaggggtagc |
| dsGαqT7F | Gcgtaatacgactcactataggtcggaggaggcgaaggag |
| dsGαqT7R | Gcgtaatacgactcactatagggtgttcgccgcagtcaaa |
| dsCDK10T7F | Gcgtaatacgactcactataggttgaacggcagcatagtgg |
| dsCDK10T7R | gcgtaatacgactcactataggagagtgacgacgcgaggag |
| DopEcR-qRTF1 | Tgacggaaagcaggttgag |
| DopEcR-qRTR1 | gaagccagcagaagacgaa |
| Gαq-qRTF1 | Ggcagttgcgaaaggac |
| Gαq-qRTR1 | Tctgagttggacggatt |
| EcRB1-qRTF1 | Aattgcccgtcagtacga |
| EcRB1-qRTR1 | Tgagcttctcattgagga |
| USP1-qRTF1 | Ggtcctgacagcaatgtt |
| USP1-qRTR1 | Ttccagctccagctgactgaag |
| HR3-qRTF1 | Tcaagcacctcaacagcagcccta |
| HR3-qRTR1 | Gactttgctgatgtcaccctccgc |
| BrZ2-qRTF1 | Ggtgactgtccttactgcggcat |
| BrZ2-qRTR1 | Ttaattcctttgaccatgact |
Hormone Treatment of HaEpi Cells and Larvae
HaEpi cells were maintained under the normal growth conditions described in a previous study until 90% confluence was obtained (21). The cells were cultured for 1, 3, 6, 12, and 24 h after 1 μm 20E was added to the cells. The control cells received an equal volume of dimethyl sulfoxide (DMSO), which was used as a solvent for 20E. To investigate the hormonal regulation of PLCG1 expression in the larval midgut, the sixth instar 6-h larvae were injected with 20E (500 ng/larva). The untreated controls were injected with an equal volume of DMSO. After hormone induction, the total RNA was extracted, and qRT-PCR was then performed.
RNA Interference (RNAi) in HaEpi Cells
The MEGAscriptTM RNAi kit (Ambion, Austin, TX) was used to generate dsRNAs corresponding to H. armigera PLCG1 (dsPLCG1), dsDopEcR, dsErGPCR, dsGαq, and dsCDK10, according to the manufacturer's instructions. The dsRNA of GFP (dsGFP) was synthesized and used as a nonspecific RNA interference control. The primers for the production of dsRNAs are listed in Table 1. The concentration of dsRNA was determined by spectrophotometry at 260 nm. HaEpi cells were seeded in 6-well plates at a density of 5 × 105 per well. A lipophilic transfection reagent, Lipofectamine 2000 (Invitrogen), was employed for dsRNA transfection according to the manufacturer's instructions. The final concentration of dsRNA was 2 μg/ml in the medium. After incubation at 26 °C for 6 h after dsRNA transfection, the cells were rinsed and then cultured for 24–48 h with Grace's medium (Invitrogen) containing 10% fetal bovine serum (Medgenics, St. Louis, MO). Then 1 μm 20E was added to the cells, and the cells were incubated for 12 h. The RNA or total protein was extracted from the cells for qRT-PCR or Western blot analysis.
RNAi in Larvae via Injection of dsRNA
The depletion of PLCG1 in the insects was performed through the injection of PLCG1 dsRNA (dsPLCG1). First, 1 μg of dsPLCG1 was injected into a fifth instar larva, and 2 μg of dsPLCG1 was injected into a sixth instar larva. Ninety larvae were injected for the RNAi of PLCG1. The control larvae received an equal amount of dsGFP. To determine the function of PLCG1 in the 20E-induced metamorphosis, 500 ng of 20E was smeared on the food administered to H. armigera (1 × 1 × 0.3 cm) to feed the PLCG1-depleted insects. An equivalent amount of DMSO was used as a control.
Overexpression of Genes in HaEpi Cells
GFP or RFP was cloned into the pIEx-4 plasmid (EMD Millipore, Billerica, MA). After the efficiency of pIEx-GFP or -RFP was examined by observing the expressed fluorescence protein, the full-length PLCG1 was then subcloned into the construct to produce PLCG1-GFP. The wild-type PLCG1 or USP1 was also cloned into pIEx-4 to generate His-tagged PLCG1-His or USP1-His, respectively. For the deletion mutant (PLCG1ΔSH2-His), the two SH2 domains were deleted and then inserted into pIEx-4. For the point mutation (USP1S21A), an alanine replaced the serine at position 21. HaEpi cells were seeded in 6-well plates at a density of 1 × 106 per well. Five micrograms of DNA per well were used for the transfection, as delineated in the manufacturer's instructions (Cellfectin Reagent, Invitrogen). After incubation at 26 °C for 24 to 48 h post-transfection, the cells were treated for the subsequent experiments. For immunocytochemistry, 4′,6-diamidino-2-phenylindole (1 μg/ml in phosphate-buffered saline (PBS), 140.0 mm NaCl, 2.7 mm KCl, 10.0 mm Na2HPO4, and 1.8 mm KH2PO4) was used to stain the nuclei. The fluorescence was observed using an Olympus BX51 fluorescence microscope (Shinjuku-ku, Tokyo, Japan) or a Zeiss LSM 700 laser confocal microscope (Zeiss, Thornwood, NY).
Analysis of the Phosphorylation of PLCG1
The HaEpi cells overexpressing PLCG1 due to the transfection of pIEx-PLCG1-His (PLCG1-His) were induced with 1 μm 20E for 15 min. Subsequently, the cellular proteins were extracted and purified using the His-Bind resin in a tube with phosphatase inhibitors (Roche Diagnostics) according to the manufacturer's instructions. The anti-His tag antibody was used to determine the purified PLCG1-His through Western blot assay. The anti-phosphotyrosine mouse monoclonal antibody (Tyr(P)-102) was used to recognize the tyrosine-phosphorylated PLCG1. For the identification of the phosphorylated sites, the two SH2 domains were deleted from the wide-type PLCG1, and the mutant PLCG1 (PLCG1ΔSH2-His) was overexpressed.
Detection of Calcium Flux in HaEpi Cells
HaEpi cells were seeded and cultured for 48 h in a 6-well tissue culture plate according to the above-described protocol. Then 3 μm AM ester Calcium CrimsonTM dye (Invitrogen) was added to the cells, and the cells were incubated in Grace's medium for 30 min at 27 °C. The cells were washed with Dulbecco's phosphate-buffered saline (DPBS, 2.7 mm KCl, 1.5 mm KH2PO4, and 8 mm Na2HPO4) without calcium ions and exposed to 1 μm 20E in DPBS for 2 min for the detection of the intracellular calcium flux. Subsequently, 1 mm CaCl2 was used to detect the extracellular calcium influx. The fluorescence was detected at 555 nm every 6 s for a period of 360 s using a Zeiss LSM 700 laser confocal microscope. The data were analyzed using the Image Pro-Plus software. For the PLCG1 RNAi and overexpression experiments, the above-described method was used to transfect the cells with the dsRNA against PLCG1 or the pIEx-PLCG1-GFP plasmids in advance. For the inhibition experiments, the cells were pretreated with different inhibitors for 30 min prior to stimulation with 20E.
Detection of 20E-induced Transcriptional Activation
The total genomic DNA was extracted from the cells using a DNA isolation kit (MagExtractor Genome, Toyobo, Osaka, Japan). Using the GenomeWalker method (24), a 1084-bp DNA fragment was cloned from the 5′ upstream region of H. armigera HR3. This fragment contains a conserved ecdysone-response element (EcRE, 5′-ggggtcaatgaactg-3′), which has been identified in Manduca sexta, that is an EcRB1-USP1 complex specifically binding element (EcRE1, 5′-ggggtcaatgaaccg-3′) (25). For the 20E-induced transcriptional activation experiments, this DNA fragment was cloned into the pIEx-4 plasmid upstream of RFP to form pIEx-HR3pro-RFP after replacing the original hr5 enhancer and IE promoter (14). Twenty four hours after the transfection of HaEpi cells with pIEx-HR3pro-RFP, 1 μm 20E was added to cell culture medium, and the cells were incubated for 18 h. The RFP expression was then assessed by Western blot to determine the 20E-stimulated transcriptional activation. An equivalent DMSO induction was used as a negative control.
USP1 Phosphorylation Levels Detection
USP1-His or USP1S21A was overexpressed in HaEpi cells using pIEx-USP1-His or pIEx-USP1S21A-His plasmid transfection and then purified by His-Bind resin (50 μl) after a 1-h treatment of 1 μm 20E. Equal volumes of DMSO were used for the negative control. The number of moles of phosphorus per mol of USP1-His or USP1S21A-His was determined using the phosphoprotein phosphate estimation assay kit (Pierce) based on the alkaline hydrolysis of phosphate from seryl and threonyl residues in phosphoproteins and then quantifying the released phosphate in a 96-well microplate using Malachite Green and ammonium molybdate. Lyophilized phosvitin was used as the phosphorylated protein standard. Detailed procedures were described in the manufacturer's instructions.
USP1-His Overexpression and Electrophoretic Mobility Shift Assay (EMSA)
USP1-His was overexpressed in HaEpi cells through their transfection with the pIEx-USP1-His plasmid. After 1 h of stimulation with DMSO or 20E (1 μm), the cell proteins were extracted. USP1-His was then purified using the His-Bind resin in a tube according to the manufacturer's instructions. The same protocol was used for the expression and purification of USP1S21A-His. Probes labeled with digoxigenin or not labeled (sense, 5′-ggtcccggggtcaatgaactgctgtga-3′; antisense, 5′-tcacagcagttcattgaccccgggacc-3′), which were used in EMSA and contain EcRE, were synthesized by Sangon Co. (Shanghai, China). In brief, 100 fmol of digoxigenin-labeled (Dig-labeled) probe were incubated with 5 μg (5 μl) of purified proteins in binding buffer (Beyotime Institute of Biotechnology, Shanghai, China). For the competition experiments, in addition to the Dig-labeled probe, a 50-fold excess of unlabeled probe was preincubated with the proteins. The reaction mixture was run on a 6% polyacrylamide gel at 100 V and then transferred onto a nylon membrane (Immobilon-Ny+, Millipore, Milford, MA). The nonspecific binding was then blocked by incubation with 1% (w/v) blocking reagent (Roche Diagnostics) for 30 min. The membrane was then incubated with anti-Dig-phosphatase antibody (1:5000) for 1 h, and the signal was visualized using 5-bromo-4-chloro-3-indolyl phosphate and p-nitro blue tetrazolium chloride.
RESULTS
PLCG1 mRNA Is Highly Transcribed during the Molting and Metamorphic Stages
The PLCG1 expression sequence tag was obtained through random sequencing of the transcriptome of the H. armigera epidermal cell line (HaEpi) (Figs. 1 and 2). To verify the tissue specificity of PLCG1 expression during larval development, the expression profile of PLCG1 was detected from the fifth instar larvae to pupae. The quantitative real time PCR (qRT-PCR) results showed that PLCG1 was expressed in integument, midgut, fat body, and hemocytes. In all of the tissues, a low transcript level of PLCG1 was detected at the fifth instar feeding stage (5–24 h), the sixth instar feeding stages from 6–0 to 6–48 h, and 2 days after pupation. However, the transcript level of PLCG1 reached a maximum at the fifth instar molting stage (5–36 h) and the sixth instar metamorphic stage (6–72 to 6–120 h) (Fig. 3). The data suggest that PLCG1 may play roles in molting and metamorphosis.
FIGURE 1.

Full-length cDNA sequence and deduced amino acid sequence of PLCG1 gene and phylogenetic analyses. 3766-bp cDNA was isolated from H. armigera epidermis cells by transcriptome sequencing. This mRNA contains a 3606-bp open read frame and encodes 1201 amino acids. The theoretical molecular mass is 138 kDa. Underlines show the two SH2 domains, which were deleted to generate tyrosine phosphorylation site deletion mutants of PLCG1. The shaded Y indicates putative tyrosine phosphorylation sites predicted by NetphosK 2.0.
FIGURE 2.

Phylogenetic analysis. The numbers above the branches denote the bootstrap values that are shown as a percentage.
FIGURE 3.

qRT-PCR shows increased mRNA levels of PLCG1 during molting and metamorphosis. A, integument; B, midgut; C, fat body; and D, hemocytes. F, feeding; M, molting; MM, metamorphic molting; P, pupae; 5F, fifth instar feeding larvae 24 h after ecdysis; 5M, fifth instar molting larvae; 6–0, 6–24, 6–48, 6–72, 6–96, and 6–120 represent sixth-instar larvae at the corresponding times. P0 and P2 represent 0- and 2-day-old pupae. H. armigera β-actin was used for internal standardization. The relative expression was calculated using the 2−ΔΔCT method. The bars indicate the means ± S.D. of three independent experiments.
Knockdown of PLCG1 Leads to Larvae Death and Pupation Defects
To investigate the function of PLCG1 in metamorphosis, PLCG1 was knocked down in larvae through their injection with dsRNA against PLCG1. The dsPLCG1 injection caused death before pupation or pupation defects in normal and 20E-treated larvae (Fig. 4, A and B). Approximately 65–69% of the larvae died or formed abnormal pupae after PLCG1 knockdown. However, only 3–5% death or abnormal pupation was observed in the larvae injected with dsGFP (GFP, green fluorescent protein) (Fig. 4C). In the surviving pupae, dsPLCG1 injection delayed pupation for 3 days compared with the dsGFP-injected larvae. The injection of dsGFP followed by 20E induction promoted pupation 2 days earlier compared with that obtained with the larvae that were only injected with dsGFP. In contrast, dsPLCG1 followed by 20E injection delayed pupation for 6 days compared with dsGFP followed by 20E injection control (Fig. 4D). These data suggest that PLCG1 functions in 20E-regulated pupation.
FIGURE 4.
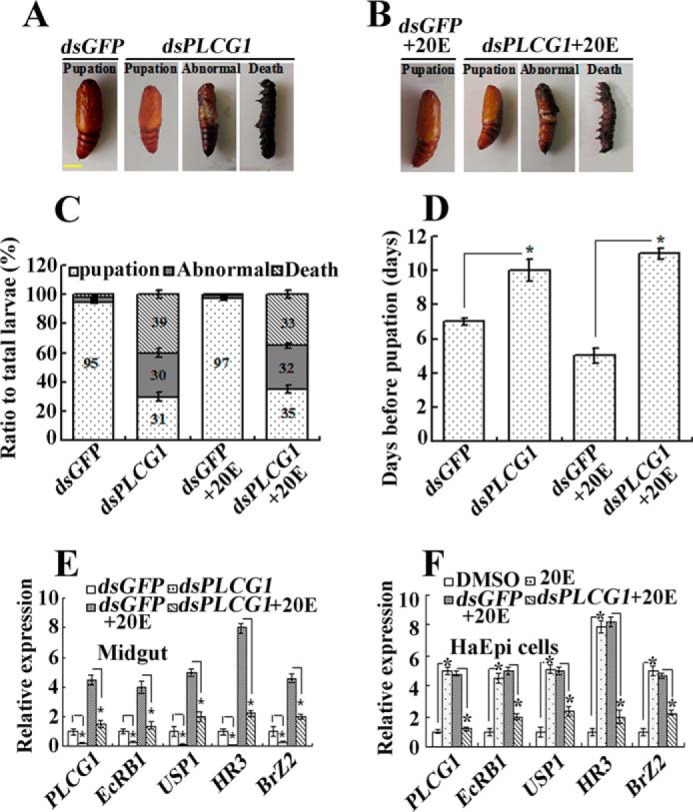
PLCG1 knockdown leads to larvae death and pupation defects and suppresses 20E-inducible gene transcription. A and B, phenotypes of normal and 20E-fed larvae after PLCG1 knockdown using dsRNA injection (1 μg for a fifth instar larva, 2 μg for a sixth instar larva). The scale bar denotes 0.5 cm. C and D, statistical analysis of the phenotypes and pupation time using Student's t test (n = 30 × 3). Abnormal indicates surviving atypical pupae. Death suggests that the larvae died before pupation. E and F, qRT-PCR analysis of the efficacy of PLCG1 knockdown and its effect on the expression of 20E-inducible genes in larvae (4 days after injection of PLCG1 dsRNA, the larvae were injected with 500 ng of 20E for a 12-h induction) and in HaEpi cells (24 h after transfection of dsPLCG1, the cells were incubated with 1 μm 20E for 12 h). GFP dsRNA was used as a control. The primers of the genes are described in Table 1. The significant differences, as determined through Student's t test based on three biologically independent repeats, are indicated: *, p < 0.05.
To determine the mechanism through which PLCG1 regulates larval pupation, the 20E-induced gene transcriptional profile was examined in larvae and in an H. armigera epidermal cell line (HaEpi) after PLCG1 knockdown. The overall transcript levels of various genes, including EcRB1, USP1, HR3, and BrZ2, were decreased when PLCG1 was depleted by RNAi (Fig. 4, E and F). Therefore, PLCG1 may control metamorphosis by regulating 20E-induced gene expression.
20E Up-regulates the Transcript Level and Subcellular Location of PLCG1
The transcript level of PLCG1 was detected in the 20E-treated midgut of larvae to investigate the regulation of PLCG1 expression by 20E. The qRT-PCR data showed that the transcript level of 20E-induced HR3 was apparently up-regulated from 3 to 24 h after induction with 20E, which suggests that the midgut responds successfully to 20E (Fig. 5A). In addition, the PLCG1 mRNA reached a maximum from 6 to 12 h after 20E induction (Fig. 5B), confirming that PLCG1 expression is up-regulated by 20E. These data are correlated to the high expression levels of PLCG1 during molting and metamorphosis because the 20E level is higher during these stages in vivo (26).
FIGURE 5.
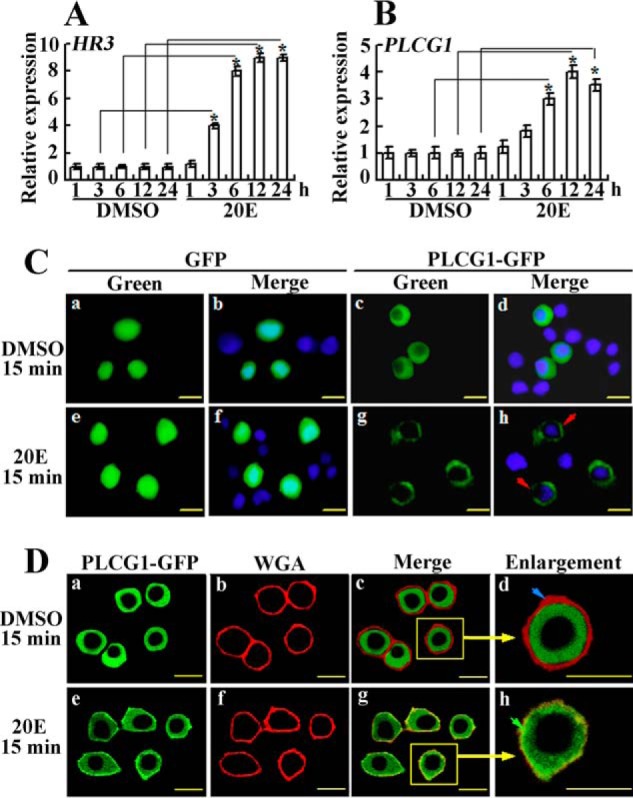
PLCG1 transcript level in larval midgut and PLCG1 protein migration toward the cell membrane in HaEpi cells depend on 20E stimulation. A and B, qRT-PCR analysis of the PLCG1 mRNA levels in the midgut of sixth instar 6-h larva after the injection of 500 ng of 20E. The negative control received an equivalent volume of DMSO. HR3 was used as a positive control for 20E induction. The statistical significance between samples was calculated using Student's t test from three repeats (*, p < 0.05). C, immunocytochemistry analysis of the response of the overexpression of PLCG1-GFP (green color) to 20E treatment. The observations were made under a fluorescence microscope. The red arrows denote PLCG1-GFP migrating toward the cell membrane after 15 min of treatment with 1 μm 20E. Cells overexpressing GFP were used as a control. The blue fluorescence indicates the cell nuclei stained with DAPI. D, migration of the overexpressed PLCG1-GFP toward the cell membrane was confirmed by a Zeiss LSM 700 laser confocal microscope. Then 1 μg/ml Alexa Fluor 594-conjugated WGA was incubated with the cells in DPBS for 15 min at room temperature to label the plasma membrane. The blue arrow indicates the cell membrane, and the green arrow indicates the superposition of the membrane and PLCG1-GFP. The yellow bars denote 20 μm.
We then tested the subcellular localization of PLCG1 in HaEpi cells to determine its response to 20E stimulation. The GFP was uniformly distributed in the cytoplasm and nucleus (Fig. 5C, panels a and b); in contrast, the overexpressed PLCG1-GFP fusion protein was generally localized in the cytoplasm in the DMSO treatment control (Fig. 5C, panels c and d). GFP did not change its uniform location in the cells after induction with 20E for 15 min (Fig. 5C, panels e and f). However, the addition of 20E to the cells induced PLCG1-GFP trafficking toward the plasma membrane within 15 min (Fig. 5C, panels g and h). To confirm the cell membrane trafficking of PLCG1, the overexpressed PLCG1-GFP was observed by confocal laser scanning microscopy (Fig. 5D, panel a). In addition, Alexa Fluor 594-conjugated wheat germ agglutinin (WGA, red) was used as an indicator of the cell membrane (Fig. 5D, panel b). PLCG1-GFP did not traffic toward the cell membrane in response to DMSO treatment for 15 min (Fig. 5D, panels c and d); however, PLCG1-GFP trafficked toward the cell membrane and was superimposed on WGA after 15 min of 20E treatment (Fig. 5D, panels g and h). These results suggest that PLCG1 responds to 20E stimulation by moving toward the plasma membrane.
Tyrosine Phosphorylation of PLCG1 Determines Its Membrane Migration and the 20E-induced Increase in the Cytosolic Ca2+ Levels
We then sought to elucidate the mechanism through which PLCG1 responds to 20E and traffics toward the membrane. Because 14 tyrosine phosphorylation sites were predicted to be found in PLCG1, as determined by NetphosK 2.0 (Fig. 1), we detected the tyrosine phosphorylation of PLCG1 through the overexpression of PLCG1-His and an SH2 two-domain deletion mutation, namely PLCG1ΔSH2-His, in HaEpi cells and using an anti-phosphotyrosine mouse monoclonal antibody (Tyr(P)-102). The Western blot analysis showed that PLCG1-His and PLCG1ΔSH2-His were successfully overexpressed, as determined through anti-His antibody detection. The tyrosine phosphorylation of PLCG1-His was detected in the cells treated with 20E for 15 min but not in the DMSO control. The GPCR inhibitor suramin (27, 28) suppressed the 20E-induced tyrosine phosphorylation of PLCG1-His (Fig. 6A, panel a). DopEcR, ErGPCR, and Gαq were knocked down by RNAi to verify the involvement of GPCR signaling in the regulation of PLCG1 phosphorylation. The depletion of DopEcR had no effect on PLCG1 phosphorylation, whereas silencing of ErGPCR and Gαq decreased the tyrosine phosphorylation of PLCG1 (Fig. 6A, panels b and c). To investigate the tyrosine kinase regulating PLCG1 phosphorylation, RTK inhibitor SU6668 (29) and Src inhibitor PP2 (30) were used. The results showed that SU6668 did not suppress PLCG1 phosphorylation, but PP2 restrained the tyrosine phosphorylation of PLCG1 (Fig. 6A, panel b). Thus, it is likely to be that ErGPCR activates Src family kinases to phosphorylate PLCG1. After the deletion of the two SH2 domains in PLCG1ΔSH2-His, tyrosine phosphorylation was not detected, even after 20E induction (Fig. 6A, panel a), suggesting that the tyrosine phosphorylation of PLCG1 involves the SH2 domains. In addition, PLCG1ΔSH2-GFP was unable to move toward the plasma membrane after a 20E induction (Fig. 6B). Therefore, with the involvement of ErGPCR, Gαq, and Src family kinases, 20E-regulated tyrosine phosphorylation of PLCG1 determined the migration of PLCG1 toward the cell membrane.
FIGURE 6.

20E-regulated tyrosine phosphorylation of PLCG1 determines its membrane migration and the 20E-induced increase in the cytosolic Ca2+ levels. A, Western blot assay for the detection of the tyrosine phosphorylation of PLCG1 in HaEpi cells. Panel a, suramin restrained the SH2 domain phosphorylation of PLCG1. PLCG1-His (with 6 histidine tags) represents the wild-type PLCG1; PLCG1ΔSH2-His is PLCG1 with the two SH2 domains deleted (195 amino acids were deleted); PLCG1-P indicates the tyrosine-phosphorylated PLCG1; and suramin is a GPCR inhibitor (the cells were incubated with 50 μm suramin for 30 min before the treatment with 20E). The cell proteins were extracted after treatment with 20E (1 μm) or an equal volume of the DMSO control for 15 min, and PLCG1-His and PLCG1ΔSH2-His were purified using the His-Bind resin in a tube (50 μl of resin per tube) and detected using an antibody against His tag. The anti-phosphotyrosine mouse monoclonal antibody (Tyr(P)-102) was used to assess the tyrosine phosphorylation of PLCG1. Panel b, inhibition of PLCG1 phosphorylation by the silencing of DopEcR, ErGPCR, and Gαq and the addition of inhibitors of RTK and Src. 5 μm SU6668 (RTK inhibitor) and 20 μm PP2 (Src inhibitor) were added to the cells for 30 min treatment before the 20E stimulation. Panel c, qRT-PCR analysis of the RNAi effect after 48 h of dsRNAs transfection. B, PLCG1ΔSH2-GFP was overexpressed in HaEpi cells to test the blockage of membrane trafficking after 15 min of treatment with 20E using a confocal fluorescence microscope. WGA was used as a cell membrane marker. The blue arrow indicates the gap between the membrane and PLCG1ΔSH2-GFP. The yellow bars denote 20 μm. C, increase in the cytosolic Ca2+ levels by 20E induction. 1 mm calcium chloride was added to the cells after treatment with 1 μm 20E for 2 min. Suramin (50 μm) or U73122 (10 μm) was added to the medium for 30 min before 20E induction. F, fluorescence of cells after treatment; F0, average fluorescence of cells before treatment. The fluorescence was detected every 6 s for a period 6 min using a laser scanning confocal microscope (Carl Zeiss LSM 700) at 555 nm and was analyzed using the Image Pro-Plus software. D, inhibition of 20E-induced increase in the cytosolic Ca2+ levels. The IP3R blocker XeC (3 μm), the T-type calcium channel blocker FL (50 μm), and the TRPC3 channel inhibitor Pyr3 (10 μm) were added to the medium 30 min before 20E induction. E, effect of the knockdown of PLCG1 by dsRNA transfection (5 μg/ml) on the Ca2+ levels. PLCG1 RNAi was detected by qRT-PCR. Statistical significance (*, p < 0.05) was based on three biologically independent repeats. F, effect of the overexpression of PLCG1-GFP and PLCG1ΔSH2-GFP on the Ca2+ levels.
To study the function of PLCG1 in the 20E pathway, we examined the involvement of PLCG1 in the 20E-triggered calcium flux in HaEpi cells. The DMSO control had no effect on the intracellular calcium signal. The first peak of the intracellular calcium signal appeared 1 min after 20E induction without Ca2+ in the cell culture medium, suggesting the release of Ca2+ from the intracellular calcium stock. The addition of 1 mm CaCl2 to the cell culture medium resulted in a second peak in the Ca2+ signal, suggesting an influx of extracellular Ca2+ from the medium to the cells. The GPCR inhibitor suramin (27, 28) and the PLC inhibitor U73122 (31) repressed both intracellular calcium release and extracellular Ca2+ influx (Fig. 6C). The IP3 receptor (IP3R) inhibitor xestospongin C (XeC) (32) also repressed both calcium peaks. However, two calcium channel inhibitors, namely the transient receptor potential calcium 3 (TRPC3) channel inhibitor pyrazole compound (Pyr3) (33) and the T-type voltage-gated calcium channel inhibitor flunarizine dihydrochloride (FL) (34), only suppressed the second peak corresponding to extracellular Ca2+ influx but did not repress the first peak corresponding to intracellular calcium release (Fig. 6D). The 20E-induced Ca2+ mobilization data largely confirm similar findings reported in our recent paper (15). In addition, we further verified that the IP3 receptor regulated intracellular calcium release. Taken together, these findings suggest that 20E triggers both intracellular calcium release and extracellular Ca2+ influx.
After depletion of PLCG1 through transfection of dsPLCG1, 20E was unable to induce neither intracellular calcium release nor extracellular Ca2+ influx compared with the dsGFP control (Fig. 6E). Inversely, the overexpression of PLCG1-GFP promoted the more rapid appearance of the calcium peaks. However, the overexpression of PLCG1ΔSH2-His did not promote the earlier appearance of the calcium peaks (Fig. 6F). These results indicate that PLCG1 participates in the 20E-triggered intracellular calcium release and extracellular Ca2+ influx and that the tyrosine phosphorylation of PLCG1 at SH2 domains is essential for this function.
PLCG1 Promotes 20E-induced Transcriptional Activity via Calcium Signaling
To address the output of 20E-induced Ca2+ influx, we constructed a pIEx-HR3pro-RFP plasmid by replacing the original hr5 enhancer and IE promoter in the pIEx-4 plasmid with the H. armigera HR3 promoter containing the EcRB1-USP1-binding element EcRE (20E response element) (14, 25). The Western blot assay showed that 20E activates the HR3 promoter to drive RFP expression in a concentration- and time-dependent manner (Fig. 7A). The RNAi transfection of dsPLCG1 decreased the 20E-activated RFP expression compared with the 20E- or dsGFP-treated cells (Fig. 7B, panels a and c). In contrast, the overexpression of PLCG1-His promoted 20E-activated RFP expression, and the overexpression of PLCG1ΔSH2-His did not result in the up-regulation of this transcriptional activation compared with the 20E-treated cells (Fig. 7B, panels b and d). These data suggest that PLCG1 regulates 20E-induced transcription and that the tyrosine phosphorylation of the SH2 domains of PLCG1 is necessary for PLCG1 activity.
FIGURE 7.

PLCG1 regulates 20E-induced transcriptional activity through ErGPCR and calcium signaling, as assessed through the detection of EcRE-driven RFP expression via Western blot. All of the cells were transfected with 2.5 μg/ml pIEx-HR3pro-RFP plasmids. A, 20E activates EcRE to express RFP in a concentration-dependent (0.1–5 μm 20E for 18 h) and time-dependent (1–48 h at 1 μm 20E) manner, as determined by Western blot analysis using an anti-RFP antibody. Bar graph shows the mean ratios of the abundance of induced RFP to that of β-actin in each sample relative to DMSO control cells quantified by Quantity One (Bio-Rad). B, effects of the knockdown or overexpression of PLCG1 on 20E-induced RFP expression. Panel a, PLCG1 knockdown through transfection with dsPLCG1 and induction with 1 μm 20E for 18 h. Panel b, overexpression of PLCG1-His and PLCG1ΔSH2-His. Panel c, qRT-PCR analysis of PLCG1 RNAi. The dsGFP was used as a control. Panel d, detection of the overexpression of PLCG1-His and PLCG1ΔSH2-His using Western blot. C and D, cells transfected with pIEx-HR3pro-RFP were pretreated with different inhibitors for 30 min, and 1 μm 20E was then added to culture medium for 18 h. E, effect of the depletions of DopEcR, ErGPCR, and Gαq on the RFP expression. F, DAG analog PMA and calcium ionophore ionomycin mimicked 20E signal for RFP up-regulation. Cells were incubated with 50 ng/ml PMA and 5 μm ionomycin for 18 h. The asterisk indicates the significant difference (*, p < 0.05) statistically analyzed by Student's t test based on three independent replicate experiments.
Given the regulation of PLCG1 by GPCRs and its role in calcium increase, some inhibitors were used to determine whether PLCG1-regulated transcriptional activation driven by EcRE relies on GPCR, calcium, and PKC signaling. The heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin (35) repressed the 20E-induced RFP expression compared with the control induced with 20E. Similarly, the GPCR inhibitor suramin and the PLC inhibitor U73122 suppressed RFP expression compared with 20E induction (Fig. 7C). Moreover, calcium signal inhibitors (including the IP3R inhibitor XeC, the T-type voltage-gated calcium channel inhibitor FL, and the TRPC3 channel inhibitor Pyr3) and a PKC inhibitor (chelerythrine chloride, CC) (36) restrained the 20E-activated RFP expression compared with the 20E-treated control (Fig. 7D). The depletions of ErGPCR and Gαq also inhibited RFP expression (Fig. 7E), indicating that ErGPCR and Gαq might be the upstream signaling of PLCG1 and calcium signals.
To verify the roles of DAG and calcium in PKC activation, the DAG analog PMA (37) and calcium ionophore ionomycin (38) were used to mimic the 20E-induced increase of DAG and calcium. The combination of PMA and ionomycin significantly induced expression of RFP compared with the individual use of the two chemicals (Fig. 7F). Therefore, through DAG and calcium, 20E activates PKC.
PLCG1 Regulates PKC Phosphorylation of USP1 via Calcium Signaling
Because 20E regulates Drosophila USP PKC phosphorylation for gene transcription in the 20E pathway (18),we investigated whether PLCG1 and its triggered calcium signaling regulate USP1 PKC phosphorylation. USP1 tagged with His6 was expressed in HaEpi cells (USP1-His). Compared with the negative control (DMSO), treatment with 0.1 μm 20E for 15 min induced USP phosphorylation, which appeared as an upper band, and 20E induced USP phosphorylation in a time- and concentration-dependent manner (Fig. 8A). Treatment with λ protein phosphatase and the PKC inhibitor CC confirmed that the upper band was the result of PKC-mediated phosphorylation (Fig. 8B). Further results showed that GPCR, PLC, IP3R, and calcium channel inhibitors restrained the 20E-induced USP1 phosphorylation (Fig. 8C, panel a). Moreover, PLCG1 knockdown repressed the phosphorylation of USP1, whereas the overexpression of PLCG1-His promoted USP1 phosphorylation. However, the PLCG1ΔSH2-His mutant, which does not contain the two SH2 domains, did not promote USP1 phosphorylation (Fig. 8C, panel b). In addition, the depletions of ErGPCR and Gαq by dsRNAs transfection suppressed USP1 phosphorylation; however, DopEcR knockdown had no effect on this phosphorylation (Fig. 8D). Compared with the addition of PMA or ionomycin individually, the combination of these two chemicals more significantly mimicked the 20E signal for USP1 phosphorylation (Fig. 8E). Therefore, 20E regulates PKC phosphorylation of USP1 via ErGPCR, Gαq, PLCG1, and calcium signaling pathways.
FIGURE 8.

USP1 phosphorylation is induced by 20E regulation via PLCG1-triggered calcium signaling. A, Western blot assay for the detection of 20E-induced USP1-His phosphorylation in HaEpi cells using an antibody against the His tag. USP1-P, phosphorylated USP1. The cells were transfected with pIEx-USP1-His plasmids (2.5 μg/ml) and treated with 20E at different concentrations (0.1–5 μm 20E for 1 h) and different times (0.25–24 h with 1 μm). Bar graphs indicate the mean ratios of the abundance of phosphorylated USP1 (USP1-P) to that of nonphosphorylated USP1 quantified by Quantity One (Bio-Rad). B, USP1-overexpressing cells were induced with 1 μm 20E for 1 h, and the protein was extracted and incubated with λ protein phosphatase (λPPase) (5 μm) for 30 min. The PKC inhibitor CC (5 μm) was added before induction with 20E to determine the PKC-mediated USP1 phosphorylation. C, effects of inhibitors and PLCG1 on 20E-induced USP1 phosphorylation. Panel a, 50 μm suramin, 10 μm U73122, 3 μm XeC, 50 μm FL, and 10 μm Pyr3 were used to pretreat the cells for 30 min before 20E induction (1 μm for 1 h). Panel b, PLCG1 depletion by RNAi and overexpression of PLCG1-His or PLCG1ΔSH2-His. D, silencing of DopEcR, ErGPCR, and Gαq by dsRNAs transfection determining 20E-induced USP1 phosphorylation. E, PMA (50 ng/ml) or ionomycin (5 μm) was used to treat cells for 1 h to mimic 20E induction for USP1 phosphorylation. The asterisk indicates the significant difference (*, p < 0.05) statistically analyzed by Student's t test based on three independent replicate experiments.
20E-mediated PKC Phosphorylation of USP1 at Ser-21 Is Necessary for Its Binding to EcRE
To identify the site of 20E-induced PKC phosphorylation of USP1, we compared the possible PKC phosphorylation site in USP1 through sequence alignment with the protein sequences in different insect orders. H. armigera USP1 does not have the PKC phosphorylation site Ser-35 identified in Drosophila (18) but does have a Ser at position 21, which is conserved in various orders (Fig. 9). The prediction of phosphorylation sites using NetphosK 2.0 indicated that this Ser-21 site has a high PKC phosphorylation score in different insect orders (Table 2). Therefore, USP1S21A-His, which has a point mutation by replacing Ser-21 with Ala, was overexpressed in HaEpi cells. 20E induced wild-type USP1-His phosphorylation at concentrations ranging from 0.1 to 2 μm; however, 20E could not induce the phosphorylation of USP1S21A-His at any of the tested concentrations (Fig. 10A, panel a). Phosphorylation level analysis experiments conformed to this result (Fig. 10A, panel b). USP1-His overexpression enhanced the 20E-induced RFP expression through the activation of EcRE in the pIEx-HR3pro-RFP plasmid, whereas USP1S21A-His did not promote the RFP up-regulation induced by 20E (Fig. 10B). These results confirm that 20E induces PKC phosphorylation of USP1 at Ser-21, which is critical to its function in the activation of EcRE.
FIGURE 9.

Alignment of H. armigera USP1 with USP1 from other insects. USP1 amino acids sequences were performed alignment from different insects, including H. armigera (ACD74808.1), Tribolium castaneum (NP_001107766.2), Tenebrio molitor (CAB75361.1), Leptinotarsa decemlineata (BAD99298.1), Apis mellifera (NP_001011634.1), Melipona scutellaris (AAW02952.1), Scaptotrigona depilis (ABB00308.1), Polyrhachis vicina (AGF50212.1), Gryllus firmus (ADL09403.1), Locusta migratoria (AAQ55293.1), Nizara viridula (ADQ43369.1), Acyrthosiphon pisum (NP_001155140.1), Blattella germanica (CAH69897.1), Periplaneta americana (BAM63276.2M.), Aedes aegypti (AAG24886.1), Anopheles gambiae (XP_320944.5), Culex quinquefasciatus (XP_001866328.1), D. melanogaster (AAF45707.1). The green box indicates the phosphorylation site identified in this study in H. armigera USP1 at Ser-21, and the blue box indicates the USP phosphorylation site Ser-35 in D. melanogaster. These data just show the alignment of N-terminal amino acids.
TABLE 2.
USP1 PKC phosphorylation sites scores in different insects
Compared with the H. armigera USP1 putative PKC phosphorylation site at Ser-21, the homologous sites of USP1 in different insects was analyzed via phosphorylation sites prediction by NetphosK 2.0.
| Species | GenBankTM no. | Sequence | Position | Score |
|---|---|---|---|---|
| H. armigera | ACD74808.1 | MSPMSPPEM | 21 | 0.996 |
| Spodoptera exigua | ACD39740.1 | MSPMSPPEM | 21 | 0.996 |
| Spodoptera frugiperda | AFX60116.1 | MSPMSPPEM | 73 | 0.996 |
| B. mori | NP_001037470.1 | MSPMSPPEM | 74 | 0.996 |
| Omphisa fuscidentalis | AGT02382.1 | MSPMSPPEM | 77 | 0.996 |
| Danaus plexippus | EHJ73160.1 | MSPMSPPEM | 76 | 0.996 |
| Plodia interpunctella | AAT44330.1 | MSPMSPPEM | 21 | 0.996 |
| Manduca sexta | AAB64234.1 | MSPMSPPEM | 73 | 0.996 |
| C. quinquefasciatus | XP_001866328.1 | MGPLSPPDM | 16 | 0.990 |
| A. aegypti | AAG24886.1 | MGPVSPQDM | 45 | 0.988 |
| A, gambiae | XP_320944.5 | MGPLSPPDL | 51 | 0.991 |
| T. castaneum | NP_001107766.2 | GAPQSPLDM | 24 | 0.934 |
| T. molitor | CAB75361.1 | GAPHSPLDM | 26 | 0.989 |
| L. decemlineata | BAD99298.1 | LGPQSPMDI | 8 | 0.996 |
| A. mellifera | NP_001011634.1 | VGPQSPLDM | 46 | 0.995 |
| Melipona scutellaris | AAW02952.1 | VGPQSPLDM | 46 | 0.995 |
| S. depilis | ABB00308.1 | VGPQSPLDM | 46 | 0.995 |
| P. vicina | AGF50212.1 | VGPQSPLDM | 41 | 0.995 |
| G. firmus | ADL09403.1 | GGPQSPLDM | 20 | 0.991 |
| L. migratoria | AAQ55293.1 | MGPQSPLDM | 23 | 0.995 |
| N. viridula | ADQ43369.1 | VGPQSPLDM | 21 | 0.986 |
| A. pisum | NP_001155140.1 | MGPQSPLDL | 43 | 0.995 |
| B. germanica | CAH69897.1 | MEPQSPLDM | 25 | 0.996 |
| P. americana | BAM63276.2 | MEPQSPLDM | 25 | 0.986 |
FIGURE 10.

20E induces USP1 phosphorylation at Ser-21 to regulate the binding of USP1 to EcRE. A, identification of USP1 phosphorylation site at Ser-21. Panel a, phosphorylated USP1-His and nonphosphorylated USP1S21A-His (Ser-21 to Ala-21) after 20E induction (0.1 or 2 μm). Panel b, number of moles of phosphorus per mol of purified USP1-His or USP1S21A-His was determined using the phosphoprotein phosphate estimation assay kit. USP1-His or USP1S21A-His was overexpressed in HaEpi cells and treated with 1 μm 20E or equivalent DMSO for 1 h. B, pIEx-HR3pro-RFP and pIEx-USP1-His or pIEx-USP1S21A-His were co-transfected in HaEpi cells to test the 20E-induced RFP expression driven by EcRE. C, EMSA was performed to test the binding ability of USP1-His and USP1S21A-His to EcRE. USP1-His and USP1S21A-His were overexpressed in HaEpi cells, and these cells were incubated with 1 μm 20E for 1 h. The protein was purified using a His-Bind resin in a tube and incubated with Dig-labeled EcRE. A 50-fold excess of the amount of unlabeled EcRE was used for the competition binding experiment. The protein-overexpressing cells received equivalent DMSO as a control. D, Western blot showing the CDK10 phosphorylation variation after the knockdown of DopEcR, ErGPCR, and Gαq by dsRNAs transfection. The cells transfected with dsRNAs for 48 h were induced by 1 μm 20E for additional 1 h. Western blot was performed by using H. armigera CDK10 antibody. The PKC inhibitor CC (5 μm) was used to treat the cells before induction with 20E to detect the PKC-mediated CDK10 phosphorylation. Bar graphs indicate the mean ratios of the abundance of phosphorylated CDK10 (CDK10-P) to that of nonphosphorylated CDK10 quantified by Quantity One (Bio-Rad). E, silencing of CDK10 had no effect on the 20E-induced USP1 phosphorylation. The cells expressing USP1-His were transfected with dsCDK10 for 24 h and were then induced by 1 μm 20E for an additional 1 h. The asterisk indicates the significant difference (*, p < 0.05) statistically analyzed by Student's t test based on three independent replicate experiments.
To verify the function of PKC-phosphorylated USP1 in the activation of EcRE, the EcRE binding ability of USP1 was assayed through an EMSA using HaEpi cells overexpressing wild-type USP1-His and mutant USP1S21A-His after being purified using a His-Bind resin in a tube. 20E induced a shift band in the incubation of USP1-His with the Dig-labeled EcRE probe compared with the DMSO control, and this shift band disappeared in response to competition with the unlabeled EcRE probe. In contrast, 20E could not induce a shift band through the incubation of USP1S21A-His with the Dig-labeled EcRE probe (Fig. 10C). These results suggest that PKC-phosphorylated USP1 binds to EcRE for subsequent gene transcription.
Given that 20E could induce the rapid phosphorylation of CDK10 to regulate gene expression via the GPCR-PLC-Ca2+ signal pathway (14), we detected whether this ErGPCR-PLCG1-Ca2+ pathway could mediate CDK10 phosphorylation and the relationship between CDK10 and USP1 phosphorylation. RNAi results showed that the depletions of ErGPCR and Gαq repressed CDK10 phosphorylation, but DopEcR knockdown did not inhibit the phosphorylation. In the meantime, PKC inhibitor CC restrained the phosphorylation of CDK10 (Fig. 10D). However, silencing of CDK10 had no effect on USP1 phosphorylation (Fig. 10E). Therefore, through the ErGPCR-, Gαq-, PLCG1-, Ca2+-, and PKC-regulated pathways, 20E mediates the independent phosphorylation of CDK10 and USP1.
DISCUSSION
It is known that 20E regulates gene expression by forming an EcR-USP heterodimeric transcription complex in the genomic pathway (26). Through PKC, 20E regulates USP phosphorylation to mediate gene expression in the 20E signaling pathway in Drosophila (18). PKC activation relies on calcium signaling through the cell membrane (3), and 20E can transmit signals through GPCRs in the cell membrane to induce calcium influx (11, 39). However, the connection between the nuclear receptor-regulated genomic pathway and the membrane-mediated nongenomic pathway in 20E signaling is not clear. This study demonstrates that 20E regulates PLCG1 tyrosine phosphorylation through ErGPCR, Gαq, and Src family kinases, which results in the triggering of calcium influx and that PKC mediates USP1 phosphorylation to promote 20E-inducible gene expression to modulate insect metamorphosis.
PLCG1 Responds to 20E Induction by Increasing Transcription and Migrating toward the Cell Membrane
In human T cells, steroid hormone vitamin D induces the expression of PLCG1 to positively regulate T cell antigen receptor signaling and the activation of human T cells (40). We found that PLCG1 exhibits high expression levels during molting and metamorphosis and is up-regulated by steroid hormone 20E. These studies suggest a common phenomenon that the PLCG1 expression level is up-regulated by steroid hormones.
In chick skeletal muscle cells, steroid hormone 1α,25(OH)2-vitamin D3 induces the tyrosine phosphorylation and membrane translocation of PLCG through the tyrosine kinase c-Src, and this effect is dependent on the SH2 domain (41). The data associated with the mechanism underlying PLCG1 phosphorylation indicate that the two SH2 domains in PLCG1 are essential (4). Studies of the T cell antigen receptor signaling pathway have shown that the T cell antigen receptor regulates PLCG1 phosphorylation by promoting the association of the PLCG1 SH2 domains with a tyrosine kinase (42). We found that 20E induces the rapid migration of PLCG1 toward the cell membrane through the rapid regulation of the tyrosine phosphorylation of the SH2 domains in the protein. Deletion of the SH2 domains results in the loss of tyrosine phosphorylation, inhibits the 20E-induced PLCG1 migration toward the cell membrane, and decreases its function in the triggering of calcium influx and transcriptional activation. By disrupting receptor-G protein coupling and inhibiting guanine nucleotide release (27), suramin blocks different types of GPCRs, including bovine rhodopsin (43), adrenergic receptor (44), δ-opioid receptors (45), A1-adenosine, and D2 dopamine receptors (28, 46). In this study, we found that suramin suppressed the 20E-induced calcium increase, PLCG1 tyrosine phosphorylation, RFP expression driven by EcRE, and USP1 phosphorylation. Moreover, the depletions of ErGPCR and Gαq also restrain these 20E-regulated nongenomic and genomic pathways. Taken together, we believe that GPCR, particularly ErGPCR, participates in 20E signaling.
ErGPCR takes part in 20E signaling on the plasma membrane, but it does not bind to 20E under our experimental conditions (15). This study presents further evidence that ErGPCR is the upstream molecule of PLCG1 in 20E signaling. However, the depletion of H. armigera DopEcR does not suppress PLCG1 phosphorylation, although 20E binds to DopEcR to regulate nongenomic action in Drosophila (11). The reason might be the difference of the insect species or the involvement of various GPCRs in 20E signaling.
Studies show that GPCR regulates PLCG1 by activating Src or RTK, and the tyrosine kinase has a more direct effect on PLCG1 (5, 6). We found that the RTK inhibitor SU6668 cannot restrain the tyrosine phosphorylation of PLCG1, whereas Src inhibitor PP2 suppressed the phosphorylation. Therefore, the insect steroid hormone 20E induces the PLCG1 tyrosine phosphorylation at SH2 domains through ErGPCR and Src family kinases to direct PLCG1 migrating toward membrane.
PLCG1 Participates in 20E-induced Ca2+ Influx
In response to steroid binding to a GPCR, the cytosolic Ca2+ levels are up-regulated through the release of Ca2+ from the endoplasmic reticulum through IP3R and/or the influx of extracellular Ca2+ through calcium channels in mammals (47). Tyrosine-phosphorylated PLCG1 hydrolyzes phosphatidylinositol 4,5-bisphosphate to produce IP3 and DAG (48), and an intracellular calcium signal is then induced by the binding of IP3 to IP3R in the endoplasmic reticulum (49) and/or the opening of plasma membrane calcium channels (50). The 20E-induced calcium influx has been described in mammal skeletal muscle cells (39) and prothoracic glands of Bombyx mori (51). In the silkworm anterior silk glands, the T-type Ca2+ channel inhibitor restrains 20E-induced programmed cell death (13). These data suggest that 20E may regulate calcium signaling through GPCR, IP3R, and calcium channels.
Our results show that inhibitors of GPCR, PLC, IP3R, and calcium channels decrease the calcium influx, indicating the possible involvement of GPCR-, PLC-, IP3R-, and calcium channel-related pathways in the 20E-induced calcium influx. In fact, ErGPCR participates in the regulation of calcium signal (15), and Gαq may also take part in the process (data not shown). In this work, we found PLCG1 is critical for the 20E-induced Ca2+ release from the endoplasmic reticulum and influx from the cell environment. Interestingly, both the calcium channel inhibitors FL and Pyr3 block the 20E-induced Ca2+ influx, which implies the involvement of T-type channels and TRP channels in the 20E-induced Ca2+ influx. This is not a special case, because TRP channel family members share functional roles with the T-type Ca2+ channel and also that both are expressed in cancer and hypertension (52, 53). Given that SKF96365, a blocker of TRP channels, also inhibits the T-type channel (54), we do not exclude that the results that both Pyr3 and FL block the 20E-induced influx are due to the nonspecific inhibitors. The IP3R inhibitor XeC represses intracellular Ca2+ release and extracellular Ca2+ influx, suggesting that the 20E-induced extracellular Ca2+ influx relies on the stored Ca2+ in the cells. These data reveal the axis of 20E-induced Ca2+ influx via GPCR, PLCG1, IP3R, and calcium channels.
PLCG1-regulated Ca2+ Influx Modulates USP1 PKC Phosphorylation at Ser-21 for Gene Transcription
In insects, 20E is able to induce USP phosphorylation for 20E-induced gene transcription in a PKC-dependent manner (18), but the mechanism is unclear. Ca2+ and DAG activate PKC by binding to the PKC C1 and C2 domains (49), and the downstream signals, including transcription, immune responses, and cell growth, are then modulated (55). In addition, calcium and PKC activation also act as parallel signaling cascades downstream of PLCG (38). We found that applying the DAG analog PMA or the calcium ionophore ionomycin alone is unable to activate 20E-induced RFP expression and USP1 phosphorylation. However, costimulation with both PMA and ionomycin is required for mimicking the USP1 phosphorylation and RFP up-regulation responses to 20E. Thus, it is possible that increased Ca2+ and PKC activation act as linearly in 20E signaling. Moreover, we found that USP1 is phosphorylated at Ser-21 after 20E induction via the ErGPCR-, Gαq-, Src-, PLCG1-, IP3R-, Ca2+-, and PKC-axis. The inhibition of one of these steps blocks the 20E-induced USP1 PKC phosphorylation. Therefore, PLCG1 links ErGPCR, Ca2+ influx, and USP1 PKC phosphorylation in 20E signaling.
USP and EcR form a heterodimeric transcription complex that binds to EcRE for gene transcription (25). Drosophila USP and EcR may use similar 20E-responsive EcREs to establish protein-protein contacts, suggesting that the binding of USP to EcRE is necessary for gene transcription (56). In Drosophila, the PKC-mediated USP phosphorylation at Ser-35 regulates 20E-inducible gene expression (18). We found that H. armigera USP1 is phosphorylated via PKC mediation at Ser-21 and that this phosphorylation is necessary for its transcriptional activation driven by EcRE. The EMSA results suggest that the binding of USP1 to EcRE requires the phosphorylation of USP1 at Ser-21. Our data indicate that the regulation of 20E-induced gene transcription depends on the binding of PKC-phosphorylated USP1 to EcRE.
In a recent study, we reported that 20E induced the rapid phosphorylation of CDK10 through GPCR/PLC/Ca2+ signaling to promote gene expression (14). In this work we further found that the 20E-regulated CDK10 phosphorylation depends on the ErGPCR-, Gαq-, Src-, PLCG1-, IP3R-, Ca2+-, and PKC-axis. Although both the phosphorylation of CDK10 and USP1 contribute to the 20E-mediated gene expression, CDK10 does not regulate USP1 phosphorylation. Our results suggest that 20E may regulate gene transcription via a typical nongenomic pathway.
In summary, the data presented in this work indicate that PLCG1 connects 20E signaling in the cell membrane to the gene transcription in the nucleus. We propose a model to outline the function of PLCG1 in the 20E signaling pathway (Fig. 11). Through ErGPCR, Gαq, and Src family kinases, 20E activates PLCG1 by mediating the tyrosine phosphorylation at its SH2 domains. The activated PLCG1 migrates toward the cell membrane to initiate intracellular Ca2+ signaling and calcium channel-controlled Ca2+ influx, which trigger PKC-mediated USP1 phosphorylation to modulate USP1 binding to EcRE for subsequent gene transcription. This work provides evidence that 20E regulates the genomic pathway for gene transcription through a PLCG1-dependent nongenomic pathway.
FIGURE 11.

Schematic representation of how PLCG1 regulates USP1 phosphorylation to mediate 20E-induced gene expression for metamorphosis. Through ErGPCR and Gαq, 20E promotes Src family kinases to up-regulate the tyrosine phosphorylation of PLCG1 and induces the migration of phosphorylated PLCG1 toward the cell membrane to produce IP3 and DAG. Then cytosolic calcium signals a fast increase, including the release of the intracellular Ca2+ and the influx of the extracellular Ca2+ via calcium channels (T-type calcium channels and/or TRP channels). The increased Ca2+ and DAG activate PKC to regulate the phosphorylation of USP1 and CDK10 for gene transcription and insect metamorphosis. This chart is based on the models reported by our laboratory, including the ErGPCR-regulated nongenomic action (15), the interaction between Hsc70 and USP1 (57), and the participation of CDK10-Hsp90-Hsc70-EcRB1 in the 20E transcriptional complex (14).
This work was supported by National Natural Science Foundation of China Grant 31230067, National Basic Research Program of China 973 Program, Grant 2012CB114101, and Ph.D. Programs Foundation of Ministry of Education of China Grant 20120131110025.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) KF800735
- PLC
- phospholipase C
- IP3
- inositol 1,4,5-triphosphate
- IP3R
- IP3 receptor
- CC
- chelerythrine chloride
- CDK10
- cyclin-dependent kinase 10
- DAG
- diacylglycerol
- EcR
- ecdysone receptor
- EcRE
- ecdysone-response element
- GPCR
- G-protein-coupled receptor
- ErGPCR
- ecdysone-responsible GPCR
- DopEcR
- dopamine/ecdysteroid receptor
- FL
- flunarizine dihydrochloride
- HaEpi
- H. armigera epidermal cell line
- HR3
- hormone receptor 3
- PLCG1
- phospholipase Cγ1
- PMA
- phorbol 12-myristate 13-acetate
- Pyr3
- pyrazole compound
- RFP
- red fluorescent protein
- Src
- cytosolic tyrosine kinase
- SH2 domain
- Src homology 2 domain
- 20E
- 20-hydroxyecdysone
- USP
- ultraspiracle
- WGA
- wheat germ agglutinin
- XeC
- xestospongin C
- qRT
- quantitative RT
- RTK
- receptor tyrosine kinase
- Dig
- digoxigenin
- CC
- chelerythrine chloride
- TRP
- transient receptor potential.
REFERENCES
- 1. Janetopoulos C., Devreotes P. (2006) Phosphoinositide signaling plays a key role in cytokinesis. J. Cell Biol. 174, 485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kadamur G., Ross E. M. (2013) Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154 [DOI] [PubMed] [Google Scholar]
- 3. Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 [DOI] [PubMed] [Google Scholar]
- 4. Rhee S. G. (2001) Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70, 281–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haendeler J., Yin G., Hojo Y., Saito Y., Melaragno M., Yan C., Sharma V. K., Heller M., Aebersold R., Berk B. C. (2003) GIT1 mediates Src-dependent activation of phospholipase Cγ by angiotensin II and epidermal growth factor. J. Biol. Chem. 278, 49936–49944 [DOI] [PubMed] [Google Scholar]
- 6. Luttrell L. M., Daaka Y., Lefkowitz R. J. (1999) Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr. Opin. Cell Biol. 11, 177–183 [DOI] [PubMed] [Google Scholar]
- 7. Prossnitz E. R., Arterburn J. B., Smith H. O., Oprea T. I., Sklar L. A., Hathaway H. J. (2008) Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 70, 165–190 [DOI] [PubMed] [Google Scholar]
- 8. Schauer S., Callender J., Henrich V. C., Spindler-Barth M. (2011) The N terminus of ecdysteroid receptor isoforms and ultraspiracle interacts with different ecdysteroid response elements in a sequence-specific manner to modulate transcriptional activity. J. Steroid Biochem. Mol. Biol. 124, 84–92 [DOI] [PubMed] [Google Scholar]
- 9. Yamanaka N., Rewitz K. F., O'Connor M. B. (2013) Ecdysone control of developmental transitions: lessons from Drosophila research. Annu. Rev. Entomol. 58, 497–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riddiford L. M., Hiruma K., Zhou X., Nelson C. A. (2003) Insights into the molecular basis of the hormonal control of molting and metamorphosis from Manduca sexta and Drosophila melanogaster. Insect Biochem. Mol. Biol. 33, 1327–1338 [DOI] [PubMed] [Google Scholar]
- 11. Srivastava D. P., Yu E. J., Kennedy K., Chatwin H., Reale V., Hamon M., Smith T., Evans P. D. (2005) Rapid, nongenomic responses to ecdysteroids and catecholamines mediated by a novel Drosophila G-protein-coupled receptor. J. Neurosci. 25, 6145–6155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iga M., Iwami M., Sakurai S. (2007) Nongenomic action of an insect steroid hormone in steroid-induced programmed cell death. Mol. Cell. Endocrinol. 263, 18–28 [DOI] [PubMed] [Google Scholar]
- 13. Manaboon M., Iga M., Iwami M., Sakurai S. (2009) Intracellular mobilization of Ca2+ by the insect steroid hormone 20-hydroxyecdysone during programmed cell death in silkworm anterior silk glands. J. Insect Physiol. 55, 122–128 [DOI] [PubMed] [Google Scholar]
- 14. Liu W., Cai M. J., Wang J. X., Zhao X. F. (2014) In a non-genomic action, steroid hormone 20-hydroxyecdysone induces phosphorylation of cyclin-dependent kinase 10 to promote gene transcription. Endocrinology 10.1210/en.2013-2020 [DOI] [PubMed] [Google Scholar]
- 15. Cai M. J., Dong D. J., Wang Y., Liu P. C., Liu W., Wang J. X., Zhao X. F. (2014) G-protein-coupled receptor participates in 20-hydroxyecdysone signaling on the plasma membrane. Cell Commun. Signal. 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicolaï M., Bouhin H., Quennedey B., Delachambre J. (2000) Molecular cloning and expression of Tenebrio molitor ultraspiracle during metamorphosis and in vivo induction of its phosphorylation by 20-hydroxyecdysone. Insect Mol. Biol. 9, 241–249 [DOI] [PubMed] [Google Scholar]
- 17. Rauch P., Grebe M., Elke C., Spindler K. D., Spindler-Barth M. (1998) Ecdysteroid receptor and ultraspiracle from Chironomus tentans (Insecta) are phosphoproteins and are regulated differently by molting hormone. Insect Biochem. Mol. Biol. 28, 265–275 [DOI] [PubMed] [Google Scholar]
- 18. Wang S., Wang J., Sun Y., Song Q., Li S. (2012) PKC-mediated USP phosphorylation at Ser-35 modulates 20-hydroxyecdysone signaling in Drosophila. J. Proteome Res. 11, 6187–6196 [DOI] [PubMed] [Google Scholar]
- 19. Wu K. M., Lu Y. H., Feng H. Q., Jiang Y. Y., Zhao J. Z. (2008) Suppression of cotton bollworm in multiple crops in China in areas with Bt toxin-containing cotton. Science 321, 1676–1678 [DOI] [PubMed] [Google Scholar]
- 20. Zhao X. F., Wang J. X., Wang Y. C. (1998) Purification and characterization of a cysteine proteinase from eggs of the cotton boll worm, Helicoverpa armigera. Insect Biochem. Mol. Biol. 28, 259–264 [Google Scholar]
- 21. Shao H. L., Zheng W. W., Liu P. C., Wang Q., Wang J. X., Zhao X. F. (2008) Establishment of a new cell line from lepidopteran epidermis and hormonal regulation on the genes. PLoS One 3, e3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 23. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 24. Morris D. D., Gibbs M. D., Chin C. W., Koh M. H., Wong K. K., Allison R. W., Nelson P. J., Bergquist P. L. (1998) Cloning of the xynB gene from Dictyoglomus thermophilum Rt46B. 1 and action of the gene product on kraft pulp. Appl. Environ. Microbiol. 64, 1759–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lan Q., Hiruma K., Hu X., Jindra M., Riddiford L. M. (1999) Activation of a delayed-early gene encoding MHR3 by the ecdysone receptor heterodimer EcR-B1-USP-1 but not by EcR-B1-USP-2. Mol. Cell. Biol. 19, 4897–4906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hiruma K., Riddiford L. M. (2009) The molecular mechanisms of cuticular melanization: the ecdysone cascade leading to dopa decarboxylase expression in Manduca sexta. Insect Biochem. Mol. Biol. 39, 245–253 [DOI] [PubMed] [Google Scholar]
- 27. Chung W. C., Kermode J. C. (2005) Suramin disrupts receptor-G protein coupling by blocking association of G protein α and βγ subunits. J. Pharmacol. Exp. Ther. 313, 191–198 [DOI] [PubMed] [Google Scholar]
- 28. Waldhoer M., Bofill-Cardona E., Milligan G., Freissmuth M., Nanoff C. (1998) Differential uncoupling of A1 adenosine and D2 dopamine receptors by suramin and didemethylated suramin (NF037). Mol. Pharmacol. 53, 808–818 [PubMed] [Google Scholar]
- 29. Abdollahi A., Lipson K. E., Han X., Krempien R., Trinh T., Weber K. J., Hahnfeldt P., Hlatky L., Debus J., Howlett A. R., Huber P. E. (2003) SU5416 and SU6668 attenuate the angiogenic effects of radiation-induced tumor cell growth factor production and amplify the direct anti-endothelial action of radiation in vitro. Cancer Res. 63, 3755–3763 [PubMed] [Google Scholar]
- 30. Selbach M., Moese S., Hauck C. R., Meyer T. F., Backert S. (2002) Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem. 277, 6775–6778 [DOI] [PubMed] [Google Scholar]
- 31. Yule D. I., Williams J. A. (1992) U73122 inhibits Ca2+ oscillations in response to cholecystokinin and carbachol but not to JMV-180 in rat pancreatic acinar cells. J. Biol. Chem. 267, 13830–13835 [PubMed] [Google Scholar]
- 32. De Smet P., Parys J. B., Callewaert G., Weidema A. F., Hill E., De Smedt H., Erneux C., Sorrentino V., Missiaen L. (1999) Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca2+ pumps. Cell Calcium 26, 9–13 [DOI] [PubMed] [Google Scholar]
- 33. Kiyonaka S., Kato K., Nishida M., Mio K., Numaga T., Sawaguchi Y., Yoshida T., Wakamori M., Mori E., Numata T., Ishii M., Takemoto H., Ojida A., Watanabe K., Uemura A., Kurose H., Morii T., Kobayashi T., Sato Y., Sato C., Hamachi I., Mori Y. (2009) Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. U.S.A. 106, 5400–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Terland O., Flatmark T. (1999) Drug-induced parkinsonism: cinnarizine and flunarizine are potent uncouplers of the vacuolar H+-ATPase in catecholamine storage vesicles. Neuropharmacology 38, 879–882 [DOI] [PubMed] [Google Scholar]
- 35. Liu W., Zhang F. X., Cai M. J., Zhao W. L., Li X. R., Wang J. X., Zhao X. F. (2013) The hormone-dependent function of Hsp90 in the crosstalk between 20-hydroxyecdysone and juvenile hormone signaling pathways in insects is determined by differential phosphorylation and protein interactions. Biochim. Biophys. Acta 1830, 5184–5192 [DOI] [PubMed] [Google Scholar]
- 36. Chmura S. J., Dolan M. E., Cha A., Mauceri H. J., Kufe D. W., Weichselbaum R. R. (2000) In vitro and in vivo activity of protein kinase C inhibitor chelerythrine chloride induces tumor cell toxicity and growth delay in vivo. Clin. Cancer Res. 6, 737–742 [PubMed] [Google Scholar]
- 37. Favaron M., Manev H., Siman R., Bertolino M., Szekely A. M., DeErausquin G., Guidotti A., Costa E. (1990) Down-regulation of protein kinase C protects cerebellar granule neurons in primary culture from glutamate-induced neuronal death. Proc. Natl. Acad. Sci. U.S.A. 87, 1983–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chatila T., Silverman L., Miller R., Geha R. (1989) Mechanisms of T cell activation by the calcium ionophore ionomycin. J. Immunol. 143, 1283–1289 [PubMed] [Google Scholar]
- 39. Gorelick-Feldman J., Cohick W., Raskin I. (2010) Ecdysteroids elicit a rapid Ca2+ flux leading to Akt activation and increased protein synthesis in skeletal muscle cells. Steroids 75, 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. von Essen M. R., Kongsbak M., Schjerling P., Olgaard K., Odum N., Geisler C. (2010) Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 11, 344–349 [DOI] [PubMed] [Google Scholar]
- 41. Buitrago C., González Pardo V., de Boland A. R. (2002) Nongenomic action of 1 α,25(OH)2-vitamin D3. Activation of muscle cell PLC γ through the tyrosine kinase c-Src and PtdIns 3-kinase. Eur. J. Biochem. 269, 2506–2515 [DOI] [PubMed] [Google Scholar]
- 42. Stoica B., DeBell K. E., Graham L., Rellahan B. L., Alava M. A., Laborda J., Bonvini E. (1998) The amino-terminal Src homology 2 domain of phospholipase Cγ1 is essential for TCR-induced tyrosine phosphorylation of phospholipase Cγ1. J. Immunol. 160, 1059–1066 [PubMed] [Google Scholar]
- 43. Lehmann N., Krishna Aradhyam G., Fahmy K. (2002) Suramin affects coupling of rhodopsin to transducin. Biophys. J. 82, 793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang R. R., Dehaven R. N., Cheung A. H., Diehl R. E., Dixon R. A., Strader C. D. (1990) Identification of allosteric antagonists of receptor-guanine nucleotide-binding protein interactions. Mol. Pharmacol. 37, 304–310 [PubMed] [Google Scholar]
- 45. Butler S. J., Kelly E. C., McKenzie F. R., Guild S. B., Wakelam M. J., Milligan G. (1988) Differential effects of suramin on the coupling of receptors to individual species of pertussis-toxin-sensitive guanine-nucleotide-binding proteins. Biochem. J. 251, 201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beindl W., Mitterauer T., Hohenegger M., Ijzerman A. P., Nanoff C., Freissmuth M. (1996) Inhibition of receptor/G protein coupling by suramin analogues. Mol. Pharmacol. 50, 415–423 [PubMed] [Google Scholar]
- 47. Marinissen M. J., Gutkind J. S. (2001) G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 22, 368–376 [DOI] [PubMed] [Google Scholar]
- 48. Nishibe S., Wahl M. I., Hernández-Sotomayor S. M., Tonks N. K., Rhee S. G., Carpenter G. (1990) Increase of the catalytic activity of phospholipase C-γ 1 by tyrosine phosphorylation. Science 250, 1253–1256 [DOI] [PubMed] [Google Scholar]
- 49. Rhee S. G., Bae Y. S. (1997) Regulation of phosphoinositide-specific phospholipase C isozymes. J. Biol. Chem. 272, 15045–15048 [DOI] [PubMed] [Google Scholar]
- 50. Putney J. W. (2002) PLC-γ: an old player has a new role. Nat. Cell Biol. 4, E280–281 [DOI] [PubMed] [Google Scholar]
- 51. Dedos S. G., Fugo H. (1999) Interactions between Ca2+ and cAMP in ecdysteroid secretion from the prothoracic glands of Bombyx mori. Mol. Cell. Endocrinol. 154, 63–70 [DOI] [PubMed] [Google Scholar]
- 52. Panner A., Cribbs L. L., Zainelli G. M., Origitano T. C., Singh S., Wurster R. D. (2005) Variation of T-type calcium channel protein expression affects cell division of cultured tumor cells. Cell Calcium 37, 105–119 [DOI] [PubMed] [Google Scholar]
- 53. Liu D., Yang D., He H., Chen X., Cao T., Feng X., Ma L., Luo Z., Wang L., Yan Z., Zhu Z., Tepel M. (2009) Increased transient receptor potential canonical type 3 channels in vasculature from hypertensive rats. Hypertension 53, 70–76 [DOI] [PubMed] [Google Scholar]
- 54. Singh A., Hildebrand M. E., Garcia E., Snutch T. P. (2010) The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 160, 1464–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Newton A. C. (2010) Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Devarakonda S., Harp J. M., Kim Y., Ozyhar A., Rastinejad F. (2003) Structure of the heterodimeric ecdysone receptor DNA-binding complex. EMBO J. 22, 5827–5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zheng W. W., Yang D. T., Wang J. X., Song Q. S., Gilbert L. I., Zhao X. F. (2010) Hsc70 binds to ultraspiracle resulting in the upregulation of 20-hydroxyecdsone-responsive genes in Helicoverpa armigera. Mol. Cell. Endocrinol. 315, 282–291 [DOI] [PubMed] [Google Scholar]