

Background: LRRK2 has emerged as one of the most relevant players in Parkinson disease pathogenesis.
Results: Enzyme kinetic and modeling studies elucidate the effect of the LRRK2 I2020T mutant on kinase activity.
Conclusion: The I2020T mutant stabilizes the active conformation and leads to increased kinase activity. As a result, it binds to type II inhibitors competitively.
Significance: This study may contribute to the development of new classes of inhibitors of LRRK2.
Keywords: Enzyme Inhibitors, Enzyme Kinetics, LRRK2, Molecular modeling, Parkinson Disease
Abstract
The effect of leucine-rich repeat kinase 2 (LRRK2) mutation I2020T on its kinase activity has been controversial, with both increased and decreased effects being reported. We conducted steady-state and pre-steady-state kinetic studies on LRRKtide and its analog LRRKtideS. Their phosphorylation differs by the rate-limiting steps: product release is rate-limiting for LRRKtide and phosphoryl transfer is rate-limiting for LRRKtideS. As a result, we observed that the I2020T mutant is more active than wild type (WT) LRRK2 for LRRKtideS phosphorylation, whereas it is less active than WT for LRRKtide phosphorylation. Our pre-steady-state kinetic data suggest that (i) the I2020T mutant accelerates the rates of phosphoryl transfer of both reactions by 3–7-fold; (ii) this increase is masked by a rate-limiting product release step for LRRKtide phosphorylation; and (iii) the observed lower activity of the mutant for LRRKtide phosphorylation is a consequence of its instability: the concentration of the active form of the mutant is 3-fold lower than WT. The I2020T mutant has a dramatically low KATP and therefore leads to resistance to ATP competitive inhibitors. Two well known DFG-out or type II inhibitors are also weaker toward the mutant because they inhibit the mutant in an unexpected ATP competitive mechanism. The I2020 residue lies next to the DYG motif of the activation loop of the LRRK2 kinase domain. Our modeling and metadynamic simulations suggest that the I2020T mutant stabilizes the DYG-in active conformation and creates an unusual allosteric pocket that can bind type II inhibitors but in an ATP competitive fashion.
Introduction
Parkinson disease (PD),2 characterized by tremor, rigidity, bradykinesia, and postural instability, is the second most common neurodegenerative disorder after Alzheimer disease. It affects over 1 million Americans and more than 60,000 patients are newly diagnosed each year. PD is caused by a loss of dopaminergic neurons in a part of the brain called substantia nigra. Once damaged, these neurons stop its normal function of producing dopamine, an essential neurotransmitter, and compromise the ability of the brain to control movement. Mutations in several genes have been genetically linked to PD in recent years (1). Among them, leucine-rich repeat kinase 2 (LRRK2) has emerged as one of the most relevant players in PD pathogenesis. At least 40 mutations in LRRK2 have been found in both familial and sporadic forms of PD, and have been associated with typical idiopathic, late-onset PD (2–6).
LRRK2 is a large and complex protein containing several independent domains, including a leucine-rich repeat (LRR) domain, a Roc domain followed by its associated COR domain, a kinase domain, and a C-terminal WD40 domain (2, 7). LRRK2 is unusual in that it encodes two distinct but functionally linked enzymes: a protein kinase and a GTPase (7–11). Several mutations demonstrate increased kinase activity that is correlated with increased neurotoxicity in central neurons (7, 12, 13). However, most of the LRRK2 mutations do not manifest their effects by simply increasing kinase activity. Among them, the effect of the mutation I2020T on kinase activity is controversial. Depending on the phosphoryl acceptors, both decreased and increased activity for the I2020T mutant has been reported (14–16). To better understand the effect of the I2020T mutant on kinase activity, in this study we (i) characterized phosphorylation of two peptide substrates LRRKtide and LRRKtideS, which were reported previously to have different rate-limiting steps (17); (ii) integrated steady-state and pre-steady-state kinetic studies and revealed the different effects of the I2020T mutant on the phosphoryl transfer and the product release steps; (iii) performed inhibition mechanism studies and showed that DFG-out inhibitors exhibit surprising variability in their inhibition mechanisms between wild type LRRK2 and the I2020T mutant; and (iv) conducted metadynamic simulation studies and provided structural support for the observed kinetics.
EXPERIMENTAL PROCEDURES
Materials
ATP, DTT, magnesium chloride, HEPES, and bovine serum albumin were purchased from Sigma. Peptides LRRKtide (RLGRDKYKTLRQIRQ) and LRRKtideS (RLGRDKYKSLRQIRQ) were purchased from American Peptide (Sunnyvale, CA). [γ-33P]ATP was from PerkinElmer Life Sciences. The N-terminal truncated wild type (t-WT) LRRK2 (amino acids 970–2527) and the N-terminal truncated mutation I2020T (t-I2020T) (amino acids 970–2527) expressed in a baculovirus system were purchased from Invitrogen.
Kinetic Analysis of LRRK2-catalyzed LRRKtide and LRRKtideS Phosphorylation
The kinase assays for phosphorylation of LRRKtide (RLGRDKYKTLRQIRQ) or LRRKtideS (RLGRDKYKSLRQIRQ) were conducted in buffer containing 20 mm HEPES (pH 7.4), 50 mm NaCl, 10 mm MgCl2, 1 mm DTT, 0.5 mg/ml of BSA, 1 mm β-Gly-PO4, LRRKtide, ATP, and [γ-33P]ATP. β-Gly-PO4 is a phosphatase inhibitor. Peptidic substrates and ATP were used at various concentrations as indicated under ”Results“ and the ratio of ATP to [γ-33P]ATP was kept constant at all ATP concentrations (250 μm ATP, 5 μCi of [γ-33P]ATP). The reactions were conducted in duplicate, initiated by the addition of 6 nm LRRK2, and incubated at room temperature for 150 min. The reactions were stopped by the addition of 20 mm EDTA and the mixture was transferred to a multiscreen PH filtration plate (Millipore, Billerica, MA) and washed six times with 75 mm H3PO4. The plate was dried, filters were removed, and the samples were counted with a scintillation counter. Background reactions were conducted in the absence of LRRK2. In all cases, reaction progress curves for production of phospho-LRRKtide or phospho-LRRKtideS were linear over at last 240 min and allowed calculation of initial velocities.
Modeling of LRRK2 Kinase Domain and Docking
The LRRK2 kinase domain, between residues 1859 and 2138, was modeled using Modeler version 9.11 (18–21). Modeling details have been described in detail previously (22). Briefly, the main variables in homology modeling are template selection and sequence alignment between the target and the template. B-raf kinase with 33% sequence identity to LRRK2 kinase domain was used as a template for homology modeling because this enzyme had the highest sequence similarity with LRRK2 around the active site region and the ATP binding hinge compared with other kinases. In addition, B-raf and LRRK2 inhibitors can cross-inhibit each other supporting our choice of template selection for modeling (details to be published elsewhere). Recently, the kinase domain of Roco4 from Dictyostelium discoidium (PDB code 4F0G), a closely related member of the LRRK2 family, was published (23) and we constructed a model of LRRK2 with this as a template and compared with our previous model. The comparison of the two LRRK2 models revealed that the overall Cα atom root mean square deviation was less than 1.3 Å. The I2020T mutant was introduced using PyMol and the resultant structure was subjected to molecular dynamics (MD) thermalization using the OPLS.2005 force field.
Docking of ATP was carried out using Glide version 2.2 (24) against the DFG-in conformation of LRRK2 (WT and I2020T), which treats the receptor rigidly. Docking of type II inhibitors used in this study was carried out using the induced fit docking (25) protocol implemented in the Schrödinger suite (25). Briefly, in the first stage of induced fit docking 20 initial poses are generated using Glide with a softened potential to allow for clashes with the receptor. For each of the top 20 poses from the initial softened-potential docking step, a cycle of protein side chain prediction and full residue minimization is performed using Prime to generate 20 induced-fit receptor structures (26). All residues having at least one atom within 5 Å of any atom from the 20 ligand poses are refined. Finally, the ligand is re-docked using Glide with default settings into each induced-fit receptor structure and a composite score that accounts for the protein/ligand interaction energy (GlideScore) and the total energy of the system (Prime energy) is used to rank the induced-fit structures.
Metadynamics Simulations
Metadynamics (27–37) simulations for the activation loop were carried out with Desmond version 3.0 using two collective variables (CVs). To test for robustness of the simulations and choice of CVs, we chose two independent pairs of CVs (one pair with distances and the other pair with torsions) and ran separate simulations. We chose the distance between the center of mass of a collection of atoms in the β-sheet (residues 1878–1906) and the N-terminal C-helix (residues 1915–1928) as the first CV denoted (s). For the second CV denoted (z), we chose the center of mass of a subset of residues in the activation loop (Tyr-2018, Ala-2021, Cys-2024, Arg-2026, and Met-2027) and the center of mass for the ATP binding hinge comprised of residues (Met-1947, Leu-1949, and Lys-1952). The distance between the N-terminal β-sheet domain and C-helix in the N-terminal domain describes the opening and closing motion of LRRK2 around the hinge region. The center of mass of the activation loop is the primary degree of freedom of interest for this study because it directly involves the transition from active to inactive form.
For measuring the dissociation rates for ATP a root mean square deviation of CV without including the H-atoms for ATP was chosen (s). In addition, a second angular CV was chosen using center of masses for two fixed water oxygen atoms and the center of mass for ATP (z).
The default equilibration protocol in Desmond was run before the metadynamics simulations, which relaxes the system in a gradual way with progressively weaker harmonic restraints and heating of the system from 0 to 300 K. The total length of each production simulation was 250 ns for the activation loop and 50 ns for ATP dissociation at a temperature of 300 K and pressure of 1 atm. A Gaussian biasing potential with a height of 0.01 kcal/mol was injected at 0.09-ps intervals throughout the simulation and the free-energy surface (FES) was reconstructed from these Gaussians. Multiple simulations were carried out using different initial random seeds. Convergence was observed in all cases and the FES constructed using these simulations were virtually identical. For the ATP dissociation experiment, the highest energy barrier was measured and used with Eyring's equation to calculate theoretical off-rates and half-lives.
Data Analysis, Basic Equations
Data were analyzed by nonlinear least-squares using either Sigma Plot or Grafit software packages. Standard kinetic mechanisms for two-substrate reactions and their rate equations are shown below.
Ping-Pong,
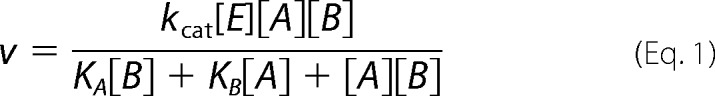 |
KA and KB are Michaelis constants.
Rapid equilibrium ordered,
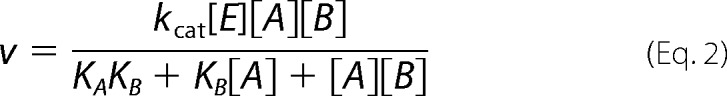 |
KA and KB are substrate dissociation constants from EA and EB, respectively.
Rapid equilibrium random/steady-state ordered,
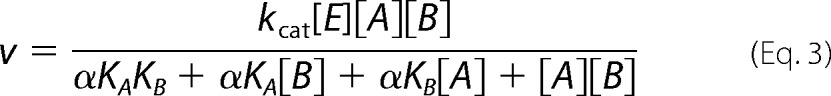 |
For rapid equilibrium systems, KA, KB, αKA, and αKB are substrate dissociation constants from EA, EB, and EAB; for steady-state systems, KA is a substrate dissociation constant from EA, αKA and αKB are Michaelis constants. See Segel (38) for definitions of mechanisms, substrate dissociation constants, and α.
RESULTS
t-WT LRRK2- and the Mutation t-I2020T-catalyzed Phosphorylation of LRRKtide, Initial Velocity Studies
In this study we used N-terminal truncated WT and the I2020T mutant, therefore they are referred to t-WT and t-I2020T, respectively. To determine the effect of the t-I2020T mutant on kinase activity, phosphorylation of LRRKtide (RLGRDKYKTLRQIRQ) was characterized. Initial velocities were measured as a function of LRRKtide concentration at several fixed concentrations of ATP for the t-I2020T mutant (data not shown). The complete data sets were subjected to global analysis by nonlinear least-squares fits to the three standard mechanisms (ping-pong, rapid equilibrium order, and random/steady-state ordered) using Equations 1–3. Statistically the data fits the random/steady-state ordered mechanism the best for the t-I2020T mutant, yielding the following estimates averaged from two independent experiments: kcat = 4.4 ± 0.1 min−1, KATP = 1.6 ± 0.4 μm, KLRRKtide = 110 ± 23 μm, and α = 7.3 ± 1.6 for the t-I2020T mutant. The same kinetic mechanism for t-WT was reported previously (17). The kinetic parameters of the t-I2020T mutant and those of t-WT are summarized in Table 1. A direct comparison of the t-I2020T mutant with t-WT LRRK2 is also shown in Fig. 1A, where the initial velocities were measured as a function of [ATP] at a saturating concentration of LRRKtide. The data reveal that (i) the t-I2020T mutant has lower kinase activity than t-WT and (ii) ATP binds to the mutant much tighter than t-WT, with a KATP value 40-fold lower than that of t-WT.
TABLE 1.
Initial velocity analysis for t-WT LRRK2 and the t-I2020T mutant
A represents ATP, B represents phosphoryl acceptors.
| LRRKtide |
LRRKtideS |
|||
|---|---|---|---|---|
| t-WTa | t-I2020T | t-WTa | t-I2020T | |
| kcat (min−1) | 8.1 ± 0.7 | 4.4 ± 0.1 | 0.31 ± 0.02 | 0.75 ± 0.03 |
| KA (μm) | 69 ± 6.6 | 1.6 ± 0.4 | 77 ± 19 | 7.6 ± 1.8 |
| KB (μm) | 87 ± 11 | 110 ± 23 | 554 ± 187 | 263 ± 65 |
| α | 1.8 ± 0.1 | 7.3 ± 1.6 | 2.6 ± 1.5 | 1.7 ± 0.1 |
a Liu et al. (17).
FIGURE 1.
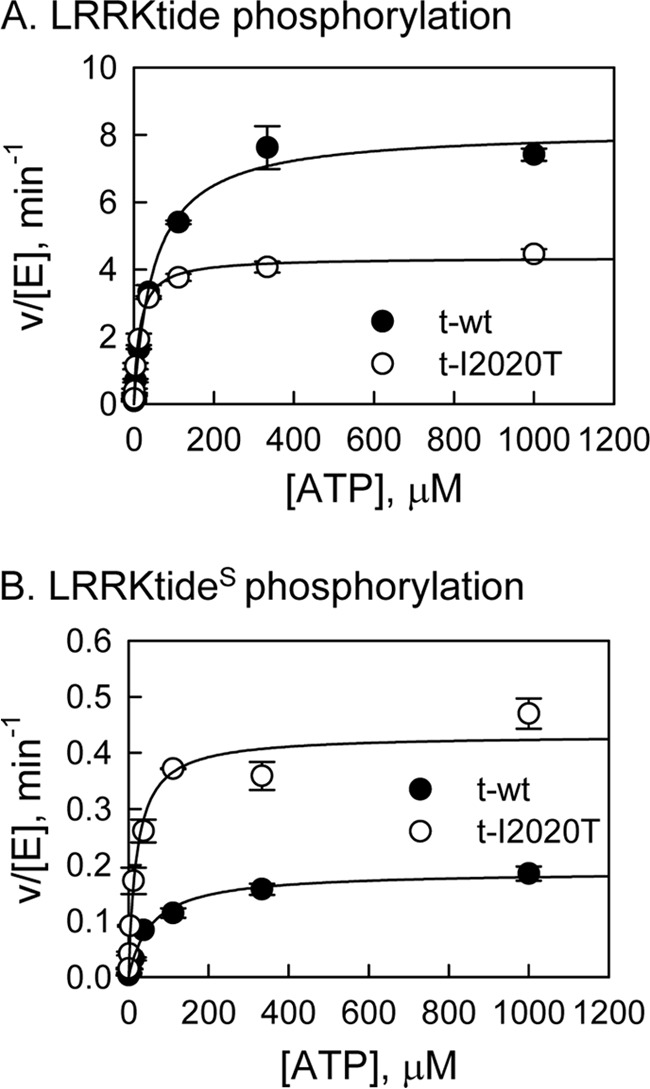
Steady-state kinetic studies of LRRK2-catalyzed phosphorylation. A, phosphorylation of LRRKtide by t-WT LRRK2 (●) and the t-I2020T mutant (○) at 1 mm LRRKtide. B, phosphorylation of LRRKtideS by t-wt LRRK2 (●) and the t-I2020T mutant (○) at 2 mm LRRKtideS.
t-WT LRRK2- and the Mutation t-I2020T-catalyzed Phosphorylation of LRRKtideS, Initial Velocity Studies
We have previously shown that the rate-limiting step for LRRKtide phosphorylation catalyzed by t-WT or the t-G2019S mutant is the product release step, whereas peptide LRRKtideS (RLGRDKYKSLRQIRQ, the phosphorylatable threonine of LRRKtide was replaced by a serine residue) is rate limited by the phosphoryl transfer step (17). To evaluate whether the t-I2020T mutant has a different effect on the phosphoryl transfer step, the initial velocity studies for LRRKtideS phosphorylation were conducted (data not shown). The data suggest a random/steady-state ordered mechanism for the t-I2020T mutant (the same kinetic mechanism for t-WT was reported previously, Ref. 17). The kinetic parameter estimates for the t-I2020T mutant and those for t-WT are summarized in Table 1. A direct comparison of the t-I2020T mutant with t-WT LRRK2 is shown in Fig. 1B, where the initial velocities were measured as a function of [ATP] at a saturating concentration of LRRKtideS for t-WT and the t-I2020T mutant. Unlike LRRKtide, when LRRKtideS is the phosphoryl acceptor, the t-I2020T mutant is more active than t-WT.
pL-dependence of Steady-state Kinetic Parameters and SKIE
We have seen both decreased and increased kinase activity for the t-I2020T mutant on two very similar peptide substrates. We hypothesize that it is due to the difference in the rate-limiting steps of the reactions. Although we have shown that for t-WT and the t-G2019S mutant, LRRKtide is rate limited by a product release step and LRRKtideS is associated with a slower phosphoryl transfer step, we need to make certain that those features remain for the t-I2020T mutant. Therefore, the steady-state kinetic parameter pL (pH or pD) profile of LRRKtide or LRRKtideS phosphorylation was carried out for the t-I2020T mutant in a triple buffer consisting of 50 mm MES, 100 mm Tris, and 50 mm acetic acid along with other components as described under ”Experimental Procedures.“ The combination of the three buffers has the advantage of providing a constant ionic strength for the entire pL range studied. The pH stability of the mutant was determined by incubating the enzyme at the desired pH and assaying aliquots at pH 7.5. The enzyme was determined to be stable from pH 6 to 9.3. Small activity loss occurs at pH < 6.0. However, the small activity loss does not affect the measurement of initial velocities, because enzyme was added from a stock solution at pH 7.5 to the assay mixture and assayed for 30 min in the presence of substrate, which is likely to provide some protection against denaturation. The t-I2020T-catalyzed phosphorylation of LRRKtide was monitored in a pL range of 4.9–8.5 for kcat (initial velocities were measured at saturating ATP and LRRKtide concentrations of 60 μm and 2 mm, respectively) and for kcat/Km (initial velocities were measured at ATP and LRRKtide concentrations less than the Km values, 1 and 50 μm, respectively). The pL profile of kcat for the t-I2020T mutant revealed an acidic limb (Fig. 2A) and was fit to Equation 4, yielding one apparent pKa of 6.8 and 7.3 from pH and pD profiles, respectively.
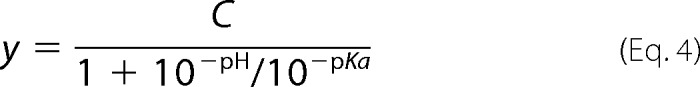 |
kcat/Km exhibited a similar pL dependence as kcat, revealing one apparent pKa of 7.2 and 7.5 from pH and pD profiles, respectively (Fig. 2B). The pL-independent steady-state parameters revealed SKIE values of 1.01 and 1.10 for kcat and kcat/Km, respectively, suggesting that like t-WT or the t-G2019S mutant, the rate-limiting step for t-I2020T-catalyzed LRRKtide phosphorylation is product release.
FIGURE 2.

pL-dependent kinetic parameters for the mutant t-I2020T-catalyzed phosphorylation. A, t-I2020T-catalzyed LRRKtide phosphorylation (panels A and B). Panel A, pH (●) and pD (○) dependences of kcat with a SKIE of 1.01. Panel B, pH (●) and pD (○) dependences of kcat/Km with a SKIE of 1.1. B, t-I2020T-catalzyed LRRKtideS phosphorylation (panels C and D). Panel C, pH (●) and pD (○) dependences of kcat with a SKIE of 0.50. Panel D, pH (●) and pD (○) dependences of kcat/Km with a SKIE of 0.61.
The same pL-dependent studies were conducted for LRRKtideS as well. The pL profiles revealed similar shape with one apparent pKa of 7.7 and 8.4 from pH and pD profiles, respectively, for kcat; and one apparent pKa value of 8.1 and 8.7 from pH and pD profiles, respectively, for kcat/Km (Fig. 2, C and D). The pL-independent steady-state parameters revealed SKIE values of 0.50 and 0.61 for kcat and kcat/Km, respectively, suggesting that the rate-limiting phosphoryl transfer step is the characteristics of the t-I2020T-catalyzed LRRKtideS phosphorylation. To directly monitor the effects of the t-I2020T mutant on phosphoryl transfer of LRRKtide phosphorylation, we conducted pre-steady-state kinetics as discussed below.
Pre-Steady-state Kinetics
LRRKtide phosphorylation was carried out at saturating concentrations of LRRKtide and ATP in the presence of 12 nm enzyme. The reaction was stopped by the rapid quench flow instrument. As shown in Fig. 3, A and B, burst kinetics or biphasic kinetics was observed for both t-WT and the mutant. Fitting data to a single exponential followed by a steady-state rate equation: y = A[1 − exp(−k1t)] + k2t, yielding values for A that represents the concentration of active enzymes, a pseudo first-order rate constant k1 for the burst phase of the reaction, and a steady-state rate k2. Averaged values of these parameters determined from two experiments are summarized in Table 2. Comparison of the parameters for t-WT and the t-I2020T mutant reveals that (i) the concentration of the active form of the mutant is significantly lower than that of t-WT; (ii) the t-I2020T mutant increases the rate of the phosphoryl transfer step of LRRKtide phosphorylation by greater than 3-fold compared with t-WT; (iii) first glance at the steady-state rate k2, the rate of product release, we found that the mutant is associated with a slower product release rate than t-WT. However, when we take into account the concentrations of the active forms of both enzymes, a product release rate constant (k2/A) similar to t-WT was revealed for the t-I2020T mutant (Table 2).
FIGURE 3.
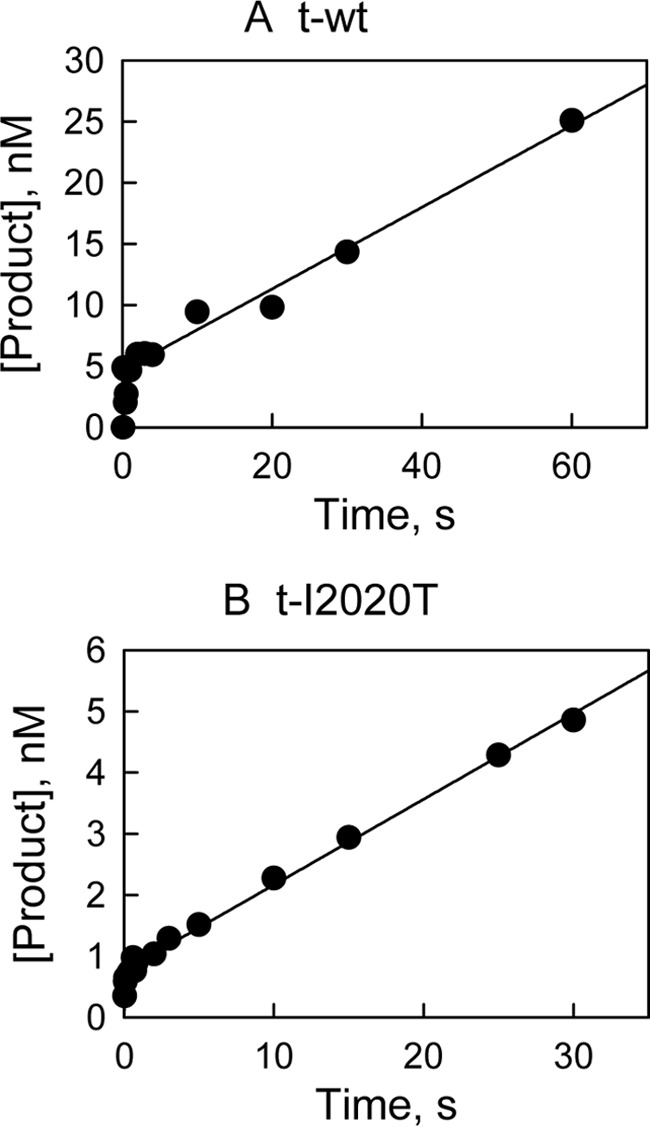
Pre-steady-state kinetic studies for LRRKtide phosphorylation. The reaction was carried out at saturating concentrations of LRRKtide and ATP in the presence of 12 nm t-WT (A) and t-I2020T mutant (B). The reaction was stopped by the rapid quench flow instrument. Data were fit to a single exponential followed by a steady-state rate equation: y = A[1 − exp(−k1t)] + k2t.
TABLE 2.
Pre-steady-state kinetics of t-WT and the t-I2020T mutant
A represents the concentration of active enzymes; k1 is a pseudo first-order rate constant for the burst phase of the reaction; k2 is a steady-state rate; and k2/A is a steady-state rate constant.
| t-WT | t-I2020T | |
|---|---|---|
| A (burst) (nm) | 4.6 ± 0.7 | 1.2 ± 0.6 |
| k1 (s−1) | 3.82 ± 0.2 | 10.4 ± 2.6 |
| k2 (nm s−1) | 0.33 ± 0.03 | 0.14 ± 0.02 |
| k2/A (s−1) | 0.07 | 0.12 |
Inhibition Studies of Two Known DFG-out Inhibitors
In an effort to develop LRRK2 inhibitors, we have identified that ponatinib and imatinib, two DFG-out inhibitors of Abl kinase (39, 40), inhibit t-WT LRRK2 with IC50 values of 160 nm and 8.3 μm, respectively; and inhibit the t-I2020T mutant with IC50 values of 800 nm and 90 μm, respectively (Table 3). Next, the mechanism of inhibition of these two type II inhibitors was studied and data were analyzed using the method of replots as described in detail previously (41). Briefly, initial velocities of LRRK2-catalyzed LRRKtide phosphorylation were measured as a function of inhibitor concentration [I] at varied ATP concentrations and at a fixed LRRKtide concentration. The shape of replots of (kcat)ATP versus [I] and (kcat/Km)ATP versus [I] were used to determine the inhibition mechanism. For ponatinib, as expected based on its mechanism of inhibition with its targeted kinase Abl (40), it shows ATP non-competitive inhibition toward t-WT; both (kcat)ATP and (kcat/Km)ATP are dependent on [ponatinib] (Fig. 4, B and C). However, surprisingly ponatinib inhibits the t-I2020T mutant via an ATP competitive mechanism, (kcat)ATP is independent of [ponatinib] (Fig. 4E). To better judge the two inhibition mechanisms, noncompetitive versus competitive inhibition, data were also globally fit into equations reflecting the two mechanisms, respectively. The goodness of fit was judged by an F test, in which a probability value lower than 0.05 indicates that one of the two equations fits the data better. F test suggests that data for t-WT fit better to the noncompetitive inhibition (probability value ≪ 0.001), and data for the mutation t-I2020T fit better to the competitive inhibition (probability value ≪ 0.001), consistent with the method of replots. The data for imatinib were analyzed the same way using both methods of replots and global fitting (data not shown). The same inhibition patterns were also observed for imatinib, it inhibits the t-I2020T mutant in an ATP competitive manner, whereas it inhibits t-WT in an ATP noncompetitive mechanism. As a comparison, an ATP competitive inhibitor GSK3-XIII shows competitive inhibition against both t-WT and the t-I2020T mutant (data not shown). Its IC50 values are summarized in Table 3.
TABLE 3.
Inhibition of t-WT LRRK2 and the t-I2020T mutant
IC50 was determined at 50 μm LRRKtide and ATP concentrations around its Km values for t-WT and the t-I2020T mutant, respectively.
| IC50 |
||
|---|---|---|
| t-WT | t-I2020T | |
| μm | ||
| Ponatinib | 0.16 ± 0.01 NCa | 0.8 ± 0.21 Cb |
| Imatinib | 8.3 ± 1.2 NC | 90 ± 10.3 C |
| GSK3-XIII | 0.2 ± 0.03 C | 1.4 ± 0.2 C |
a NC represents ATP noncompetitive inhibition.
b C represents ATP competitive inhibition.
FIGURE 4.

Inhibition study of phosphorylation of LRRKtide by ponatinib. A, inhibition study of the t-WT-catalyzed phosphorylation of LRRKtide by ponatinib (panels A–C). Panel A, plot of initial velocities versus [ATP] at [ponatinib] = 1 (●), 0.3 (○), 0.1 (▾), 0.04 (▿), 0.01 (■), and 0 μm (□) all at a fixed LRRKtide concentration of 50 μm. Panels B and C, ponatinib concentration dependences of (kcat)ATP and (kcat/Km)ATP apparent values derived from analysis of the data of panel A. B, inhibition study of the t-I2020T mutant-catalyzed phosphorylation of LRRKtide by ponatinib (panels D–F). Panel D, plot of initial velocities versus [ATP] at [ponatinib] = 3 (●), 1.5 (○), 0.8 (▾), 0.4 (▿), 0.2 (■), and 0 μm (□) all at a fixed LRRKtide concentration of 50 μm. Panels E and F, ponatinib concentration dependences of (kcat)ATP and (kcat/Km)ATP apparent values derived from analysis of the data of panel D.
Effects of the I2020T Mutant on the Structure of LRRK2 and ATP Binding
A structural model for LRRK2 in the DYG-out form (inactive conformation) has been previously published (22). In the DYG-out form of LRRK2 (DFG-out in many other kinases), the tyrosine side chain (Phe in other kinases) moves to a position that would otherwise be occupied by ATP. The kinase in this form therefore no longer binds ATP. However, in this conformation, the kinase structure still remains competent to bind hinge-binding elements of type II inhibitors such as ponatinib. Our model is consistent with x-ray structures of ponatinib bound to other kinases such as cAbl in their DFG-out state (40). In our LRRK2 model, residue Ile-2020 lies in the activation loop adjacent to the DYG motif as shown in Fig. 5A. The isoleucine residue at position 2020 was mutated to threonine using PyMol and the resultant I2020T mutant could be accommodated within the LRRK2 structure as evidenced by its stability after MD thermalization. Analysis of the energy minimized structure reveals that the side chain of Thr-2020 makes a hydrogen bond with the backbone carbonyl of Asp-2017 (Fig. 5B). This in turn results in the side chain of Asp-2017 making a salt-bridge network with Lys-1906 and Glu-1920 (Fig. 5B). To fully understand the thermodynamic consequence of this mutation, we carried out metadynamic simulations to explore the free energy surface of the activation loop. The dynamics of a protein kinase when it transitions between active and inactive conformations involves two major motions: (a) the movement of the center of mass of N-terminal β-sheet region and C-helix and (b) the movement of the activation loop bearing the DYG motif as the enzyme switches between the active and inactive form. Because these motions occur on microsecond or millisecond time scales and are not readily accessible using conventional MD simulations, we used metadynamic simulation to explore the structural fluctuations of LRRK2. We utilized a set of collective variables using the active DYG-in (closed) form as the input structure. The change in the center of mass for residues of the N-terminal β-sheet domain (residues 1878–1906) and C-helix in the N-terminal domain (residues 1915–1928) was an effective descriptor of the opening and closing motion of LRRK2 and represented the first collective variable (s) in our calculations. The details of these collective variables have been published previously by our group (22). The change in the center of mass for the Cα atoms of residues Tyr-2018, Ala-2021, Cys-2024, Arg-2026, and Met-2027 was an effective descriptor for the motion of the activation loop and represented the second collective variable (z) in our calculation. These final set of atoms used to define the collective variables for these calculations were reached after several pilot calculations to ensure that the MD path leads to both activation-loop movement and flip of the DYG motif from DYG-in to DYG-out occurred in a correlated fashion. The results of the metadynamic simulation are plotted as a FES, which also represents the barriers involved in the motion of the kinase as it switches between the DYG-in and DYG-out states. The FES for WT LRRK2 shows a smooth transition between DYG-in and DYG-out, whereas the FES for I2020T shows two clear energy minima representing the two states (Fig. 6). There is an average 4.7 kcal/mol energy barrier for the mutant that separates the two states from each other.
FIGURE 5.
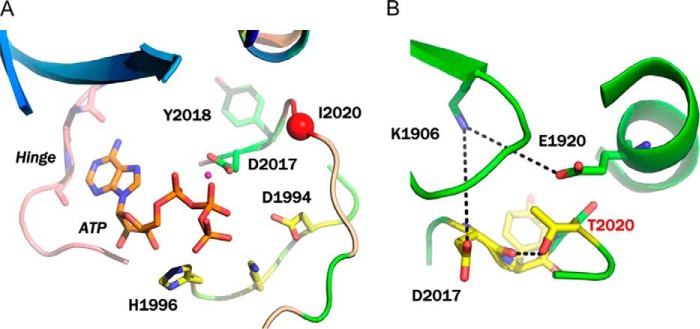
Modeling of LRRK2. A, modeling of ATP binding site of WT LRRK2. The key interacting residues and the position of Ile-2020 residue on the activation loop are highlighted. B, modeling of the I2020T mutant after MD thermalization. The side chain of I2020T participates in a network of hydrogen bond interactions that stabilizes the DYG-in conformation of the mutant.
FIGURE 6.

FES from metadynamics simulation generated from the collective variables (s) and (z) that describe the transition of the activation loop from DYG-in to DYG-out. A, the FES of WT LRRK2 shows a contiguous low energy path (blue) that connects the two conformations forms. B, the FES of the I2020T mutant shows a deeper low energy minima corresponding to the DYG-in state and a much higher barrier separating the DYG-in and DYG-out states.
To measure the affinity of ATP for the binding site of LRRK2, we performed 50-ns metadynamic simulations with the ATP-bound form of WT LRRK2 and the I2020T mutant. In this form of MD, a root mean square deviation type collective variable was used in conjunction with an angular collective variable between ATP and two fixed water oxygen atoms in the bulk solvent. Through several trial simulations, we found that this set of collective variables is optimal because it encourages ATP to move away from the binding site. The type and number of barriers encountered during this process estimates the energy required to dissociate from the binding site, which is a measurement of Km for ATP. The FES for dissociation of ATP from WT LRRK2 and I2020T are shown in Fig. 7. The FES of WT LRRK2 shows that the ATP dissociation path has an average barrier of 3.8 kcal/mol, which results in half-life of the ATP binding site at the active site for 3 s. The mutant I2020T shows an average barrier of 5.5 kcal/mol, which results in a half-life of ATP at the binding site for 75 s. A key distinction between the ATP dissociation mechanism of WT and I2020T is the number of intermediate steps involved. WT LRRK2 shows four distinct states including bound/unbound states and two intermediates (Fig. 7A). The mutant on the other hand shows more of a two-state dissociation with a deep distinctive low energy minima corresponding to the bound conformation and a second one corresponding to the unbound form (Fig. 7B).
FIGURE 7.

A, the FES of the undocking process of ATP in WT LRRK2 as a function of the angle and root mean square deviations of CVs. The FES shows an internal, partially bound and an external energy minimas. A detailed description of the various intermediate steps is provided in text. Briefly, the dissociation path of ATP involves two metastable states (states 2 and 3) with a relatively smooth transition between bound and unbound states. B, the FES of the undocking process of ATP in I2020T as a function of the angle and root mean square deviations of CVs. Unlike WT LRRK2, the dissociation appears to follow a two-step-like mechanism with a highly stable bound state that persists much longer compared with WT and then dissociate very quickly to an unbound external energy minima.
DISCUSSION
Unlike the most common PD-linked LRRK2 mutation G2019S, which has been consistently reported to increase kinase activity (7, 8, 11, 17, 43), the effect of the I2020T mutant on kinase activity is controversial. It was reported to have a lower kinase activity in assays measuring LRRKtide phosphorylation (14, 16) but has either higher or unchanged activity in assays monitoring autophosphorylation depending on the autophosphorylation sites (12, 15, 44). To better understand the substrate-dependent effects of the I2020T mutant, we chose two peptide substrates, LRRKtide and LRRKtideS. They differ only by the phosphorylatable sites, threonine in LRRKtide versus serine in LRRKtideS. However, as a result, the rate-limiting steps of their phosphorylation are quite different as reported previously for t-WT LRRK2 and the t-G2019S mutant (17). In this study, we found that these features remained for the t-I2020T mutant, LRRKtide phosphorylation is rate-limited by a slow product release step as evidenced by a SKIE value close to 1 for kcat, whereas a slow phosphoryl transfer step is associated with LRRKtideS phosphorylation as evidenced by an inverse SKIE value of 0.5 for kcat. Using these two peptide substrates, we could directly evaluate the effects of the t-I2020T mutant on the two steps, phosphoryl transfer and product release steps even in steady-state kinetic studies.
Initial velocity studies suggest that the t-I2020T mutant has a lower kinase activity in catalyzing LRRKtide phosphorylation than t-WT LRRK2. However, for LRRKtideS phosphorylation, we surprisingly observed a higher kinase activity for the t-I2020T mutant, 2 times t-WT kinase activity. These steady-state kinetic studies suggest that the t-I2020T mutant increases phosphoryl transfer but it seems to slow down product release. Pre-steady-state kinetic studies for LRRKtide phosphorylation also revealed a faster phosphoryl transfer rate for the t-I2020T mutant, greater than 2 times that for t-WT. Although we were well aware that the I2020T mutant degrades faster than WT in cells (45), we were surprised to see that the concentration of the active form of the mutant is significantly lower than that of t-WT. Taken into account the real concentrations of the active forms of both enzymes, we found that (i) for LRRKtide phosphorylation, the t-I2020T mutant releases products with a rate similar to t-WT and (ii) for LRRKtideS phosphorylation, the mutant increases kinase activity or the rate of phosphoryl transfer by 7-fold compared with t-WT. Combined results from our steady-state and pre-steady-state kinetic studies suggest that the t-I2020T mutant increases kinase activity by accelerating the phosphoryl transfer rate. However, this increase was masked in some reactions in which the rate-limiting step is product release, like in LRRKtide phosphorylation.
The LRRK2 research community has put in extensive efforts in identifying LRRK2 substrates since LRRK2 was linked to PD in 2004. As a result, a number of proteins have been reported to be phosphorylated by LRRK2 in vitro although none of them have been demonstrated as LRRK2 natural substrates. The first such potential LRRK2 substrate is moesin, a member of the ERM (ezrin/radixin/moesin) family that anchors the actin cytoskeleton to the plasma membrane. It was reported in 2007 by Jaleel et al. (14) that LRRK2 phosphorylated moesin at Thr558. LRRK2 also phosphorylates ezrin and radixin at the residues equivalent to Thr558.
Although ezrin/radixin/moesin as real substrates of LRRK2 is debatable in the LRRK2 field, a peptide encompassing Thr558 of moesin was derived and named LRRKtide. LRRKtide has since been widely used in the LRRK2 field for activity and inhibition studies. Inhibitors evaluated using LRRKtide in vitro shows consistent potency in cells. Our previous publication (22) on the G2019S mutant based on LRRKtide phosphorylation and modeling studies made the same conclusion as a crystal structural study on Roco4, the D. discoideum version of LRRK2 (23). All these observations make our assessment of the I2020T mutant based on LRRKtide and its analog reliable.
Currently the physiological substrate of LRRK2 is unknown. Recent studies reported that LRRK2 phosphorylates Tau (46, 47) and MKK6 (48). However, due to the slow kinetics of the phosphorylation of these proteins catalyzed by LRRK2, we were unable to conduct the pre-steady-state studies using MKK6 or Tau proteins and were unable to determine whether the I2020T mutant increases the phosphoryl transfer rate of protein substrate phosphorylation. In the future, with the hope that LRRK2 natural substrate would be identified, we will re-evaluate this mutant.
It has been reported that the I2020T mutant exhibits resistance toward ATP competitive inhibitors due to its small KATP (49). We were able to replicate this finding using an ATP competitive inhibitor GSK3-XIII (Table 3). We rationalize that DFG-out or type II inhibitors could overcome this issue, because they are known to bind noncompetitively with ATP by interacting with an allosteric site that exists when the kinase switches to an inactive DFG-out form (39, 40, 50–52). However, two known DFG-out inhibitors, ponatinib and imatinib, inhibit the t-I2020T mutant with a potency 5–10-fold weaker than they inhibit t-WT (Table 3). Inhibition mechanism studies revealed that ponatinib and imatinib exhibit a surprising difference in their inhibition mechanisms between t-WT and t-I2020T mutant. Both ponatinib and imatinib inhibit t-WT in an expected ATP non-competitive mechanism, whereas they inhibit the t-I2020T mutant in an ATP competitive mechanism.
To explain the observed inhibition mechanism and kinetics, we compared homology models of LRRK2 in the DYG-in (active) and DYG-out (inactive) forms for both WT and the I2020T mutant. Almost in all kinases, there are two synergistic motions that occur as a result of kinase activation. At the N-terminal of the flexible activation loop there exists a conserved three-residue motif, DYG for LRRK2 (DFG for most kinases). The Gly residue acts as a bipositional switch, which allows the Asp and Tyr/Phe to flip and exchange positions during the activation process. In addition, the activated configuration of DYG flip facilitates an inward movement of C-helix making a salt-bridge with a conserved Lys residue in the N-terminal β-sheet domain (53, 54). In the case of LRRK2, this salt bridge is between Lys-1906 and Glu-1920. The motions of two lobes thus become coordinated for precisely positioning ATP bound to the N-lobe with respect to the catalytic site on the C lobe. Homology modeling of LRRK2 suggests that the mutation I2020T is in fact right next to this critical regulatory conserved DYG motif. The threonine side chain of the mutant makes a hydrogen bond with the backbone carbonyl of Asp-2017. This would in fact rigidify the DYG-motif and have a stabilizing effect on the DYG-in conformation. The FES associated with the activation loop transition indicates that the I2020T mutant has a barrier of 4.7 kcal/mol that separates the DYG-in active state from the DYG-out inactive state. On the other hand, WT LRRK2 has no such barrier. In other words, the I2020T mutant stabilizes the DYG-in active form and may not easily switch to the DYG-out inactive form. As a result, the I2020T mutant leads to increased kinase activity (increased phosphoryl transfer rate). In addition, the I2020T mutant is able to bind traditional type II DFG-out inhibitors but in an unusual type I-like ATP competitive inhibitor binding mode. A detailed study of the binding mode for these inhibitors has been previously published from our research group (42).
To measure the effect of the energy barrier of the activation loop transition associated with the mutant on ATP binding, we carried out metadynamic simulation, in which the bound ATP molecule is encouraged to move away from the active site during the course of the simulation. As the ATP molecule dissociates, the energy barriers of dissociation are measured and the highest energy barrier can be converted to off-rates or residence time using Eyring's equation. Analysis of the FES from metadynamic simulations for WT and the I2020T mutant indicates a similar but not identical path for ATP dissociation but with slightly different barriers. The majority of barriers in ATP dissociation from WT LRRK2 involve a series of hydrogen bonds between ATP and the hinge region, glycine-rich loop, and Arg-2026. In the case of the I2020T mutant, the side chain of Thr-2020 makes a hydrogen bond with the backbone and side chain of Asp-2017. This rigidifies the activation loop region leading to a less mobile DYG region (part of ATP binding pocket), whose motion is necessary for dissociation of ATP. We used a simulation interaction diagram, which measures the contribution of each amino acid and water molecule toward the motion of the ligand to analyze the MD trajectories for WT LRRK2 and I2020T mutant (Fig. 8). The analysis shows that the rigidified DYG region of I2020T makes strong and persistent hydrogen bonds with the phosphate groups of ATP thereby contributing toward prolonged residence time of ATP. These interactions are absent from the WT LRRK2 simulation trajectory. Metadynamic simulations estimated a half-life of 3 s at 37 °C for ATP bound to WT LRRK2, whereas a half-life of 75 s for ATP bound to the I2020T mutant. This prolonged ATP-I2020T complex explains the kinetic observation that the I2020T mutant has an unusually high binding affinity toward ATP.
FIGURE 8.

A, a simulation interaction diagram showing prolonged interactions between ATP, amino acid residues of WT LRRK2, and water molecules over the course of simulation. The simulation interaction diagram for WT LRRK2 shows several interactions between ATP phosphate groups and water molecules. This indicates that the ATP molecule moved fairly rapidly from the binding site to other non-native sites. The only strong persistent interaction (47%) is observed between the γ-phosphate of ATP and Arg-110 (structurally equivalent to R2026 in native LRRK2 sequence). B, a simulation interaction diagram showing prolonged interactions between ATP, amino acid residues of I2020T mutant, and water molecules over the course of simulation. Unlike WT LRRK2, the simulation interaction diagram for I2020T shows direct interaction between the ATP molecule and protein side chains that make up the hinge region and the ATP binding cavity. This indicates that the molecule has higher residence time in the ATP binding site compared with WT. In addition to the strong interaction with Arg-110 (structurally equivalent to Arg-2026 in native LRRK2 sequence), the ATP molecule also makes strong and persistent (51% total) water-mediated hydrogen bonds with Asp-170 (structurally equivalent to Asp-2017) of the DYG motif.
CONCLUSION
Although the x-ray structure for LRKK2 is yet to be determined (a closely related Roco4 kinase domain is now available (23)), enzyme mechanism studies and molecular modeling have provided significant insights into the nature of LRRK2 and the PD-linked mutation I2020T. Given the unique insights gained in this work with traditional type II kinase inhibitors binding to the I2020T mutant in an ATP-competitive way, it is conceivable that new classes of inhibitors will be needed to gain the desired affinity and selectivity to target the I2020T mutant in the pursuit to treat Parkinson disease. The presence of two exposed cysteine residues (Cys-2024 and Cys-2025) close to the ATP binding site opens up the possibility of developing covalent or irreversible inhibitors for LRRK2. Such inhibitors could provide a sustained duration of inhibition for this resistance-conferring mutant. In addition, with further studies, possibly including x-ray crystallography or long time scale molecular dynamics simulations, it may eventually be possible to understand the nature of the allosteric pocket in the I2020T mutant enzyme and develop highly selective inhibitors that will also overcome the resistant issue.
Acknowledgments
We thank Dr. Dimitri Luypan and Dr. Woody Sherman (Schrodinger LLC) for discussions around the metadynamics simulations.
This work was supported, in whole or in part, by National Institutes of Health Grant R21 NS 072519 and the Harvard Milton fund foundation.
- PD
- Parkinson disease
- LRRK2
- leucine-rich repeat kinase 2
- MD
- molecular dynamics
- CV
- collective variable
- FES
- free-energy surface
- t-WT
- truncated wild type.
REFERENCES
- 1. Moore D. J., West A. B., Dawson V. L., Dawson T. M. (2005) Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 28, 57–87 [DOI] [PubMed] [Google Scholar]
- 2. Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., de Silva R., Lees A., Martí-Massó J. F., Pérez-Tur J., Wood N. W., Singleton A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 [DOI] [PubMed] [Google Scholar]
- 3. Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 [DOI] [PubMed] [Google Scholar]
- 4. Berg D., Schweitzer K. J., Leitner P., Zimprich A., Lichtner P., Belcredi P., Brüssel T., Schulte C., Maass S., Nägele T., Wszolek Z. K., Gasser T. (2005) Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain 128, 3000–3011 [DOI] [PubMed] [Google Scholar]
- 5. Khan N. L., Jain S., Lynch J. M., Pavese N., Abou-Sleiman P., Holton J. L., Healy D. G., Gilks W. P., Sweeney M. G., Ganguly M., Gibbons V., Gandhi S., Vaughan J., Eunson L. H., Katzenschlager R., Gayton J., Lennox G., Revesz T., Nicholl D., Bhatia K. P., Quinn N., Brooks D., Lees A. J., Davis M. B., Piccini P., Singleton A. B., Wood N. W. (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796 [DOI] [PubMed] [Google Scholar]
- 6. Mata I. F., Taylor J. P., Kachergus J., Hulihan M., Huerta C., Lahoz C., Blazquez M., Guisasola L. M., Salvador C., Ribacoba R., Martinez C., Farrer M., Alvarez V. (2005) LRRK2 R1441G in Spanish patients with Parkinson's disease. Neurosci. Lett. 382, 309–311 [DOI] [PubMed] [Google Scholar]
- 7. Smith W. W., Pei Z., Jiang H., Dawson V. L., Dawson T. M., Ross C. A. (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 9, 1231–1233 [DOI] [PubMed] [Google Scholar]
- 8. Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M. P., Beilina A., Blackinton J., Thomas K. J., Ahmad R., Miller D. W., Kesavapany S., Singleton A., Lees A., Harvey R. J., Harvey K., Cookson M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341 [DOI] [PubMed] [Google Scholar]
- 9. Guo L., Gandhi P. N., Wang W., Petersen R. B., Wilson-Delfosse A. L., Chen S. G. (2007) The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp. Cell Res. 313, 3658–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., Iwatsubo T. (2007) GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry 46, 1380–1388 [DOI] [PubMed] [Google Scholar]
- 11. West A. B., Moore D. J., Biskup S., Bugayenko A., Smith W. W., Ross C. A., Dawson V. L., Dawson T. M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. West A. B., Moore D. J., Choi C., Andrabi S. A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K. L., Dawson V. L., Dawson T. M. (2007) Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 16, 223–232 [DOI] [PubMed] [Google Scholar]
- 13. Xiong Y., Coombes C. E., Kilaru A., Li X., Gitler A. D., Bowers W. J., Dawson V. L., Dawson T. M., Moore D. J. (2010) GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 6, e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaleel M., Nichols R. J., Deak M., Campbell D. G., Gillardon F., Knebel A., Alessi D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem. J. 405, 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gloeckner C. J., Kinkl N., Schumacher A., Braun R. J., O'Neill E., Meitinger T., Kolch W., Prokisch H., Ueffing M. (2006) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 15, 223–232 [DOI] [PubMed] [Google Scholar]
- 16. Anand V. S., Reichling L. J., Lipinski K., Stochaj W., Duan W., Kelleher K., Pungaliya P., Brown E. L., Reinhart P. H., Somberg R., Hirst W. D., Riddle S. M., Braithwaite S. P. (2009) Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 276, 466–478 [DOI] [PubMed] [Google Scholar]
- 17. Liu M., Kang S., Ray S., Jackson J., Zaitsev A. D., Gerber S. A., Cuny G. D., Glicksman M. A. (2011) Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry 50, 9399–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fiser A., Sali A. (2003) ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 [DOI] [PubMed] [Google Scholar]
- 19. John B., Sali A. (2003) Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 31, 3982–3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V. A., Pieper U., Stuart A. C., Marti-Renom M. A., Madhusudhan M. S., Yerkovich B., Sali A. (2003) Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sánchez R., Sali A. (1997) Evaluation of comparative protein structure modeling by MODELLER-3. Proteins 1, 50–58 [DOI] [PubMed] [Google Scholar]
- 22. Liu M., Bender S. A., Cuny G. D., Sherman W., Glicksman M., Ray S. S. (2013) Type II kinase inhibitors show an unexpected inhibition mode against Parkinson's disease-linked LRRK2 mutant G2019S. Biochemistry 52, 1725–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gilsbach B. K., Ho F. Y., Vetter I. R., van Haastert P. J., Wittinghofer A., Kortholt A. (2012) Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. U.S.A. 109, 10322–10327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 25. Sherman W., Day T., Jacobson M. P., Friesner R. A., Farid R. (2006) Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 49, 534–553 [DOI] [PubMed] [Google Scholar]
- 26. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., Friesner R. A. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 [DOI] [PubMed] [Google Scholar]
- 27. Barducci A., Bonomi M., Parrinello M. (2010) Linking well tempered metadynamics simulations with experiments. Biophys. J. 98, L44–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barducci A., Bussi G., Parrinello M. (2008) Well tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 100, 020603 [DOI] [PubMed] [Google Scholar]
- 29. Barducci A., Chelli R., Procacci P., Schettino V., Gervasio F. L., Parrinello M. (2006) Metadynamics simulation of prion protein: β-structure stability and the early stages of misfolding. J. Am. Chem. Soc. 128, 2705–2710 [DOI] [PubMed] [Google Scholar]
- 30. Berteotti A., Barducci A., Parrinello M. (2011) Effect of urea on the β-hairpin conformational ensemble and protein denaturation mechanism. J. Am. Chem. Soc. 133, 17200–17206 [DOI] [PubMed] [Google Scholar]
- 31. Bonomi M., Barducci A., Gervasio F. L., Parrinello M. (2010) Multiple routes and milestones in the folding of HIV-1 protease monomer. PLoS One 5, e13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonomi M., Parrinello M. (2010) Enhanced sampling in the well tempered ensemble. Phys. Rev. Lett. 104, 190601. [DOI] [PubMed] [Google Scholar]
- 33. Branduardi D., Gervasio F. L., Parrinello M. (2007) From A to B in free energy space. J. Chem. Phys. 126, 054103 [DOI] [PubMed] [Google Scholar]
- 34. Bussi G., Laio A., Parrinello M. (2006) Equilibrium free energies from nonequilibrium metadynamics. Phys. Rev. Lett. 96, 090601 [DOI] [PubMed] [Google Scholar]
- 35. Leone V., Marinelli F., Carloni P., Parrinello M. (2010) Targeting biomolecular flexibility with metadynamics. Curr. Opin. Struct. Biol. 20, 148–154 [DOI] [PubMed] [Google Scholar]
- 36. Limongelli V., Bonomi M., Marinelli L., Gervasio F. L., Cavalli A., Novellino E., Parrinello M. (2010) Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 5411–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Limongelli V., Marinelli L., Cosconati S., La Motta C., Sartini S., Mugnaini L., Da Settimo F., Novellino E., Parrinello M. (2012) Sampling protein motion and solvent effect during ligand binding. Proc. Natl. Acad. Sci. U.S.A. 109, 1467–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Segel I. H. (1975) Enzyme Kinetics, John Wiley & Sons, Inc., New York [Google Scholar]
- 39. Schindler T., Bornmann W., Pellicena P., Miller W. T., Clarkson B., Kuriyan J. (2000) Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942 [DOI] [PubMed] [Google Scholar]
- 40. Zhou T., Commodore L., Huang W. S., Wang Y., Thomas M., Keats J., Xu Q., Rivera V. M., Shakespeare W. C., Clackson T., Dalgarno D. C., Zhu X. (2011) Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem. Biol. Drug Des. 77, 1–11 [DOI] [PubMed] [Google Scholar]
- 41. Liu M., Dobson B., Glicksman M. A., Yue Z., Stein R. L. (2010) Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry 49, 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ray S., Liu M. (2012) Current understanding of LRRK2 in Parkinson's disease: biochemical and structural features and inhibitor design. Future Med. Chem. 4, 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nichols R. J., Dzamko N., Morrice N. A., Campbell D. G., Deak M., Ordureau A., Macartney T., Tong Y., Shen J., Prescott A. R., Alessi D. R. (2010) 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 430, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sheng Z., Zhang S., Bustos D., Kleinheinz T., Le Pichon C. E., Dominguez S. L., Solanoy H. O., Drummond J., Zhang X., Ding X., Cai F., Song Q., Li X., Yue Z., van der Brug M. P., Burdick D. J., Gunzner-Toste J., Chen H., Liu X., Estrada A. A., Sweeney Z. K., Scearce-Levie K., Moffat J. G., Kirkpatrick D. S., Zhu H. (2012) Ser-1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 4, 164ra161. [DOI] [PubMed] [Google Scholar]
- 45. Ohta E., Katayama Y., Kawakami F., Yamamoto M., Tajima K., Maekawa T., Iida N., Hattori S., Obata F. (2009) I(2020)T leucine-rich repeat kinase 2, the causative mutant molecule of familial Parkinson's disease, has a higher intracellular degradation rate than the wild-type molecule. Biochem. Biophys. Res. Commun. 390, 710–715 [DOI] [PubMed] [Google Scholar]
- 46. Kawakami F., Yabata T., Ohta E., Maekawa T., Shimada N., Suzuki M., Maruyama H., Ichikawa T., Obata F. (2012) LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS One 7, e30834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bailey R. M., Covy J. P., Melrose H. L., Rousseau L., Watkinson R., Knight J., Miles S., Farrer M. J., Dickson D. W., Giasson B. I., Lewis J. (2013) LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol. 126, 809–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gloeckner C. J., Schumacher A., Boldt K., Ueffing M. (2009) The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J. Neurochem. 109, 959–968 [DOI] [PubMed] [Google Scholar]
- 49. Reichling L. J., Riddle S. M. (2009) Leucine-rich repeat kinase 2 mutants I2020T and G2019S exhibit altered kinase inhibitor sensitivity. Biochem. Biophys. Res. Commun. 384, 255–258 [DOI] [PubMed] [Google Scholar]
- 50. Garuti L., Roberti M., Bottegoni G. (2010) Non-ATP competitive protein kinase inhibitors. Curr. Med. Chem. 17, 2804–2821 [DOI] [PubMed] [Google Scholar]
- 51. Choi H. G., Zhang J., Weisberg E., Griffin J. D., Sim T., Gray N. S. (2012) Development of ”DFG-out“ inhibitors of gatekeeper mutant kinases. Bioorg. Med. Chem. Lett. 22, 5297–5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang J., Yang P. L., Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PubMed] [Google Scholar]
- 53. Cox S., Radzio-Andzelm E., Taylor S. S. (1994) Domain movements in protein kinases. Curr. Opin. Struct. Biol. 4, 893–901 [DOI] [PubMed] [Google Scholar]
- 54. Johnson D. A., Akamine P., Radzio-Andzelm E., Madhusudan M., Taylor S. S. (2001) Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101, 2243–2270 [DOI] [PubMed] [Google Scholar]