

Background: Phosphoenolpyruvate carboxykinase (Pck) catalyzes the interconversion of phosphoenolpyruvate and oxaloacetate but typically prefers gluconeogenic formation of phosphoenolpyruvate.
Results: Interactions of Mycobacterium tuberculosis (MTb) Pck with thioredoxin and reducing environments favor the anaplerotic oxaloacetate synthesis.
Conclusion: A mechanism explaining the regulation of Pck functions is proposed.
Significance: Regulation of Pck is important for the MTb non-replicative state associated with latent tuberculosis infection.
Keywords: Enzyme Kinetics, Hypoxia, Metabolism, Mycobacterium Tuberculosis, Oxidation-Reduction, Thioredoxin, Phosphoenolpyruvate Carboxykinase
Abstract
Tuberculosis remains a major health concern worldwide. Eradication of its causative agent, the bacterial pathogen Mycobacterium tuberculosis, is particularly challenging due to a vast reservoir of latent carriers of the disease. Despite the misleading terminology of a so-called dormant state associated with latent infections, the bacteria have to maintain basic metabolic activities. Hypoxic conditions have been widely used as an in vitro system to study this dormancy. Such studies identified a rearrangement of central carbon metabolism to exploit fermentative processes caused by the lack of oxygen. Phosphoenolpyruvate carboxykinase (Pck; EC 4.1.1.32) is the enzyme at the center of these metabolic rearrangements. Although Pck is associated with gluconeogenesis under standard growth conditions, the enzyme can catalyze the reverse reaction, supporting anaplerosis of the tricarboxylic acid cycle, under conditions leading to slowed or stopped bacterial replication. To study the mechanisms that regulate the switch between two Pck functions, we systematically investigated factors influencing the gluconeogenic and anaplerotic reaction kinetics. We demonstrate that a reducing environment, as found under hypoxia-triggered non-replicating conditions, accelerates the reaction in the anaplerotic direction. Furthermore, we identified proteins that interact with Pck. The interaction between Pck and the reduced form of mycobacterial thioredoxin, gene expression of which is increased under hypoxic conditions, also increased the Pck anaplerotic activity. We thus propose that a reducing environment and the protein-protein interaction with thioredoxin in particular enable the Pck anaplerotic function under fermentative growth conditions.
Introduction
Mycobacterium tuberculosis (MTb)2 is the causative agent of tuberculosis, a disease that causes ∼1.3 million deaths each year worldwide (1). In addition to the active, symptomatic form of the disease, inactive, symptom-free tuberculosis is widespread, affecting an estimated one-third of the world's population. Such latency develops due to the ability of MTb to escape host defense mechanisms, such as reactive oxygen species (2–4). With progressing infection, the surviving bacteria assemble in lung granulomas characteristic of latent tuberculosis infection. In this state, the bacteria can survive for up to several decades. Measurement of low oxygen tensions in granulomas has led to the hypothesis that the lack of oxygen might be a major trigger for the persistency program in this obligate aerobic pathogen (5). In fact, hypoxia induces widespread changes in MTb metabolism due to the induction of the DosR/S two-component system (6) and the KstR repressor (7). During the transition to the hypoxia-induced non-replicating state of the bacteria, the intracellular ATP level decreases, and the absence of oxygen as a terminal electron acceptor leads to the accumulation of reduced cofactors (8, 9). Transcriptional and metabolomics studies of MTb under hypoxic conditions have indicated a rearrangement of central carbon metabolism, leading to the synthesis of oxaloacetate (OAA) by an anaplerotic CO2-fixing reaction and to the secretion of succinic acid (9, 10). The main pathways of central metabolism (glycolysis, gluconeogenesis, and the tricarboxylic acid cycle) are interconnected via the phosphoenolpyruvate (PEP)-pyruvate-OAA node. The reactions within this node are catalyzed by a set of enzymes that can be regulated under different conditions by various factors, including enzyme activities and specificities, substrate availability, and product inhibition. This flexible node can switch the carbon flux distribution within the central metabolism (11). Pck catalyzes the interconversion between PEP and OAA. In most bacteria, Pck is primarily associated with gluconeogenesis because the thermodynamics of the reaction and concentrations of associated metabolites favor the reaction from OAA to PEP to build sugar backbones for storage or biosynthesis of nucleotides and several of the proteinogenic amino acids. Anaplerotic fixation of CO2 to OAA in bacteria is usually mediated by pyruvate carboxylase, PEP carboxylase, and malic enzyme. Likewise, the Pck of MTb is thought to operate in vivo in the gluconeogenic PEP formation direction because (i) its presence is essential for growth on lipid substrates and (ii) the reported intracellular metabolite concentrations strongly favor this direction. Furthermore, infection of primary macrophages and mice with a Pck knock-out MTb strain were impaired, demonstrating the gene's pivotal role for MTb infection and for persistence of MTb in mice (12, 13). Although the requirement for Pck was linked to the enzyme's gluconeogenic (anabolic) function, in vitro studies under hypoxic and growth-limiting conditions demonstrated its anaplerotic function in MTb (9, 14). Additionally, a recent study applying 13C labeling during THP1 cell infection with a Pck knock-out strain demonstrated that Pck contributes to carbon fixation in MTb (15). Pck seems to be able to play a role in the gluconeogenic and, unexpectedly, also in the anaplerotic route of the central carbon metabolism of MTb.
To address which regulatory mechanisms affect the direction of Pck catalysis, we systematically investigated the influence of various physiological effectors on the specificity and activity of both the gluconeogenic and the anaplerotic reaction directions of MTb Pck. We found that highly reducing conditions, as have been reported for hypoxia-triggered non-replicating mycobacteria, drive MTb Pck into the anaplerotic direction and thus lead to the formation of OAA. Furthermore, we applied protein pull-down assays to identify physical interactions between Pck and protein partners that could play a regulatory role. We demonstrate that mycobacterial thioredoxins serve a regulatory function as disulfide reductants, which upon interaction with Pck also lead to increased anaplerotic activity. We show that the kinetics of the anabolic and catabolic reactions catalyzed by MTb Pck are significantly influenced by the intracellular conditions.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of MTb Pck and Thioredoxins TrxC and TrxB
The coding region for MTb Pck (Rv0248) was amplified by PCR using the MTb DNA library as a template, gene-specific primers (forward, 5′-GGCGGCCATATGCATCATCACCATCACCACATGACCTCAGCGACCATC-3′; reverse, 5′-GGCGGCAAGCTTCTAACCTAGGCGCTCCTTC-3′), and Pfu turbo DNA polymerase (Stratagene). The DNA product was cloned into pET26b vector (Novagen) via NdeI and HindIII restriction sites. The coding regions for MTb thioredoxins B (Rv1471) and C (Rv3914) were amplified by PCR, using gene-specific primers (thioredoxin B: forward, 5′-GTACGCTCATATGGTGACTACCCGAGAC; reverse, 5′-CTACGGGATCCTCAGGCTTGTTGGGCTCG; thioredoxin C: forward, 5′-GCTAGTCCATATGACCGATTCCGAGAAG; reverse, 5′-CTACGGGATCCCTAGTTGAGGTTGGGAAC) and H37RvcDNA library as a template. The amplicons were cloned into pET15b vector cleaved with NdeI and BamHI restriction enzymes. Both Pck and thioredoxins contained an N-terminal His tag.
Pck was expressed in Escherichia coli BL21(DE3) cells that were cultivated in LB medium supplemented with kanamycin (100 μg/ml) at 30 °C for 12 h. When the culture reached A600 3.0–4.0, Pck expression was induced by the addition of isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 1 mm, and the cells were further incubated at 20 °C for 24 h. The harvested cells were resuspended in 20 mm Tris-HCl, pH 7.4, containing 500 mm NaCl and protease inhibitor mixture (LaRoche), and frozen at −70 °C. The cell walls were lysed by multiple freeze-thaw cycles and by the addition of lysozyme (0.5 mg/1 ml of supernatant). Pck was precipitated from the supernatant by the addition of ammonium sulfate (final concentration, 35% (w/v)). Following centrifugation (20,000 × g, 20 min), the supernatant was loaded onto a butyl-Sepharose column equilibrated with 50 mm sodium phosphate, pH 7.4, containing 1.7 m ammonium sulfate. The protein was eluted from the chromatography column by a decreasing ammonium sulfate gradient (buffer A: 50 mm sodium phosphate, pH 7.4, 1.7 m ammonium sulfate; buffer B: 50 mm sodium phosphate, pH 7.4). Pck eluted as a broad peak, and the fractions containing the majority of the protein were pooled and subsequently dialyzed against 20 mm Tris-HCl, pH 7.4, 500 mm NaCl, 5 mm 2-mercaptoethanol, 1% Triton X-100 buffer. The sample was loaded onto Talon® chromatography resin (Stratagene), and after quantitative washes with the same buffer supplemented with 10 mm imidazole, the protein was eluted from the column using buffers containing 50, 150, 300, 500, and 800 mm imidazole. The fractions containing Pck were identified by interaction of the N-terminal His tag of Pck with monoclonal anti-polyhistidine antibodies (mouse) conjugated with horseradish peroxidase (Sigma-Aldrich) by Western blot analysis. The pooled fractions were dialyzed against 50 mm Tris-HCl, pH 7.4, 300 mm NaCl, 5 mm 2-mercaptoethanol, concentrated in an Amicon Ultra centrifugal filter unit (Millipore), and stored at 4, −20, and −70 °C. Protein concentrations were determined by Bradford assay (16) with bovine serum albumin as a standard or by quantification of amino acid content after complete hydrolysis.
TrxC and TrxB also were expressed in E. coli BL21 (DE3) cells grown in LB medium containing ampicillin (100 μg/ml) at 30 °C until the A600 reached 4–5. Then isopropyl 1-thio-β-d-galactopyranoside (1 mm) was added to induce expression of the target proteins, and cells were subsequently incubated at 20 °C for 20 h. The cells were lysed similarly as described above. After repeated centrifugation at 20,000 × g for 30 min at 4 °C, the supernatant was loaded onto Talon® resin. Proteins were eluted with 20 mm to 0.5 m imidazole using stepwise gradients. The collected fractions were dialyzed against 20 mm Tris-HCl, pH 7.4, containing 100 mm NaCl. The reduced forms of thioredoxins were obtained by incubating the proteins in the presence of 5 mm dithiothreitol (DTT) for 2 h. The reduced state of thioredoxin samples was verified by titration with DTNB (17).
Site-directed Mutagenesis
Site-directed mutagenesis was performed according to the QuikChange protocol by StratageneTM with minor modifications. The complementary primers (1 μg/ml) (forward, GCATTCCCGTCGGCGTCTGGCAAGACCAACCTG; reverse, CAGGTTGGTCTTGCCAGACGCCGACGGGAATGC) were mixed in a Pfu polymerase reaction buffer, 0.2 mm dNTPs, 50 ng of plasmid template containing Pck coding sequence. Upon adding Pfu polymerase (2 units/reaction) the cycling reaction (linear amplification reaction) was started with the following program: denaturation at 94 °C (50 s), annealing at 55 °C (50 s), and extension at 72 °C (8 min), with 19 repeats. After the cycling reaction, DpnI restriction enzyme was added, and the final mixture was subsequently incubated at 37 °C for 12 h. The digest is crucial because enzyme only cleaves at methylated sites. The final product was transformed into DH5α ultracompetent cells, and grown colonies were used for sequencing.
Assay Measuring OAA Formation (Anaplerotic Reaction)
All enzyme assays of Pck were performed at 37 °C. To monitor the reaction rate in the OAA formation direction, the Pck-catalyzed reaction was coupled with the subsequent malate dehydrogenase (MDH) reaction using a modification of methods described previously (18, 19). We used MDH from Thermus flavus (Sigma-Aldrich), which is sufficiently active at 37 °C. The reaction progress was followed by monitoring the decrease in absorbance at 340 nm due to NADH oxidation to NAD+. The standard reaction mixture contained 100 mm HEPES-NaOH, pH 7.2, 100 mm KHCO3, 37 mm DTT, 2 mm PEP, 1 mm GDP, 2 mm MgCl2, 0.1 mm MnCl2, 2 units/ml MDH, and 0.25 mm NADH. Each reaction was started by the addition of essential divalent cations (Mg2+ and Mn2+). The control experiments confirming that the coupled reaction is dependent only on Pck concentration were performed at fixed concentration of 2 mm PEP, 1 mm GDP, 0.25 mm NADH, and 2 units/ml MDH.
Assay Measuring PEP Formation (Gluconeogenic Reaction)
To monitor the reaction rate in the PEP formation direction, the Pck-catalyzed reaction was coupled to pyruvate kinase (PK) (Roche Applied Science) and lactate dehydrogenase (LDH) (Roche Applied Science) reactions. Briefly, OAA was converted to PEP by Pck, PEP was dephosphorylated to pyruvate by PK, and pyruvate was reduced by LDH to lactate. In this set-up, the reaction rate corresponds to the decrease in absorbance at 340 nm due to the oxidation of NADH to NAD+. The typical reaction mixture was composed of 100 mm HEPES-NaOH, pH 7.2, 0.3 mm OAA, 0.2 mm GTP, 2 mm MgCl2, 0.2 mm MnCl2, 10 mm DTT, LDH (10 units/ml), PK (3 units/ml), and 0.2 mm NADH. To obtain correct values for the Pck reaction rates, it was necessary to take into account the decrease in absorbance due to the spontaneous decarboxylation of OAA to pyruvate from the total decrease in absorbance for each experimental data point. The control experiments confirming that coupled reaction is dependent only on Pck concentration were performed at fixed concentrations of 0.3 mm OAA, 0.2 mm GTP, 10 units/ml LDH, and 3 units/ml PK.
Activation of Pck by Thioredoxins
Pck was preincubated with increasing concentrations of thioredoxin B or C (80, 120, or 150 μm) in 50 mm Tris-HCl, pH 7.4, containing 150 mm NaCl, at room temperature for 0.5–8.5 h. Each incubation reaction contained 1 mm DTT for reduction of thioredoxin. Aliquots of incubation mixtures, taken at different time points, were transferred into the anaplerotic reaction buffer (100 mm HEPES-NaOH, pH 7.2, 100 mm KHCO3, 2 mm PEP, 1 mm GDP, 2 units/ml MDH, and 0.25 mm NADH). The final concentration of Pck in the reactions was 15 nm, and reactions were initiated by the addition of 0.1 mm MnCl2 and 2 mm MgCl2.
Mass Spectrometric Determination of Enzyme Activities
Mass spectrometry-based enzyme assays were performed as described above without the respective coupling enzymes and NADH. Samples were collected at 0, 3, 6, 9, 12, 15, and 18 min after starting the gluconeogenic reaction and at 0, 10, 20, 30, 40, 50, and 60 min after starting the anaplerotic reaction. At each time point, 20 μl of reaction mixture was removed and stopped by the addition of an equivalent volume of cold acetonitrile. Samples were diluted in water 3- and 20-fold, respectively. Fully 13C-labeled metabolite extract derived from Saccharomyces cerevisiae cultured on d-[U-13C]glucose was added to the samples as an internal standard solution, allowing absolute quantification of the metabolite concentration using a calibration curve with chemical standards ranging from 0.14 to 100 μm. After drying and resuspension in water, the samples were analyzed by UPLC-MS/MS as described previously (20).
Detection of SH Groups in Proteins
Detection of cysteine residues in the reduced state was performed using two methods described previously (17, 21). In the first method, ∼1 nmol of enzyme was mixed in a cuvette with 1 mm DTNB (5,5′-dithiobis(2-nitrobenzoic acid)) in the presence of 4 m guanidine HCl, pH 8.0, and the absorbance at 412 nm was measured. The reported extinction coefficient (13,600 cm−1 m−1) was used to calculate the number of moles of nitromercaptobenzoate anion released upon reaction of DTNB with the enzyme, which corresponds to the total amount of reduced Cys in the protein. The exact concentration of the enzyme was calculated from the quantification of individual amino acids after complete hydrolysis.
According to the protocol for the second method, 1 nmol of enzyme was incubated with 1 mm DTNB in 100 mm Tris-HCl, pH 8.0, containing 1 mm EDTA and 0.9% SDS for 5 min. The absorbance at 412 nm was measured, and the total amount of reduced Cys was determined according to the procedure described above.
Affinity Purification of Proteins Interacting with Pck
The Rv0211 locus from the MTb Gateway Entry Clone Library was amplified (forward primer, GCTCGCTACTCTCATCGTGGAATCCTGACAGGATCCGCGATATCAAGCTTAAGGCCGCCGACATCACCTC; reverse primer, CTCTAGGGTCCCCAATTAATTAGCTAAAGCTCACTTGTACAGCTCGTCCA) and cloned into a pKW08 vector using the SLIC (sequence- and ligation-independent cloning) method (22). The Pck-containing construct was transformed into Mycobacterium bovis BCG Danish strain 1331 by electroporation. BCG cells were cultured in Middlebrook 7H9 broth supplemented with sodium chloride, albumin, dextrose, and catalase at 37 °C with gentle shaking. Cells were then collected by centrifugation (15 min, 4800 × g, 4 °C) and resuspended in 9 ml of cold sonication buffer containing 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm DTT, 2 mm phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich), 25 units/ml benzonase (Sigma-Aldrich), and 0.5% Triton X-100 (Sigma-Aldrich) supplemented with protease inhibitors (2 μm pepstatin A, 2 μg/ml chymostatin, 0.6 μm leupeptin, 1 mm benzamidine HCl, and 0.1 m PMSF). Cells were sonicated (Diagonade) at 4 °C with a power output of 300 W for 90 cycles with 45 s on and 30 s off for each cycle. Cell debris was removed by centrifugation (20 min, 4800 × g, 4 °C), and the clarified cell lysate was transferred into a 15-ml conical tube containing 40 μl of anti-GFP-Sepharose.
The anti-GFP-Sepharose was prepared by coupling anti-GFP nanobodies with cyanogen bromide-activated Sepharose 4 Fast Flow beads (Sigma-Aldrich). The anti-GFP nanobodies were prepared as described elsewhere (23). Briefly, DNA coding sequences were cloned in frame into pET28PP, and a His6 tag was added at the C terminus for ease of purification. The construct was transformed into E. coli BL21-CodonPlus®-RIL and expressed in autoinduction medium (Formidium Super Broth Base including trace elements). Then cells were lysed by sonication in 20 mm Tris, pH 8.0, containing 500 mm NaCl, 20 mm imidazole, and 10 mm 2-mercaptoethanol. The nanobodies were subsequently purified using a Superdex 75 column (GE Healthcare).
Samples containing anti-GFP Sepharose were incubated for 2 h in a cold room with slow (6–8 rpm) end-to-end rotation. The beads were recovered on a polypropylene Poly-Prep chromatography column (Bio-Rad), and the columns were washed twice with 10 ml of buffer containing 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.1% Triton X-100 (Sigma-Aldrich), followed by two washes with TEV buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.5 mm EDTA, 1 mm DTT). Twenty microliters of TEV protease (cloned, expressed, purified, and successfully used in our laboratory (24)) was added to 430 μl of TEV buffer and applied to the columns to cleave the bait protein from the beads, leaving the GFP tag on the column. The cleavage was performed at 4 °C overnight. Purified proteins were collected into Eppendorf tubes. The bait protein and its interacting partners were precipitated by adding one-fourth of the original volume of pyrogallol red-molybdate, PRM reagent (0.05 mm pyrogallol red, 0.16 mm sodium molybdate, 1 mm sodium oxylate, 50 mm succinic acid, pH 2.5; all from Sigma-Aldrich) and vigorously mixed for 30 s followed by incubation at room temperature for at least 1 h. Precipitated proteins were collected by centrifugation (25 min, 21,000 × g, room temperature), and the pellets were submitted for LC-MS/MS analysis. Protein pellets were subjected to a standard procedure of trypsin digestion, and peptide mixtures were applied to RP-18 precolumns of a Waters HPLC system using water containing 0.1% trifluoroacetic acid as the mobile phase and transferred onto a nano-HPLC RP-18 column (internal diameter 75 μm; Waters) using an acetonitrile gradient (0–35% ACN in 160 min) in the presence of 0.1% trifluoroacetic acid at a flow rate of 250 nl/min. The column outlet was coupled directly to the ion source of an Orbitrap Velos mass spectrometer (Thermo Scientific). Blank runs were performed to ensure the absence of cross-contamination from preceding samples.
Peak lists in MGF format were generated from the acquired raw spectra using Mascot Distiller software (Matrix Science, version 2.4) with default Orbitrap_low_res_MS2 parameters. Protein identification was performed by searching the data against the M. bovis protein database (see the Pathosystems Resource Integration Center Web site; NC_008769, 3952 entries). The databases were modified in house to also contain randomized sequences of all entries as a control of false positive identifications during analysis (total number of entries in the database: 13,756). We used Mascot Daemon for multiple search submission on a local Mascot server (Matrix Science, version 2.3). The following search parameters were used: enzyme specificity, trypsin/with no proline restriction; maximum missed cleavages, 1; carbamidomethyl (Cys) as fixed modification; oxidation (Met) as variable modification; precursor ion mass tolerance, 40 ppm; and MS/MS mass tolerance, 0.8 Da. Using the randomized database entries, the Mascot score threshold was computed for each Mascot search in order for the individual false discovery rate to be lower than 0.01. Only peptides exceeding the threshold were accepted.
Structural Change Determination by NMR, Circular Dichroism (CD), and Raman Spectroscopy
The one-dimensional 1H NMR spectra of 100 μm reduced (in the presence of 30 mm DTT) and oxidized Pck were collected at 25 °C on a 600-MHz Bruker Avance spectrometer equipped with a triple-resonance (15N/13C/1H) cryoprobe.
The circular dichroism (CD) spectra of reduced MTb Pck containing 5 mm DTT and oxidized Pck, prepared by dialysis into buffer without DTT, slow oxidation using atmospheric oxygen for 1 day, or chemical oxidation using 10 mm hydrogen peroxide, were measured separately in the UV (190–260 nm) and near UV (250 nm-320 nm) spectral regions in standard experimental setups: UV, 0.1-cm quartz cells, protein concentration of 41 μm, 2 scans, 0.5-nm steps, 10 nm/min speed, 15 s time constant, 1-nm spectral bandwidth; near UV, 0.2-cm quartz cells, protein concentration of 411 μm, 2 scans, 0.5-nm steps, 10 nm/min speed, 16 s time constant, 1-nm spectral bandwidth. Following baseline correction, the final spectra were expressed as molar ellipticity, θ (deg·cm2·dmol−1) per residue. Numerical analysis of the secondary structure and secondary structure assignment was performed using an online circular dichroism analysis program (Dichroweb).
RESULTS
MTb Pck Is a Reversible GTP-dependent Enzyme
To characterize the properties of MTb Pck, we cloned, expressed, and purified Pck (Rv0211 gene product) by ammonium sulfate precipitation, reverse-phase chromatography (butyl-Sepharose), and subsequent affinity chromatography on Talon® resin (Fig. 1). To detect Pck enzymatic activities, we followed the oxidation of NADH to NAD+ in coupling reactions. To set up the optimal arrangement of coupled reactions, we first used fixed concentrations of substrates and cofactors in reactions and showed that concentration of Pck is the limiting factor (Fig. 2). The kinetic data obtained in the presence of 15 nm Pck indicated that Pck can catalyze reactions in both directions (PEP formation or OAA formation) with different efficiencies that depend on multiple factors (see Fig. 3 for reaction schemes) (25, 26). The concentration dependences of initial reaction velocities for individual components of the reaction mixtures followed typical Michaelis-Menten curves (Fig. 3). The enzyme exhibited a 10-fold lower Km value for OAA in the gluconeogenic direction than for PEP in the anaplerotic direction. The reactions operated with the highest efficiencies with GDP and GTP substrates, respectively. MTb Pck also accepted inosine diphosphate (IDP) and inosine triphosphate (ITP) as substrates in the anaplerotic reaction and gluconeogenic reactions, respectively. The apparent Km value for IDP was 3-fold higher than that for GDP (Table 1). Neither the purine nucleotide ADP nor the pyrimidine CDP was processed by Pck in an anaplerotic reaction. We observed a substrate inhibition effect for HCO3− in the anaplerotic direction (Fig. 4A). The addition of the highest concentration of KHCO3 (400 mm) into the reaction solution increased the pH to 8.1. To assess the influence of higher pH on the inhibitory effect of HCO3− on Pck activity, we measured this dependence also at pH 8.1. We did not observe any difference (Fig. 4B), which confirmed that Pck can be inhibited by higher concentration of CO2. However, the inhibiting concentrations of CO2 or hydrogen carbonate (over 100 mm) are much higher than the typical physiological concentrations of 10–50 mm (27). Comparison of the kinetic parameters for Pck forward and reverse reactions clearly indicates that the enzyme belongs to the GTP-dependent enzyme family, as does the homologous enzyme in Mycobacterium smegmatis (25) and Pcks from higher eukaryotic species, such as mice, rats, chickens, and humans. The results from kinetic measurements showed that MTb Pck can operate in both directions in vitro. However, Pck prefers the gluconeogenic reaction under standard conditions. This result corresponds well to the thermodynamics of the gluconeogenic reaction catalyzed by Pck. Under standard conditions, the ΔrG′ value for the gluconeogenic reaction calculated from MTb metabolic data was −8.0 kJ/mol (data not shown).
FIGURE 1.
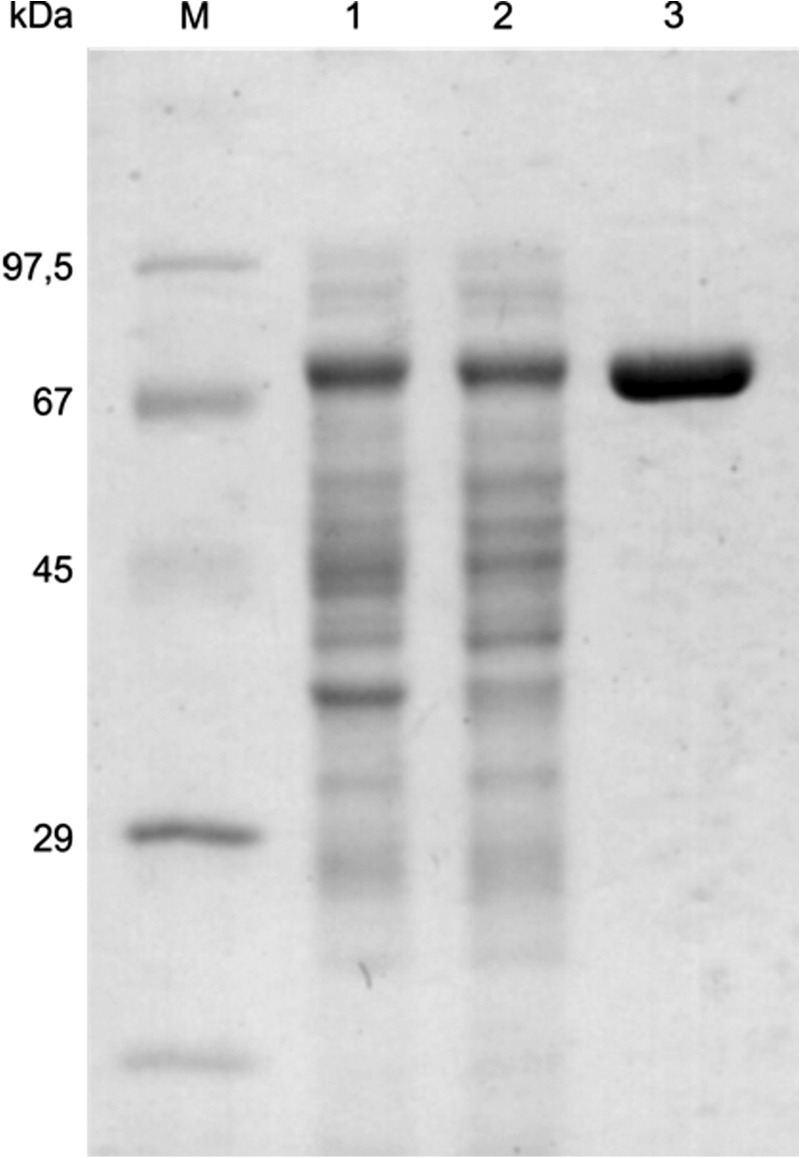
SDS-PAGE analysis of MTb Pck purification. M, molecular mass standards; lane 1, lysate of crude extract of E. coli (BL21) expressing Pck; lane 2, cell supernatant after centrifugation; lane 3, purified MTb Pck after ammonium sulfate precipitation and chromatography on butyl-Sepharose and Talon® resin.
FIGURE 2.
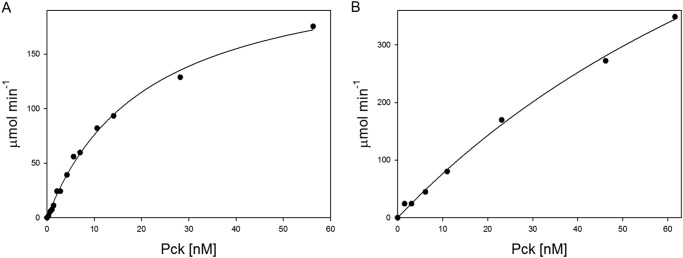
Dependence of reaction velocity on Pck concentration in anaplerotic (A) and gluconeogenic (B) reactions. The assays were performed as described under “Experimental Procedures”. The concentrations of individual components were as follows: 1 mm GDP, 2 mm PEP, and 2 units/ml MDH for the anaplerotic reaction; 0.3 mm OAA, 0.2 mm GTP, 10 units/ml LDH, and 3 units/ml PK for the gluconeogenic reaction.
FIGURE 3.

Substrate kinetics for OAA synthesis by anaplerotic reaction and PEP synthesis by gluconeogenic reaction. The anaplerotic conversion of PEP into OAA by Pck in the presence of GDP was coupled with conversion of OAA into malate in a reaction catalyzed by malate dehydrogenase. Oxidation of NADH to NAD+ was monitored at 340 nm. Synthesis of PEP from OAA catalyzed by Pck in the presence of GTP was coupled with dephosphorylation of PEP into pyruvate by pyruvate kinase and with conversion into lactate in a reaction catalyzed by lactate dehydrogenase. Oxidation of NADH to NAD+ was monitored at 340 nm.
TABLE 1.
Values of kinetic constants for MTb Pck
The reactions were carried out in the standard reaction mixtures described under “Experimental Procedures” in the presence of different concentrations of substrates and cofactors.
| Substrate (Concentration range) | Km | Vmax | Vmax/Km |
|---|---|---|---|
| μm | μmol min−1 mg−1 | ||
| Anaplerotic reaction | |||
| PEP (0.05–6 mm) | 213.2 ± 51.4 | 89.5 ± 4.8 | 0.4 |
| GDP (0.05–4 mm) | 72.9 ± 20.4 | 97.6 ± 4.7 | 1.3 |
| IDP (0.05–4 mm) | 246.8 ± 13.7 | 116.2 ± 1.5 | 0.5 |
| Gluconeogenic reaction | |||
| OAA (0.01–1 mm) | 17.8 ± 2.8 | 142.8 ± 6.6 | 8 |
| GTP (0.02–6 mm) | 209.1 ± 59.0 | 292.2 ± 19.2 | 1.4 |
| ITP (0.02–6 mm) | 177.5 ± 57.6 | 274.0 ± 20.3 | 1.5 |
FIGURE 4.
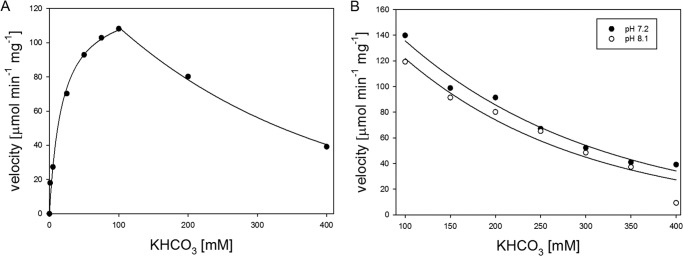
A, the substrate inhibition effect of HCO3− in the anaplerotic direction. The reaction was performed using a standard assay mixture for anaplerotic reaction with concentration of KHCO3 ranging from 0 to 400 mm. The data were analyzed by two non-linear regression methods. The first part of the curve was obtained using a standard Michaelis-Menten curve equation, and the second part was obtained by exponential regression. B, dependence of the initial reaction velocities on different concentration of KHCO3 in the anaplerotic reaction at different pH values. The individual reaction mixtures contained 100 mm HEPES-NaOH (final pH 7.2), 37 mm DTT, 2 mm PEP, 1 mm GDP, 2 mm MgCl2, 0.1 mm MnCl2, 2 units/ml MDH, 0.25 mm NADH, and different concentrations of KHCO3.
Anaplerotic Formation of Oxalacetate Requires Reducing Conditions
Pck has been suggested to operate in the anaplerotic direction in slowly growing MTb, which has intracellular redox conditions different from those of active bacteria. The ratio of NADH/NAD+ is almost 70-fold greater in slowly growing MTb (9). The presence of higher concentrations of reduced cofactors can influence the properties and functions of a number of enzymes, including Pck. Therefore, we further investigated the influence of reducing agents on both the anaplerotic and gluconeogenic reactions catalyzed by Pck. Whereas the anaplerotic reaction was found to be stimulated by and strictly dependent on the presence of DTT (Fig. 5A, black dots), the gluconeogenic reaction was almost independent of the presence of reducing agents (Fig. 5B, black dots). Similarly, the reducing agents 2-mercaptoethanol, DTT, tris(2-carboxyethyl)phosphine, and reduced glutathione exclusively increased the anaplerotic formation of OAA from PEP (Fig. 5C). Because we used Pck activity assays with coupled reactions, which might themselves be influenced by the conditions tested, we used mass spectrometry to directly quantify the reactants and products (PEP, GTP, and GDP) over the course of the Pck enzymatic reactions without the presence of coupled enzymes. These alternative data confirmed the activity profiles and dependences on reducing conditions that resulted from the coupled biochemical reactions exploiting the oxidation of NADH and its decrease in absorbance at 340 nm. These results indicate that ambient conditions significantly influence MTb Pck substrate specificity and preference for metabolites.
FIGURE 5.

Effect of DTT on activity of wild type Pck (black dots) and Pck C273S (empty dots) mutant in anaplerotic and gluconeogenic reactions. Shown is the effect of increasing concentrations of DTT on anaplerotic (A) and gluconeogenic Pck activities (B). C, influence of different reducing agents on activity of Pck in the anaplerotic reaction. The relative activity is expressed as a ratio of maximal gluconeogenic Pck activity in the presence of 100 mm DTT and Pck activity in the presence of a particular DTT concentration.
Reducing Conditions Do Not Alter the Pck Structure
Having established that reducing conditions differentially favor the anaplerotic activity of the enzyme, we further investigated the impact of reducing conditions on the molecular structure of Pck.
We collected one-dimensional 1H NMR spectra from 100 μm Pck free of reducing agents and containing 30 mm DTT, conditions under which MTb Pck anaplerotic activity is clearly stimulated. The collected spectra of both Pck samples did not differ significantly (Fig. 6), which suggests that reducing conditions do not cause significant changes in the protein structure. To support our conclusion and to investigate the influence of reducing conditions on Pck conformation, we analyzed reduced and oxidized Pck samples using CD spectroscopy. Pck oxidation was performed either by atmospheric oxygen or by chemical oxidation using 10 mm hydrogen peroxide for 20 min. We observed only small changes in the secondary structure of Pck oxidized by H2O2, in which the α-helical content was slightly suppressed in favor of β-sheets and random structures. The near UV CD spectra showed changes in the interval from 260 to 290 nm, which reflects the molecular movements of aromatic side chains (Phe, Tyr, and Trp) (Fig. 7, A and B). In summary, the collected spectroscopic data show that reducing conditions do not cause any substantial conformational changes in the protein structure.
FIGURE 6.
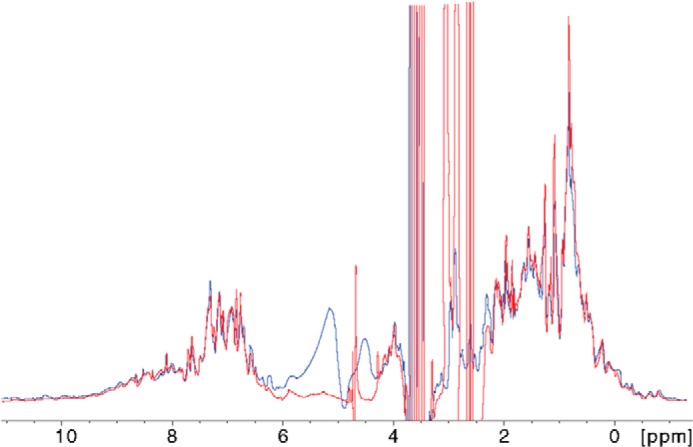
One-dimensional 1H NMR spectra of Pck in the absence (blue) and presence (red) of 30 mm DTT. The signals in the relatively well dispersed spectral regions from backbone and side-chain (Trp) amide protons (7.5–11.0 ppm) and side-chain methyls (−1.0–0.5 ppm) were not affected by the presence of reducing agent.
FIGURE 7.

A and B, far UV CD (A) and near UV CD (B) spectra of WT Pck reduced by 5 mm DTT (MTb; Pck-red), in the absence of any reducing agents (MTb; Pck-ox), and after oxidation with hydrogen peroxide (MTb; Pck-H2O2). C, far-UV CD spectra of WT MTb Pck and C273S Pck mutant in the presence of 5 mm DTT.
Hyperreactive Cys Contributes to the Switch between the Anaplerotic and Gluconeogenic Functions of Pck
The sequence alignment of MTb and rat and human Pcks (Fig. 8) indicated that MTb Pck shares 51% identity and 66% similarity with human and rat Pcks. The three-dimensional structures of these Pck orthologues are very similar, as illustrated by the MTb Pck model prepared using the crystal structure of rat Pck as a template (Fig. 9). MTb Pck contains nine cysteine residues that might co-determine the redox state and conformation of Pck under different conditions. The structural alignment showed that Cys-273, located within the putative P loop of MTb Pck, is probably the hyperreactive cysteine residue, which is typical for the GTP-dependent Pck family and coordinates binding of Mn2+ in the active site (28). DTNB titration of total free sulfhydryl groups in Pck in buffer without DTT indicated the presence of seven cysteines in the reduced state (data not shown), suggesting that only one disulfide bridge can be formed in the enzyme. Changes in reducing conditions could potentially influence the formation of this disulfide bridge.
FIGURE 8.

Structure-based sequence alignment of MTb, rat cytosolic, and human cytosolic Pcks created by ClustalW2. UniProt accession numbers of sequences are as follows: P65686, MTb; P07379, rat; and P35558, human. Black and gray backgrounds denote identical and similar amino acid residues, respectively. The black arrow indicates the conserved reactive cysteine residue. The flexible R, P, and Ω loops are indicated.
FIGURE 9.

A, superimposition of MTb Pck (yellow) and rat Pck (blue; Swiss Prot entry PCKGC_RAT). The homology modeling of the primary structure of MTb Pck was carried out using the program Modeler. Phosphoenolpyruvate and GDP in the active site are shown in red. B, model of MTb Pck. The P loop is shown in gray, cysteine 273 in black, R loop in blue, and Ω loop in green.
To elucidate the role of the putative hyperreactive Cys residue in the gluconeogenic and anaplerotic Pck activities, we mutated Cys-273 into Ser (C273S). Comparison of kinetic data from WT and C273S Pcks revealed that PEP bound into the active site of C273S with a Km value almost 6-fold lower than that for WT Pck, which led to more efficient catalysis and production of OAA by the C273S mutant in the anaplerotic reaction. In contrast, the catalytic efficiency of C273S in the gluconeogenic reaction was about 3.5-fold lower than that of WT (Table 2). Interestingly, both reactions catalyzed by the C273S mutant strictly required the presence of a reducing environment and were stimulated by increasing DTT concentrations (Fig. 5, A and B). In contrast, the gluconeogenic reaction catalyzed by WT Pck is not affected by reducing agents. CD spectra of the C273S mutant showed that the content of α-helices was reduced in favor of β-sheets (see Fig. 7C). These results show that Cys-273 has a regulatory role for MTb Pck activities but is not essential for the enzymatic activities of Pck.
TABLE 2.
Values of kinetic constants for MTb Pck C273S mutant
The reactions were carried out in the standard reaction mixtures described under “Experimental Procedures” in the presence of different concentrations of substrates and cofactors.
| Substrate (Concentration range) | Km | Vmax | Vmax/Km |
|---|---|---|---|
| μm | μmol min−1 mg−1 | ||
| Anaplerotic reaction | |||
| PEP (0.05–6 mm) | 36.2 ± 17.1 | 59.6 ± 4.7 | 1.6 |
| GDP (0.05–4 mm) | 19.7 ± 6.4 | 57.6 ± 1.8 | 2.9 |
| IDP (0.05–4 mm) | 20.3 ± 5.1 | 42.1 ± 1.1 | 2.1 |
| Gluconeogenic reaction | |||
| OAA (0.01–1.2 mm) | 21.4 ± 5.2 | 48.4 ± 2.4 | 2.3 |
| GTP (0.02–3 mm) | 128 ± 13.6 | 125.6 ± 3.2 | 1 |
| ITP (0.02–3 mm) | 118.8 ± 13.7 | 132.6 ± 3.6 | 1.1 |
MTb Pck Interacts with Proteins Involved in Oxidative Stress Protection
To identify physiological partners of MTb Pck that might be involved in the regulation of its enzymatic activities, we analyzed the formation of complexes of MTb Pck with M. bovis (BCG) cellular proteins using tandem affinity chromatography and mass spectrometry analysis. The MTb Pck-interacting proteins are summarized in Table 3. Purifications of Pck-protein complexes were performed in three independent experiments. The parental BCG Danish 1331 strain transformed with GFP vector (without any target coding sequence) was used as a background control. Cell lysate from the control strain was purified and analyzed in the same manner as experimental samples to determine nonspecific binding and false positives. All proteins detected in the control samples were subtracted from experimental samples. Additionally, as described under “Experimental Procedures,” the database used for this experiment was randomized to avoid false positive identifications during analysis.
TABLE 3.
Proteins interacting with MTb Pck
Proteins known as “common contaminants” (hypothetical proteins, ribosomal proteins, chaperonins, heat-shock proteins, transcription factors) were filtered out. All presented proteins were detected in at least two experiments, Mascot score <100, number of unique peptides <2. Expt., experiment; ND, not detected; *, tagged protein (bait); boldface type, proteins involved in oxidative stress protection.
| Protein ID/Gene ID | Protein description | Mascot score |
||
|---|---|---|---|---|
| Expt. 1 | Expt. 2 | Expt. 3 | ||
| BCG_0248* | Phosphoenolpyruvate carboxykinase (M. bovis BCG str. Pasteur 1173P2) | 57359 | 21556 | 37844 |
| BCG_2545c | Putative fatty acid synthase fas (M. bovis BCG str. Pasteur 1173P2) | 2217 | 4496 | 5536 |
| BCG_0048c | Serine/threonine phosphatase PPP (M. bovis BCG str. Pasteur 1173P2) | 568 | 175 | ND |
| BCG_2024c | Putative ferredoxin fdxA (M. bovis BCG str. Pasteur 1173P2) | 446 | 833 | 910 |
| BCG_0020/BCG_3972 | Thioredoxin TrxC (M. bovis BCG str. Pasteur 1173P2) | 250 | ND | 125 |
| BCG_2962c | Acyl-CoA synthetase (M. bovis BCG str. Pasteur 1173P2) | ND | 2851 | 4391 |
| BCG_2875 | Mycothione reductase (M. bovis BCG str. Pasteur 1173P2) | ND | 1862 | 4135 |
| BCG_1908c | Putative l-lactate dehydrogenase (cytochrome) lldD2 (M. bovis BCG str. Pasteur 1173P2) | ND | 1750 | 2503 |
| BCG_2371c | Glycyl-tRNA synthetase (M.bovis BCG str. Pasteur 1173P2) | ND | 1090 | 1588 |
| BCG_2953 | Phenolpthiocerol synthesis type-I polyketide synthase ppsA (M. bovis BCG str. Pasteur 1173P2) | ND | 850 | 1385 |
| BCG_3481c | Inosine 5′-monophosphate dehydrogenase (M. bovis BCG str. Pasteur 1173P2) | ND | 740 | 409 |
| BCG_2969c | Acyl-CoA synthetase (M. bovis BCG str. Pasteur 1173P2) | ND | 719 | 1352 |
| BCG_2882c | Putative glutamine synthetase glnA4 (M. bovis BCG str. Pasteur 1173P2) | ND | 660 | 749 |
| BCG_2515c | Branched-chain α-keto acid dehydrogenase subunit E2 (M. bovis BCG str. Pasteur 1173P2) | ND | 634 | 276 |
| BCG_2447 | Alkyl hydroperoxide reductase C protein ahpC (M. bovis BCG str. Pasteur 1173P2) | ND | 560 | 278 |
| BCG_3075c | Ribonucleotide-diphosphate reductase subunitα (M. bovis BCG str. Pasteur 1173P2) | ND | 553 | 1139 |
| BCG_1370 | F0F1-ATP synthase subunitβ (M. bovis BCG str. Pasteur 1173P2) | ND | 552 | 1146 |
| BCG_1964 | Acyl-CoA synthetase (M. bovis BCG str. Pasteur 1173P2) | ND | 462 | 1042 |
| BCG_1637 | Histidinol dehydrogenase (M. bovis BCG str. Pasteur 1173P2) | ND | 395 | 656 |
| BCG_3863c | Acyl-CoA synthetase (M. bovis BCG str. Pasteur 1173P2) | ND | 394 | 935 |
| BCG_3302 | Putative transmembrane carbonic anhydrase (M. bovis BCG str. Pasteur 1173P2) | ND | 393 | 354 |
Intriguingly, the redox-regulating proteins ferredoxin A (FdxA), thioredoxin C (TrxC), alkyl hydroperoxide reductase C (AhpC), and mycothione reductase (Mtr) were among the identified interacting proteins. This suggests that Pck function is connected to the cellular redox state and its regulatory machinery. Thioredoxin has recently been proposed to coordinate the oxidative stress responses for Candida albicans survival in macrophages (29). Thioredoxin is responsible for maintaining a reducing intracellular environment by reversible reduction of protein disulfides. The major substrates of thioredoxin are peroxiredoxin enzymes, which are oxidized upon reduction of H2O2 and utilize thioredoxin during their catalysis. AhpC is representative of typical two-Cys peroxiredoxins and thus might participate in protecting Pck from reactive oxygen species (30). Both TrxC (BCG_0020) and AhpC (BCG_2447) of BCG are identical to MTb TrxC (Rv3914) and AhpC (Rv2428), respectively. This alkyl hydroperoxide reductase very likely serves as a ferroactivator, which can remove peroxide formed by autoactivation of Fe2+ in bacteria.
Thioredoxin Stimulates the Anaplerotic Pck Activity
To validate our pull-down results, we investigated the influence of thioredoxin on Pck activities. We cloned, expressed, and purified MTb TrxB (Rv1471) and TrxC (Rv3914). The function of both thioredoxins in reduction of disulfide bridges was first tested with a model substrate: human insulin. The time dependence curves of insulin β-chain formation had comparable profiles, which reflect the similar abilities of these thioredoxins to reduce insulin (Fig. 10C). To account for the contribution of 1 mm DTT to Pck activity in the presence of thioredoxins, the parallel Pck anaplerotic reactions without thioredoxins were performed, and the measured activities were subtracted from the Pck anaplerotic activities detected in the presence of both thioredoxin and 1 mm DTT (Fig. 10, A and B). We then tested the effect of MTb TrxB and TrxC on anaplerotic reactions catalyzed by Pck (Fig. 11, A and B). We observed time- and dose-dependent increases in Pck anaplerotic activity upon the addition of both redox proteins. Their effects were comparable at higher concentrations, suggesting that the thioredoxin system participates in the regulation of MTb Pck activity. Importantly, TrxB and TrxC did not activate the anaplerotic activity of the C273S mutant (data not shown), which indicates an important role for Cys-273 in the regulation of MTb Pck by the cellular redox machinery.
FIGURE 10.

A and B, anaplerotic activity of Pck in the presence of thioredoxin B (A) and thioredoxin C (B). The activities of Pck after the incubation of the enzyme with thioredoxins and DTT (open circles) or 1 mm DTT only (full circles) were measured for each incubation time data point. C, reduction of insulin β-chains catalyzed by TrxB and TrxC. Insulin (50 μm) was incubated in 100 mm potassium phosphate buffer, pH 7, containing 1 mm DTT. Upon the addition of thioredoxins (200 μm), the turbidity measured at 650 nm increased over time.
FIGURE 11.
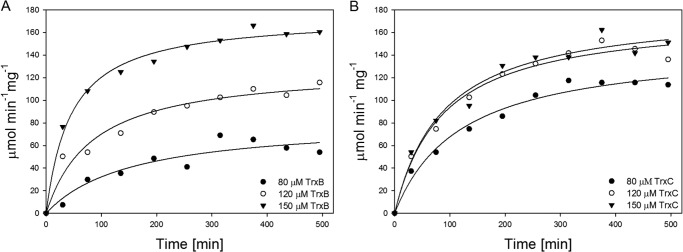
Influence of TrxB (A) and TrxC (B) on anaplerotic reactions catalyzed by Pck. The individual reaction mixtures contained 100 mm HEPES-NaOH, 100 mm KHCO3, 2 mm PEP, 1 mm GDP, 2 mm MgCl2, 0.1 mm MnCl2, 2 units/ml MDH, and 0.25 mm NADH.
DISCUSSION
Previous analyses of MTb metabolism indicated that MTb Pck can be involved in both anaplerotic and gluconeogenic routes, depending on the bacterial growth conditions. This suggests that Pck might contribute to regulation of metabolic adaptations of MTb central metabolism in response to changing environments (9, 12, 14). In this study, we set out to systematically investigate in vitro reaction conditions that influence the activity of the Pck anaplerotic and gluconeogenic reactions and could thus regulate the PEP-OAA node in vivo. Hypoxia arrest, which has been associated with the anaplerotic reaction of Pck, induces changes in the MTb intracellular milieu. The NADH/NAD+ ratio increases, and because the terminal electron acceptor is reduced, the ATP level is decreased, and the pH is slightly acidic in slowly growing MTb (9, 31). We found that reducing environments significantly influence MTb Pck substrate preferences and the direction of catalysis. Whereas gluconeogenic synthesis of PEP from OAA catalyzed by MTb Pck was completely independent of the presence of reducing agents, the anaplerotic synthesis of OAA from PEP proceeded only in the presence of reducing agents. The addition of 10 mm DTT to the Pck reaction mixture increased anaplerotic synthesis of OAA by 80%, but the presence of even 60 mm DTT did not increase synthesis of PEP. Importantly, the gluconeogenic reaction was not inhibited by a reducing environment, suggesting that the availability of substrate under reducing conditions can also influence the direction of catalysis.
The overall structure and fold of MTb Pck do not change during a transition to reducing conditions; the NMR and CD spectra collected from oxidized and reduced forms of MTb Pck were very similar. We cannot, however, rule out the possibility that some movements of short flexible loops in the MTb Pck structure might play a role in correct positioning of substrates in the active site and protection of substrate intermediates during catalysis, as has been shown for the R, Ω, and P loops of GTP-dependent rat cytosolic Pck (32). The structure of MTb Pck is similar to that of rat Pck, as indicated by homology modeling of MTb Pck using a crystal structure of rat Pck as a template (Fig. 9). Therefore, we can speculate that similar loops might play a role in correct substrate positioning in MTb Pck under various redox conditions. Further detailed structural study of MTb Pck in complex with different substrates, metal ions, and GTP/GDP under various conditions would help to elucidate the structure-function dependences of this enzyme.
MTb possesses several machineries that can potentially influence intracellular redox homeostasis. In addition to NADH/NAD+ and NADPH/NADP+, the oxidized-reduced low molecular weight redox couple mycothiol (MSSM/2MSH) and mycothione reductase (Mtr) and/or the thioredoxin redox couple (TrxSS/TrxSH2) reduce disulfide bonds in interacting proteins and maintain a reducing intracellular environment. Trx also regenerates the free SH groups in oxidoreductases and peroxiredoxins that remove H2O2 and reactive O2− in different organisms and tissues and thus contribute to their detoxification. Our pull-down experiments indicated that MTb Pck interacts with several proteins involved in the cellular redox balance and antioxidant defense. Thioredoxin C was detected as one of the candidates. To decrease the false interactions during pull-down experiments, we used rigorous controls, described in detail under “Experimental Procedures,” during the affinity-based purifications. Because the monothiol mutants of MTb thioredoxins that are often used for identification of interacting partners (33) were not applied in our study, we verified the influence of thioredoxins on MTb Pck by in vitro testing of recombinant TrxC on Pck activities. In addition, we prepared TrxB (Rv1471) and confirmed that both TrxB and TrxC notably increase the Pck anaplerotic reaction rate. Similar effects of both Trx forms on Pck activity reflect their similar redox potentials (E0(TrxB) = −262 mV; E0(TrxC) = −269 mV) (34).
Thioredoxins may contribute to the reduced state of the hyperreactive Cys-273 and also to the proper state of other cysteines. Thus, they may play a regulatory role in determining the catalytic state of Pck. The reactive Cys has been found to be important but not essential for both enzymatic activities of Pck. Rather, its presence seems to be critical for the correct regulation of Pck gluconeogenic activity. TrxC and/or TrxB very likely also participate in reduction of the thiol-dependent peroxidase AhpC, which is highly expressed during hypoxic conditions (35). Our results from pull-down experiments suggest that the reducing system composed of thiol-dependent peroxidase AhpC, TrxC, and thioredoxin reductase participates and promotes the anaplerotic reaction of Pck.
Taken together, the results of this study provide evidence that reaction conditions help to determine the preference of Pck for anaplerotic and gluconeogenic directions of catalysis and help to explain previously published data from transcriptomic and metabolomics studies. Our results show that MTb Pck operating in the anaplerotic direction can fully profit from increased intracellular reducing conditions and from up-regulation of proteins maintaining the intracellular reduced state and antioxidant defense.
Acknowledgments
We thank Dr. V. Veverka (Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic) for NMR experiments, Dr. L. Bednarova for CD experiments, D. Grundova for excellent technical assistance, and H. Hoffman for editing of the manuscript.
This work was supported by EU-PF7 SysteMtb Collaborative Project 241587, RVO 61388963, and by Operační program Praha-Konkurenceschopnost (OPPK) Project CZ.2.16/3.1.00/24016.
- MTb
- M. tuberculosis
- OAA
- oxaloacetate
- PEP
- phosphoenolpyruvate
- Pck
- phosphoenolpyruvate carboxykinase
- MDH
- malate dehydrogenase
- PK
- pyruvate kinase
- LDH
- lactate dehydrogenase.
REFERENCES
- 1. World Health Organization (2013) Global Tuberculosis Report 2013, World Health Organization, Geneva [Google Scholar]
- 2. Rohde K. H., Abramovitch R. B., Russell D. G. (2007) Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2, 352–364 [DOI] [PubMed] [Google Scholar]
- 3. Waddell S. J., Butcher P. D. (2007) Microarray analysis of whole genome expression of intracellular Mycobacterium tuberculosis. Curr. Mol. Med. 7, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berney M., Cook G. M. (2010) Unique flexibility in energy metabolism allows mycobacteria to combat starvation and hypoxia. PLoS One 5, e8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Via L. E., Lin P. L., Ray S. M., Carrillo J., Allen S. S., Eum S. Y., Taylor K., Klein E., Manjunatha U., Gonzales J., Lee E. G., Park S. K., Raleigh J. A., Cho S. N., McMurray D. N., Flynn J. L., Barry C. E. (2008) Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 76, 2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rohde K. H., Veiga D. F., Caldwell S., Balázsi G., Russell D. G. (2012) Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 8, e1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kendall S. L., Withers M., Soffair C. N., Moreland N. J., Gurcha S., Sidders B., Frita R., Ten Bokum A., Besra G. S., Lott J. S., Stoker N. G. (2007) A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol. Microbiol. 65, 684–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Srinivasan V., Morowitz H. J. (2006) Ancient genes in contemporary persistent microbial pathogens. Biol. Bull. 210, 1–9 [DOI] [PubMed] [Google Scholar]
- 9. Watanabe S., Zimmermann M., Goodwin M. B., Sauer U., Barry C. E., 3rd, Boshoff H. I. (2011) Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 7, e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shi L., Sohaskey C. D., Pfeiffer C., Datta P., Parks M., McFadden J., North R. J., Gennaro M. L. (2010) Carbon flux rerouting during Mycobacterium tuberculosis growth arrest. Mol. Microbiol. 78, 1199–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sauer U., Eikmanns B. J. (2005) The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol. Rev. 29, 765–794 [DOI] [PubMed] [Google Scholar]
- 12. Marrero J., Rhee K. Y., Schnappinger D., Pethe K., Ehrt S. (2010) Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. U.S.A. 107, 9819–9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu K., Yu J., Russell D. G. (2003) pckA-deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophages. Microbiology 149, 1829–1835 [DOI] [PubMed] [Google Scholar]
- 14. Beste D. J., Bonde B., Hawkins N., Ward J. L., Beale M. H., Noack S., Nöh K., Kruger N. J., Ratcliffe R. G., McFadden J. (2011) 13C metabolic flux analysis identifies an unusual route for pyruvate dissimilation in mycobacteria which requires isocitrate lyase and carbon dioxide fixation. PLoS Pathog. 7, e1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beste D. J., Nöh K., Niedenführ S., Mendum T. A., Hawkins N. D., Ward J. L., Beale M. H., Wiechert W., McFadden J. (2013) 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem. Biol. 20, 1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 17. Ríos S. E., Nowak T. (2002) Role of cysteine 306 in the catalytic mechanism of Ascaris suum phosphoenolpyruvate carboxykinase. Arch. Biochem. Biophys. 404, 25–37 [DOI] [PubMed] [Google Scholar]
- 18. Ballard F. J. (1970) Kinetic studies with cytosol and mitochondrial phosphoenolpyruvate carboxykinases. Biochem. J. 120, 809–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dharmarajan L., Case C. L., Dunten P., Mukhopadhyay B. (2008) Tyr235 of human cytosolic phosphoenolpyruvate carboxykinase influences catalysis through an anion-quadrupole interaction with phosphoenolpyruvate carboxylate. FEBS J. 275, 5810–5819 [DOI] [PubMed] [Google Scholar]
- 20. Buescher J. M., Moco S., Sauer U., Zamboni N. (2010) Ultrahigh performance liquid chromatography-tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Anal. Chem. 82, 4403–4412 [DOI] [PubMed] [Google Scholar]
- 21. Makinen A. L., Nowak T. (1989) A reactive cysteine in avian liver phosphoenolpyruvate carboxykinase. J. Biol. Chem. 264, 12148–12157 [PubMed] [Google Scholar]
- 22. Li M. Z., Elledge S. J. (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256 [DOI] [PubMed] [Google Scholar]
- 23. Płocinski P., Laubitz D., Cysewski D., Stodus K., Kowalskaand K., Dziembowski A. (2014) Identification of protein partners in Mycobacteria using a single step affinity purification method. PLoS One 9, e91380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomecki R., Kristiansen M. S., Lykke-Andersen S., Chlebowski A., Larsen K. M., Szczesny R. J., Drazkowska K., Pastula A., Andersen J. S., Stepien P. P., Dziembowski A., Jensen T. H. (2010) The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 29, 2342–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mukhopadhyay B., Concar E. M., Wolfe R. S. (2001) A GTP-dependent vertebrate-type phosphoenolpyruvate carboxykinase from Mycobacterium smegmatis. J. Biol. Chem. 276, 16137–16145 [DOI] [PubMed] [Google Scholar]
- 26. Zamboni N., Maaheimo H., Szyperski T., Hohmann H. P., Sauer U. (2004) The phosphoenolpyruvate carboxykinase also catalyzes C3 carboxylation at the interface of glycolysis and the TCA cycle of Bacillus subtilis. Metab. Eng. 6, 277–284 [DOI] [PubMed] [Google Scholar]
- 27. Dyson J. E., Anderson W. B., Nordlie R. C. (1969) Inhibitory effect of physiological bicarbonate ion levels on the activities of glucose 6-phosphate phosphohydrolase. J. Biol. Chem. 244, 560–566 [PubMed] [Google Scholar]
- 28. Holyoak T., Sullivan S. M., Nowak T. (2006) Structural insights into the mechanism of PEPCK catalysis. Biochemistry 45, 8254–8263 [DOI] [PubMed] [Google Scholar]
- 29. da Silva Dantas A., Patterson M. J., Smith D. A., Maccallum D. M., Erwig L. P., Morgan B. A., Quinn J. (2010) Thioredoxin regulates multiple hydrogen peroxide-induced signaling pathways in Candida albicans. Mol. Cell. Biol. 30, 4550–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jaeger T., Budde H., Flohé L., Menge U., Singh M., Trujillo M., Radi R. (2004) Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch. Biochem. Biophys. 423, 182–191 [DOI] [PubMed] [Google Scholar]
- 31. Abramovitch R. B., Rohde K. H., Hsu F. F., Russell D. G. (2011) aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol. Microbiol. 80, 678–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson T. A., Holyoak T. (2012) The Ω-loop lid domain of phosphoenolpyruvate carboxykinase is essential for catalytic function. Biochemistry 51, 9547–9559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sturm N., Jortzik E., Mailu B. M., Koncarevic S., Deponte M., Forchhammer K., Rahlfs S., Becker K. (2009) Identification of proteins targeted by the thioredoxin superfamily in Plasmodium falciparum. PLoS Pathog. 5, e1000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akif M., Khare G., Tyagi A. K., Mande S. C., Sardesai A. A. (2008) Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J. Bacteriol. 190, 7087–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sherman D. R., Voskuil M., Schnappinger D., Liao R., Harrell M. I., Schoolnik G. K. (2001) Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding α-crystallin. Proc. Nat. Acad. Sci. 98, 7534–7539 [DOI] [PMC free article] [PubMed] [Google Scholar]