

Background: The mTOR complex 1 (mTORC1) phosphorylates ribosomal S6 kinase (S6K1) on Thr-389, leading to S6K1 activation.
Results: Inhibition of SIRT1/2, members of the sirtuin family of proteins, induces S6K1 acetylation and inhibits mTORC1-dependent S6K1 phosphorylation.
Conclusion: The SIRT1/2 support mTORC1-induced S6K1 activation by inhibiting S6K1 acetylation.
Significance: The study provides a novel mechanistic insight into the cross-talk between sirtuins and mTORC1 signaling.
Keywords: mTOR, mTOR Complex (mTORC), Phosphorylation, S6 Kinase, Sirt1, Sirtuins, Acetylation, Rapamycin
Abstract
p70 ribosomal S6 kinase (S6K1), a major substrate of the mammalian target of rapamycin (mTOR) kinase, regulates diverse cellular processes including protein synthesis, cell growth, and survival. Although it is well known that the activity of S6K1 is tightly coupled to its phosphorylation status, the regulation of S6K1 activity by other post-translational modifications such as acetylation has not been well understood. Here we show that the acetylation of the C-terminal region (CTR) of S6K1 blocks mTORC1-dependent Thr-389 phosphorylation, an essential phosphorylation site for S6K1 activity. The acetylation of the CTR of S6K1 is inhibited by the class III histone deacetylases, SIRT1 and SIRT2. An S6K1 mutant lacking acetylation sites in its CTR shows enhanced Thr-389 phosphorylation and kinase activity, whereas the acetylation-mimetic S6K1 mutant exhibits decreased Thr-389 phosphorylation and kinase activity. Interestingly, relative to the acetylation-mimetic S6K1 mutant, the acetylation-defective mutant displays higher affinity toward Raptor, an essential scaffolding component of mTORC1 that recruits mTORC1 substrates. These observations indicate that sirtuin-mediated regulation of S6K1 acetylation is an additional important regulatory modification that impinges on the mechanisms underlying mTORC1-dependent S6K1 activation.
Introduction
The evolutionarily conserved ribosomal protein S6 kinases belong to the AGC (PKA, PKG, and PKC) kinase family and play important roles in the regulation of cell growth and metabolism (1, 2). In mammals, two separate genes encode S6Ks:2 S6K1 (or S6Kα) and S6K2 (or S6Kβ). The use of alternative ATG start codons makes two protein isoforms for each S6 kinase (for S6K1, p85 and p70; for S6K2, p56 and p54) (3–5). Among these S6Ks, the p70 S6K1 (S6Kα2), termed S6K1 in this study, is the best characterized isoform for its function and regulation. In response to environmental cues such as growth factors and nutrients, S6K1 is phosphorylated and activated by two upstream serine/threonine kinases, mTOR and phosphoinositide-dependent kinase 1 (PDK1) (6–8). Importantly, S6K1 is specifically phosphorylated by mTOR kinase forming a complex with Raptor (mTORC1), which is a rapamycin-sensitive complex (9–11). mTORC1 first phosphorylates Thr-389 on the hydrophobic motif of S6K1, which creates a PDK1 binding motif, and subsequently induces PDK1-dependent Thr-229 phosphorylation on the activation loop of S6K1 (12–14). These cooperative and sequential phosphorylations by mTORC1 and PDK1 are essential for the activation of S6K1.
It has been demonstrated that both N-terminal and C-terminal regions of S6K1 play important roles in mTORC1-mediated Thr-389 phosphorylation in the linker region (15–17). The N terminus of S6K1 contains unique short regulatory sequences termed TOR signaling (TOS) motifs, which are also found in other mTORC1 substrates such as 4EBPs and PRAS40 (18, 19). Mutation or deletion of the TOS motif of S6K1 abolishes mTORC1-mediated Thr-389 phosphorylation. Although it has been postulated that the TOS motif of S6K1 interacts with Raptor, an essential scaffold protein of mTORC1 (20, 21), mutation of the autoinhibitory motif in the C-terminal region (CTR) of S6K1 restores mTORC1-mediated Thr-389 phosphorylation of the S6K1 TOS motif mutant (22). These observations suggest that the TOS motif may play a role in modulating the regulatory mechanisms of the S6K1 CTR for mTORC1-mediated Thr-389 phosphorylation.
The CTR is unique to S6Ks among other AGC family members and contains a pseudo-substrate domain with an autoinhibitory motif. The pseudo-substrate domain has been proposed to interact with the kinase domain of S6K1 and inhibit mTORC1-dependent phosphorylation of Thr-389 (23–25). In addition to its inhibitory role for S6K1 activation, the CTR determines the dependence of S6K1 regulation by mTORC1. Thr-389 phosphorylation of the S6K1 lacking the CTR (ΔCT S6K1) becomes partially rapamycin-resistant (26). Consistent with this observation, genetic and biochemical studies have demonstrated that Thr-389 of the ΔCT S6K1 can be phosphorylated by both mTORC1 and rapamycin-insensitive mTORC2 (26). These observations indicated that the intact CTR is required for mTORC1-specific and -dependent Thr-389 phosphorylation of S6K1. The CTR can be subjected to multiple post-translational modifications including phosphorylation and acetylation. Although phosphorylation of the CTR (Ser-411, Ser-418, Thr-421, and Ser-424) has been proposed to play a role in supporting mTORC1-dependent Thr-389 phosphorylation by exposing the linker region to mTORC1 (23, 25), the regulatory role of CTR acetylation for S6K1 activation has not been well understood.
Here we show that S6K1 is acetylated on three lysine residues in its CTR. Acetylation of S6K1 suppresses mTORC1-dependent Thr-389 phosphorylation and results in an inhibition of its activity. We found that S6K1 acetylation is suppressed by members of the sirtuin family of deacetylases, SIRT1 and SIRT2, indicating that the sirtuin pathway impinges on the regulation of mTORC1-dependent S6K1 activation. Furthermore, acetylation-defective S6K1 shows greater affinity for Raptor in vivo. Our study thus provides evidence for a previously unrecognized cross-talk between the sirtuins and the mTOR pathways at the level of S6K1 activation.
EXPERIMENTAL PROCEDURES
Reagents, Antibodies, and Plasmids
Nicotinamide, trichostatin A, and rapamycin were obtained from Sigma-Aldrich. Torin 1 was purchased from TOCRIS Bioscience. Protein G-Sepharose and ECL were purchased from GE Healthcare. [γ-32P]ATP was obtained from MP Biomedicals. Antibodies against Myc and HA tags were purchased from Covance. Acetyl Lys-516 (Lys-493)-S6K1 antibody was obtained from Abcam. S6K1 antibody for immunoprecipitation was described previously (27). All other antibodies were purchased from Cell Signaling Technology. S6K1 and Raptor constructs were kindly provided by Drs. John Blenis (Harvard University) and David Sabatini (Massachusetts Institute of Technology (MIT)), respectively. HA-SIRT1 and SIRT2 constructs were gifts from Dr. Roland Kwok (University of Michigan). All the HA-S6K1 mutants were generated using QuikChange site-directed mutagenesis kit (Stratagene).
Cell Culture and Treatments
COS-7 or HEK293T cells were cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (FBS, HyClone Laboratories) and penicillin/streptomycin (Invitrogen). To inhibit histone deacetylases, COS-7 cells were treated with 5 nm trichostatin A (TSA), 5 mm nicotinamide (NAM), or both NAM + TSA (NT). 100 nm Torin 1 was used for inhibiting mTOR activity.
Immunoblot and Immunoprecipitation
Cells were lysed on ice for 10 min in Nonidet P-40 lysis buffer (10 mm Tris, pH 7.5, 2 mm EDTA, 100 mm NaCl, 1% Nonidet P-40, 50 mm NaF, 10 mm sodium pyrophosphate, 10 mm glycerophosphate, and protease inhibitor cocktail (Roche Applied Science)). Cleared lysates were collected by spinning at 15,000 rpm for 15 min and then denatured with SDS sample buffer for immunoblotting. For immunoprecipitations, cleared lysates were incubated with 1 μg of antibody for 2 h and with protein G-Sepharose beads for an additional hour with gentle rocking at 4 °C. Immunoprecipitates were denatured in SDS sample buffer and further analyzed by immunoblotting.
Small Interfering RNAs
siGENOME SMARTpool small interfering RNAs (siRNAs) targeting SIRT1, SIRT2, HDAC6, and GFP (control) were purchased from Dharmacon. Gene knockdown efficiency by siRNAs on each target was examined by quantitative reverse transcription-PCR (RT-PCR).
S6K1 in Vitro Kinase Assay
COS-7 cells were transfected with 10 ng of the indicated Myc-S6K1 construct using Lipofectamine (Invitrogen) by following the manufacturer's instructions. 48 h after transfection, Myc-S6K1 was immunoprecipitated (IPed) and washed once with buffer A (10 mm Tris, pH 7.2, 1% Nonidet P-40, 0.5% sodium deoxycholic acid, 100 mm NaCl, 1 mm EDTA), once with buffer B (10 mm Tris pH 7.2, 0.1% Nonidet P-40, 1 m NaCl), and once with buffer ST (5 mm Tris-base, 50 mm Tris-Cl, 150 mm NaCl, pH 7.2). After washing with buffer ST, beads were washed with 1× S6K1 kinase reaction buffer without β-mercaptoethanol (20 mm Hepes, pH 7.2, 10 mm MgCl2, 1 mg/ml BSA). S6K1 kinase assay was performed in 1× S6K1 kinase reaction buffer with 2 mm β-mercaptoethanol, 2 μg of GST-S6, 0.5 mm cold ATP, and 5 μCi of [γ-32P]ATP for 10 min at 30 °C. The reactions were terminated by denaturation in SDS sample buffer at 100 °C for 5 min, and the reaction samples were separated by SDS-PAGE and transferred to PVDF membrane. Autoradiographs were visualized and quantified by a PhosphorImager and ImageQuant (GE Healthcare).
Statistical Analysis
All data were analyzed by analysis of variance with Scheffe's post hoc test. Asterisks represent statistically significant differences. p values less than 0.05 were considered significant.
RESULTS
Nicotinamide, but Not Trichostatin A-sensitive, Regulation of Thr-389 S6K1 Phosphorylation
Recent studies demonstrated a cross-talk between sirtuins and the mTORC1 signaling pathway in various types of cells and organs (28–33). However, the role of sirtuins in the regulation of mTORC1 activity determined by measuring Thr-389 phosphorylation of S6K1 has been controversial. Conflicting conclusions of these studies may be due to the use of different approaches including overexpression and knock-out strategies of single sirtuins, such as SIRT1, under various experimental conditions. Mammalian sirtuins, consisting of seven different members, represent class III histone deacetylases and are specifically inhibited by high concentrations of NAM (34–36). To examine the effect of acute inhibition of sirtuins or other HDACs on mTORC1 activity, COS-7 cells were treated with NAM and/or TSA, an inhibitor of class I/II HDACs, and phosphorylation levels of S6K1 (Thr-389) as well as 4EBP1 (Thr-37/46 and Ser-65), an established phosphorylation substrate of mTORC1, were monitored. Levels of cellular acetylated proteins were enhanced by the treatment of NAM or TSA, and they were further enhanced by co-treatment with NT (Fig. 1A). As expected, treatment of COS-7 cells with Torin 1, a highly potent and selective ATP-competitive inhibitor of mTOR kinase (37, 38), abolished phosphorylation of both S6K1 (Thr-389) and 4EBP1 (Thr-37/46 and Ser-65) in a similar time-dependent manner (Fig. 1B). Interestingly, NAM treatment significantly decreased S6K1 and S6 phosphorylation but did not inhibit 4EBP1 phosphorylation on Thr-37/46 and Ser-65, although 4EBP1 protein was observed to migrate slightly faster by SDS-PAGE (Fig. 1C). These observations indicate that NAM dominantly attenuates S6K1 Thr-389 phosphorylation through an unknown mechanism rather than by inhibiting mTOR kinase activity. In contrast, TSA treatment showed little effect on the phosphorylation of either S6K1 or 4EBP1 (Fig. 1D). These data suggest that among all HDACs, the sirtuin family of proteins may have a specific effect on S6K1 phosphorylation. It has been demonstrated that treatment with NT shows greater acetylation of proteins that are targeted by sirtuin-dependent deacetylation (39, 40). This reaction may be due to a shift of acetyltransferase activity toward sirtuin-targeted proteins under conditions in which class I/II HDAC-targeted proteins are highly acetylated. Importantly, co-treatment with NT resulted in a further decrease in phosphorylation of S6K1 (Fig. 1E). Consistently, 4EBP1 phosphorylation was resistant to NT treatment. These observations suggest a possibility that the acetylation status of S6K may affect mTORC1-dependent Thr-389 phosphorylation.
FIGURE 1.

Nicotinamide specifically inhibits S6K1 phosphorylation. COS-7 cells were treated with 100 nm Torin 1, 5 mm NAM, 5 nm TSA, or NT for the indicated times. Total cell lysates were prepared and assessed by immunoblotting (WB) with the indicated antibodies. A, NAM, TSA, and NT induce cellular protein acetylation. Samples treated for 3 h from C–E were analyzed by immunoblotting with anti-acetyl-lysine and actin antibodies. B, Torin 1, a specific mTOR kinase inhibitor, suppresses both Thr-389 phosphorylation of S6K1 (pT389-S6K1) and Thr-37/46 and Ser-65 phosphorylation of 4EBP1 (pT37/46–4EBP1 and pS65–4EBP1) in a similar dose-dependent manner. C, NAM specifically attenuates S6K phosphorylation. pS235/236-S6, Ser-235/236 phosphorylation of S6. D, inhibition of Class I/II HDACs by TSA does not affect S6K1 phosphorylation. E, inhibitory effect of nicotinamide on S6K1 phosphorylation is augmented by TSA co-treatment.
S6K1 Acetylation Is Regulated by the Sirtuin Pathway
We next assessed whether inhibition of sirtuins with NAM or NT induces acetylation of S6K1. HA-S6K1 or HA-S6K2 was transiently expressed in COS-7 cells and treated with NT. HA-S6Ks were IPed with HA antibody, and levels of acetylation were determined by immunoblotting with a pan-acetyl-lysine antibody. Inhibition of deacetylases with NT treatment robustly induced S6K1, but not S6K2, acetylation (Fig. 2A). To further examine NAM-mediated S6K1 acetylation, COS-7 cells expressing HA-S6K1 were treated with NAM alone, TSA alone, or both (NT). Consistent with the effect of these treatments on S6K1 phosphorylation, NAM, but not TSA treatment, increased S6K1 acetylation (Fig. 2B). Moreover, NT treatment further enhanced S6K1 acetylation as compared with NAM treatment alone. These results indicate that S6K1 acetylation was induced upon NAM treatment, which inhibits sirtuin deacetylases. We also performed these experiments in HEK293T cells and observed essentially the same results where NAM and NT, but not TSA treatment, increased S6K1 acetylation (Fig. 2C). Taken together, these data suggest that S6K1 acetylation is regulated by sirtuins in a cell type-independent manner. To examine whether these regulations could be seen on endogenous S6K1, endogenous S6K1 was IPed from HEK293T and COS-7 cells, and acetylation levels were determined. As predicted, NT treatment significantly increased acetylation of endogenous S6K1 (Fig. 2, D and E). Collectively, our results suggest that S6K1 is acetylated in response to the inhibition of sirtuins and that this acetylation may be constantly suppressed by sirtuins under normal culture conditions.
FIGURE 2.
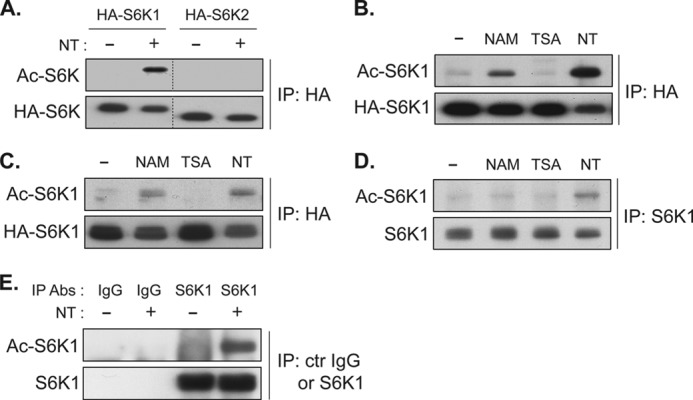
Inhibition of class III HDACs, the sirtuins, by nicotinamide induces acetylation of S6K1. A, S6K1 but not S6K2 acetylation (Ac-S6K) is induced by NT treatment. COS-7 cells were transfected with HA-S6K1 or HA-S6K2 plasmid. 48 h after transfection, the cells were treated with NT for 3 h. HA-S6Ks were IPed, and levels of acetylation were analyzed by immunoblotting with anti-acetyl-lysine antibody. B and C, NAM, but not TSA, induces S6K1 acetylation. HA-S6K1 was expressed in COS-7 (B) or HEK293T cells (C). Cells were treated with NAM, TSA, or NT for 3 h. Levels of S6K1 acetylation were monitored as in A. D and E, acetylation of endogenous S6K1 was enhanced by NT treatment. HEK293T (D) or COS-7 cells (E) were treated with or without NT for 3 h. Endogenous S6K1 was IPed, and levels of S6K1 acetylation were monitored as in A. Abs, antibodies; ctr IgG, control IgG.
SIRT1 and SIRT2 of the Sirtuin Class III HDAC Family Are Responsible for Deacetylation of S6K1
To determine whether sirtuins directly inhibit acetylation of p70 S6K1, which is predominantly expressed in the cytosol, the interaction between S6K1 and the cytoplasmic sirtuins (SIRT1 and SIRT2) and the effect of overexpression of these sirtuins on S6K1 acetylation were examined.
Co-immunoprecipitation assays showed that both HA-tagged SIRT1 and HA-tagged SIRT2 were well co-IPed with FLAG-tagged S6K1 in COS-7 cells (Fig. 3A). NT treatment had little effect on these interactions. We also observed weak interactions between endogenous SIRT1 or SIRT2 with endogenous S6K1 when cells were treated with NT (Fig. 3B), suggesting that both SIRT1 and SIRT2 may function as deacetylases for S6K1. To examine whether S6K1 is a direct substrate for SIRT1 or SIRT2 deacetylase activity, S6K1 was co-expressed with wild type or catalytically inactive SIRT1 or SIRT2 and treated with NT in COS-7 cells (41–43). Accordingly, inhibition of sirtuins upon NT treatment increased S6K1 acetylation, whereas overexpression of either SIRT1 or SIRT2 reduced NT-induced S6K1 acetylation (Fig. 3C). Importantly, the catalytically inactive SIRT1 (SIRT1 HY) failed to suppress S6K1 acetylation. Similarly, inactive SIRT2 HY was also less effective in inhibiting S6K1 acetylation, although higher expression of SIRT2 HY displayed a moderate effect on reducing the levels of S6K1 acetylation possibly due to the reduction of cellular histone acetyltransferase activity such as p300 (44, 45). These results indicated that deacetylase activity of the overexpressed SIRT1 or SIRT2 was still maintained in the cells treated with NT, and both SIRT1 and SIRT2 may be responsible for the deacetylation of S6K1. To further investigate the role of SIRT1 and SIRT2 in the regulation of S6K1 acetylation, we examined the effect of transient knockdown of endogenous SIRT1 or/and SIRT2 on S6K1 acetylation. A single knockdown of SIRT1 or SIRT2 slightly induced S6K1 acetylation, whereas simultaneous knockdown of SIRT1 and SIRT2 further enhanced S6K1 acetylation, suggesting that both endogenous SIRT1 and SIRT2 contribute to deacetylation of S6K1 (Fig. 3, D and E). Consistent with the observations that co-treatment with TSA augmented S6K1 acetylation with NAM treatment, TSA treatment increased S6K1 acetylation induced by the double knockdown of SIRT1 and SIRT2. To examine the possible role of endogenous class I/II deacetylase activity in the regulation of S6K1 acetylation, we inhibited HDAC6, a major cytoplasmic class I/II HDAC, and assessed S6K acetylation. Consistent with the effect of TSA treatment alone, HDAC6 knockdown failed to induce S6K1 acetylation even under conditions in which both SIRT1 and SIRT2 are silenced (Fig. 3, E and F). Furthermore, overexpression of HDAC6 has little effect on NT-induced S6K1 acetylation (data not shown). Taken together, our observations suggest that SIRT1 and SIRT2 both act to regulate S6K1 deacetylation in a redundant manner.
FIGURE 3.

Both SIRT1 and SIRT2 are responsible for deacetylation of S6K1. A, overexpressed S6K1 interacts with exogenous SIRT1 and SIRT2. COS-7 cells were transfected with either HA-SIRT1 or HA-SIRT2 along with FLAG-S6K1. After 48 h, cells were treated with NT for 3 h, and FLAG-S6K1 was IPed. Immunoprecipitates were analyzed by immunoblotting using FLAG and HA antibodies. B, endogenous S6K1 interacts with endogenous SIRT1 and SIRT2. COS-7 cells were treated with NT for 3 h, and endogenous S6K1 was IPed. Immunoprecipitates were analyzed by immunoblotting using the indicated antibodies (Abs). Arrows indicate SIRT1 or SIRT2. C, S6K1 acetylation (Ac-S6K1) induced by NT treatment was suppressed by overexpression of SIRT1 or SIRT2. COS-7 cells were transfected with Myc-S6K1 together with HA-SIRT1 or HA-SIRT2 as indicated. SIRT1 HY and SIRT2 HY denote the deacetylase-inactive mutants of SIRT1 and SIRT2, respectively. Cells were treated with NT for 3 h and Myc-S6K1 was IPed. Levels of acetylation of Myc-S6K1 were determined. D, both endogenous SIRT1 and endogenous SIRT2 are responsible for S6K1 deacetylation. COS-7 cells were transfected with HA-S6K1 together with the indicated siRNAs against SIRT1, SIRT2, or GFP (Control). TSA treatment was for 3 h, as indicated. E, the effect of cytoplasmic HDAC6 knockdown on S6K1 acetylation. The indicated siRNAs were co-transfected with HA-S6K1. NT treatment (3 h) was as indicated. F, knockdown efficiency of the indicated siRNAs in COS-7 cells was examined by quantitative RT-PCR and immunoblotting. Error bars indicate mean ± S.D.
Identification of Novel Sirtuin-regulated Lysine Residues (Lys-484 and Lys-485)
It has been demonstrated that Lys-493 of S6K1 (Lys-516 in S6K1 isoform α1) is acetylated by overexpression of p300 acetyltransferase (46, 47). However, the regulatory role and functional importance of Lys-493 acetylation for S6K1 activity have remained unclear. A point mutation of Lys-493 to arginine (K493R) did not eliminate NT-induced S6K1 acetylation (Fig. 4B), whereas a deletion of 85 amino acids from the C terminus (S6K1 1–417 amino acids) abolished NT-induced S6K1 acetylation (Fig. 4A). These observations suggest that additional sirtuin-sensitive acetylation sites may exist in the CTR of S6K1. To identify novel acetylation sites, mutation of lysine residues within the CTR of S6K1 were examined for their effects on S6K1 acetylation. The K484R mutation dramatically but incompletely suppressed NT-induced S6K1 acetylation. Double mutations of Lys-484/485 to arginines (hereafter called DKR) further decreased acetylation of S6K1 relative to 484R alone. Although DKR showed significant reduction of NT-induced S6K1 acetylation (Fig. 4B), it failed to completely abolish the acetylation signal (Fig. 4, B and C). While we were performing this study, a specific antibody against acetyl-lysine 493 of S6K1 (acetyl Lys-493-S6K1) became commercially available. In agreement with the earlier study of Lys-493, we could detect Lys-493 acetylation in the DKR S6K1 but not in TKR, which has the K493R mutation in addition to DKR (Fig. 4D). Taken together, these results indicate that at least three lysines in the CTR of S6K1 are acetylated.
FIGURE 4.
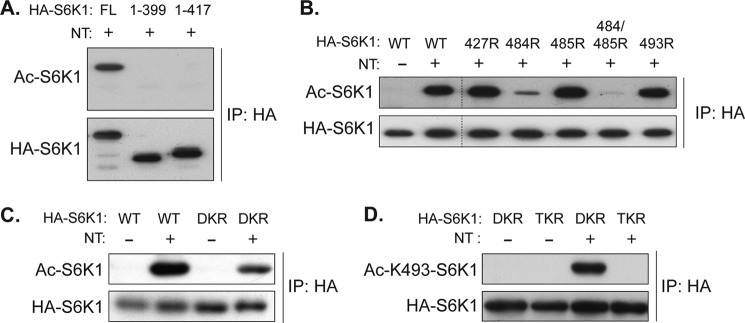
Inhibition of sirtuins induces acetylation of lysines 484, 485, and 493 of S6K1. A, the CTR of S6K1 possesses NT-sensitive acetylation sites. COS-7 cells were transfected with full-length wild type (FL 1–502 amino acids) and two C-terminal truncation dC-S6K1 mutants (1–399 amino acids and 1–417 amino acids), and treated with NT for 3 h. Levels of acetylation (Ac) in the indicated S6K1s were determined. B and C, mutations of lysine 484 and 485 to arginine (DKR) largely inhibit NT-induced S6K1 acetylation. COS-7 cells were transfected with the indicated S6K1 KR mutants, and levels of acetylation induced by NT were monitored. 427R, 484R, 485R, 484/485R, and 493R denote KR mutation of the indicated residues. D, mutations of lysine 484, 485, and 493 to arginine (TKR) abolished NT-induced S6K1 acetylation. COS-7 cells were transfected with DKR or TKR and treated with NT for 3 h. An anti-acetyl lysine 493 antibody was used to determine the level of S6K1 acetylation.
SIRT1 and SIRT2 Enhance S6K1 Phosphorylation
Our studies demonstrated that NAM, a sirtuin inhibitor, enhanced S6K1 acetylation and suppressed S6K1 phosphorylation (Figs. 1, C and E, and Fig. 2B). Furthermore, both SIRT1 and SIRT2 were able to reduce S6K1 acetylation (Fig. 3, C–E). These data suggest that cytoplasmic sirtuins may have a positive role in the regulation of S6K1 phosphorylation. Consistent with this hypothesis, transient knockdown of both SIRT1 and SIRT2 reduced serum/amino acid-induced Thr-389 phosphorylation of endogenous S6K1 (Fig. 5A). In contrast, overexpression of SIRT1 or SIRT2 significantly enhanced Thr-389 phosphorylation and moderately increased Thr-229 (activation loop) and other phosphorylations of S6K1 (Fig. 5B). Consistently, Myc-S6K1 co-expressed with SIRT1 or SIRT2 increased its kinase activity as measured by in vitro kinase assays (Fig. 5C). However, transient overexpression of SIRT1 or SIRT2 showed little effect on the levels of AKT (Ser-473) or AMP-activated protein kinase (AMPK)α (Thr-172) phosphorylation, which reflect the activity of two important upstream regulators of mTORC1 in COS-7 cells (Fig. 5, D and E). These observations further support the idea that SIRT1 and SIRT2 may have direct roles in modulating mTORC1-dependent S6K1 phosphorylation (Thr-389). It should be noted that SIRT1 or SIRT2 overexpression only moderately enhanced Thr-229 phosphorylation (activation loop) and the kinase activity of S6K1 despite their substantial effect on enhancing Thr-389 phosphorylation. Although our data indicate that SIRT1 or SIRT2 overexpression has little effect on mTORC2-dependent Ser-473 phosphorylation of AKT, it is possible that overexpression of SIRT1/SIRT2 may reduce cytoplasmic PDK1 activity, which is responsible for Thr-229 phosphorylation of S6K1, by increasing the levels of plasma membrane-associated PDK1 (48, 49). To further test the role of sirtuins in the regulation of mTORC1-mediated S6K1 phosphorylation, we examined the effect of endogenous sirtuin activation on S6K1 phosphorylation by stimulating the biosynthetic pathway of NAD, which is an essential cofactor for sirtuin deacetylase activity (50). Overexpression of nicotinamide phosphoribosyltransferase (NAMPT) or nicotinamide mononucleotide adenylyltransferase (NMNAT), which stimulates NAD biosynthesis, enhances S6K1 phosphorylation but has little effect on AKT phosphorylation (Fig. 5, F and G). Together, these observations suggest that SIRT1 and SIRT2 enhance S6K1 phosphorylation possibly through deacetylation of S6K1.
FIGURE 5.

SIRT1 and SIRT2 enhance S6K1 phosphorylation and its kinase activity. A, COS-7 cells were transfected with the siRNAs against SIRT1 and SIRT2 or GFP (Control). 72 h after transfection, cells were starved with growth factors and nutrients with Hanks' balanced salt solution for 1 h and stimulated with the culture medium (10% FBS in DMEM) for the indicated times. Total cell lysates were prepared, and acetylated S6K1 was IPed by Ac-Lys-493-S6K1 antibody (Ac-K493-S6K1). Immunoprecipitates and lysates were analyzed by immunoblotting with the indicated antibodies. Levels of Thr-389 phosphorylation (pT389, left panel) were quantified with ImageJ (right panel). *, p < 0.05, versus 90 min control; mean ± S.D., n = 3. A. U., arbitrary units. B, transient overexpression of SIRT1 or SIRT2 increases S6K1 phosphorylation. COS-7 cells were co-transfected with HA-S6K1 and HA-SIRT1 or HA-SIRT2. After 48 h, total cell lysates were prepared, and levels of S6K1 phosphorylation were monitored by immunoblotting with phospho-specific Thr-389 (pT389), Thr-229 (pT229), and Thr-421/Ser-424 (pT421/S424) S6K1 antibodies. C, SIRT1 and SIRT2 overexpression enhances S6K1 kinase activity. COS-7 cells were co-transfected with Myc-S6K1 and HA-SIRT1 or HA-SIRT2. After 48 h, Myc-S6K1 was IPed and subjected to an in vitro kinase assay. GST-S6 was prepared from bacteria and used for the in vitro kinase assay as a substrate. The incorporated radioactivity into GST-S6 was quantified. Data were expressed as mean ± S.D., *, p < 0.05 versus control (without sirtuins) (n = 3). D and E, the effect of SIRT1 or SIRT2 on AKT or AMP-activated protein kinase (AMPK) phosphorylation. COS-7 cells were transfected with HA-S6K1 and GST-AKT or Myc-S6K1 and HA-AMP-activated protein kinase together with either HA-SIRT1 or HA-SIRT2. Experiments were performed as in panel A. Levels of AKT and AMP-activated protein kinase phosphorylation were monitored by immunoblotting with the indicated phospho-specific antibodies. pS473, Ser-473 phosphorylation; pT172, Thr-172 phosphorylation. F and G, activation of the NAD biosynthetic pathway stimulates S6K1 phosphorylation. Myc-nicotinamide phosphoribosyltransferase (Myc-NAMPT) (F) or Myc-nicotinamide mononucleotide adenylyltransferase (Myc-NMNAT) (G) was co-expressed with HA-S6K1 (upper panels) or GST-AKT (lower panels) in COS-7 cells. Levels of S6K1 or AKT phosphorylation were monitored by immunoblotting with the indicated phospho-specific antibodies.
Acetylation-defective S6K1 Shows Higher S6K1 Phosphorylation
To investigate the direct role of S6K1 acetylation in the regulation of its phosphorylation, we created a series of lysine point mutations to generate acetylation-defective (KR)3 or acetylation-mimetic (KQ, KA, or KT) S6K1 mutants, as has been reported in previous studies (51, 52). Consistent with the hypothesis that S6K1 acetylation suppresses mTORC1-mediated S6K1 phosphorylation, the S6K1 acetylation-defective mutant (TKR),4 in which three lysine residues (484, 485, and 493) were all mutated to arginine, showed higher Thr-389 phosphorylation levels as compared with wild type S6K1 (Fig. 6A). Importantly, the reduction of S6K1 phosphorylation by NAM or NT treatment was significantly attenuated in the TKR S6K1 mutant (Fig. 6A). In contrast, S6K1 phosphorylation in the acetylation-mimetic mutants (TKQ, TKA, and TKT) showed lower S6K1 phosphorylation levels than that in the wild type or TKR mutant (Fig. 6B). In accordance with the observations that the TKA S6K1 mutant has reduced Thr-389 phosphorylation as compared with the TKR S6K1 mutant, the TKA mutant showed lower kinase activity than the TKR mutant (Fig. 6C). These observations indicate that S6K1 acetylation plays an important role in inhibiting mTORC1-induced S6K1 phosphorylation and its kinase activity. Interestingly, the above reciprocal correlation between S6K1 acetylation and phosphorylation can also be seen in cells treated with a proteasome inhibitor. It has been reported that heat shock factor-1 (HSF1), a master transcriptional factor that stimulates the transcription of a large number of anti-stress genes, is acetylated in response to multiple cellular stresses including heat shock, hypoxia, and proteasome inhibition (53). The acetylation of HSF1 terminates its transcriptional activity by blocking its association with the promoter elements of its target genes. Notably, SIRT1 deacetylates HSF1 and maintains its DNA binding activity, thereby enhancing HSF1 function to further induce anti-stress gene expressions. We observed that proteasome inhibition induced S6K acetylation in a time-dependent manner. In contrast, S6K phosphorylation (Thr-389 and Thr-229) was reciprocally reduced in a similar time-dependent manner (Fig. 6D). In addition, levels of Thr-421/Ser-424 phosphorylation in the CTR of S6K1 were not inhibited by proteasome inhibition. Importantly, this time-dependent inhibition of S6K1 phosphorylation by proteasome inhibition was not observed in the acetylation-defective S6K1 mutant (TKR). Taken together, these observations indicate that the acetylation of S6K1 and its negative role for S6K1 phosphorylation are dynamic, and suggest that the inhibition of S6K1 phosphorylation stemming from proteasome inhibition is a consequence of S6K1 acetylation.
FIGURE 6.

S6K1 acetylation attenuates mTORC1-dependent S6K1 phosphorylation. A, acetylation-defective mutant (TKR) showed higher levels of S6K1 phosphorylation. Wild type or TKR S6K1 mutant was expressed in COS-7 cells. After 48 h, cells were treated with either NAM or NT for 3 h before harvest. Levels of Thr-389 phosphorylation (pT389) were monitored by immunoblotting (left panel) and quantified by ImageJ (right panel). (*, p < 0.05, **, p < 0.01 versus control (Ctr), N.S. indicates not significant; mean ± S.D., n = 3). A. U., absorbance units. B, characterization of the acetylation-defective mutant (TKR) and acetylation-mimetic mutants (TKA, TKQ, and TKT) of S6K1. COS-7 cells were transfected with wild type, TKR, TKA, TKQ, or TKT S6K1. Levels of Thr-389 phosphorylation of S6K1 were monitored (left panel) and quantified (right panel) (*, p < 0.05 versus WT; mean ± S.D., n = 3). C, acetylation-defective mutant (TKR) shows higher kinase activity as compared with that of acetylation-mimetic mutant (TKA) in vitro. COS-7 cells were transfected with either HA-S6K1 TKR or TKA mutant. After 48 h, the HA-S6K1 mutants were IPed and subjected to an in vitro kinase assay. GST-S6 was used as a substrate. The incorporated radioactivity into GST-S6 was quantified. Data were expressed as mean ± S.D., *, p < 0.05 versus TKR (n = 3). D, proteasome inhibition induces acetylation of S6K1 concomitantly decreasing S6K1 phosphorylation. COS-7 cells were transfected with either wild type HA-S6K1 or acetylation-defective mutant (TKR). Cells were treated with 10 μm MG132 for the indicated times, and total cell lysates were analyzed by immunoblotting using the indicated antibodies. Ac-K493-S6K1, Ac-Lys-493-S6K1 antibody; pT421/S424, phosphorylation of Thr-421 and Ser-424. E, Thr-389 phosphorylation status does not affect S6K1 acetylation. Wild type, T389E (phosphorylation-mimetic mutant), or T389A S6K1 (phosphorylation-defective mutant) was expressed in COS-7 cells. 48 h after transfection, cells were treated with NT for 3 h. HA-S6K1s were IPed with HA antibody, and immunoprecipitates were analyzed by immunoblotting with the indicated antibodies (top panel). Levels of S6K1 acetylation were quantified (right panel). N.S. indicates not significant versus WT treated with NT; mean ± S.D., n = 3. F, TKR S6K1 shows higher affinity for mTORC1 as compared with TKA S6K1. COS-7 cells were co-transfected with Myc-Raptor together with either wild type HA-S6K1, HA-TKR, or HA-TKA S6K1 mutant. Levels of co-IPed HA-S6K1 were determined. G, schematic model of the effect of sirtuins on S6K1 phosphorylation by mTORC1. Deacetylation of S6K1 by sirtuins enhances mTORC1-dependent S6K1 phosphorylation (Thr-389). HATs indicate histone acetyltransferases.
To further investigate the interrelationship between S6K acetylation and its phosphorylation status, we examined and compared the levels of S6K1 acetylation in wild type and T389E (phospho-mimetic) and T389A (phospho-defective) mutants in the presence or absence of deacetylase inhibitors. Upon deacetylase inhibition, similar levels of acetylation were detected in all of these S6K1 constructs, suggesting that S6K1 is acetylated in a manner independent of the phosphorylation status of Thr-389 (Fig. 6E). Note that the phospho-Thr-389 S6K1 antibody strongly recognizes the S6K1 T389E mutant even under nutrient starvation or rapamycin treatment conditions (data not shown), suggesting that the substitution of Thr-389 with a glutamic acid well mimics the structure of phosphorylated Thr-389. Thus, the preferred model based on above observations is that once nonphosphorylated S6K1 is acetylated, mTORC1-dependent Thr-389 phosphorylation does not efficiently occur.
Given that lysine acetylation blocks its positive charge, our results suggest that the inhibitory effect of S6K1 acetylation on Thr-389 phosphorylation may be due to a decrease in the positive charge of the three lysine residues in the CTR of S6K1. Because the CTR of S6K1 determines the dependence of S6K1 regulation by mTORC1, the affinity of these S6K1 CTR mutants for Raptor was evaluated. Raptor is an essential scaffolding protein of the mTORC1 complex and recruits S6K1 to mTORC1. Interestingly, the affinity of the TKR mutant with Raptor was much higher than that of the TKA mutant (Fig. 6F). Thus, our data support a model where the acetylation of S6K1, which reduces positive charge on three lysine residues of S6K1, weakens the interaction between S6K1 and mTORC1, leading to a decrease in S6K1 phosphorylation by mTORC1 (Fig. 6G).
DISCUSSION
This study establishes cross-talk between the mTORC1 and the sirtuin signaling pathways in the regulation of S6K1 phosphorylation. Our biochemical data postulate that S6K1 is constantly deacetylated by SIRT1 and SIRT2 in growing cells, which supports mTORC1-mediated Thr-389 phosphorylation, a key phosphorylation event that stimulates S6K1 activity. The results also suggest an important role for positive charges at the C terminus of S6K1 in its association with mTORC1 through Raptor. Collectively, these results promote a model where SIRT1 and SIRT2 play a role in maintaining positive charges at the C terminus of S6K1, which eventually makes S6K1 a better substrate for mTORC1. It has been demonstrated that the CTR of S6K1 designates mTORC1 as a principal kinase for S6K1 phosphorylation as S6K1 lacking the CTR can be phosphorylated by both mTORC1 and mTORC2 (26). These observations imply that not only the TOS motif in the N terminus but also the positive charge of the CTR of S6K1, which is inhibited by acetylation, plays an important role in the recognition of mTORC1 as a specific kinase for S6K1. Although further study will be required to determine the precise molecular mechanism by which these CTR lysine residues of S6K1 engage Raptor in the mTORC1 complex, we posit that these S6K1 positive charges may play a crucial role in interacting with a certain domain of mTORC1.
The expanding interest in sirtuin function largely stems from previous studies that link Sir2 to lifespan regulation in model organisms. Increased Sir2 gene dosage has been proposed to be sufficient to extend lifespan (54, 55), which is genetically opposed to the effect of TORC1 signaling in the regulation of lifespan (56, 57). More recently, the attractive consensus of Sir2 function in longevity has been scrutinized, and Sir2 appears to be dispensable in extending lifespan in both Caenorhabditis elegans and Drosophila melanogaster (58). However, it should be noted that overexpression of mammalian SIRT1 in the brain increases longevity in mice. Thus, the role of Sir2/SIRT1 in the regulation of longevity continues to be an area of active discussion. Accordingly, recent studies in mammalian cells and mouse models have also revealed that both SIRT1 and SIRT2 exert bidirectional roles in suppressing and promoting cancer cell proliferation and survival (59). There are also mixed reports regarding the functional role of SIRT1 in the regulation of insulin-mTOR signaling. SIRT1 deacetylates the insulin receptor substrate 2 (IRS2), AKT, and PDK1 and stimulates insulin signaling (30, 48). SIRT1 also suppresses the expression of the PTP1B (60), a negative regulator of the insulin-mTORC1 pathway, and also directly deacetylates PTEN (phosphatase and tensin homolog) protein, although the functional importance of PTEN acetylation for its lipid phosphatase activity remains elusive (61). Furthermore, SIRT1 activation in human diploid fibroblasts promotes S6K phosphorylation (28), and ablation of SIRT1 in a polycystic kidney disease mouse model (PKD1−/−) attenuates cystic formation and renal epithelial cell proliferation that are promoted by mTORC1 activity (31, 62). Moreover, physiological exercise- or angiotensin II-induced cardiac hypertrophy is attenuated in SIRT1-deficient mice (63). In contrast, it has also been reported that loss of SIRT1 in mouse embryonic fibroblasts (SIRT1−/− mouse embryonic fibroblasts) increases mTORC1 activity as compared with that in wild type mouse embryonic fibroblasts (33). SIRT1 and SIRT2 have many targets even within the insulin signaling pathway and play important roles in the regulation of not only stress/energy-related gene expression but also signal transduction molecules that may directly or indirectly affect mTORC1 activity. Therefore, the inconsistent or bifunctional roles of SIRT1 in the regulation of the insulin-mTOR pathway and cell proliferation/survival may reflect a cell or tissue type specificity, differential activity of SIRT2, or the distinct experimental contexts under which it has been investigated. Although the physiological role of SIRT1 and SIRT2 in the regulation of S6K1 remains to be addressed, our biochemical studies demonstrate a clear association of the sirtuin pathway and mTORC1 signaling at the level of S6K1 phosphorylation. We speculate that this regulatory axis may play important roles in the quality control of cellular proteins under certain stress conditions and in sirtuin-mediated cell survival and proliferation.
Acknowledgments
We appreciate the gift of reagents from Drs. John Blenis (Harvard University), David Sabatini (MIT), and Roland Kwok (University of Michigan). We thank Dr. Kun-Liang Guan for critical discussions and Dr. John Kim for comments on this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK083491 (to K. I.) and GM101171 and CA177925 (to D. B. L.).
Throughout this study, the following designations were used for mutants: KR, acetylation-defective mutation by replacing lysine with arginine; KQ, acetylation-mimetic mutation by replacing lysine with glutamine; KA, acetylation-mimetic mutation by replacing lysine with alanine; KT, acetylation-mimetic mutation by replacing lysine with threonine.
Throughout this study, the following designations were used for multiple mutants: TKR, K484R/K485R/K493R; TKQ, K484Q/K485Q/K493Q; TKA, K484A/K485A/K493A; TKT, K484T/K485T/K493T.
- S6K
- S6 kinase
- mTOR
- mammalian target of rapamycin
- mTORC1
- mammalian target of rapamycin complex 1
- 4EBP1
- 4E-binding protein 1
- TOS
- TOR signaling
- PDK1
- phosphoinositide-dependent kinase 1
- CTR
- C-terminal region
- TSA
- trichostatin A
- NAM
- nicotinamide
- NT
- NAM + TSA
- HSF1
- heat shock factor-1
- HDAC
- histone deacetylase
- IPed
- immunoprecipitated.
REFERENCES
- 1. Su B., Jacinto E. (2011) Mammalian TOR signaling to the AGC kinases. Crit. Rev. Biochem. Mol. Biol. 46, 527–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Magnuson B., Ekim B., Fingar D. C. (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441, 1–21 [DOI] [PubMed] [Google Scholar]
- 3. Grove J. R., Banerjee P., Balasubramanyam A., Coffer P. J., Price D. J., Avruch J., Woodgett J. R. (1991) Cloning and expression of two human p70 S6 kinase polypeptides differing only at their amino termini. Mol. Cell. Biol. 11, 5541–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saitoh M., ten Dijke P., Miyazono K., Ichijo H. (1998) Cloning and characterization of p70S6Kβ defines a novel family of p70 S6 kinases. Biochem. Biophys. Res. Commun. 253, 470–476 [DOI] [PubMed] [Google Scholar]
- 5. Lee-Fruman K. K., Kuo C. J., Lippincott J., Terada N., Blenis J. (1999) Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 18, 5108–5114 [DOI] [PubMed] [Google Scholar]
- 6. Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., Sabatini D. M. (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U.S.A. 95, 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pullen N., Dennis P. B., Andjelkovic M., Dufner A., Kozma S. C., Hemmings B. A., Thomas G. (1998) Phosphorylation and activation of p70s6k by PDK1. Science 279, 707–710 [DOI] [PubMed] [Google Scholar]
- 8. Alessi D. R., Kozlowski M. T., Weng Q. P., Morrice N., Avruch J. (1998) 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 8, 69–81 [DOI] [PubMed] [Google Scholar]
- 9. Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., Hall M. N. (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 [DOI] [PubMed] [Google Scholar]
- 10. Hara K., Maruki Y., Long X., Yoshino K., Oshiro N., Hidayat S., Tokunaga C., Avruch J., Yonezawa K. (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 [DOI] [PubMed] [Google Scholar]
- 11. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 12. Biondi R. M., Kieloch A., Currie R. A., Deak M., Alessi D. R. (2001) The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 20, 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Williams M. R., Arthur J. S., Balendran A., van der Kaay J., Poli V., Cohen P., Alessi D. R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10, 439–448 [DOI] [PubMed] [Google Scholar]
- 14. Wang J., Zhong C., Wang F., Qu F., Ding J. (2013) Crystal structures of S6K1 provide insights into the regulation mechanism of S6K1 by the hydrophobic motif. Biochem. J. 454, 39–47 [DOI] [PubMed] [Google Scholar]
- 15. Cheatham L., Monfar M., Chou M. M., Blenis J. (1995) Structural and functional analysis of pp70S6k. Proc. Natl. Acad. Sci. U.S.A. 92, 11696–11700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dennis P. B., Pullen N., Kozma S. C., Thomas G. (1996) The principal rapamycin-sensitive p70s6k phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol. Cell. Biol. 16, 6242–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weng Q. P., Andrabi K., Kozlowski M. T., Grove J. R., Avruch J. (1995) Multiple independent inputs are required for activation of the p70 S6 kinase. Mol. Cell. Biol. 15, 2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schalm S. S., Blenis J. (2002) Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639 [DOI] [PubMed] [Google Scholar]
- 19. Oshiro N., Takahashi R., Yoshino K., Tanimura K., Nakashima A., Eguchi S., Miyamoto T., Hara K., Takehana K., Avruch J., Kikkawa U., Yonezawa K. (2007) The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem. 282, 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nojima H., Tokunaga C., Eguchi S., Oshiro N., Hidayat S., Yoshino K., Hara K., Tanaka N., Avruch J., Yonezawa K. (2003) The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 278, 15461–15464 [DOI] [PubMed] [Google Scholar]
- 21. Schalm S. S., Fingar D. C., Sabatini D. M., Blenis J. (2003) TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 13, 797–806 [DOI] [PubMed] [Google Scholar]
- 22. Schalm S. S., Tee A. R., Blenis J. (2005) Characterization of a conserved C-terminal motif (RSPRR) in ribosomal protein S6 kinase 1 required for its mammalian target of rapamycin-dependent regulation. J. Biol. Chem. 280, 11101–11106 [DOI] [PubMed] [Google Scholar]
- 23. Dennis P. B., Pullen N., Pearson R. B., Kozma S. C., Thomas G. (1998) Phosphorylation sites in the autoinhibitory domain participate in p70s6k activation loop phosphorylation. J. Biol. Chem. 273, 14845–14852 [DOI] [PubMed] [Google Scholar]
- 24. Banerjee P., Ahmad M. F., Grove J. R., Kozlosky C., Price D. J., Avruch J. (1990) Molecular structure of a major insulin/mitogen-activated 70-kDa S6 protein kinase. Proc. Natl. Acad. Sci. U.S.A. 87, 8550–8554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price D. J., Mukhopadhyay N. K., Avruch J. (1991) Insulin-activated protein kinases phosphorylate a pseudosubstrate synthetic peptide inhibitor of the p70 S6 kinase. J. Biol. Chem. 266, 16281–16284 [PubMed] [Google Scholar]
- 26. Ali S. M., Sabatini D. M. (2005) Structure of S6 kinase 1 determines whether raptor-mTOR or rictor-mTOR phosphorylates its hydrophobic motif site. J. Biol. Chem. 280, 19445–19448 [DOI] [PubMed] [Google Scholar]
- 27. Acosta-Jaquez H. A., Keller J. A., Foster K. G., Ekim B., Soliman G. A., Feener E. P., Ballif B. A., Fingar D. C. (2009) Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol. Cell. Biol. 29, 4308–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang J., Gan Q., Han L., Li J., Zhang H., Sun Y., Zhang Z., Tong T. (2008) SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS One 3, e1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gagarina V., Gabay O., Dvir-Ginzberg M., Lee E. J., Brady J. K., Quon M. J., Hall D. J. (2010) SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 62, 1383–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J. (2007) The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem. 282, 34356–34364 [DOI] [PubMed] [Google Scholar]
- 31. Zhou X., Fan L. X., Sweeney W. E., Jr., Denu J. M., Avner E. D., Li X. (2013) Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Invest. 123, 3084–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo W., Qian L., Zhang J., Zhang W., Morrison A., Hayes P., Wilson S., Chen T., Zhao J. (2011) Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res. 89, 1723–1736 [DOI] [PubMed] [Google Scholar]
- 33. Ghosh H. S., McBurney M., Robbins P. D. (2010) SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One 5, e9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Avalos J. L., Bever K. M., Wolberger C. (2005) Mechanism of sirtuin inhibition by nicotinamide: altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Mol. Cell 17, 855–868 [DOI] [PubMed] [Google Scholar]
- 35. Imai S., Guarente L. (2010) Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol. Sci. 31, 212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Satoh A., Stein L., Imai S. (2011) The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb. Exp. Pharmacol. 206, 125–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Q., Chang J. W., Wang J., Kang S. A., Thoreen C. C., Markhard A., Hur W., Zhang J., Sim T., Sabatini D. M., Gray N. S. (2010) Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 53, 7146–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S. (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., Alt F. W., Greenberg M. E. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 40. Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 41. Kim D., Nguyen M. D., Dobbin M. M., Fischer A., Sananbenesi F., Rodgers J. T., Delalle I., Baur J. A., Sui G., Armour S. M., Puigserver P., Sinclair D. A., Tsai L. H. (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Imai S., Armstrong C. M., Kaeberlein M., Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 43. Tanny J. C., Kirkpatrick D. S., Gerber S. A., Gygi S. P., Moazed D. (2004) Budding yeast silencing complexes and regulation of Sir2 activity by protein-protein interactions. Mol. Cell. Biol. 24, 6931–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Black J. C., Mosley A., Kitada T., Washburn M., Carey M. (2008) The SIRT2 deacetylase regulates autoacetylation of p300. Mol. Cell 32, 449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Y., Matsumori H., Nakayama Y., Osaki M., Kojima H., Kurimasa A., Ito H., Mori S., Katoh M., Oshimura M., Inoue T. (2011) SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 16, 34–45 [DOI] [PubMed] [Google Scholar]
- 46. Fenton T. R., Gwalter J., Cramer R., Gout I. T. (2010) S6K1 is acetylated at lysine 516 in response to growth factor stimulation. Biochem. Biophys. Res. Commun. 398, 400–405 [DOI] [PubMed] [Google Scholar]
- 47. Fenton T. R., Gwalter J., Ericsson J., Gout I. T. (2010) Histone acetyltransferases interact with and acetylate p70 ribosomal S6 kinases in vitro and in vivo. Int. J. Biochem. Cell Biol. 42, 359–366 [DOI] [PubMed] [Google Scholar]
- 48. Sundaresan N. R., Pillai V. B., Wolfgeher D., Samant S., Vasudevan P., Parekh V., Raghuraman H., Cunningham J. M., Gupta M., Gupta M. P. (2011) The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 4, ra46. [DOI] [PubMed] [Google Scholar]
- 49. McManus E. J., Collins B. J., Ashby P. R., Prescott A. R., Murray-Tait V., Armit L. J., Arthur J. S., Alessi D. R. (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 23, 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Imai S. (2011) Dissecting systemic control of metabolism and aging in the NAD World: the importance of SIRT1 and NAMPT-mediated NAD biosynthesis. FEBS Lett. 585, 1657–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y. H., Tsay Y. G., Tan B. C., Lo W. Y., Lee S. C. (2003) Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem. 278, 25568–25576 [DOI] [PubMed] [Google Scholar]
- 52. Scroggins B. T., Robzyk K., Wang D., Marcu M. G., Tsutsumi S., Beebe K., Cotter R. J., Felts S., Toft D., Karnitz L., Rosen N., Neckers L. (2007) An acetylation site in the middle domain of Hsp90 regulates chaperone function. Molecular cell 25, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Westerheide S. D., Anckar J., Stevens S. M., Jr., Sistonen L., Morimoto R. I. (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323, 1063–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaeberlein M., McVey M., Guarente L. (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rogina B., Helfand S. L., Frankel S. (2002) Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science 298, 1745. [DOI] [PubMed] [Google Scholar]
- 56. Vellai T., Takacs-Vellai K., Zhang Y., Kovacs A. L., Orosz L., Muller F. (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620. [DOI] [PubMed] [Google Scholar]
- 57. Harrison D. E., Strong R., Sharp Z. D., Nelson J. F., Astle C. M., Flurkey K., Nadon N. L., Wilkinson J. E., Frenkel K., Carter C. S., Pahor M., Javors M. A., Fernandez E., Miller R. A. (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burnett C., Valentini S., Cabreiro F., Goss M., Somogyvári M., Piper M. D., Hoddinott M., Sutphin G. L., Leko V., McElwee J. J., Vazquez-Manrique R. P., Orfila A. M., Ackerman D., Au C., Vinti G., Riesen M., Howard K., Neri C., Bedalov A., Kaeberlein M., Soti C., Partridge L., Gems D. (2011) Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477, 482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roth M., Chen W. Y. (2014) Sorting out functions of sirtuins in cancer. Oncogene 33, 1609–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sun C., Zhang F., Ge X., Yan T., Chen X., Shi X., Zhai Q. (2007) SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 6, 307–319 [DOI] [PubMed] [Google Scholar]
- 61. Ikenoue T., Inoki K., Zhao B., Guan K. L. (2008) PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 68, 6908–6912 [DOI] [PubMed] [Google Scholar]
- 62. Shillingford J. M., Piontek K. B., Germino G. G., Weimbs T. (2010) Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 21, 489–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Satoh A., Brace C. S., Rensing N., Cliften P., Wozniak D. F., Herzog E. D., Yamada K. A., Imai S. (2013) Sirt1 Extends Life Span and Delays Aging in Mice through the Regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430 [DOI] [PMC free article] [PubMed] [Google Scholar]