

Background: A cardinal feature of many neurological disorders is mitochondrial dysfunction.
Results: Knocking down neutral ceramidase reduces mitochondrial sphingosine, preserves mitochondrial function, and improves brain function recovery after trauma.
Conclusion: Activation of the sphingosine-generating pathway plays a significant role in promoting mitochondrial injury.
Significance: This is the first direct evidence of endogenous sphingosine involvement in regulation of mitochondrial function.
Keywords: Brain, Cell Death, Mitochondria, Respiratory Chain, Sphingolipid
Abstract
In addition to immediate brain damage, traumatic brain injury (TBI) initiates a cascade of pathophysiological events producing secondary injury. The biochemical and cellular mechanisms that comprise secondary injury are not entirely understood. Herein, we report a substantial deregulation of cerebral sphingolipid metabolism in a mouse model of TBI. Sphingolipid profile analysis demonstrated increases in sphingomyelin species and sphingosine concurrently with up-regulation of intermediates of de novo sphingolipid biosynthesis in the brain. Investigation of intracellular sites of sphingosine accumulation revealed an elevation of sphingosine in mitochondria due to the activation of neutral ceramidase (NCDase) and the reduced activity of sphingosine kinase 2 (SphK2). The lack of change in gene expression suggested that post-translational mechanisms are responsible for the shift in the activities of both enzymes. Immunoprecipitation studies revealed that SphK2 is complexed with NCDase and cytochrome oxidase (COX) subunit 1 in mitochondria and that brain injury hindered SphK2 association with the complex. Functional studies showed that sphingosine accumulation resulted in a decreased activity of COX, a rate-limiting enzyme of the mitochondrial electron transport chain. Knocking down NCDase reduced sphingosine accumulation in mitochondria and preserved COX activity after the brain injury. Also, NCDase knockdown improved brain function recovery and lessened brain contusion volume after trauma. These studies highlight a novel mechanism of secondary TBI involving a disturbance of sphingolipid-metabolizing enzymes in mitochondria and suggest a critical role for mitochondrial sphingosine in promoting brain injury after trauma.
Introduction
TBI3 is a result of both immediate mechanical disruption of brain tissue (the primary injury) and delayed (secondary) injury mechanisms. A complex cascade of processes is activated and generates continued endogenous changes affecting cellular systems and the overall outcome from the initial insult to the brain (1). Homeostatic cellular processes governing synaptic neurotransmission, cytoskeletal structure, redox balance, calcium influx, and mitochondrial function often become dysfunctional after TBI. A secondary phase of damage clinically presents as patients who regain consciousness after head injury and demonstrate the ability to speak and obey commands die days later or develop chronic neurological deficits. Current therapies of TBI do not address secondary injury mainly because the molecular mechanisms underlying the secondary brain injury are not fully uncovered, although mitochondrial dysfunction appears to be crucial (2).
The majority of the previous animal studies of mitochondrial function in TBI have shown that mitochondrial damage occurs rapidly as early as 1 h postinjury (3, 4), with peak mitochondrial respiratory chain dysfunction at 12–24 h (5). However, the effects of TBI on mitochondrial function at later stages of secondary brain damage remain unclear. Although a few studies have provided evidence for reduced respiratory chain activity at 7 days post-TBI, which can persist for up to 14 days postinjury (6, 7), there is a gap in our knowledge as to what biochemical mechanisms underlie chronic brain dysfunction after TBI. Likewise, an impairment of the mitochondrial respiratory chain enzyme COX has been implicated in secondary brain damage following TBI, but the mechanisms remain unresolved (8).
Sphingolipids are essential structural components of cellular membranes, playing prominent roles in signal transduction governing cell proliferation, differentiation, migration, and apoptosis (9). The building block of many complex sphingolipids is ceramide, which has numerous cellular signaling functions and plays a key role in promoting apoptosis (10, 11). Ceramides, a family of distinct molecular species characterized by various acyl chains, are synthesized de novo at the cytosolic side of the endoplasmic reticulum (12, 13), serving as precursors for the biosynthesis of complex sphingolipids and sphingomyelin (SM) in the Golgi (14, 15). Ceramide levels are tightly controlled in the cells via rapid conversion into less harmful sphingolipids, including SM and sphingosine 1-phosphate (S1P) (16).
Recent studies provide strong clues to a second messenger role of another bioactive sphingolipid, sphingosine, and identify signaling pathways recruited by sphingosine to induce cell death (17, 18), which sometimes differ from those activated by ceramide (19, 20). In mammalian cells, sphingosine is not formed de novo, but it is produced via deacylation of ceramide, catalyzed by a family of ceramidases localized in lysosomes, Golgi, plasma membrane, and mitochondria (17, 21). Five members of the ceramidase family encoded by five distinct genes (acid ceramidase, NCDase, and alkaline ceramidase 1–3) have maximal activities in acidic, neutral, and alkaline environments, respectively (17).
Mitochondria emerged as a novel specialized compartment of sphingolipid metabolism with their own subset of sphingolipid-generating and -degrading enzymes (22, 23). Thus, NCDase (24, 25), a novel neutral sphingomyelinase (26), (dihydro)ceramide synthase (27, 28), and SphK2 (29) have been found associated with mitochondria. In mitochondria, ceramide can be converted by NCDase into sphingosine, which, in turn, could be phosphorylated by SphK2, yielding S1P.
Our studies described here were aimed to elucidate the impact of TBI on mitochondrial sphingolipid metabolism and the role of sphingolipids in mitochondrial dysfunction. We report that brain trauma resulted in a substantial increase of sphingosine levels in brain tissue. Investigation of intracellular sites of sphingosine accumulation revealed the elevation of sphingosine in mitochondria due to the brain injury-induced disturbance of mitochondrial sphingosine metabolism. Thus, enhanced generation of sphingosine by NCDase was accompanied and exacerbated by reduced sphingosine utilization by SphK2. Furthermore, NCDase and SphK2 appear to form a complex with the inner mitochondrial membrane-resident protein COX-1, and SphK2 association with the complex is diminished after TBI.
Functional analysis exposed a significant decrease in respiratory chain enzyme COX activity in mitochondria from an injured brain. Given that exogenously added sphingosine selectively inhibited COX activity in baseline mitochondria, the accumulation of sphingosine seems to be a plausible cause of mitochondrial respiratory chain defect in an injured brain.
Gene knockdown experiments revealed a critical role for NCDase in promoting secondary brain damage. Thus, knocking down NCDase rescued COX activity defect and reduced brain damage after trauma. Moreover, NCDase-deficient mice demonstrated greatly improved recovery of behavioral deficits after brain trauma. These studies emphasize sphingolipid-metabolizing enzymes as novel regulators of mitochondrial respiratory chain function and identify the sphingolipid responsible for mitochondrial dysfunction after brain trauma.
EXPERIMENTAL PROCEDURES
Animals and Reagents
Male C57BL6 (8-week-old) mice (Jackson Laboratory, Bar Harbor, ME) were acclimated for 1 week prior to experimentation. NCDase knock-out (KO) mice were originally developed in the laboratory of Dr. Richard L. Proia (NIDDK, National Institutes of Health) (30) and transferred to the animal facility at the Veterans Affairs Medical Center (Charleston, SC). The mice were C57BL6 genetic background and back-crossed for at least 20 additional generations using the wild-type (WT) C57BL6 mice. Complete protease inhibitor mixture and PhoStop phosphatase inhibitor mixture were from Roche Applied Science. C17-C18-ceramide and C17-sphingosine were from Avanti Polar Lipids (Alabaster, AL). Trypsin and trypsin inhibitor were from Worthington. All other chemicals were purchased from Sigma-Aldrich.
Controlled Cortical Impact (CCI) Model of TBI
Experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Ralph H. Johnson Veterans Affairs Medical Center (Charleston, SC) and followed the National Institutes of Health guidelines for experimental animal use. Mice were anesthetized with isoflurane and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA). During the surgery, mice maintained a 37 °C body temperature by a heating pad. The skull was exposed, and the bite bar was adjusted to level bregma and lambda in the horizontal plane. The bone over cortex was removed, exposing a 3.5-mm region on one side. The impact was delivered by a computer-controlled head impactor (Precision Systems, Fairfax Station, VA) using a 3-mm flat circular impactor tip at a speed of 4 m/s and a depth of 1.5 mm below the cortical surface for 0.1 s. The parameter set was to provide a moderate contusive brain injury in mice (31, 32). Sham-injured animals received craniotomy only. After the impact, the wound was sutured, followed by application of analgesic. Mice were placed on the heating pad until ambulatory and then returned to the cage.
Analysis of Contusion Size
Mice were deeply anesthetized and transcardially perfused with chilled (4 °C) PBS plus 0.01% heparin followed by 4% paraformaldehyde in PBS. Brains were removed, postfixed in paraformaldehyde overnight at 4 °C, cryoprotected in 30% sucrose, and sliced coronally (20 μm) using a cryostat at 400-μm intervals throughout the brain hemispheres. Four sections were obtained at each interval, mounted onto a glass slide, and stained with 0.1% cresyl violet (33). Histological lesion areas were quantified with a standard computer-assisted National Institutes of Health ImageJ analysis program, and lesion areas were then integrated to obtain total lesion volume in cubic millimeters. A single operator blinded to treatment status performed all measurements.
Rotarod Test
To assess behavioral deficits after TBI, the standard rotarod test was performed because it has been shown to be effective and reliable in rodent brain trauma experiments (34, 35). The rotarod is a motorized cylinder that accelerates linearly until the animal falls off. Rotarod training and measurement were carried out using a four-lane rotarod apparatus (AccuScan Instruments, Columbus, OH). Each day for 3 days prior to injury, animals were trained on the rotarod at a speed of 18 rpm in the acceleration mode (0–18 rpm/90 s). Animals were tested using three trials in each training or measurement session, with a minimum of 5 min resting between trials. One hour before the injury, mice were assessed on the rotarod to obtain preinjury baselines. Scores were measured as the latency or time successfully spent running on the rotarod. After the injury, each animal performance was measured at three time points: 1 day, 2 days, and 7 days after CCI. Postinjury scores were normalized using preinjury means to control for variability in preinjury performance.
Isolation of Mouse Brain Mitochondria
All procedures were performed at 4 °C as described (28, 36). Briefly, tissue was placed immediately in ice-cold isolation medium containing 230 mm mannitol, 70 mm sucrose, 10 mm HEPES, and 1 mm EDTA, pH 7.4. Brain tissue (∼1 g) was homogenized in 10 ml of isolation medium using a Teflon-glass homogenizer. The homogenate was centrifuged at 900 × g for 10 min. The supernatant was then centrifuged at 12,000 × g for 10 min. The pellet was resuspended in 2 ml of 15% Percoll-Plus (GE Healthcare) and placed atop a discontinuous Percoll gradient consisting of a bottom layer of 4 ml of 40% Percoll and a top layer of 4 ml of 23% Percoll. The gradient was spun at 31,000 × g for 20 min in a SW-Ti40 rotor in a Beckman LE80K centrifuge. The fraction at the 23–40% interface that contained mitochondria was washed three times with isolation medium by centrifugation at 12,000 × g for 10 min. Protein concentration was measured with a bicinchoninic acid assay (Sigma) using bovine serum albumin (BSA) as a standard. Typically, the contamination of mitochondria with endoplasmic reticulum was <1% by activity measurements of the endoplasmic reticulum-specific marker enzyme, NADPH-cytochrome c reductase (28, 36). Western blot analysis showed no contamination of mitochondria with Na+/K+ ATPase (plasma membrane marker), LAMP-2 (lysosomal marker), calnexin (endoplasmic reticulum marker), and myelin basic protein (myelin marker) (36).
Preparation of Mitoplasts
Mitoplasts (i.e. mitochondria devoid of the outer mitochondrial membrane) were prepared by the treatment of mitochondria with nonionic detergent digitonin (37). Mitochondria (5 mg/ml) were incubated in isolation medium with 0.5% digitonin for 10 min on ice with gentle stirring. The mitochondrial suspension was diluted with 3 volumes of the medium to terminate the treatment and centrifuged at 20,000 × g for 20 min. The pellet (mitoplasts) was resuspended in the isolation medium and incubated with trypsin (50 μg/ml) for 10 min on ice. Trypsin was inactivated by the addition of a 30-fold excess of trypsin inhibitor. Proteins were precipitated with 9 volumes of ice-cold acetone and subjected to SDS-PAGE and immunoblotting.
Mitochondrial Respiratory Chain Activity
Mitochondrial respiration was measured by recording oxygen consumption at 25 °C in a chamber equipped with a Clark-type oxygen electrode (Instech Laboratories, Plymouth Meeting, PA) as described previously (28, 36). Briefly, mitochondria were incubated in the medium containing 125 mm KCl, 10 mm HEPES, 2 mm KH2PO4, 5 mm MgCl2, and 0.5 mg/ml mitochondrial protein supplemented with either Complex I substrate (mixture of 5 mm glutamate and 5 mm malate) or Complex II substrate (5 mm succinate) in the presence of 1 μm rotenone or Complex IV (COX) substrate (1 mm ascorbate in the presence of 250 μm N,N,N′,N′-tetramethyl-p-phenelenediamine (TMPD) and 1 μm antimycin). A respiratory control ratio was measured as the oxygen consumption rate in the presence of the substrate and 500 μm ADP (state 3) divided by the rate in the resting state (state 4) in the presence of 2 μg/ml oligomycin. Uncoupler-stimulated (state 3u) respiration was measured in the presence of 50 μm 2,4-dinitrophenol (2,4-DNP). COX activity was measured as the oxygen consumption rate supported by COX substrate 1 mm ascorbate with 250 TMPD in the presence of the Complex III inhibitor 1 μm antimycin to block the endogenous oxygen consumption.
Antibodies
Rabbit monoclonal antibody against the voltage-dependent anion channel (VDAC) was supplied by Cell Signaling Technology (Danvers, MA). Rabbit polyclonal anti-SphK2 antibody was purchased from Abcam (Cambridge, MA). Goat polyclonal antibodies against SphK2, COX-1, and neutral ceramidase were obtained from Santa Cruz Biotechnology, Inc. Rabbit polyclonal anti-NCDase antibody was raised by Bethyl Laboratories (Montgomery, TX) and originally characterized by Dr. Proia's laboratory (30). Secondary horseradish peroxidase-conjugated antibodies were supplied by Jackson ImmunoResearch (West Grove, PA).
Immunoprecipitation
Immunoprecipitations were performed as described previously (36, 38, 39). For immunoprecipitation, mitochondrial lysates (500 μg) were precleared in buffer A (0.15 m NaCl, 0.5 mm EDTA, 1% Triton X-100, protease and phosphatase inhibitor mixture, 0.05 m Tris, pH 7.5, 0.2% BSA) by incubation with appropriate species-specific, IgG-conjugated magnetic beads (Dynabeads, Invitrogen/Dynal) for 1 h. Antibodies were then added. After incubation at 4 °C overnight with gentle mixing, antibody-antigen complexes were captured with Dynabeads and washed two times with buffer A (without BSA) and then washed twice with Tris-buffered saline, pH 7.5. The immunoprecipitates were eluted by boiling in SDS-sample buffer. As a control, the same immunoprecipitation procedure was performed except for the primary antibody application.
Western Blot
Proteins were analyzed by Western blot as described previously (36, 38, 39). Proteins were separated by 4–15% SDS-PAGE, blotted to PVDF membrane, blocked with 5% nonfat dry milk (Bio-Rad) or 5% BSA in TBS-T buffer (10 mm Tris, 150 mm NaCl, and 0.2% Tween 20, pH 8.0), and subsequently probed with the appropriate primary antibody. Immunoreactive bands were visualized using a SuperSignal West Dura substrate (Thermo Scientific).
Analysis of Sphingolipids by Tandem Mass Spectrometry (MS)
Mitochondria and tissue samples were analyzed by reverse-phase high pressure liquid chromatography coupled to electrospray ionization followed by separation by MS as described previously (28, 38, 39). Sphingolipid analysis was performed in the Lipidomics Analytical Core Facility at MUSC using a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a multiple reaction-monitoring positive ionization mode (40). The peaks for the target analytes and internal standards were collected and processed with the Xcalibur software system. The target analyte peak area ratios from the samples were similarly normalized to their respective internal standard and compared with the calibration curves using a linear regression model. Each sample was normalized to its respective total protein levels.
Sphingolipid-metabolizing Enzyme Array
A custom PCR array of the sphingolipid-metabolizing gene network was developed by SA Biosciences (Valencia, CA) in collaboration with Dr. Christopher J. Clarke (Stony Brook University). Primers were designed against the cDNA regions of the genes. Actin and GAPDH were utilized as reference genes. mRNA was extracted from tissue samples using RNA Easy kit (Qiagen, Germantown, MD). 0.5–1 μg of RNA was used to synthesize cDNA with the SuperScript II kit for the first strand synthesis. Real-time RT-PCR was performed on a Bio-Rad MiniOpticon detection system. To run the assay, a MasterMix of 637.5 μl of SYBR Green, 51 μl of cDNA, and 586.5 μl of distilled water was prepared for each cDNA template. 25 μl was loaded per well. The RT-PCR protocol consisted of 10 min at 95 °C and 60 s at 60 °C for annealing and extension. After the cycles were run, a melt curve was performed to confirm a single product for each primer pair. The data were analyzed utilizing the PCR array data analysis portal at the SA Biosciences Web site.
Ceramidase Activity
NCDase is an enzyme that cleaves the N-acyl linkage of ceramide to form sphingosine and free fatty acid. Detection of NCDase enzymatic activity was performed according to methods described previously (41, 42) with slight modifications. To enhance the sensitivity of the assay, we employed a synthetic substrate C17-C18-ceramide. NCDase cleaves C17-C18-ceramide, yielding C17-sphingosine that could be easily discriminated from the endogenous C18-sphingosine. Briefly, the substrate C17-C18-ceramide was solubilized by sonication in 0.2 m HEPES, pH 7.0, containing 0.1% Triton X-100. Each sample of mitochondria (500 μg) was assayed for NCDase activity by incubation with 50 μm C17-C18-ceramide in 0.2 m HEPES (pH 7.0) containing 0.25% Triton X-100 for 2 h at 37 °C. The reaction was terminated; sphingolipids were extracted by the addition of 1 ml of an ethyl acetate/isopropyl alcohol/water (60:30:10%, v/v/v) solvent system and subjected to analysis by tandem MS. NCDase-specific activity was expressed in pmol of C17-sphingosine/min/mg of protein.
Sphingosine Kinase Activity
SphK1 and SphK2 activities were determined by isotype-specific assays (43, 44). Mitochondria (500 μg) were incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm EDTA, 20% glycerol, 40 mm β-glycerophosphate, 1 mm DTT, 10 mm ATP, and 10 μm C17-sphingosine. To measure SphK1 activity, the buffer was supplemented with 0.25% Triton X-100, which inhibits SphK2. To determine SphK2 activity, the buffer was supplemented with 4 mg/ml BSA and 1 m KCl, conditions in which SphK1 activity is inhibited (29). The reaction was performed at 37 °C for 1 h, stopped by the addition of 1 ml of an ethyl acetate/isopropyl alcohol/water (60:30:10%, v/v/v) solvent system, and subjected to analysis by tandem MS. A specific activity of sphingosine kinase was expressed in pmol of C17-S1P/min/mg of protein.
Statistical Analysis
All experiments and assays were performed three or more times. Data were collected, and the mean value of the treatment groups and the S.E. were calculated. Data were analyzed for statistically significant differences between groups by one-way analysis of variance with a post hoc Bonferroni test, which adjusts for multiple simultaneous comparisons (SigmaPlot software version 12.0). Statistical significance was ascribed to the data when p was <0.05.
RESULTS
Brain Sphingolipid Metabolism Is Disturbed after TBI
A sphingolipid profile was determined in brains of mice at 4 h, 1 day, 2 days, and 7 days post-CCI. Tissue lysates were prepared from a mouse brain hemisphere ipsilateral to the injury or sham-injured animal brain (control) and analyzed by tandem MS. SM was significantly elevated at 2 and 7 days after brain injury (Fig. 1A). The content of SM species characterized by long-chain fatty acid, including C14:0-SM and C16:0-SM, was augmented at 1, 2, and 7 days post-TBI. In contrast, C18:0-SM and C18:1-SM were not changed (Fig. 1B). SM species containing very long-chain fatty acid, such as C24:0-SM and C24:1-SM, were elevated at 2 and 7 days after brain trauma, whereas C20:0-SM and C22:0-SM did not change (Fig. 1C). The data are presented as percentage increases of brain SM species content shown in Table 1.
FIGURE 1.

SM and dihydrosphingosine changes in brain tissue after TBI. Sphingolipids were analyzed in brain tissue lysate made from the brain hemisphere ipsilateral to the injury imposed by a control cortical impact device as described under “Experimental Procedures.” Sham-injured animal brain was used as a control (con). A, total SM content was increased in the injured brain tissue. B and C, SM species changes in the injured tissue. D, dihydrosphingosine (DHsphingosine) was increased in the injured brain. Data are means ± S.E. (error bars); *, p < 0.05, n = 16. Each sample was normalized to its respective total protein levels.
TABLE 1.
Sphingolipid species content of a mouse brain tissue
SM and ceramide species (pmol/mg protein) were determined in the mouse brain after sham injury. Values are means ± S.E., n = 16.
| SM | Ceramide | |
|---|---|---|
| C14:0 | 7.9 ± 1.4 | 8.4 ± 0.9 |
| C16:0 | 652.5 ± 44.2 | 89.1 ± 6.4 |
| C18:0 | 10,792.0 ± 354.3 | 1023.6 ± 46.7 |
| C18:1 | 1002.4 ± 59.8 | 612.5 ± 24.1 |
| C20:0 | 618.8 ± 45.1 | 119.8 ± 15.3 |
| C24:0 | 574.6 ± 37.6 | 98.7 ± 7.5 |
| C24:1 | 994.7 ± 49.3 | 284.3 ± 19.2 |
An enhanced accumulation of SM seems to result from a stimulation of de novo biosynthesis of ceramide, a precursor of SM. Thus, the levels of dihydrosphingosine (Fig. 1D) and dihydroceramide (Fig. 2A) were up-regulated in the brain at 1, 2, and 7 days postinjury. Remarkably, ceramide was significantly attenuated at 2 and 7 days after the brain injury (Fig. 2B). All ceramide species were decreased at days 2 and 7 postinjury (Fig. 2C), which is in line with the activation of ceramide utilization for SM synthesis. The data are presented as percentage decreases of brain ceramide species content shown in Table 1.
FIGURE 2.

Dihydroceramide and ceramide changes in brain tissue after TBI. Brain tissue samples were prepared from the mouse brain hemisphere ipsilateral to the damage. Sham-injured animal brain was used as a control (con). Dihydroceramide (DHceramide) (A), total ceramide (B), and ceramide species (C) were measured in the injured brain tissue after TBI as described under “Experimental Procedures.” Data are means ± S.E. (error bars); *, p < 0.05, n = 16. Each sample was normalized to its respective total protein levels.
To determine if ceramide is converting into sphingosine and S1P, through another ceramide-utilizing pathway that could be activated after TBI, sphingosine and S1P levels were assessed in injured brain. Consistent with an enhanced ceramide deacylation into sphingosine, sphingosine levels were markedly elevated in the brain at 2 and 7 days after the injury (Fig. 3A). There were no changes in brain S1P levels (Fig. 3B). The data suggest that TBI triggered the activation of de novo ceramide biosynthesis concurrently with activation of ceramide utilization pathways yielding elevations in SM and sphingosine.
FIGURE 3.

Sphingosine and S1P changes in brain tissue and mitochondria after TBI. Brain tissue samples and mitochondria were prepared from the mouse brain hemisphere ipsilateral to the damage. Sham-injured animal brain was used as a control (con). Sphingosine content was up-regulated (A), and S1P content was unchanged (B) in the brain during the first week post-TBI. Data are means ± S.E. (error bars); *, p < 0.05; n = 16. Each sample was normalized to its respective total protein levels. C, sphingosine was gradually elevated in mitochondria after TBI. D, S1P was reduced in brain mitochondria after TBI, whereas ceramide was increased at 1 day, returning to control levels at 2 days post-TBI. The ceramide content of control mitochondria was 1318 ± 94.7 pmol/mg of protein, whereas S1P content was 8.7 ± 0.9 pmol/mg of protein. Data are means ± S.E.; *, p < 0.05, n = 12. Each sample was normalized to its respective total protein levels.
Sphingosine Is Accumulated in Mitochondria after Brain Trauma
Specific subcellular localization of most sphingolipid-metabolizing enzymes coupled with high hydrophobicity of key sphingolipids imposes clear restrictions on subcellular distribution of sphingolipids (16). The generation site of a bioactive sphingolipid is believed to dictate the mechanism of its action and cell functional responses (11). Recently, mitochondria emerged as a specialized compartment of sphingolipid metabolism (22), which could play an important role in mitochondrial response to TBI. The impact of brain injury on sphingolipid profile was elucidated in mitochondria purified from injured mouse brain at various times after TBI. Sham-injured animal brain was used to isolate control mitochondria. There was a gradual building up of sphingosine level in mitochondria that reached about 286% of the control mitochondria at day 7 after the brain injury (Fig. 3C). Ceramide was slightly elevated at 1 day (up to 135%) and returned to control level at 2 days postinjury. S1P was considerably down-regulated in cerebral mitochondria after the brain trauma during the first week postinjury (Fig. 3D). Sphingosine could be generated in mitochondria by NCDase from ceramide and further metabolized by SphK2, forming S1P. The data suggest TBI-induced activation of NCDase as a possible source of mitochondrial sphingosine accumulation. The reduced levels of S1P in mitochondria from injured brain indicate that sphingosine accumulation could be in part due to its diminished conversion into S1P by SphK2.
TBI Affects Sphingolipid-metabolizing Enzymes via Post-translational Mechanisms
The levels and turnover of sphingolipids are regulated by sphingolipid-metabolizing enzymes that play essential roles in the shaping of sphingolipid signaling effects and cell responses (16). To facilitate analysis of gene expression for the wider sphingolipid network, a custom PCR array from SA Biosciences was successfully used for evaluation of cell stress response (45). The effects of TBI on the sphingolipid enzyme gene expression network were examined using the PCR array in tissue samples from brain exposed to TBI. Fig. 4 shows that of 41 genes investigated, only a mitochondria-associated neutral sphingomyelinase (Smpd5) gene exhibited a significant up-regulation of >3-fold (Smpd5) at day 2 post-TBI. This suggests a possible physiological role of the mitochondria-associated neutral sphingomyelinase in the brain response following TBI. There were no changes in the gene expression of NCDase (Asah2) and SphK2 (Sphk2), suggesting that secondary TBI proceeds through post-translational mechanisms of enzyme modulation.
FIGURE 4.

Sphingolipid-metabolizing enzyme gene expression after TBI. Brain samples were prepared from the ipsilateral hemisphere after TBI and sham-injured brain (control). RNA was extracted and converted to cDNA for analysis by real-time PCR using a PCR array as described under “Experimental Procedures.” The data are presented as -fold change TBI/sham injury and are means of three independent experiments. The dashed line represents a 3-fold change. *, p < 0.05; n = 6. Error bars, S.E.
NCDase Activation Contributes to Mitochondrial Sphingosine Accumulation after TBI
To gain insight into the mechanism of mitochondrial sphingosine accumulation, the NCDase activity was measured using synthetic C17-C18:0-ceramide as a substrate. NCDase catalyzes the cleavage of C17-C18-ceramide, yielding C17-sphingosine, which is easily discriminated from the endogenous C18-sphingosine by tandem MS. Consistent with the brain injury-induced activation of the sphingosine-generating pathway, the activity of NCDase was increased (up to 138%) in mitochondria isolated from brain at 7 days postinjury (Fig. 5A).
FIGURE 5.
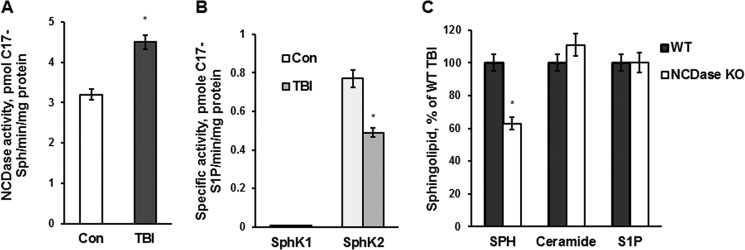
TBI-induced mitochondrial sphingosine accumulation is due to an activation of NCDase and could be partially rescued by knocking down NCDase. Mitochondria were purified from the injured hemisphere of a mouse brain (TBI) and sham-injured mouse brain (Con) at 7 days post-TBI. A, specific NCDase activity was measured with C17-C18-ceramide as a substrate (42), yielding C17-sphingosine (C17-Sph) as described under “Experimental Procedures.” Data are means ± S.E. (error bars); *, p < 0.05; n = 8. B, specific SphK2 and SphK1 activities were measured with C17-sphingosine as a substrate, yielding C17-S1P, as described under “Experimental Procedures.” The specific activities of SphK1 and SphK2 in brain homogenate were 1.10 ± 0.12 and 0.68 ± 0.14 pmol of C17-S1P/min/mg of protein, respectively. Data are means ± S.E.; *, p < 0.05; n = 8. C, cerebral mitochondria were purified from WT and NCDase KO mice at day 7 post-TBI. Sphingolipid content was measured by tandem MS. Data are means ± S.E.; *, p < 0.05; n = 12. Each sample was normalized to its respective total protein levels.
Next, TBI impact on the sphingosine utilization pathway in mitochondria was investigated. The activity of SphK2 was measured in mitochondria using synthetic C17-sphingosine as a substrate. SphK2 activity was reduced by 35% in mitochondria from injured brain at 7 days post-TBI (Fig. 5B). Another isoform of sphingosine kinase, SphK1, is a cytosolic protein; however, SphK1 could translocate to a membrane compartment in response to growth factors and cytokines(46). To rule out the involvement of SphK1 in mitochondrial sphingosine phosphorylation after brain trauma, SphK1 activity was measured in mitochondria (Fig. 5B). The brain homogenate was used as a positive control. Although the SphK1 activity was present in the brain homogenate, there was no detectable SphK1 activity in mitochondria. The data suggest that TBI-triggered activation of sphingosine generation by NCDase alongside with diminishing sphingosine utilization by SphK2 led to profound accumulation of sphingosine in mitochondria.
To confirm that NCDase is required for the increase in mitochondrial sphingosine after the brain injury, NCDase KO mice were employed. The sphingolipid profile was determined in mitochondria from WT and NCDase-deficient mouse brain at 7 days post-TBI (Fig. 5C). Sphingosine accumulation in mitochondria from NCDase KO mice was significantly reduced (about 37%) as compared with WT mice. Knocking down NCDase did not affect the levels of ceramide and S1P in the injured mitochondria. These results suggest that activation of mitochondrial NCDase is in part responsible for sphingosine accumulation in mitochondria of injured brain.
SphK2 Association with NCDase and COX Is Decreased after TBI
To examine the mechanisms of TBI-induced modulation of NCDase and SphK2 activity, mitochondria were analyzed by Western blotting. Consistent with the results of the PCR array studies, Fig. 6A shows no changes in protein expression levels of either NCDase or SphK2, indicating the involvement of post-translational mechanisms regulating the enzyme activity. An inner mitochondrial protein, COX-1, and an outer mitochondrial membrane protein-marker, VDAC, were used as protein loading controls (Fig. 6A).
FIGURE 6.

TBI hindered SphK2 association with NCDase and COX. Mitochondria were purified from the injured hemisphere of mouse brain and sham-injured brain (con) at 7 days post-TBI. A and B, mitochondrial lysate was loaded into the lane (30 μg/lane) (A). To investigate submitochondrial localization of NCDase and SphK2, equal volume samples of untreated mitochondria (MT), mitoplasts (MP), or mitoplasts treated with trypsin (MPT) were loaded into the lane (B). Blots were analyzed using specific anti-NCDase (Bethyl Laboratories), anti-SphK2 (Abcam), anti-COX-1 (Santa Cruz Biotechnology), and anti-VDAC (Cell Signaling) antibodies. C and D, SphK2 association with NCDase and COX-1 was detected in reciprocal immunoprecipitation (IP) experiments. Mitochondria were immunoprecipitated using anti-SphK2 (Abcam) antibodies, and the blots were probed with anti-NCDase (Bethyl Laboratories), anti-COX-1 (Santa Cruz Biotechnology), or anti-SphK2 (Santa Cruz Biotechnology) antibodies. Input load was 30 mg/lane (C). Mitochondria were immunoprecipitated with anti-NCDase antibody (Bethyl Laboratories) and probed using anti-SphK2 (Santa Cruz Biotechnology), anti-COX-1 (Santa Cruz Biotechnology), or anti-NCDase (Santa Cruz Biotechnology) antibodies. Input load was 30 μg/lane (D). As a control, the same immunoprecipitation procedure was performed except for primary antibody application (IgG).
In mitochondria, TBI appears to instigate both elevated generation of sphingosine by NCDase and decreased sphingosine utilization by SphK2 (Fig. 5). This suggests that these enzymes could be functionally organized in a novel metabolic pathway for generating and utilizing sphingosine in mitochondria.
The spatial organization of sphingolipid-metabolizing enzymes in mitochondria has not yet been determined, which prompted our investigation of possible physical interaction of the sphingosine-metabolizing enzymes. To investigate a submitochondrial localization of SphK2 and NCDase, mitoplasts (i.e. mitochondria devoid of the outer mitochondrial membrane) were prepared from baseline cerebral mitochondria. Fig. 6B shows that both SphK2 and NCDase are associated with mitoplasts, not with the outer mitochondrial membrane. Moreover, the enzymes were still associated with the mitoplasts following the brief treatment with the protease trypsin. However, there was slightly less SphK2 in mitoplasts treated with the protease, suggesting that SphK2 could be localized on the outer side of the inner mitochondrial membrane.
To explore whether SphK2 is associated with NCDase in mitochondria, co-immunoprecipitation studies were performed using anti-SphK2 antibodies. Fig. 6C shows that SphK2 is associated with NCDase in control mitochondria and that there is less NCDase complexed with SphK2 after TBI. Further analysis of the SphK2 immunoprecipitates revealed that a major catalytic subunit of COX, COX-1, is associated with SphK2 in control mitochondria. However, there was less COX-1 complexed with SphK2 in mitochondria after TBI (Fig. 6C). The association of SphK2, NCDase, and COX-1 was confirmed in reciprocal immunoprecipitation experiments using anti-NCDase antibodies (Fig. 6D). Both SphK2 and COX-1 were found in a complex with NCDase in control mitochondria. Again, there was less NCDase complexed with SphK2 after TBI. In contrast, TBI did not have any impact on COX-1 association with NCDase. The results of the immunoprecipitation studies indicate that SphK2 exists in a complex with NCDase in the vicinity of the inner mitochondrial membrane protein COX-1. The data suggest that TBI disrupts SphK2 association with NCDase, whereas NCDase association with COX-1 is still intact. TBI-induced disturbance of SphK2 association with NCDase could interfere with the substrate (sphingosine) flow from NCDase to SphK2, thus decreasing the sphingosine utilization and contributing to sphingosine elevation in mitochondria.
Mitochondrial COX Activity Is Reduced after TBI
To elucidate the impact of TBI on mitochondrial function, oxidative phosphorylation was assessed by measuring the oxygen consumption rate coupled to ATP synthesis. The respiratory chain enzymes transport electrons from electron donors at Complex I or Complex II to Complex III and then to Complex IV (COX), where the electrons reach their acceptor oxygen. With Complex I substrates glutamate plus malate, the respiration rates determined in the presence of ADP (state 3) were about 29% lower in cerebral mitochondria purified from the injured brain compared with sham (Fig. 7A). Similar decreases in respiration rates were observed when Complex II substrate succinate was used. Importantly, oxygen consumption rates supported by the substrate of COX, ascorbate, plus TMPD, measured with ADP, were also 29% lower in mitochondria from the injured brain, suggesting that TBI compromised the oxidative phosphorylation at the level of the terminal enzyme of the electron transport chain COX (Fig. 7A). The respiration rates measured in the presence of the uncoupler of oxidative phosphorylation, 2,4-DNP (state 3u) were not different from the respiration rates in the presence of ADP (state 3), indicating that the TBI-induced defect is at the level of the electron transport chain, not in the ATP synthesis apparatus, including ATP synthase. The data suggest that the electron transport chain activity is suppressed at the level of COX in cerebral mitochondria after TBI.
FIGURE 7.

Respiratory chain activity was reduced at the level of COX after TBI and sphingosine inhibited the COX activity in baseline mitochondria. A, mitochondria were purified from the ipsilateral hemisphere of WT mouse brain at day 7 post-TBI. Sham-injured animal brain was used as a control (Con). Mitochondrial respiration was measured by recording oxygen consumption with a Clark-type oxygen electrode in the presence of Complex I substrate 5 mm glutamate plus 5 mm malate (glutamate + malate), Complex II substrate 10 mm succinate (succinate), and Complex IV (COX) substrate 1 mm ascorbate plus 250 μm TMPD (ascorbate + TMPD) in the presence of 100 μm ADP (state 3). Data are means ± S.E. (error bars); *, p < 0.05, n = 8. B, mitochondrial COX activity was measured by recording oxygen consumption of baseline mitochondria in the presence of COX substrate (1 mm ascorbate plus 250 μm TMPD), 1 μm antimycin, and 50 μm 2,4-DNP. Sphingosine (Sph) and dihydrosphingosine (DHSph) were delivered in ethanol. C16:0-ceramide and C18:0-ceramide were delivered in ethanol/dodecane (98:2, v/v). Data are means ± S.E.; *, p < 0.05, n = 8.
Sphingosine Inhibits an Electron Transport Chain Activity at the Level of COX
To investigate whether sphingosine accumulated in mitochondria after brain injury could cause the electron transport chain defect at the level of COX, the COX activity of baseline mitochondria was assessed in the presence of sphingosine. The COX activity was measured as an oxygen consumption rate supported by the COX substrate ascorbate plus TMPD in the presence of the uncoupler 2,4-DNP. To inhibit the oxygen consumption supported by endogenous substrates, the inhibitor of Complex III antimycin was used. Fig. 7B shows that exogenous sphingosine inhibited the respiration supported by COX substrate in the concentration-dependent manner. To assess the efficiency of sphingosine delivery to mitochondria following incubation with exogenous sphingosine, mitochondria were incubated with 20 μm C17-sphingosine in an oxygraph chamber for the duration of the COX activity measurement. Mitochondria were centrifuged at 10,000 × g for 10 min, washed twice with the incubation medium without C17-sphingosine, and subjected to analysis by tandem MS. There was 212.7 ± 3.5 pmol of C17-sphingosine/mg of protein bound to mitochondria. In line with the findings in liver mitochondria (47), the sphingosine binding site on COX in cerebral mitochondria was somewhat nonspecific. Thus, a close analog of sphingosine, dihydrosphingosine (sphinganine), inhibited the COX activity, similar to sphingosine (Fig. 7B). In contrast to the liver mitochondria (47), C16:0-ceramide or C18:0-ceramide did not have any effect on COX activity in isolated brain mitochondria (Fig. 7B), which is consistent with previous reports in cerebral mitochondria (36, 48). Although sphinganine could inhibit the COX activity as well as sphingosine, the sphinganine was unlikely to cause the suppression of COX activity after TBI. The concentration of sphinganine in mitochondria was about 30 times lower than the concentration of sphingosine (Fig. 3C). Thus, there was 1.74 ± 0.15 pmol/mg of protein of sphinganine in control mitochondria (sham) and 1.81 ± 0.14 pmol/mg of protein of sphinganine in mitochondria at day 7 after TBI. The concentration of sphingosine was 59.8 ± 3.3 pmol/mg of protein at day 7 post-TBI. This suggests that the accumulation of sphingosine in the cerebral mitochondria after the brain injury is a likely cause of the decreased COX activity after TBI.
Although endogenous mitochondrial sphingosine levels (Fig. 3C) are much lower than exogenous sphingosine concentrations required for modulating respiratory chain function, it is conceivable that endogenous hydrophobic sphingosine is generated inside the membrane near its target COX; therefore, the effective concentration of the sphingolipid in the vicinity of the target could be much higher.
Knocking Down NCDase Rescues the TBI-induced Defect in COX Activity
To ascertain whether the mitochondrial sphingosine responsible for the respiratory chain defect is generated by NCDase, COX activity was measured in cerebral mitochondria purified from WT and NCDase KO mouse brain at day 7 after TBI. Sham-injured mouse brain was used as a control. COX activity was 29% lower in mitochondria from the WT mouse injured brain compared with control (Fig. 8A). Furthermore, NCDase knockdown preserved the COX activity (Fig. 8A). The data indicate that TBI-induced activation of NCDase leading to mitochondrial sphingosine accumulation is the primary cause of the decreased COX activity in mitochondria.
FIGURE 8.

NCDase knockdown rescued COX activity defect, attenuated brain damage, and improved sensorimotor deficit recovery after TBI. A, mitochondria were purified from the ipsilateral hemisphere of WT and NCDase KO mouse brain at day 7 post-TBI. Sham-injured animal brain was used as a control (Con). Mitochondrial COX activity was measured by recording oxygen consumption in the presence of COX substrate (1 mm ascorbate plus 250 μm TMPD), 1 μm antimycin, and 50 μm 2,4-DNP (state 3u). Data are means ± S.E. (error bars); *, p < 0.05; n = 8. B, lesion cavity volume was measured at day 28 post-TBI by staining of brain sections with 0.1% cresyl violet and image analysis as described under “Experimental Procedures.” Data are means ± S.E.; *, p < 0.05; n = 12. C, sensorimotor deficits were assessed using a standard rotarod test. Each day for 3 days prior to injury, animals were trained on the rotarod at a speed of 18 rpm in the acceleration mode (0–18 rpm/90 s). Animals were tested with the rotarod apparatus using three trials in session, with a minimum of 5 min resting between trials. Data are means ± S.E.; *, p < 0.05; n = 16.
Knocking Down NCDase Reduces Brain Damage and Improves Sensorimotor Deficit Recovery after TBI
To assess the role of NCDase in promoting secondary brain injury after trauma, the contusion volume was measured in WT and NCDase KO mice after TBI. Brain injury after TBI was significantly attenuated in NCDase-deficient mice, indicating the important role of the mitochondrial enzyme in promoting secondary brain injury (Fig. 8B). To examine the impact of NCDase on brain function recovery after the injury, behavioral deficits were assessed in WT and NCDase KO mice after TBI. A standard rotarod test was used because it has been shown to be effective and reliable in rodent brain trauma experiments (34, 35). Consistent with previous reports (35), the WT mice had significant impairments in sensorimotor functions, which were gradually recovered up to 57% compared with sham-injured mice on day 7 postinjury (Fig. 8C). NCDase knockdown significantly improved the sensorimotor function recovery after TBI, highlighting the critical role of NCDase in promoting behavioral deficits after brain trauma.
DISCUSSION
The present studies are unique in establishing a novel role of sphingolipid sphingosine and its metabolizing enzymes in mitochondrial dysfunction after TBI. We have shown that TBI stimulates the utilization of ceramide by NCDase, which results in the accumulation of mitochondrial sphingosine and mitochondrial dysfunction promoting secondary brain injury. Furthermore, TBI induced a decrease in mitochondrial SphK2 activity, thus limiting sphingosine conversion into S1P, which contributed to excessive sphingosine accumulation in mitochondria. This is the first demonstration of the essential role of sphingosine in mitochondrial dysfunction in the brain after trauma. Given the significance of mitochondria in regulation of neural cell function and survival, the characterization of a sphingosine-mediated mitochondrial defect triggered by experimental TBI is a valuable contribution to our understanding of the pathophysiological mechanisms of long term brain impairment after trauma.
Our studies identify the disruption of sphingolipid metabolism as a novel determinant of the secondary brain injury after TBI. Here, we show that TBI stimulated the de novo biosynthesis of ceramide, as indicated by increases in the intermediates of the pathway, dihydrosphingosine and dihydroceramide (Figs. 1D and 2A). Surprisingly, activation of the ceramide biosynthetic pathway did not result in enhanced levels of ceramide. In fact, ceramide gradually decreased during the first week after TBI (Fig. 2, B and C), suggesting its increased conversion into the other sphingolipids, such as SM (Fig. 1A) and sphingosine (Fig. 3A). We have previously reported that an activation of the de novo ceramide biosynthesis after transient brain ischemia results in an accumulation of several ceramide species (C16:0-ceramide, C18:0-ceramide, and C18:1-ceramide) at 1 day of reperfusion, returning to baseline levels at 2 days of reperfusion (28). Altogether, these studies provide further support for the concept that secondary brain injury is produced by a dynamic interplay of multiple pathophysiological cascades, not limited to ischemic insult and the disruption of cerebral circulation (49). However, the cornerstones of current therapy of TBI are treatment of elevated intracranial pressure and optimization of cerebral perfusion (50). These studies extend the experimental evidence base for refining the current TBI therapies, with greater emphasis on long term metabolic and functional changes.
Our studies showed the continued excessive accumulation of sphingosine in brain tissue and, specifically, in mitochondria during the first week after brain injury. To determine the impact of sphingosine on mitochondrial functions, the respiratory chain activity of baseline mitochondria was measured in the presence of various concentrations of exogenous sphingosine (Fig. 7B). These studies revealed COX as a novel target of sphingosine, leading to a reduced enzyme activity in brain mitochondria after TBI (Figs. 7A and 8A). This is in line with a recent report describing sphinganine- and sphingosine-mediated inhibition of COX activity in liver mitochondria (47). However, the effect of sphingosine on COX activity was somewhat nonspecific in liver mitochondria because ceramide was also capable of reducing COX activity. In cerebral mitochondria, sphingosine selectively inhibited COX activity, whereas ceramide did not have any effect (Fig. 7B). These data support the notion that brain mitochondria are functionally different from liver mitochondria (51).
Our studies point to an important role of the NCDase as a source of excessive sphingosine accumulation in cerebral mitochondria after TBI. The NCDase activity was elevated in mitochondria isolated from injured brain (Fig. 5A). Knocking down NCDase decreased mitochondrial sphingosine after TBI (Fig. 5C), indicating NCDase involvement in the TBI-induced accumulation of sphingosine within cerebral mitochondria. Furthermore, the TBI-induced defect in COX activity was rescued in NCDase-deficient mice (Fig. 8A). The results from this study implicate NCDase as an upstream regulator of COX activity in the mitochondrial response to TBI.
The NCDase was first purified from rat brain, and then the human isoforms were cloned (24, 52). On the basis of confocal microscopy data, this activity was ascribed to mitochondria (24) and plasma membrane (53), and it was also demonstrated in purified mitochondria (25, 27). The ceramide-cleaving activity of purified rat NCDase, a glycosylated protein with a molecular mass 116 kDa, was not affected by Ca2+, Mg2+, or Mn2+, but was inhibited by Zn2+, Fe2+, and Cu2+ (54). NCDase could also function in the reverse direction, e.g. catalyzing formation of ceramide as a result of condensation of palmitate and sphingosine (25, 55). Evidence has accumulated demonstrating the importance of NCDase in regulating ceramide levels in response to cytokine and growth factor-mediated signaling. Cytokines, including TNF-α, IL-1β, and interferon-γ, could up-regulate the mRNA and protein expression of NCDase as a part of the mechanism protecting the cell against cytokine-induced ceramide toxicity (56–58). Our studies are the first demonstration of the pathophysiological role of NCDase in promoting mitochondrial dysfunction and brain damage after TBI. TBI-induced activation of NCDase resulted in elevated sphingosine in mitochondria, leading to mitochondrial dysfunction and brain damage. The results of mRNA (Fig. 4) and protein expression analysis (Fig. 6A) suggest a post-translational mechanism of NCDase activation after TBI.
In the present study, we explored the spatial organization of the sphingolipid-metabolizing enzymes NCDase and SphK2 in mitochondria by immunoprecipitation (Fig. 6, C and D). It appears that SphK2 and NCDase are complexed with COX-1, forming a tripartite complex associated with the inner mitochondrial membrane. Furthermore, SphK2 association with the NCDase·COX-1 complex was hindered by TBI.
The mammalian COX (EC 1.9.3.1.), the terminal enzyme of the mitochondrial electron transport chain, is composed of 13 subunits with a molecular mass of 205 kDa (59). The biogenesis of COX involves the coordinated assembly of three subunits encoded by mitochondrial DNA and 10 by nuclear DNA. The mitochondria-encoded COX subunits 1–3 contain three catalytic centers of the enzyme and are essential and sufficient for the catalytic COX activity (60). Ten peripheral COX subunits are required for the stability of the catalytic core and the regulation of the enzyme activity (61). COX catalyzes the oxidation of its substrate, cytochrome c, and the reduction of molecular oxygen to water. It assists in the pumping the protons across the inner mitochondrial membrane to create the electrochemical proton gradient that drives the synthesis of ATP. Inactivation of COX by inhibitors, such as cyanide and carbon monoxide, is incompatible with life. In the brain and some other highly oxidative organs, such as the heart, liver, and kidney, COX represents the rate-limiting enzyme of the respiratory chain, which emphasizes the impact of regulating COX as a central site for the regulation of the energy metabolism (62).
Our studies provide further evidence for the critical involvement of sphingolipids in the regulation of COX. Recently, it has been reported that S1P generated by SphK2 in mitochondria binds to prohibitin 2, which regulates the assembly and function of COX in the electron transport chain (29). Typically, SphK2 is localized in the nucleus of many types of cells (63) and has been shown to produce S1P, which binds to and inhibits histone deacetylases HDAC1 and HDAC2, resulting in enhanced local histone acetylation and increased transcription of specific genes (64). Our studies indicate mitochondrial localization of SphK2 in the brain and suggest a novel role of SphK2 in modulating the activity of COX after TBI. The present study implicates another sphingolipid, sphingosine, as a critical determinant regulating the activity of the respiratory chain rate-limiting enzyme COX. There are two well known regulatory mechanisms of COX: the allosteric feedback inhibition of the enzyme by its direct product ATP and the expression of COX subunits (62). These studies suggest a novel mechanism of regulation of COX activity by sphingolipid sphingosine and identify the metabolic pathway involved in generation and utilization of sphingosine in mitochondria.
Our data suggest that TBI-induced coordinated activation of NCDase and inactivation of SphK2 (Fig. 5, A and B) contributed to the excessive accumulation of mitochondrial sphingosine, which is a potent inhibitor of COX activity (Fig. 7B). Remarkably, the NCDase·SphK2 protein complex is associated with the core catalytic subunit 1 of COX (Fig. 6, C and D). As a result, the elevated sphingosine brought about a reduction in COX activity after TBI (Figs. 7A and 8A). Consistent with previous studies (29), our data further implicate SphK2 as an important regulator of COX function in mitochondria. These studies suggest a novel role for the NCDase·sphingosine·SphK2 axis in governing COX activity in brain mitochondria.
In summary, this study provides experimental evidence that TBI disturbs the activity of NCDase and SphK2, leading to accumulation of mitochondrial sphingosine, which results in mitochondrial dysfunction promoting brain damage, and the data shed more light on the role of the compartmentalization of sphingolipid metabolism and accentuate the function of sphingolipid-metabolizing enzymes in cerebral mitochondria.
Acknowledgments
We are very grateful to Dr. Lina M. Obeid for stimulating discussions regarding the manuscript. We thank Dr. Christopher J. Clarke for helping with PCR array studies. We thank Alexander S. Novgorodov for help with preparation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant P30 GM103339 (to S. A. N.). This work was also supported by Veterans Affairs Merit Awards I01RX000206 (to T. I. G.), I01BX001104 (to T. I. G.), and I01RX000331 (to M. S. K.). The Lipidomics Core Facility at Medical University of South Carolina is supported in part by National Institutes of Health Grant P30 GM103339.
- TBI
- traumatic brain injury
- CCI
- controlled cortical impact
- COX
- cytochrome oxidase
- COX-1
- cytochrome oxidase subunit 1
- 2,4-DNP
- 2,4-dinitrophenol
- NCDase
- neutral ceramidase
- KO
- knockout
- SphK2
- sphingosine kinase 2
- SM
- sphingomyelin
- S1P
- sphingosine 1-phosphate
- TMPD
- N,N,N′,N′-tetramethyl-p-phenelenediamine
- VDAC
- voltage-dependent anion channel.
REFERENCES
- 1. Spaethling J. M., Geddes-Klein D. M., Miller W. J., von Reyn C. R., Singh P., Mesfin M., Bernstein S. J., Meaney D. F. (2007) Linking impact to cellular and molecular sequelae of CNS injury: modeling in vivo complexity with in vitro simplicity. Prog. Brain Res. 161, 27–39 [DOI] [PubMed] [Google Scholar]
- 2. Robertson C. L., Scafidi S., McKenna M. C., Fiskum G. (2009) Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 218, 371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan P. G., Keller J. N., Mattson M. P., Scheff S. W. (1998) Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J. Neurotrauma 15, 789–798 [DOI] [PubMed] [Google Scholar]
- 4. Gilmer L. K., Roberts K. N., Joy K., Sullivan P. G., Scheff S. W. (2009) Early mitochondrial dysfunction after cortical contusion injury. J. Neurotrauma 26, 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh I. N., Sullivan P. G., Hall E. D. (2007) Peroxynitrite-mediated oxidative damage to brain mitochondria: protective effects of peroxynitrite scavengers. J. Neurosci. Res. 85, 2216–2223 [DOI] [PubMed] [Google Scholar]
- 6. Harris L. K., Black R. T., Golden K. M., Reeves T. M., Povlishock J. T., Phillips L. L. (2001) Traumatic brain injury-induced changes in gene expression and functional activity of mitochondrial cytochrome c oxidase. J. Neurotrauma 18, 993–1009 [DOI] [PubMed] [Google Scholar]
- 7. Hovda D. A., Yoshino A., Kawamata T., Katayama Y., Becker D. P. (1991) Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain Res. 567, 1–10 [DOI] [PubMed] [Google Scholar]
- 8. Opii W. O., Nukala V. N., Sultana R., Pandya J. D., Day K. M., Merchant M. L., Klein J. B., Sullivan P. G., Butterfield D. A. (2007) Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma 24, 772–789 [DOI] [PubMed] [Google Scholar]
- 9. Merrill A. H., Jr. (2011) Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 111, 6387–6422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stancevic B., Kolesnick R. (2010) Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 584, 1728–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hannun Y. A., Obeid L. M. (2011) Many ceramides. J. Biol. Chem. 286, 27855–27862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merrill A. H., Jr. (2002) De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J. Biol. Chem. 277, 25843–25846 [DOI] [PubMed] [Google Scholar]
- 13. Mandon E. C., Ehses I., Rother J., van Echten G., Sandhoff K. (1992) Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 267, 11144–11148 [PubMed] [Google Scholar]
- 14. Kolter T., Proia R. L., Sandhoff K. (2002) Combinatorial ganglioside biosynthesis. J. Biol. Chem. 277, 25859–25862 [DOI] [PubMed] [Google Scholar]
- 15. Futerman A. H., Stieger B., Hubbard A. L., Pagano R. E. (1990) Sphingomyelin synthesis in rat liver occurs predominantly at the cis and medial cisternae of the Golgi apparatus. J. Biol. Chem. 265, 8650–8657 [PubMed] [Google Scholar]
- 16. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 17. Mao C., Obeid L. M. (2008) Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 1781, 424–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuvillier O. (2002) Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 1585, 153–162 [DOI] [PubMed] [Google Scholar]
- 19. Woodcock J. (2006) Sphingosine and ceramide signalling in apoptosis. IUBMB Life 58, 462–466 [DOI] [PubMed] [Google Scholar]
- 20. Rotstein N. P., Miranda G. E., Abrahan C. E., German O. L. (2010) Regulating survival and development in the retina: key roles for simple sphingolipids. J. Lipid Res. 51, 1247–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tani M., Ito M., Igarashi Y. (2007) Ceramide/sphingosine/sphingosine 1-phosphate metabolism on the cell surface and in the extracellular space. Cell. Signal. 19, 229–237 [DOI] [PubMed] [Google Scholar]
- 22. Novgorodov S. A., Gudz T. I. (2011) Ceramide and mitochondria in ischemic brain injury. Int. J. Biochem. Mol. Biol. 2, 347–361 [PMC free article] [PubMed] [Google Scholar]
- 23. Novgorodov S. A., Gudz T. I. (2009) Ceramide and mitochondria in ischemia/reperfusion. J. Cardiovasc. Pharmacol. 53, 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El Bawab S., Roddy P., Qian T., Bielawska A., Lemasters J. J., Hannun Y. A. (2000) Molecular cloning and characterization of a human mitochondrial ceramidase. J. Biol. Chem. 275, 21508–21513 [DOI] [PubMed] [Google Scholar]
- 25. Novgorodov S. A., Wu B. X., Gudz T. I., Bielawski J., Ovchinnikova T. V., Hannun Y. A., Obeid L. M. (2011) Novel pathway of ceramide production in mitochondria: thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J. Biol. Chem. 286, 25352–25362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu B. X., Rajagopalan V., Roddy P. L., Clarke C. J., Hannun Y. A. (2010) Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 285, 17993–18002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bionda C., Portoukalian J., Schmitt D., Rodriguez-Lafrasse C., Ardail D. (2004) Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 382, 527–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu J., Novgorodov S. A., Chudakova D., Zhu H., Bielawska A., Bielawski J., Obeid L. M., Kindy M. S., Gudz T. I. (2007) JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J. Biol. Chem. 282, 25940–25949 [DOI] [PubMed] [Google Scholar]
- 29. Strub G. M., Paillard M., Liang J., Gomez L., Allegood J. C., Hait N. C., Maceyka M., Price M. M., Chen Q., Simpson D. C., Kordula T., Milstien S., Lesnefsky E. J., Spiegel S. (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kono M., Dreier J. L., Ellis J. M., Allende M. L., Kalkofen D. N., Sanders K. M., Bielawski J., Bielawska A., Hannun Y. A., Proia R. L. (2006) Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J. Biol. Chem. 281, 7324–7331 [DOI] [PubMed] [Google Scholar]
- 31. Saatman K. E., Feeko K. J., Pape R. L., Raghupathi R. (2006) Differential behavioral and histopathological responses to graded cortical impact injury in mice. J. Neurotrauma 23, 1241–1253 [DOI] [PubMed] [Google Scholar]
- 32. Washington P. M., Forcelli P. A., Wilkins T., Zapple D. N., Parsadanian M., Burns M. P. (2012) The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J. Neurotrauma 29, 2283–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mori T., Wang X., Jung J. C., Sumii T., Singhal A. B., Fini M. E., Dixon C. E., Alessandrini A., Lo E. H. (2002) Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J. Cereb. Blood Flow Metab. 22, 444–452 [DOI] [PubMed] [Google Scholar]
- 34. Hamm R. J., Pike B. R., O'Dell D. M., Lyeth B. G., Jenkins L. W. (1994) The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma 11, 187–196 [DOI] [PubMed] [Google Scholar]
- 35. Fujimoto S. T., Longhi L., Saatman K. E., Conte V., Stocchetti N., McIntosh T. K. (2004) Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci. Biobehav. Rev. 28, 365–378 [DOI] [PubMed] [Google Scholar]
- 36. Novgorodov S. A., Chudakova D. A., Wheeler B. W., Bielawski J., Kindy M. S., Obeid L. M., Gudz T. I. (2011) Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J. Biol. Chem. 286, 4644–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hartl F. U., Schmidt B., Wachter E., Weiss H., Neupert W. (1986) Transport into mitochondria and intramitochondrial sorting of the Fe/S protein of ubiquinol-cytochrome c reductase. Cell 47, 939–951 [DOI] [PubMed] [Google Scholar]
- 38. Novgorodov A. S., El-Alwani M., Bielawski J., Obeid L. M., Gudz T. I. (2007) Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 21, 1503–1514 [DOI] [PubMed] [Google Scholar]
- 39. Chudakova D. A., Zeidan Y. H., Wheeler B. W., Yu J., Novgorodov S. A., Kindy M. S., Hannun Y. A., Gudz T. I. (2008) Integrin-associated Lyn kinase promotes cell survival by suppressing acid sphingomyelinase activity. J. Biol. Chem. 283, 28806–28816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bielawski J., Szulc Z. M., Hannun Y. A., Bielawska A. (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39, 82–91 [DOI] [PubMed] [Google Scholar]
- 41. Geoffroy K., Wiernsperger N., Lagarde M., El Bawab S. (2004) Bimodal effect of advanced glycation end products on mesangial cell proliferation is mediated by neutral ceramidase regulation and endogenous sphingolipids. J. Biol. Chem. 279, 34343–34352 [DOI] [PubMed] [Google Scholar]
- 42. Tani M., Okino N., Mitsutake S., Ito M. (1999) Specific and sensitive assay for alkaline and neutral ceramidases involving C12-NBD-ceramide. J. Biochem. 125, 746–749 [DOI] [PubMed] [Google Scholar]
- 43. Liu H., Sugiura M., Nava V. E., Edsall L. C., Kono K., Poulton S., Milstien S., Kohama T., Spiegel S. (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275, 19513–19520 [DOI] [PubMed] [Google Scholar]
- 44. Olivera A., Kohama T., Tu Z., Milstien S., Spiegel S. (1998) Purification and characterization of rat kidney sphingosine kinase. J. Biol. Chem. 273, 12576–12583 [DOI] [PubMed] [Google Scholar]
- 45. Clarke C. J., Mediwala K., Jenkins R. W., Sutton C. A., Tholanikunnel B. G., Hannun Y. A. (2011) Neutral sphingomyelinase-2 mediates growth arrest by retinoic acid through modulation of ribosomal S6 kinase. J. Biol. Chem. 286, 21565–21576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chan H., Pitson S. M. (2013) Post-translational regulation of sphingosine kinases. Biochim. Biophys. Acta 1831, 147–156 [DOI] [PubMed] [Google Scholar]
- 47. Zigdon H., Kogot-Levin A., Park J. W., Goldschmidt R., Kelly S., Merrill A. H., Jr., Scherz A., Pewzner-Jung Y., Saada A., Futerman A. H. (2013) Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J. Biol. Chem. 288, 4947–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gudz T. I., Tserng K. Y., Hoppel C. L. (1997) Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 272, 24154–24158 [DOI] [PubMed] [Google Scholar]
- 49. Veenith T., Goon S. S., Burnstein R. M. (2009) Molecular mechanisms of traumatic brain injury: the missing link in management. World J. Emerg. Surg. 4, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nortje J., Menon D. K. (2004) Traumatic brain injury: physiology, mechanisms, and outcome. Curr. Opin. Neurol. 17, 711–718 [DOI] [PubMed] [Google Scholar]
- 51. Brustovetsky N., Dubinsky J. M. (2000) Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J. Neurosci. 20, 8229–8237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. El Bawab S., Bielawska A., Hannun Y. A. (1999) Purification and characterization of a membrane-bound nonlysosomal ceramidase from rat brain. J. Biol. Chem. 274, 27948–27955 [DOI] [PubMed] [Google Scholar]
- 53. Hwang Y. H., Tani M., Nakagawa T., Okino N., Ito M. (2005) Subcellular localization of human neutral ceramidase expressed in HEK293 cells. Biochem. Biophys. Res. Commun. 331, 37–42 [DOI] [PubMed] [Google Scholar]
- 54. Galadari S., Wu B. X., Mao C., Roddy P., El Bawab S., Hannun Y. A. (2006) Identification of a novel amidase motif in neutral ceramidase. Biochem. J. 393, 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. El Bawab S., Birbes H., Roddy P., Szulc Z. M., Bielawska A., Hannun Y. A. (2001) Biochemical characterization of the reverse activity of rat brain ceramidase: a CoA-independent and fumonisin B1-insensitive ceramide synthase. J. Biol. Chem. 276, 16758–16766 [DOI] [PubMed] [Google Scholar]
- 56. Zhu Q., Jin J. F., Shan X. H., Liu C. P., Mao X. D., Xu K. F., Liu C. (2008) Chronic activation of neutral ceramidase protects β-cells against cytokine-induced apoptosis. Acta Pharmacol. Sin. 29, 593–599 [DOI] [PubMed] [Google Scholar]
- 57. Franzen R., Pautz A., Bräutigam L., Geisslinger G., Pfeilschifter J., Huwiler A. (2001) Interleukin-1β induces chronic activation and de novo synthesis of neutral ceramidase in renal mesangial cells. J. Biol. Chem. 276, 35382–35389 [DOI] [PubMed] [Google Scholar]
- 58. O'Neill S. M., Houck K. L., Yun J. K., Fox T. E., Kester M. (2011) AP-1 binding transcriptionally regulates human neutral ceramidase. Arch. Biochem. Biophys. 511, 31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 272, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 60. Ferguson-Miller S., Babcock G. T. (1996) Heme/copper terminal oxidases. Chem. Rev. 96, 2889–2908 [DOI] [PubMed] [Google Scholar]
- 61. Wong-Riley M. T. (2012) Bigenomic regulation of cytochrome c oxidase in neurons and the tight coupling between neuronal activity and energy metabolism. Adv. Exp. Med. Biol. 748, 283–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Arnold S. (2012) Cytochrome c oxidase and its role in neurodegeneration and neuroprotection. Adv. Exp. Med. Biol. 748, 305–339 [DOI] [PubMed] [Google Scholar]
- 63. Igarashi N., Okada T., Hayashi S., Fujita T., Jahangeer S., Nakamura S. (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 278, 46832–46839 [DOI] [PubMed] [Google Scholar]
- 64. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]