

Background: The role of PRD in scrambling of phospholipids by hPLSCR1 is not known.
Results: The addition of PRD of hPLSCR1 to hPLSCR2 restored its PL scrambling activity, which in turn leads to aggregation of the protein.
Conclusion: PRD is crucial for PL scrambling activity and could probably mediate its function by metal ion-dependent oligomerization.
Significance: The results provide an insight in understanding the mechanism of scramblases.
Keywords: Calcium-binding Proteins, Fluorescence, Membrane Biophysics, Membrane Proteins, Membrane Reconstitution, Protein Aggregation, Human Phospholipid Scramblase, Proline-rich Domain
Abstract
Human phospholipid scramblase 1 (hPLSCR1), a type II integral class membrane protein, is known to mediate bidirectional scrambling of phospholipids in a Ca2+-dependent manner. hPLSCR2, a homolog of hPLSCR1 that lacks N-terminal proline-rich domain (PRD), did not show scramblase activity. We attribute this absence of scramblase activity of hPLSCR2 to the lack of N-terminal PRD. Hence to investigate the above hypothesis, we added the PRD of hPLSCR1 to hPLSCR2 (PRD-hPLSCR2) and checked whether scramblase activity was restored. Functional assays showed that the addition of PRD to hPLSCR2 restored scrambling activity, and deletion of PRD in hPLSCR1 (ΔPRD-hPLSCR1) resulted in a lack of activity. These results suggest that PRD is crucial for the function of the protein. The effects of the PRD deletion in hPLSCR1 and the addition of PRD to hPLSCR2 were characterized using various spectroscopic techniques. Our results clearly showed that hPLSCR1 and PRD-hPLSCR2 showed Ca2+-dependent aggregation and scrambling activity, whereas hPLSCR2 and ΔPRD-hPLSCR1 did not show aggregation and activity. Thus we conclude that scramblases exhibit Ca2+-dependent scrambling activity by aggregation of protein. Our results provide a possible mechanism for phospholipid scrambling mediated by PLSCRs and the importance of PRD in its function and cellular localization.
Introduction
The biological semipermeable membrane that separates the interior of the cell from external environment is the plasma membrane (PM).4 The unique feature of PM is the maintenance of membrane asymmetry by phosphatidylcholine and sphingomyelin, which are localized in the outer leaflet, whereas aminophospholipids such as phosphatidylserine (PS) and phosphatidylethanolamine are predominant in the inner leaflet of PM. This asymmetry is maintained by the concerted action of energy (ATP)-dependent phospholipid (PL) translocators, which include flippases and floppases (1–5). PM of eukaryotic cells is also equipped with a special class of PL translocators called human phospholipid scramblase 1(hPLSCR1). It is a type II membrane protein first identified in erythrocyte membranes that is activated upon elevated cytosolic Ca2+ levels resulting in destruction of PM asymmetry (6). Later other members of scramblases, hPLSCR2–4, were identified that were highly conserved from Caenorhabditis elegans to humans (7). Although initially identified as scramblase, hPLSCR1 was found to be involved in many signal transduction pathways like IFN-mediated antiviral activity and PKC-δ mediated pathways and is also a substrate for cellular kinases (8, 9). hPLSCR3 localizes to mitochondria and is involved in intrinsic apoptotic pathway and cardiolipin translocation in mitochondria (10). Recent evidence suggests that hPLSCR4 also mediates bidirectional translocation of PLs across PL bilayer (11). hPLSCR2 is known to be localized to the nucleus; however, the structural and functional characterization of hPLSCR2 has not been performed yet (12).
Homology studies of PLSCRs reveal that hPLSCR2, -3, and -4 share 59, 47, and 46% similarity with hPLSCR1 (5). PLSCRs are multidomain-containing proteins where each domain has distinct functions that need to be elucidated. Major domains of PLSCRs include proline-rich domain (PRD), DNA binding motif, palmitoylation motif, nuclear localization signal, putative EF-hand like calcium binding motif, and C-terminal helix (CTH) (5). Except for hPLSCR2, members of scramblase family contain an N-terminal PRD that possesses PXXP and PPXY motifs via which they interact with multiple proteins (13–17). hPLSCR1 was also reported to bind to the promoter of type I IP3 receptor gene and enhance its expression (18). A cysteine-rich palmitoylation site in the protein directs the subcellular localization to PM, and a nonclassical nuclear localization signal assists in the import of protein into the nucleus via importin α/β-mediated pathway (19–21). A highly conserved EF-hand-like Ca2+ binding motif was essential for its scrambling activity, and point mutations within this domain affected its function. It was also reported that CTH of hPLSCR1 is indeed a true transmembrane domain and is required for membrane insertion and Ca2+ binding (22, 23).
To date, the exact mechanism of scramblase activity is not known. Sims and co-workers (22) proposed that the PL translocation could be due to protein aggregation or oligomerization, which was not sufficiently validated. Recent reports have disputed that hPLSCR1 might not be a true scramblase as the members of the TMEM16 family also show Ca2+-dependent scramblase activity and have been implicated in Scott syndrome mechanism (24). Sequence homology of hPLSCR2 with hPLSCR1 and hPLSCR1 lacking PRD (ΔPRD-hPLSCR1) shows 59% and 84% homology, respectively. Even though hPLSCR2 shows such a high similarity with hPLSCR1 and retains all the major domains of hPLSCR1 except for the PRD domain, it fails to externalize PS in CHO-K1 cell lines under apoptotic conditions (12). Despite several years of research on scramblases, many questions are still unanswered. Does hPLSCR2 show Ca2+-dependent activity in vitro? What is the role of PRD in scramblase function? Is protein aggregation or oligomerization required for scramblase activity? To address the above questions, we cloned, overexpressed, and purified hPLSCR1, hPLSCR2, ΔPRD-hPLSCR1, and chimeric PRD-hPLSCR2 proteins to homogeneity, and various biochemical and biophysical studies were performed. Our results clearly showed that PRD is required for the Ca2+-dependent aggregation of protein that is required for functional activation of the protein.
EXPERIMENTAL PROCEDURES
Materials
Escherichia coli DH5α and BL21 (DE3) strains were obtained from ATCC. cDNA of hPLSCR1 and -2 was purchased from Invitrogen, and pET-28b(+) was from Novagen. Isopropyl β-d-1-thiogalactopyranoside, dithiothreitol (DTT), and EDTA were purchased from Himedia. SM-2 Biobeads and Chelex-100 resin were obtained from Bio-Rad. Nickel nitrilotriacetic acid was purchased from Qiagen. N-Lauroylsarcosine, BCA protein estimation kit, molecular biology grade CaCl2, trypsin, and egg phosphatidylcholine, phosphatidylserine, and phenylmethylsulfonyl fluoride were purchased from Sigma. 7-Nitrobenz-2-oxa-1,3-diazol-4-yl phosphatidylcholine and 7-nitrobenz-2-oxa-1,3-diazol-4-yl phosphatidylserine (NBD-PS) fluorescent-labeled lipids were purchased from Avanti Polar Lipids.
Plasmid Construction
The open reading frames of hPLSCR1 (957 bp) and hPLSCR2 (675 bp) were amplified from mammalian expression vector pCMV-SPORT6 by polymerase chain reaction using the primers PLSCR1F (5′-AGCTATCATATGATGCCAGCACCACC-3′) and PLSCR1R (5′-CGTAGTGAATTCTTACCTAGTTCTTTCAAAAAACATG-3′) with NdeI and EcoR1 sites and PLSCR2F (5′-AAACATATGCCAGCACCACCACC-3′) and PLSCR2R (5′-ATATGCGGCCGCTTACCTAGTTCTTTCAAAAAACATGTAGTCAATGAG-3′) with NdeI and Not1 sites, respectively. The amplicon was then digested and ligated into respective digested sites of pET-28b (+) vector. For generation of PRD-hPLSCR2 plasmid, we analyzed the sequence homology of hPLSCR1 and hPLSCR2 and found a unique SacI site that was used as a restriction site to fuse the PRD domain of hPLSCR1 to hPLSCR2 in-frame using the primers PLSCR2F (5′-ACTATGAGCTCCTGGAAGTTCTATTCAGTTTTGAAAGTAG-3′) and PLSCR1R (5′-ACTACTGAGCTCAATTTGCTGATGAATCAGTATCTG-3′). ΔPRD-hPLSCR1-pET-28a(+) plasmid was constructed using the forward primer (5′-TATTTAAGTCAGATAGATC-3′), and same reverse primers were used as PLSCR1R primer.
Overexpression and Purification of hPLSCR1 and -2 and Mutant Constructs
Recombinant overexpression and purification of recombinant proteins was done as described earlier (25). E. coli BL-21 (DE3) cells were transformed with the respective plasmids and grown in a selective media containing kanamycin (50 μg/ml). Post-induction, cells were pelleted and lysed in buffer A (20 mm Tris (pH 7.4), 200 mm NaCl) with 1 mm PMSF, 1 mm EDTA, and 1 mm DTT using a probe sonicator (Vibro cell ultrasonicator). Cell lysate was then clarified at 12,000 × g, and the inclusion bodies were thoroughly washed with lysis buffer. Inclusion bodies were solubilized by 0.3% N-lauroylsarcosine followed by centrifugation at 4400 × g, and the supernatant was collected. Supernatant was then pulse-dialyzed against buffer A containing 0.025% w/v Brij-35. Protein in this buffer was used for all assays, and any changes in the composition are mentioned below. His-tagged proteins were then purified to homogeneity by nickel nitrilotriacetic acid chromatography. The proteins were visualized on 12% SDS-PAGE stained with Coomassie Brilliant Blue. Protein estimation was done by the BCA method.
Biochemical Reconstitution into Proteoliposomes
Reconstitution of purified proteins into vesicles was performed, and estimation of the reconstituted proteins was done as described earlier (23, 26). 4.5 μmol of egg phosphatidylcholine and PS (9:1) were dried, and the lipids were solubilized in reconstitution buffer containing 10 mm HEPES (pH 7.5), 100 mm NaCl, 1% w/v Triton X-100 along with 60 μg of the protein. Slow detergent removal was facilitated by the addition of SM2 Bio-Beads (Bio-Rad) for proteoliposomes formation. Outside-labeled proteoliposomes were prepared by incubating unlabeled vesicles with 0.3 mol of % NBD lipids in 0.25% v/v DMSO. Unbound lipids were then removed by centrifugation at 230,000 × g, vesicles were then incubated with appropriate metal ion and EDTA, and scramblase assay was performed.
Scramblase Assay
Outside-labeled vesicles were incubated at 37 °C for 4 h, with 4 mm metal ions like Ca2+, Mg2+, and Zn2+ independently or 4 mm EDTA. Fluorescence was monitored with excitation at 470 nm and emission at 532 nm with a slit width of 3 and 5 nm, respectively, using a stirred spectrofluorimeter (PerkinElmer Life Sciences LS-55) continuously at 23 °C. Initial fluorescence was measured for 200 s, after which freshly prepared dithionite in 1 m Tris base was added, and the fluorescence was monitored for an additional 400 s. The residual difference between the non-quenchable fluorescence in the presence and absence of metal ion gives scramblase activity. Scramblase activity = Fmetal ion − FEDTA, where Fmetal ion is residual fluorescence of Ca2+ or any other metal ion-treated vesicles after dithionite addition, and FEDTA is residual fluorescence of EDTA-treated vesicles after dithionite addition.
Apoptosis Assay by FACS
HEK 293T cells were grown on 60-mm2 polylysine-coated dishes. Once the cells reached 60–70% confluency, cells were transfected with 5 μg of respective plasmids by using Lipofectamine 2000 transfection reagent (Invitrogen) as per the manufacturer's guidelines. Post-transfection cells were maintained in serum-free DMEM media for 4 h for synchronization of the cells in same phase. After 4 h, serum-free medium was replaced with fresh DMEM media with 10% FBS and incubated for 24 h at 37 °C, 5% CO2. Apoptosis was induced by serum starvation for 12 h where the cells were incubated with DMEM medium containing 0.1% serum. Cells were then washed twice with phosphate-buffered saline (PBS) and subjected to trypsinization. Trypsinized cells were collected in microcentrifuge tubes and pelleted at 1000 rpm at 4 °C for 10 min. Cells were then washed twice with PBS and then resuspended in Annexin V binding buffer (10 mm HEPES, pH 7.4, 140 mm NaCl, 2.5 mm CaCl2) at a concentration of ∼1 × 106 cells/ml. Annexin V-FITC (BD Biosciences) staining was done as per the manufacturer's instructions. Dead cells were stained by propidium iodide (PI) (50 μg/ml). Cell sorting was performed in BD FACSVerse flow cytometer (BD Biosciences) with FITC and PI dual staining. Cells transfected with only vector was used as the control. Results were analyzed using BD FACSuite software (BD Biosciences).
Localization Studies Using Confocal Microscopy
Various constructs used in this study were cloned into pEGFP-C1 vector. COS-7 cells were grown on poly d-lysine-coated coverslips and transfected with 2.5 μg of DNA for 24 h. Post-transfection cells were serum-starved by replacing the medium with serum-free media and grown for 9 h. Cells expressing the respective GFP constructs were washed with PBS and fixed with 4% formaldehyde and observed using fluorescence microscopy. The nucleus was stained with DAPI to differentiate the nucleus from cytoplasm. The images were captured using Carl Zeiss LSM 710 confocal microscope (Carl Ziess AG, Germany) with 40× objective.
Fractionation Studies
HEK293T cells were transfected with GFP-tagged constructs of various proteins used in this study to determine the subcellular localization. GFP-tagged constructs were used to differentiate between endogenous and overexpressed protein. 24 h post transfection cells were serum-starved to induce apoptosis. Cells were washed with PBS and fractionation buffer containing 20 mm Tris (pH 7.4), 100 mm NaCl, 2 mm Mg(CH3COO)2, 5 mm KCl, and protease inhibitor mixture was added and incubated on ice for 10 min. Cells were lysed by passing through a 27-gauge needle 25 times and centrifuged at 450 × g for 10 min, and the pellet (nuclear fraction) and supernatant (cytosolic + membrane fraction) were saved. Supernatant was then centrifuged at 21,000 × g for 30 min to separate the membrane and cytosolic fraction. The membrane and nuclear fraction were then solubilized using lysis buffer containing 1% Nonidet P-40 detergent and used for Western blot analysis. Equal amounts of cytosolic, membrane, and nuclear proteins (50 μg) was taken for Western blot analysis.
Western Blot Analysis
Transfected cells were lysed in lysis buffer (5 mm Tris (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm EDTA, 1 mm PMSF, and protease inhibitors). Total protein was estimated by the BCA method using BSA as the standard. 50 μg of total protein was loaded on 12% SDS-PAGE and transferred onto nitrocellulose membrane. Membrane was blocked using blocking buffer with BSA (10 mm Tris (pH 7.4), 150 mm NaCl, and 0.1% Tween 20 for 1 h at 25 °C. Immunoblotting was done using hPLSCR1 and hPLSCR2 mouse monoclonal antibodies (Santa Cruz), and detection was performed using an ECL kit (Thermo Scientific kit). To check the protein expression levels of all four constructs, HEK 293T cells were transiently transfected with GFP-tagged gene constructs. After 18 h of transfection, cells were lysed in lysis buffer, and Western blots were developed as described above with rabbit monoclonal antibodies specific to GFP (Promega) and β-actin (Sigma, mouse) with 1:5000 dilutions. The bands were visualized by Clarity Western ECL substrate (Bio-Rad).
Ca2+ Binding Studies
Stains-All, a cationic carbocyanine dye, was used to monitor the calcium binding properties of hPLSCR1 and -2 and mutant constructs. Stains-All was dissolved in 2 mm MOPS buffer (pH 7.2) containing 30% ethylene glycol. Stains-All produces a series of discrete spectra depending upon interaction and conformation of binding region (27). The free form of the dye produces two distinctive spectra at 535 nm (β-band) and 575 nm (α-band) that correspond to the monomeric and dimeric form of the dye. In the presence of EF-hand containing proteins, Stains-All dye produces a discrete band at 650 nm termed J band. For competition experiments, to the protein-Stains-All complex 2 and 4 mm Ca2+ was added, and the spectrum was monitored. Additionally Tb3+, an isomorphous luminescent replacement probe for Ca2+, was used for Ca2+ binding studies. It has a broad excitation spectrum that overlaps with the emission spectrum of tyrosine and tryptophan. This allows the energy transfer between protein and terbium, giving distinctive luminescent peaks at 490 and 547 nm (28). Protein was titrated with small aliquots of terbium chloride (TbCl3) ranging between 50 and 700 μm, and samples were excited at 295 nm, and emission spectrum was recorded between 310 and 560 nm with a scan speed of 100 nm/min and bandwidths of 5 nm each.
Intrinsic Tryptophan Fluorescence Assay
Additionally the binding affinity of all the proteins used in this study with Ca2+ and Mg2+ was determined. 2 μm concentrations of the respective proteins were titrated with small aliquots of CaCl2 and MgCl2 (5 mm increments until saturation) independently. Binding constants were determined by a Scatchard plot using nonlinear curve-fitting (Prism 5.0 GraphPad Software). Curve-fitting was done for the fraction of ligand sites occupied [(Fo − F)/(Fo − FS)] versus ligand concentration using the one-site binding nonlinear regression analysis, where Fo is fluorescence in the absence of ligand, FS is fluorescence at saturation ligand concentration, F is fluorescence in the presence of a particular ligand concentration, Bmax is the maximum specific binding, and Kd is the dissociation constant (mm).
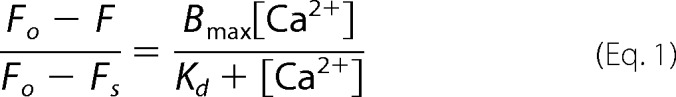 |
Protein Conformation Changes
Far UV-CD spectra were recorded using JASCO J-810 spectropolarimeter (Easton MD) at 25 °C with a thermostat cell holder. Samples were scanned using 1-mm path length cuvettes from 250 to 190 nm. Protein surface hydrophobicity changes were monitored using 8-anilinonaphthalene-1-sulfonic acid (ANS) (29). 10 μm protein in 20 mm Tris (pH 7.4) and 200 mm NaCl was used to perform the assay without the addition of Brij-35 to the buffer due to its interference with ANS emission spectra. ANS dye is weakly fluorescent in water (emission at 530 nm) and the fluorescence emission increases only when ANS is bound to protein, which in turn exposes the hydrophobic patches to the surface. The ANS binding of protein was monitored by recording the emission between 400 and 650 nm after excitation at 365 nm with a scan speed of 100 nm/min and bandwidth of 10 nm each. ANS binding to the protein alters the emission maxima from 530 to 460 nm.
Protease Protection Assay
Trypsin digestion was used to monitor conformation changes in WT hPLSCR2 and PRD hPLSCR2. The protein-lipid (1:20) ratio was incubated with and without Ca2+ at 37 °C for 30 min. Subsequently trypsin (1:75 w/w) was added to the protein lipid mixture and incubated at 37 °C for 30 min. The digested samples were analyzed on a 12% SDS-PAGE.
Light Scattering and Protein Size Studies
UV absorbance at 360 nm is a routine method to monitor the formation of protein aggregates in solution. A protein with a concentration of 0.5 mg/ml was used to monitor Ca2+-dependent aggregation. Time course measurement of absorbance at 360 nm was performed for 30 min at 25 °C using V-670 Jasco UV-visible spectrophotometer. Apoprotein absorbance at 360 nm in the buffer composed of 20 mm Tris (pH 7.4), 200 mm NaCl, and 0.025% Brij-35 was recorded in a 1-cm path length quartz cuvette with a bandwidth of 1 nm. The absorbance of protein incubated with 4 mm Ca2+ at 37 °C was measured. To determine the size of the aggregates, dynamic light scattering was performed by using Zetatrac particle size analyzer. A particle size analyzer measures the size distribution by velocity of the particles dispersed in the buffer. For particle size distribution of each sample, proteins (0.5 mg/ml) in the presence and absence of 4 mm Ca2+ was monitored with a run time of 60 s and 15 repetitions. The results were analyzed by Microtrac-FLEX software 10.5.3.
RESULTS
Scramblase Assay
hPLSCR1, hPLSCR2, PRD-hPLSCR2, and Δ100-hPLSCR1 constructs as in Fig. 1A were overexpressed and purified from inclusion bodies to homogeneity as described earlier (25). These purified proteins were reconstituted into synthetic vesicles as described earlier to perform a scramblase assay (23). The principle of the assay was to monitor the percentage of NBD-PLs sequestered inside in the presence and absence of Ca2+, Mg2+, and Zn2+ (Fig. 1B). This was achieved by quenching the outside-labeled vesicles using a membrane-impermeable reagent sodium dithionite. Our biochemical assay clearly showed that in vitro reconstituted hPLSCR2 does not show scramblase activity in the presence of Ca2+, whereas hPLSCR1 translocated 14% of PLs (Fig. 1, C and E) in the presence of Ca2+. Deletion of the PRD domain of hPLSCR1 resulted in a loss of scramblase activity similar to hPLSCR2 (Fig. 1D). Interestingly, the chimeric protein (PRD-hPLSCR2) regained its scrambling activity similarly to hPLSCR1 (Fig. 1F). This suggested that the PRD domain of scramblase is crucial for its activity. Fig. 1G shows the percentage PLs translocated by four constructs used in this study in the presence of Ca2+. Similarly, a scramblase assay in presence of Mg2+ and Zn2+ was also performed (Fig. 1, H and I). Scramblase activity was significantly low for all the four constructs in the presence of Mg2+ and Zn2+ when compared with activity obtained in the presence of Ca2+.
FIGURE 1.

Transbilayer movement of PLs assayed by scramblase assay. A, schematic representation of various plasmid constructs used in this study. These proteins contain highly conserved domains: PRD (absent in hPLSCR2), DNA binding domain (DBD), cysteine palmitoylation motif (CPM), nuclear localization signal (NLS), EF-hand like motif (EFM), CTH. B, scramblase assay scheme. Outside-labeled proteoliposomes were incubated with Ca2+ and EDTA independently. The membrane-impermeable dithionite was used to quench the outside-labeled lipids, and residual fluorescence as a result of fluorescent lipids that are flipped to the inner leaflet of proteoliposomes. Scramblase activity was determined by the difference between fluorescence intensity of metal ion-treated and EDTA-treated vesicles. a.u., arbitrary units. C, scramblase assay of hPLSCR1 reconstituted vesicles. D and E, scramblase assay of ΔPRD-hPLSCR1 and hPLSCR2. F, scramblase assay with PRD-hPLSCR2. Solid line, EGTA-treated vesicles; dashed line, Ca2+-treated vesicles. G–I, relative comparisons of scramblase activity by all the proteins in this study in the presence of different metal ions, Ca2+, Mg2+, and Zn2+, respectively. Results are representative of at least three sets of experiments. ***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05; ns, p > 0.05.
Apoptosis Study Using FACS
The effect of the addition of PRD to hPLSCR2 and deletion of PRD from hPLSCR1 on PS exposure during apoptosis was quantified by fluorescence-assisted cell sorting. Annexin V FITC and propidium iodide were used for staining. Annexin V-FITC was used to identify cells that were in early apoptotic phase, and propidium iodide was used as a marker for dead cells as it binds to DNA. When compared with vector control (Fig. 2A), cells transfected with hPLSCR1 had 10.5% more apoptotic cells (Fig. 2B). Cells expressing ΔPRD-hPLSCR1 showed a decreased number of apoptotic cells (7%), and hPLSCR2 did not show any significant effect on apoptosis (2.1%) (Fig. 2, C and D). PRD-hPLSCR2-transfected cells showed an 8.7% increase in the count of apoptotic cells (Fig. 2E). These results strongly suggest that the PRD of PLSCRs plays a crucial role in PS exposure, which is one of the major functions of scramblases.
FIGURE 2.

Apoptosis assay using FACS. HEK 293T cells expressing hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2 were stained with Annexin V FITC and PI as mentioned under “Experimental Procedures.” The dot plots were used to determine the count of the cells in quadrants that were positive for PI (Q1), positive for Annexin V-FITC (Q4), unstained cells (Q3), and cells that were stained by both FITC and PI (Q2). Q4 contains cells that were in early apoptotic phase, Q2 has cells that were in late apoptotic phase, and Q1 represent dead cells. A total of 10,000 cells were counted, and the percentage of cells that fall on each quadrant is mentioned by a value in that quadrant. PE-A (Phycoerythrin A) denotes intensity of PI. The figures show dot plots of cells transfected with vector pCDNA 3.1 (A), cells expressing hPLSCR1 (B), ΔPRD-hPLSCR1 (C), hPLSCR2 (D), and PRD-hPLSCR2 (E) respectively. F, Western blot analysis showing expression levels of all the four constructs in HEK 293T cells. The upper panel in the picture shows all four GFP constructs probed with anti-GFP monoclonal antibody. The lower panel shows the loading control probed with the β-actin monoclonal antibody. All the experiments were performed in duplicate.
To rule out the possibility that variations in the percentage of apoptosis in four constructs are not due to differences in expression levels, Western blot analysis of the GFP-tagged overexpressed proteins in HEK 293T cells were performed. The results clearly showed that expression levels of all four constructs are similar when compared with the expression of endogenous actin (control) (Fig. 2F). These results strongly suggest that the variations in the percentage of apoptosis in the four constructs are directly due to the PRD and not because of variations in the protein expression.
Localization Studies Using Confocal Microscopy
We further examined whether PRD plays any role in the subcellular distribution of PLSCRs under apoptotic conditions. Our results showed that hPLSCR1 primarily localizes to the plasma membrane and nucleus with lesser distribution to cytosol. But ΔPRD-hPLSCR1, a deletion construct of hPLSCR1, was predominantly localized in cytosol and with lesser distribution in the PM and nucleus (Fig. 3, A and B). hPLSCR2-GFP was predominantly localized to the nucleus, and punctate-like aggregation was observed (Fig. 3C). PRD-hPLSCR2 was distributed in the entire cell similarly to hPLSCR1 (Fig. 3D) but with differences in the distribution levels. These results imply that PRD domain is crucial for determining the subcellular localization of proteins.
FIGURE 3.
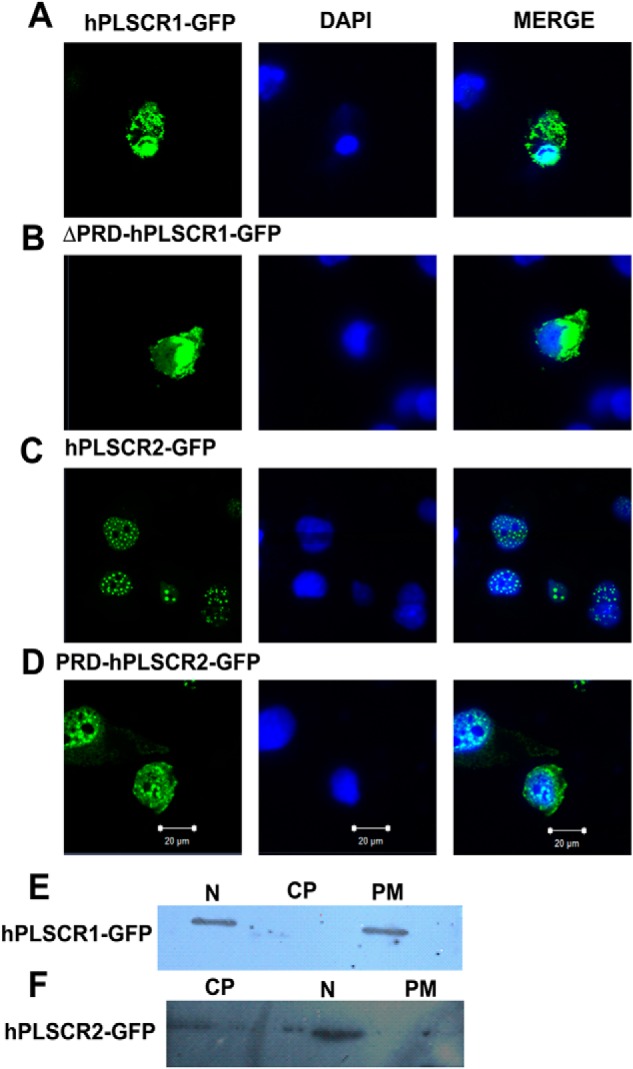
Localization studies using confocal microscopy and subcellular fractionation. A, subcellular localization of hPLSCR1-GFP. B, subcellular localization of ΔPRD-hPLSCR1-GFP. C, subcellular localization of hPLSCR2-GFP. D, subcellular localization of PRD-hPLSCR2-GFP. COS-7-transfected cells (green) were serum-starved to facilitate induction of apoptosis, and the subcellular localization was determined. Nuclear (N), cytoplasmic (CP), and PM distribution was determined using DAPI (blue) nuclear-specific dye and GFP (green). HEK cells were fractionated by differential centrifugation and probed by Western blot. E, subcellular fractionation of hPLSCR1-GFP expressing HEK293T cells; anti-PLSCR1 antibody was used to probe hPLSCR1-GFP localization. F, subcellular fractionation of hPLSCR2-GFP-expressing HEK cells; anti-PLSCR2 antibody was used to probe hPLSCR2-GFP localization. All the experiments were performed at least three times.
Subcellular Fractionation of hPLSCR1 and -2
To further confirm the subcellular localization of PLSCRs used in this study, we performed cell fractionation studies. Cytosolic, membrane, and nuclear fractions were extracted, detergent-solubilized and probed using respective antibodies, and analyzed by Western blotting. Because PLSCR1 shows constitutive gene expression, we used GFP-tagged proteins to determine the subcellular localization of proteins. hPLSCR1-GFP was predominantly localized to the nuclear and membrane fractions, and there was no detection of the protein in the cytosolic fraction, which was similar to the reported data on hPLSCR1 localization (Fig. 3, A and E). Similarly, Western blot results of hPLSCR2 localization in nucleus were in accordance with the localization studies using confocal microscopy (Fig. 3, C and F). However, subcellular fractionation studies were not performed for ΔPRD-hPLSCR1 and PRD-hPLSCR2 because of the non-availability of antibodies commercially.
Calcium Binding to hPLSCR1 and -2 and Other Mutant Proteins
Sequence analysis of hPLSCR1 and -2 revealed the presence of a single putative EF-hand-like Ca2+ binding motif, which was confirmed using Stains-All dye (27). Binding of this dye to EF-hand-containing proteins produces a distinct peak at 650 nm termed as the J-band. The spectrum for the free form of dye is shown Fig. 4, A–D, traces a. Upon binding of hPLSCR1, ΔPRD hPLSCR1, hPLSCR2, and PRD hPLSCR2 (7 μm) to the Stains-All dye, a distinct J-band (650 nm) was observed (Fig. 4, A–D, traces b). Competition experiments showed that the addition of Ca2+ to the protein-Stains-All complex displaces the Stains-All as evident from a decrease in the J-band intensity, indicating that both compete for the same binding site (Fig. 4, A–D, traces c and d). These results indicate that Ca2+ binds to the putative EF-hand-like calcium binding motif in all four constructs and the presence or absence of PRD does not affect its binding.
FIGURE 4.
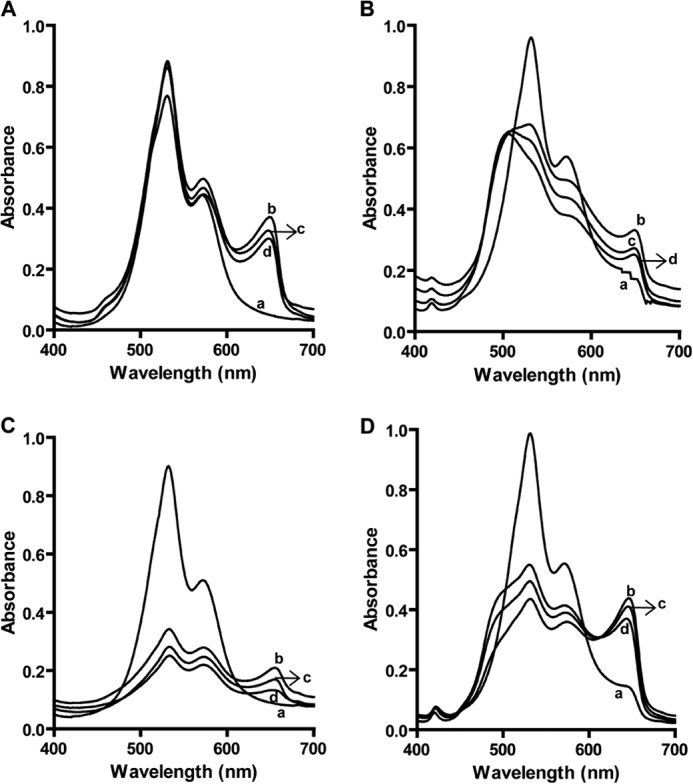
Stains-All assay to probe Ca2+ binding to hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2. Stains-All binding to hPLSCR1 (A), ΔPRD-hPLSCR1 (B), hPLSCR2 (C), and PRD-hPLSCR2 (D) is shown. Traces a, Stains-All dye in buffer; traces b, protein-Stains-All complex; traces c, protein-Stains-All complex in the presence of 2 mm Ca2+. Traces d represents the protein-Stains-All complex in the presence of 4 mm Ca2+. The presence of J-band upon Stains-All binding to the protein confirms that all the wild type and mutant proteins in the study bind to Ca2+. Decrease in the intensity of J-band by adding Ca2+ was due to the replacement of Stains-All dye by Ca2+. Results are representative of at least three set of experiments (p < 0.05).
TbCl3 FRET Assay
Binding of Ca2+ to hPLSCR1 and -2 and mutant proteins was further monitored by Tb3+ fluorescence (28). Tb3+ is weakly fluorescent in water, and the observed fluorescence at 490 and 547 nm reflects only terbium bound to protein (Fig. 5, A–D, trace a). In all the experiments a protein concentration of 4 μm was used in this assay. Upon the addition of 200 μm Tb3+ to the respective proteins, distinctive luminescent peaks at 492 and 547 nm were observed indicating the FRET between Tb3+ and the respective protein (Fig. 5, A–D, trace b). Furthermore with increasing concentrations of Tb3+, greater FRET was observed (Fig. 5, A–D, traces c and d), confirming that the presence or absence of PRD does not affect the calcium binding properties of scramblase proteins.
FIGURE 5.

Probing of Ca2+ binding to hPLSCR1 and -2, PRD-hPLSCR2, and ΔPRD-hPLSCR1 by Tb3+ FRET assay. Tb3+ FRET assay for hPLSCR1 (A), ΔPRD-hPLSCR1 (B), hPLSCR2 (C), and PRD-hPLSCR2 (D). Traces a represent the apo form of the protein; traces b–d represent the respective protein in presence of 200, 400, and 600 μm of terbium. Two peaks at 490 and 547 nm in the figure depict the binding of terbium to the protein and energy transfer. Results are representative of at least three set of experiments (p < 0.05).
Binding Affinities of hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2 with Ca2+ and Mg2+
To further confirm if the presence or absence of PRD affects the affinity of Ca2+ and Mg2+ to the four constructs, binding affinity values were determined using intrinsic tryptophan fluorescence. The apo form of proteins exhibited an emission peak at 345 nm upon titrating with Ca2+, and a dose-dependent decrease in fluorescence intensity was observed without any shift in the emission maxima of the proteins. The observed decrease in intrinsic tryptophan fluorescence due to Ca2+ binding was similar to those demonstrated in earlier reports on Ca2+ binding to synthetic EF-hand peptides of hPLSCR1–4 (5). Binding constants were determined by nonlinear curve-fitting as described under “Experimental Procedures.” These results showed that hPLSCR1 has high affinity for Ca2+ compared with hPLSCR2, and the presence or absence of PRD does not affect the binding values (Table 1). Mg2+ binding affinity values were also determined for all four constructs. The affinity values for Mg2+ were found to be lower than the values obtained for Ca2+. Similar to Ca2+ studies, the presence and absence of PRD did not affect the Mg2+ binding values (Table 1).
TABLE 1.
Ca2+ and Mg2+ binding affinities of hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2
| Protein | Kd for Ca2+ | Kd for Mg2+ |
|---|---|---|
| mm | mm | |
| hPLSCR1 | 25.4 ± 2.6 | 151.8 ± 31.0 |
| ΔPRD-hPLSCR1 | 28.0 ± 4.5 | 116.4 ± 18.1 |
| hPLSCR2 | 45.7 ± 5.3 | 134.0 ± 35.7 |
| PRD-hPLSCR2 | 40.8 ± 2.6 | 123.8 ± 22.5 |
CD Spectroscopy
Ca2+- and Mg2+-induced secondary structural changes were monitored by far UV-CD spectroscopy. It was found that all the proteins exist predominantly in the α-helical state as characterized by the presence of double negative minima at 208 and 222 nm in the spectra of the apo forms of hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2. (Fig. 6, A–H, trace a). Minor changes in the secondary structure by the addition of both 2 mm and 4 mm Ca2+ and Mg2+ were observed with hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR1 (Fig. 6, A–H, traces b and c).
FIGURE 6.

Far UV-CD spectroscopy. A and B represent the secondary structure of hPLSCR1 in the presence of Ca2+ and Mg2+, respectively. C and D, ΔPRD-hPLSCR1 secondary structure in presence of Ca2+ and Mg2+, respectively. E and F, secondary structure of hPLSCR2 with Ca2+ and Mg2+, respectively. G and H, PRD-hPLSCR2 secondary structure in the presence of Ca2+ and Mg2+, respectively. Traces a, respective protein; traces b and c, respective protein in presence of 2 and 4 mm Ca2+ or Mg2+. The presence and absence of PRD have no major changes on the secondary structure content of PLSCRs. Results are representative of at least three set of experiments (p < 0.05).
ANS Fluorescence
ANS is a fluorescent dye used to probe the surface hydrophobicity changes in protein (29). The emission maxima of the Ca2+-free proteins used in this study were centered at 460 nm (Fig. 7, A–H, trace a). In the presence of Ca2+, the increase in ANS fluorescence was observed for hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2 indicating protein conformation changes associated with surface exposure of hydrophobic patches. ANS fluorescence increased with the increase in the Ca2+ concentration (Fig. 7, A, C, E, and G, traces b, c, and d). In the case of hPLSCR1 and PRD-hPLSCR2, an 8-nm red shift in ANS fluorescence was observed in the presence of Ca2+, indicating aggregation or self-assembly of the protein (Fig. 7A, traces b, c, and d, and 7G, traces c and d). In our earlier report a similar aggregation pattern was observed for hPLSCR4 (23). In addition, experiments were also performed for these proteins in the presence of Mg2+. Similar to Ca2+, ANS fluorescence increased for all the four constructs with an increase in Mg2+ concentration. Interestingly, we found that the addition of Mg2+ did not result in any shift in wavelength as observed in the case of Ca2+ (Fig. 7). This probably suggested that Ca2+ binding might lead to aggregation of protein, which was not observed with Mg2+.
FIGURE 7.
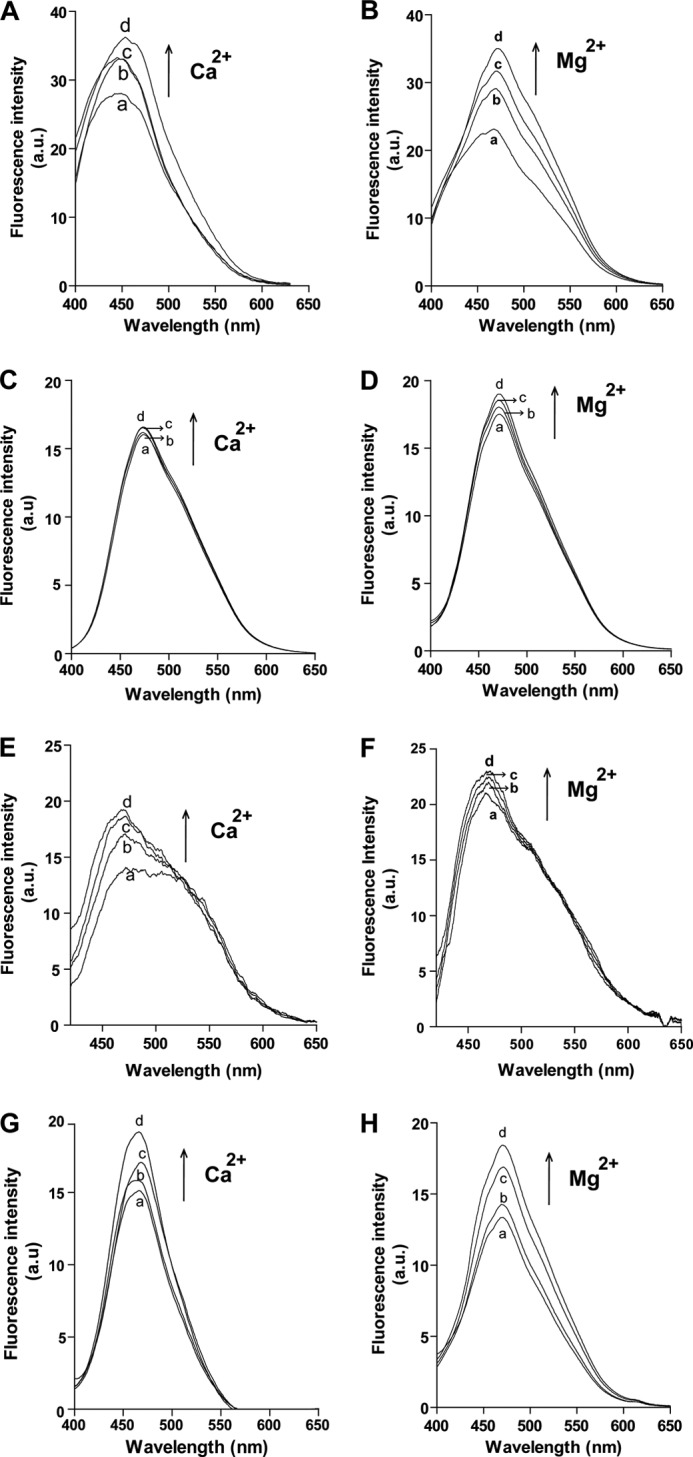
Monitoring surface hydrophobicity changes by ANS. A and B, ANS binding to hPLSCR1 in the presence of Ca2+ and Mg2+, respectively. a.u., arbitrary units. C and D, ANS binding to ΔPRD-hPLSCR1 in the presence of Ca2+ and Mg2+, respectively. E and F, ANS binding to hPLSCR2 in the presence of Ca2+ and Mg2+, respectively. G and H, ANS binding to PRD-hPLSCR2 in the presence of Ca2+ and Mg2+, respectively. Traces a, protein-ANS complex; traces b–d, respective ANS-protein complex in the presence of 2, 4, and 6 mm Ca2+or Mg2+, respectively. A red shift was observed in ANS-hPLSCR1 and ANS-PRD-hPLSCR2 fluorescence upon Ca2+ binding. Results are representative of at least three set of experiments (p < 0.05).
Protease Protection Assay
To further monitor the protein conformational changes, we used protease (trypsin) protection to compare the digestion patterns in the presence and absence of PRD. The results showed that PRD-containing proteins (PRD-hPLSCR2 and hPLSCR1, ∼37 kDa) underwent digestion in the presence and absence of Ca2+, resulting in the formation of ∼25 kDa. However, in case of hPLSCR2 and ΔPRD-hPLSCR1 (lacking PRD domain, ∼25 kDa), trypsin digestion was not observed (Fig. 8, A and B). The differences in the size of fragments obtained for PRD-containing proteins versus PRD lacking proteins suggest that the presence of PRD domain influences the structural changes in protein.
FIGURE 8.
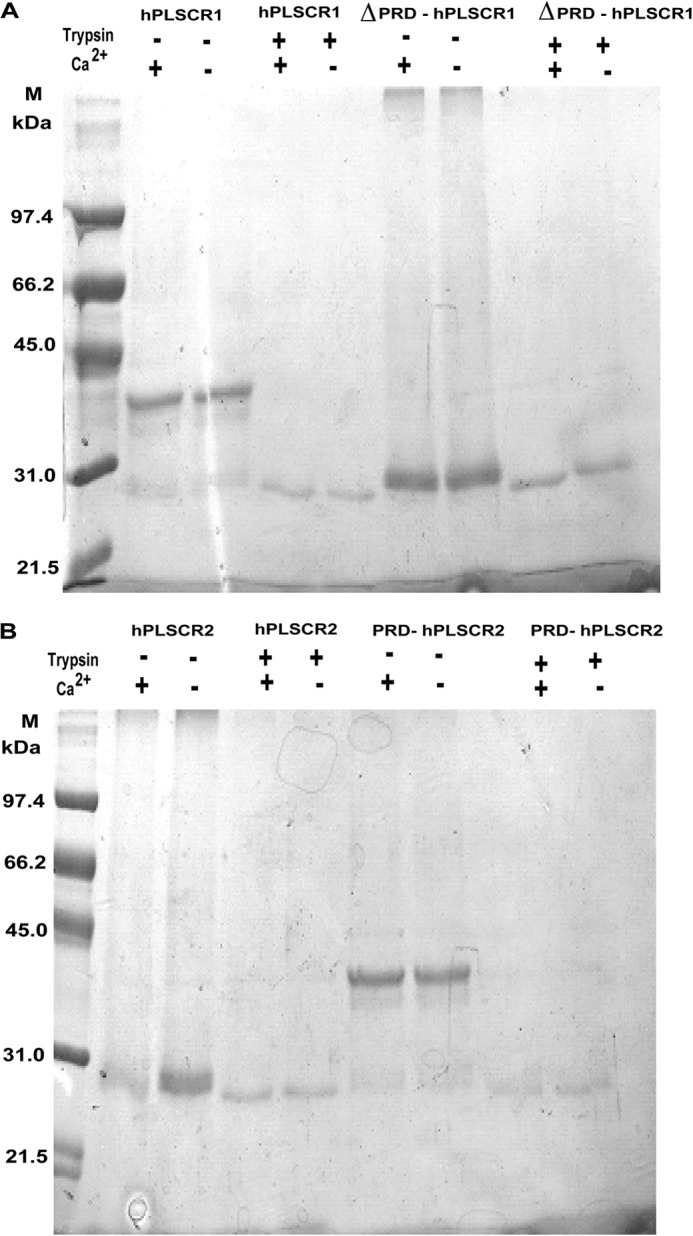
Protease protection assay to monitor metal ion induced conformation changes. Trypsin digestion of protein in the presence of liposomes was performed to check the membrane binding properties and Ca2+-dependent conformational changes in the protein as described under “Experimental Procedures.” A, trypsin-mediated digestion profile of hPLSCR1 and ΔPRD-hPLSCR1 in the presence and absence of Ca2+. B, trypsin-mediated digestion profile of hPLSCR2 and PRD-hPLSCR2 in the presence and absence of Ca2+. Control lanes represent the protein size in the absence of trypsin in both A and B.
Protein Aggregation Studies
Based on ANS studies, we hypothesize that PLSCRs undergoes self-aggregation upon binding to Ca2+ during functional activation. To prove this we determined the oligomeric state of the protein in the presence and absence of Ca2+ by measuring absorbance at 360 nm. In the absence of Ca2+, hPLSCR1 and ΔPRD-hPLSCR1 showed no increase in the absorbance spectra (Fig. 9A, traces a and c). In the presence of Ca2+, the increase in the absorbance spectra was observed only for hPLSCR1 and not found with ΔPRD- hPLSCR1 (Fig. 9A, traces b and d). Similarly, hPLSCR2 and PRD-hPLSCR2 spectra were monitored at 360 nm. In the presence and absence of Ca2+, there was no change in the absorbance spectra of hPLSCR2 (Fig. 9B, traces a and b). However, PRD-hPLSCR2 in the presence of Ca2+ showed an increase in the absorbance spectra similar to hPLSCR1 (Fig. 9B, traces c and d). The increase in absorbance of protein in the presence of Ca2+ indicates that PRD-containing PLSCRs undergo Ca2+-dependent aggregation.
FIGURE 9.

Protein aggregation detection by monitoring absorbance. A, trace a, hPLSCR1; trace b, hPLSCR1 in the presence of 4 mm Ca2+; trace c, ΔPRD-hPLSCR1; trace d, ΔPRD-hPLSCR1 in the presence of 4 mm Ca2+. B, trace a, hPLSCR2; trace b, hPLSCR2 in the presence of 4 mm Ca2+; trace c, PRD-hPLSCR2; trace d, PRD-hPLSCR2 in the presence of 4 mm Ca2+. Results are representative of at least three sets of experiments (p < 0.05).
The oligomeric size of proteins was also detected by dynamic light scattering, and the size of the protein was calculated from the mean average of the distributed particles. Fig. 10, A, C, E, and G shows that hPLSCR1, ΔPRD-hPLSCR1, hPLSCR2, and PRD-hPLSCR2 are ∼1 nm in size in the absence of Ca2+. In the presence of Ca2+, only hPLSCR1 and PRD-hPLSCR2 showed a increase in mean aggregate size (∼30–40 nm) (Fig. 10, B and H). However, no such increase in the size distribution for hPLSCR2 and ΔPRD-hPLSCR1 was observed in the presence of Ca2+ (Fig. 10, D and F). These results strongly suggest that PRD is required for aggregation of scramblases in the presence of Ca2+.
FIGURE 10.
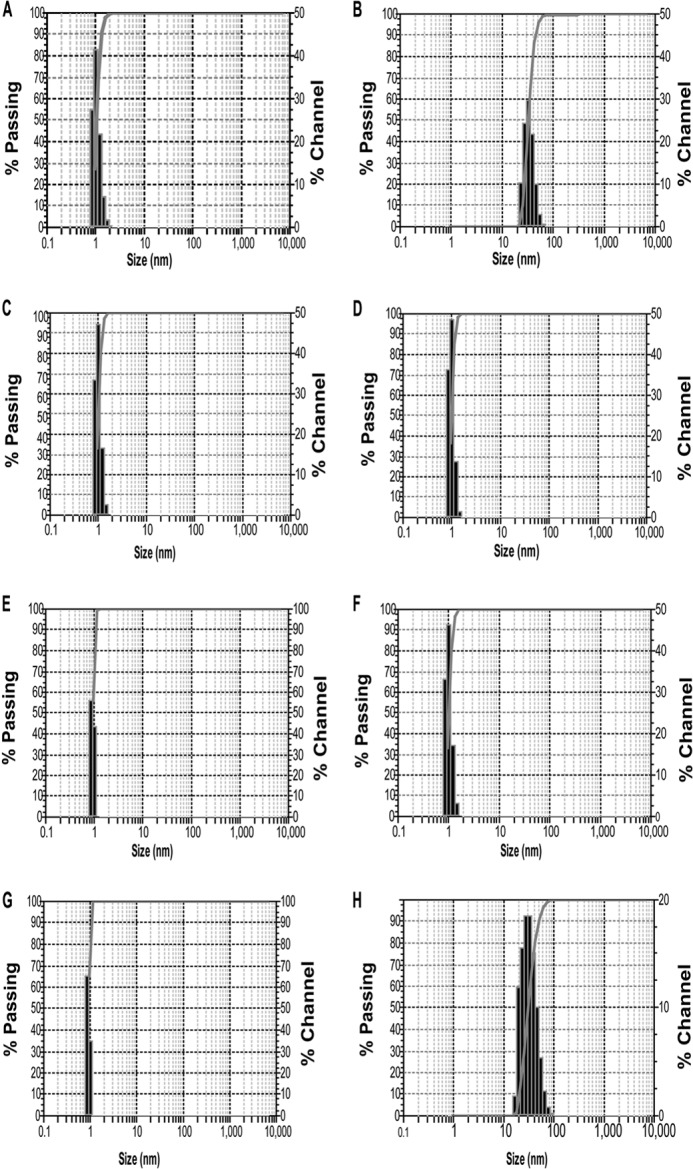
Protein size analysis by dynamic light scattering. A and B, particle size distribution of hPLSCR1 in the absence and presence of Ca2+ respectively. C and D, particle size distribution of ΔPRD- hPLSCR1 in the absence and presence of Ca2+. E and F, particle size distribution of hPLSCR2 in the absence and presence of Ca2+, respectively. G and H, particle size distribution of PRD-hPLSCR2 in the absence and presence of Ca2+. In the presence of Ca2+, only hPLSCR1 and PRD-hPLSCR2 showed an increase in mean aggregate size (∼30–40 nm). However, no such increase in the size distribution for hPLSCR2 and ΔPRD- hPLSCR1 was observed in the presence of Ca2+. These results strongly suggest that PRD is required for aggregation of scramblases in the presence of Ca2+. Results are representative of at least three sets of experiments (p < 0.05).
DISCUSSION
It has been proposed that hPLSCR1 is known to scramble PLs, but the mechanism of PL scrambling still remains unknown. To date only one homology model for hPLSCR1 exists based on the sequence similarity with Tubby-like proteins which predicted that hPLSCR1 will contain 12 β-barrel sheets with a central CTH, which is not hydrophobic in nature (30). In contrast to this theoretical model, experiments showed CTH was hydrophobic in nature and is crucial for membrane insertion and function (23). Sequence alignment revealed that hPLSCR2 retains most of the major domains of hPLSCR1 except for the PRD, and the sequence homology between hPLSCR1 with hPLSCR1 lacking PRD and hPLSCR2 showed 59 and 84% homology respectively. The N-terminal region of PLSCRs harboring the PRD has been shown to be important for the interaction of scramblase with other proteins. A recent report further underlies the importance of PRD where blocking the N-terminal region with antibody abolishes metastasis in colorectal cancer (31). Our biochemical reconstitution experiments revealed that hPLSCR2 does not exhibit scrambling activity, whereas hPLSCR1 showed maximum activity in the presence of Ca2+ rather than Mg2+ and Zn2+. We reasoned whether the lack of PL scrambling by hPLSCR2 could be due to the absence of the PRD; hence, we fused the PRD of hPLSCR1 with hPLSCR2. Our results show that Ca2+-dependent scrambling activity was restored in hPLSCR2 when the PRD of hPLSCR1 was fused. Unlike Ca2+, other metal ions, Mg2+ and Zn2+, failed to induce significant functional activity in hPLSCR1 and PRD-hPLSCR2, indicating that Ca2+ binding conformational changes are crucial for the functional activity of PLSCRs.
Even though all the reconstitution experiments were started with 60 μg of all four constructs, it is not clear whether the presence or absence of PRD affects the reconstitution efficiency. To check this, we measured protein content of vesicles after reconstitution by earlier reported methods (26). The results clearly showed that the amount of protein inserted in vesicles after reconstitution was ∼45–55% for all the four constructs (data not shown), which is consistent with other earlier published reports (11, 23, 25, 26). This suggests that the PRD does not affect the reconstitution efficiency but plays an important role in the function of the protein.
hPLSCR1 is one of the many proteins responsible for externalizing PS during apoptosis. Because it was now confirmed that PRD plays an important role in scrambling activity in vitro, we wanted to explore the biological role of PRD in hPLSCR1-mediated PS exposure during apoptosis. To achieve this, we transiently expressed the constructs in HEK 293T cell lines and induced apoptosis by serum starvation. Removal of PRD in hPLSCR1 drastically reduced the count of apoptotic cells when compared with wild type hPLSCR1. hPLSCR2 did not show any PS exposure as such, but the addition of PRD to hPLSCR2 promoted PS exposure comparable to hPLSCR1. This clearly confirms that PRD plays a significant role in PS exposure during apoptosis. The PRD of p53, a tumor suppressor protein, was reported to be necessary for p53-mediated induction of apoptosis. Mutation of key residues in PRD of p53 affected the local protein structure differently, thereby leading to a loss in activity (32).
A change in the local protein structure might have drastic effects on the localization of the protein in a cell. Prolines were found to be crucial for proper localization of many proteins. The proline-rich domain of SLP-76 is vital for association of Gad proteins that in-turn are responsible for the localization and its function (33). The PRD in phospholipase Cβ1 is responsible for the localization of the protein to the sarcolemma, and removal of the PRD caused cytoplasmic localization, attributing to the loss of function (34). A conserved proline-rich motif in the V3 domain of PKC-θ is essential for the localization of the protein to the center of immunological synapse (35). Thus it is possible that PRD in hPLSCRs might play a role in the localization of the protein to PM. To substantiate this, we performed localization studies for the four hPLSCR constructs using confocal microscopy and fractionation experiments. Our results showed that hPLSCR1 was prominently present in PM and nucleus, whereas hPLSCR2 was found only in nucleus as punctate aggregates, which were in accordance with the previous report (12). The addition of PRD to hPLSCR2 re-localized the protein to the entire cell similar to hPLSCR1 with difference in the distribution levels, thus clearly stating that PRD plays a vital role in the localization of the PLSCRs to various compartments of the cell. The loss of scramblase activity in the absence of PRD and failure to expose PS might be possibly due to the inability of the PRD lacking hPLSCRs to be localized to the PM.
The majority of the Ca2+-binding proteins contain EF-hand domain, and these proteins regulate ion transport and various signaling pathways. Earlier studies have shown that hPLSCR1 binds to Ca2+ and undergoes conformation changes (22). Even though hPLSCR2 has all the domains including the EF-hand-like domain of hPLSCR1, it failed to exhibit scramblase activity in the presence of Ca2+. We reasoned whether the lack of activity is because of its inability to bind to Ca2+, and to check this we used Stains-All and the terbium FRET assay and measured binding affinity by the intrinsic tryptophan fluorescence assay. Our results clearly showed that Ca2+ binds to hPLSCR2 with lower affinity than hPLSCR1. We reasoned that the lack of scramblase activity of hPLSCR2 is because it lacks PRD, and to further investigate on this, two different constructs PRD-hPLSCR2 and ΔPRD-hPLSCR1 were used. Stains-All and Tb3+ assays showed that Ca2+ binds to the PRD-hPLSCR2 and ΔPRD-hPLSCR1 in a similar manner to wild type proteins. No significant changes in affinities have been observed for hPLSCR1 and ΔPRD-hPLSCR1 and for hPLSCR2 and PRD-hPLSCR2, suggesting that the presence or absence of PRD does not affect the binding of Ca2+ to scramblases. To check whether Ca2+ binding to hPLSCR2 and other constructs affects the secondary structure, we performed far UV-CD spectroscopy. CD results showed that the presence and absence of PRD does not cause any significant change in secondary structures of proteins with minor changes in ellipticity. Similar changes in ellipticity pattern were also observed for hPLSCR1 and hPLSCR 4 (11, 22, 23). Conformational changes of protein upon Ca2+ binding was also confirmed by protease protection assay. Our results suggest that the presence of PRD domain influences the structural changes in protein.
ANS fluorescence studies showed that upon the addition of Ca2+ to ANS-bound hPLSCR1, hPLSCR2, PRD-hPLSCR2, and ΔPRD-hPLSCR1, an increase in fluorescence was observed, indicating movement of hydrophobic residues from the core to exterior. However, a red shift in emission maximum was observed only for ANS-hPLSCR1 and ANS-PRD-hPLSCR2 (Fig. 7), which was similar to a 10-nm red shift observed for hPLSCR4 (11). No such red shift was observed with hPLSCR1 and PRD-hPLSCR2 in the presence of Mg2+. A red shift in the presence of Ca2+ with hPLSCR1 and PRD-hPLSCR2 but not with hPLSCR2 and ΔPRD-hPLSCR1 indicated that PRD might play an important role in the Ca2+-dependent oligomerization of the protein. These results support the hypothesis that scramblases could possibly mediate their function by a Ca2+-dependent surface exposure of hydrophobic patches, thereby leading to protein oligomerization, which needs to be further confirmed. Ca2+ induced aggregation of hPLSCR1 and PRD-hPLSCR2 in buffer was detected by absorbance at 360 nm. A constant increase in the absorbance of PRD-containing proteins in the presence of Ca2+ suggests the formation of soluble aggregates, which was not observed in the case of hPLSCR2 and ΔPRD-hPLSCR1 (Figs. 9 and 10). Dynamic light scattering (DLS) experiments further confirms the increase in the protein size in the presence of Ca2+ for PRD-containing proteins hPLSCR1 and PRD-hPLSCR2. Based on our results, we strongly propose that PRD-mediated oligomerization of protein in the presence of Ca2+ is essential for the function of scramblase. This is in agreement with several published reports on the oligomerization-mediated function of proteins.
Previously proposed mechanisms for PL translocation included membrane permeabilization and oligomer-induced pore formation (36, 37). It has been reported that proteins such as mellitin and α-synuclein facilitated membrane leakage and destabilization by pore formation via oligomerization (38, 39). The synthetic peptide GALA induces vesicle leakage at pH 5.0 by pore formation and hPLSCR1 was also found to be functionally activated at acidic pH in the absence of Ca2+ (40, 41). A similar mechanism of functional activation for synaptotagmin I was observed where Ca2+-dependent oligomerization was crucial for function (42). A similar mechanism for protein function is mediated by calcyclin 21 and sorcin, which undergo oligomerization through hydrophobic contacts due to Ca2+ binding (43, 44).
Our results clearly showed that PRD of PLSCRs is not only necessary for its PL scrambling activity but also determines its subcellular localization. Additionally there was no major effect of PRD on the Ca2+ binding. Based on this study we propose that PLSCRs could mediate scramblase activity by oligomerization via surface-exposed hydrophobic patches due to Ca2+ binding, and PRD is crucial for this.
Acknowledgments
We thank the Department of Science and Technology for financial support for CD spectroscopy. We also thank Dr. Suresh Kumar Rayala, IIT Madras for providing anti-GFP monoclonal antibody.
This work was supported by a research grant from Department of Biotechnology, Government of India.
- PM
- plasma membrane
- PS
- phosphatidylserine
- PL
- phospholipid
- PLSCR
- phospholipid scramblase
- hPLSCR1
- human phospholipid scramblase 1
- hPLSCR2
- human phospholipid scramblase 2
- CTH
- C-terminal helix
- PRD
- proline rich domain
- PRD-hPLSCR2
- PRD of hPLSCR1 fused to hPLSCR2
- ΔPRD-hPLSCR1
- PRD deleted from hPLSCR1
- NBD
- 7-nitrobenz-2-oxa-1,3 diazol-4-yl
- PI
- propidium iodide
- ANS
- 8-anilino-1-naphthalenesulfonic acid.
REFERENCES
- 1. Zachowski A. (1993) Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem. J. 294, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daleke D. L. (2007) Phospholipid flippases. J. Biol. Chem. 282, 821–825 [DOI] [PubMed] [Google Scholar]
- 3. Williamson P., Bevers E. M., Smeets E. F., Comfurius P., Schlegel R. A., Zwaal R. F. (1995) Continuous analysis of the mechanism of activated transbilayer lipid movement in platelets. Biochemistry 34, 10448–10455 [DOI] [PubMed] [Google Scholar]
- 4. Bevers E. M., Williamson P. L. (2010) Phospholipid scramblase: an update. FEBS Lett. 584, 2724–2730 [DOI] [PubMed] [Google Scholar]
- 5. Sahu S. K., Gummadi S. N., Manoj N., Aradhyam G. K. (2007) Phospholipid scramblases: an overview. Arch. Biochem. Biophys. 462, 103–114 [DOI] [PubMed] [Google Scholar]
- 6. Bassé F., Stout J. G., Sims P. J., Wiedmer T. (1996) Isolation of an erythrocyte membrane protein that mediates Ca2+-dependent transbilayer movement of phospholipid. J. Biol. Chem. 271, 17205–17210 [DOI] [PubMed] [Google Scholar]
- 7. Wiedmer T., Zhou Q., Kwoh D. Y., Sims P. J. (2000) Identification of three new members of the phospholipid scramblase gene family. Biochim. Biophys. Acta 1467, 244–253 [DOI] [PubMed] [Google Scholar]
- 8. Lizak M., Yarovinsky T. O. (2012) Phospholipid scramblase 1 mediates type I interferon-induced protection against staphylococcal α-toxin. Cell Host Microbe 11, 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun J., Zhao J., Schwartz M. A., Wang J. Y., Wiedmer T., Sims P. J. (2001) c-Abl tyrosine kinase binds and phosphorylates phospholipid scramblase 1. J. Biol. Chem. 276, 28984–28990 [DOI] [PubMed] [Google Scholar]
- 10. Liu J., Dai Q., Chen J., Durrant D., Freeman A., Liu T., Grossman D., Lee R. M. (2003) Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 1, 892–902 [PubMed] [Google Scholar]
- 11. Francis V. G., Gummadi S. N. (2012) Biochemical and functional characterization of human phospholipid scramblase 4 (hPLSCR4). Biol. Chem. 393, 1173–1181 [DOI] [PubMed] [Google Scholar]
- 12. Yu A., McMaster C. R., Byers D. M., Ridgway N. D., Cook H. W. (2003) Stimulation of phosphatidyl serine biosynthesis and facilitation of UV-induced apoptosis in chinese hamster ovary cells overexpressing phospholipid scramblase 1. J. Biol. Chem. 278, 9706–9714 [DOI] [PubMed] [Google Scholar]
- 13. Talukder A. H., Bao M., Kim T. W., Facchinetti V., Hanabuchi S., Bover L., Zal T., Liu Y. J. (2012) Phospholipid Scramblase 1 regulates Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Cell Res. 22, 1129–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kametaka S., Shibata M., Moroe K., Kanamori S., Ohsawa Y., Waguri S., Sims P. J., Emoto K., Umeda M., Uchiyama Y. (2003) Identification of phospholipid scramblase 1 as a novel interacting molecule with β-secretase (β-site amyloid precursor protein (APP) cleaving enzyme (BACE). J. Biol. Chem. 278, 15239–15245 [DOI] [PubMed] [Google Scholar]
- 15. Merregaert J., Van Langen J., Hansen U., Ponsaerts P., El Ghalbzouri A., Steenackers E., Van Ostade X., Sercu S. (2010) Phospholipid scramblase 1 is secreted by a lipid raft-dependent pathway and interacts with the extracellular matrix protein 1 in the dermal epidermal junction zone of human skin. J. Biol. Chem. 285, 37823–37837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Y., Rogulski K., Zhou Q., Sims P. J., Prochownik E. V. (2006) The negative c-Myc target onzin affects proliferation and apoptosis via its obligate interaction with phospholipid scramblase 1. Mol. Cell Biol. 26, 3401–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Py B., Basmaciogullari S., Bouchet J., Zarka M., Moura I. C., Benhamou M., Monteiro R. C., Hocini H., Madrid R., Benichou S. (2009) The phospholipid scramblases 1 and 4 are cellular receptors for the secretory leukocyte protease inhibitor and interact with CD4 at the plasma membrane. PLoS ONE 4, e5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Q., Ben-Efraim I., Bigcas J. L., Junqueira D., Wiedmer T., Sims P. J. (2005) Phospholipid scramblase 1 binds to the promoter region of the inositol 1,4,5-triphosphate receptor type 1 gene to enhance its expression. J. Biol. Chem. 280, 35062–35068 [DOI] [PubMed] [Google Scholar]
- 19. Wiedmer T., Zhao J., Nanjundan M., Sims P. J. (2003) Palmitoylation of phospholipid scramblase 1 controls its distribution between nucleus and plasma membrane. Biochemistry 42, 1227–1233 [DOI] [PubMed] [Google Scholar]
- 20. Chen M. H., Ben-Efraim I., Mitrousis G., Walker-Kopp N., Sims P. J., Cingolani G. (2005) Phospholipid scramblase 1 contains a nonclassical nuclear localization signal with unique binding site in importin-α. J. Biol. Chem. 280, 10599–10606 [DOI] [PubMed] [Google Scholar]
- 21. Lott K., Bhardwaj A., Sims P. J., Cingolani G. (2011) A minimal nuclear localization signal (NLS) in human phospholipid scramblase 4 that binds only the minor NLS-binding site of importin α1. J. Biol. Chem. 286, 28160–28169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stout J. G., Zhou Q., Wiedmer T., Sims P. J. (1998) Change in conformation of plasma membrane phospholipid scramblase induced by occupancy of its Ca2+ binding site. Biochemistry 37, 14860–14866 [DOI] [PubMed] [Google Scholar]
- 23. Francis V. G., Mohammed A. M., Aradhyam G. K., Gummadi S. N. (2013) The single C-terminal helix of human phospholipid scramblase 1 is required for membrane insertion and scrambling activity. FEBS J. 280, 2855–2869 [DOI] [PubMed] [Google Scholar]
- 24. Suzuki J., Umeda M., Sims P. J., Nagata S. (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 [DOI] [PubMed] [Google Scholar]
- 25. Francis V. G., Majeed M. A., Gummadi S. N. (2012) Recovery of functionally active recombinant human phospholipid scramblase 1 from inclusion bodies using N-lauroylsarcosine. J. Ind. Microbiol. Biotechnol. 39, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 26. Gummadi S. N., Menon A. K. (2002) Transbilayer movement of diplamitoylphosphatidylcholine in proteoliposomes reconstitute from detergent extracts of endoplasmic reticulum: kinetics of transbilayer transport mediated by a single flippase and identification of proteins fractions enriched in flippase acitivity. J. Biol. Chem. 277, 25337–25343 [DOI] [PubMed] [Google Scholar]
- 27. Caday C. G., Lambooy P. K., Steiner R. F. (1986) The interaction of Ca2+-binding proteins with the carbocyanine dye stains-all. Biopolymers 25, 1579–1595 [DOI] [PubMed] [Google Scholar]
- 28. Brittain H. G., Richardson F. S., Martin R. B. (1976) Terbium (III) emission as a probe of calcium(II) binding sites in proteins. J. Am. Chem. Soc. 98, 8255–8260 [DOI] [PubMed] [Google Scholar]
- 29. Daniel E., Weber G. (1966) Cooperative effects in binding by bovine serum albumin. I. The binding of 1-anilino-8-naphthalenesulfonate. Fluorimetric titrations. Biochemistry 5, 1893–1900 [DOI] [PubMed] [Google Scholar]
- 30. Bateman A., Finn R. D., Sims P. J., Wiedmer T., Biegert A., Söding J. (2009) Phospholipid scramblases and Tubby-like proteins belong to new superfamily of membrane tethered transcription factors. Bioinformatics 25, 159–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan C. W., Chen C. Y., Chen K. T., Shen C. R., Kuo Y. B., Chen Y. S., Chou Y. P., Wei W. S., Chan E. C. (2012) Blockade of phospholipid scramblase 1 with its N-terminal domain antibody reduces tumorigenesis of colorectal carcinomas in vitro and in vivo. J. Transl. Med. 10, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edwards S. J., Hananeia L., Eccles M. R., Zhang Y. F., Braithwaite A. W. (2003) The proline-rich region of mouse p53 influences transactivation and apoptosis but is largely dispensable for these functions. Oncogene 22, 4517–4523 [DOI] [PubMed] [Google Scholar]
- 33. Singer A. L., Bunnell S. C., Obstfeld A. E., Jordan M. S., Wu J. N., Myung P. S., Samelson L. E., Koretzky G. A. (2004) Role of proline-rich domain in SLP-76 subcellular localization and T cell function. J. Biol. Chem. 279, 15481–15490 [DOI] [PubMed] [Google Scholar]
- 34. Grubb D. R., Vasilevski O., Huynh H., Woodcock E. A. (2008) The extreme C-terminal region of phospholipase cβ1 determines subcellular localization and function; the “b” splice variant mediates α1-adrenergic receptor responses in cardiomyocytes. FASEB J. 22, 2768–2774 [DOI] [PubMed] [Google Scholar]
- 35. Kong K. F., Yokosuka T., Canonigo-Balancio A. J., Isakov N., Saito T., Altman A. (2011) A novel motif in the V3 domain of the kinase PKCθ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol. 12, 1105–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imura Y., Choda N., Matsuzaki K. (2008) Magainin 2 in action: distinct modes of membrane permeabilization in living bacteria and mammalian cells. Biophys. J. 95, 5757–5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanyal S., Menon A. K. (2009) Flipping lipids: why an' what's the reason for? Chem. Biol. 4, 895–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsuzaki K., Yoneyama S., Miyajima K. (1997) Pore formation and translocation of melittin. Biophys. J. 73, 831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Rooijen B. D., Claessens M. M. A. E., Subramaniam V. (2010) Membrane permeabilization by oligomeric α-synuclein: in search of the mechanism. PLoS ONE 5, e14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li W., Nicol F., Szoka F. C. (2004) GALA: a designed synthetic pH-responsive amphipathic peptide with application in drug and gene delivery. Adv. Drug. Deliv. Rev. 56, 967–985 [DOI] [PubMed] [Google Scholar]
- 41. Stout J. G., Bassé F., Luhm R. A., Weiss H. J., Wiedmer T., Sims P. J. (1997) Scott syndrome erythrocytes contain a membrane protein capable of mediating Ca2+-dependent transbilayer migration of membrane phospholipids. J. Clin. Invest. 99, 2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Damer C. K., Creutz C. E. (1996) Calcium dependent self-association of synaptotagmin I. J. Neurochem. 67, 1661–1668 [DOI] [PubMed] [Google Scholar]
- 43. Zamparelli C., Ilari A., Verzili D., Vecchini P., Chiancone E. (1997) Calcium and pH-linked oligomerization of sorcin causing translocation from cytosol to membranes. FEBS Lett. 409, 1–6 [DOI] [PubMed] [Google Scholar]
- 44. Sudo T., Hidaka H. (1999) Characterization of the calcyclin (S100A6) binding site of annexin XI-A by site-directed mutagenesis. FEBS Lett. 444, 11–14 [DOI] [PubMed] [Google Scholar]