

Background: The Cas9 RNA-guided endonuclease has been adapted for genome manipulation and regulation.
Results: We have characterized target recognition and cleavage by Streptococcus thermophilus LMG18311 Cas9.
Conclusion: The two nuclease domains of Cas9 select their cleavage sites by different mechanisms.
Significance: These findings contribute to the molecular basis of Cas9-mediated DNA cleavage.
Keywords: DNA, DNA-binding Protein, DNA Transformation, RNA, RNA-binding Protein, CRISPR, Cas9, Genome Editing, Nuclease
Abstract
Cas9, the RNA-guided DNA endonuclease from the CRISPR-Cas (clustered regularly interspaced short palindromic repeat–CRISPR-associated) system, has been adapted for genome editing and gene regulation in multiple model organisms. Here we characterize a Cas9 ortholog from Streptococcus thermophilus LMG18311 (LMG18311 Cas9). In vitro reconstitution of this system confirms that LMG18311 Cas9 together with a trans-activating RNA (tracrRNA) and a CRISPR RNA (crRNA) cleaves double-stranded DNA with a specificity dictated by the sequence of the crRNA. Cleavage requires not only complementarity between crRNA and target but also the presence of a short motif called the PAM. Here we determine the sequence requirements of the PAM for LMG18311 Cas9. We also show that both the efficiency of DNA target cleavage and the location of the cleavage sites vary based on the position of the PAM sequence.
Introduction
A promising tool for genome manipulation (1–14) and regulation (15–18) in a wide variety of organisms has recently been identified in the RNA-guided DNA endonuclease activity of the CRISPR-Cas2 (clustered regularly interspaced short palindromic repeat–CRISPR-associated) system. CRISPR-Cas, an inheritable prokaryotic immune system, protects bacteria and archaea against mobile genetic elements via RNA-guided target silencing. CRISPR-Cas systems consist of an array of short direct repeats interspersed by variable invader-derived sequences (spacers) (19–21) and a cas operon. During invasion, small fragments of the invading DNA from phage or plasmids (protospacers) are incorporated into host CRISPR loci, transcribed, and processed to generate small CRISPR RNAs (crRNA). The invading nucleic acid is then recognized and silenced by Cas proteins guided by the crRNAs. There are three types of CRISPR-Cas system each characterized by the presence of a signature gene (22).
Programmed DNA cleavage requires the fewest components in the type II CRISPR-Cas system, requiring only crRNA, a trans-activating crRNA (tracrRNA), and the Cas9 endonuclease (23, 24), the signature gene of the type II system. The system can be further simplified by fusing the mature crRNA and tracrRNA into a single guide RNA (sgRNA) (23). In addition to its role in target cleavage, tracrRNA also mediates crRNA maturation by forming RNA hybrids with primary crRNA transcripts, leading to co-processing of both RNAs by endogenous RNase III (25). Cas9 contains two nuclease domains that together generate a double-strand (ds) break in target DNA. The HNH nuclease domain cleaves the complementary strand, and the RuvC-like nuclease domain cleaves the noncomplementary strand (23, 24).
A short signature sequence, named the protospacer adjacent motif (PAM), is characteristic of the invading DNA targeted by the type I and type II CRISPR-Cas systems. The PAM serves two functions. It has been linked to the acquisition of new spacer sequences, and it is necessary for the subsequent recognition and silencing of target DNA, reviewed in Ref. 26. The sequence, length, and position of the PAM vary depending on the CRISPR-Cas type and organism. PAMs from type II systems are located downstream of the protospacer and contain 2–5 bp of conserved sequence. A variable sequence, of up to 4 bp, separates the conserved sequence of the PAM from the protospacer. This variable region is often included in the definition of the PAM sequence, but for simplicity, we refer to this variable region as the linker and the conserved sequence as the PAM. To date, Cas9 from Streptococcus pyogenes, Cas9 from Streptococcus thermophilus DGCC7710, and Cas9 from Neisseria meningitidis have been employed as tools for genome editing or regulation. For these Cas9 orthologs, the PAMs are GG, GGNG, and GATT, and the linkers are 1, 1, and 4 bp, respectively (23, 27, 28).
The simplicity of sgRNA design and sequence-specific targeting means the RNA-guided Cas9 machinery has great potential for programmable genome engineering. Cas9 can be employed to generate mutations in cells by introducing dsDNA breaks. The capabilities of Cas9 can be expanded to various genome engineering purposes, such as transcription repression or activation, with its nickase (generated by inactivating one of its two nuclease domains) or nuclease null variants (15, 17, 18, 29). Another appealing possibility for the Cas9 system is to target different Cas9-mediated activities to multiple target sites, for example transcriptional repression of one gene but activation of another (30). To achieve this, multiple Cas9 orthologs will need to be employed as a single ortholog cannot concurrently mediate different activities at multiple sites (30). Therefore to broaden our understanding of Cas9 proteins, we have characterized the Cas9 ortholog from S. thermophilus LMG18311, which we refer to as LMG18311 Cas9. We chose to investigate Cas9 from this organism not only to increase the repertoire of Cas9 orthologs but also because it utilizes a PAM distinct from those previously characterized and its small gene size is compatible with the standard viral vectors used for delivery into exogenous systems in vivo (30).
Here we demonstrate that requirements for DNA cleavage in vitro and in vivo by LMG18311 Cas9 are the same as other Cas9 orthologs. We also reveal the sequence and linker length requirements of the PAM for LMG18311 Cas9. Finally, we show that the HNH and RuvC-like nuclease domains of Cas9 select the location of their cleavage sites via different mechanisms. The HNH domain catalyzes cleavage of the complementary strand at a fixed position, whereas the RuvC-like domain catalyzes cleavage of the noncomplementary strand using a ruler mechanism.
EXPERIMENTAL PROCEDURES
Identification of the PAM
Natural target sequences were found using the program BLAST. A single mismatch was allowed between the spacer and target sequences. Allowing more mismatches did not increase the number of sequences found. Sequences were considered unique if they were from distinct target genomes.
Cloning and Mutagenesis
The sequence encoding full-length Cas9 was PCR-amplified from S. thermophilus LMG18311 genomic DNA (American Type Culture Collection) and inserted into the pMAT expression vector (31, 32). The resulting construct encodes Cas9 fused to an N-terminal hexahistidine-maltose-binding protein (His6-MBP) tag. Cas9 mutants were created using the QuikChange site-directed mutagenesis method (Stratagene). To generate plasmid targets and RNA encoding vectors, synthetic oligonucleotides, bearing the appropriate sequence, were annealed and ligated into the pACYCDuet-1 (Novagen), pRSFDuet-1 (Novagen), or pMK (GeneArt). Primers and oligonucleotides are listed in Table 1. All constructs were verified by DNA sequencing.
TABLE 1.
Primers and oligonucleotides used in these studies

Protein Expression and Purification
Cas9 was overexpressed in T7Express Escherichia coli (New England Biolabs). Cells were grown at 37 °C in Luria-Bertani (LB) medium supplemented with ampicillin to an A600 of ∼0.3. Protein expression was induced with 0.2 mm iso-propyl-β-d-thiogalactopyranoside (IPTG) overnight at 20 °C. Cells were harvested by centrifugation and quickly frozen in liquid nitrogen.
For purification, cells were resuspended in lysis buffer (20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm imidazole, and 10% glycerol) supplemented with protease inhibitor mixture (Sigma-Aldrich) and lysed by French press. Lysate was clarified by centrifugation at 18,000 rpm at 4 °C for 45 min, and the supernatant was loaded on a 5-ml immobilized metal chromatography column (Bio-Rad) charged with nickel sulfate. The column was washed with lysis buffer, and bound protein was eluted with lysis buffer containing 250 mm imidazole. The elution was run on a HiLoad 26/60 S200 size exclusion column (GE Healthcare) pre-equilibrated with gel-filtration buffer A (20 mm Tris-HCl, pH 8.0, and 500 mm NaCl). Fractions containing His6-MBP tagged Cas9 were collected and treated with tobacco etch virus protease overnight at 4 °C to remove the His6-MBP tag. Samples were reapplied to immobilized metal affinity chromatography resin to remove the His-tagged tobacco etch virus protease, free His6-MBP, and any remaining tagged protein. The flow-through was collected, concentrated using an Ultracel 10K centrifugal filter unit (Millipore), and further purified by size exclusion chromatography in gel-filtration buffer B (20 mm Tris-HCl, pH 8.0, 200 mm KCl, and 1 mm EDTA). The final fractions containing Cas9 were concentrated to ∼16 mg/ml. Purified proteins were >95% pure as judged by SDS-PAGE and Coomassie Blue staining (see Fig. 1A). The mutant variants of Cas9 were expressed and purified in the same manner as the wild-type protein (see Fig. 1A).
FIGURE 1.
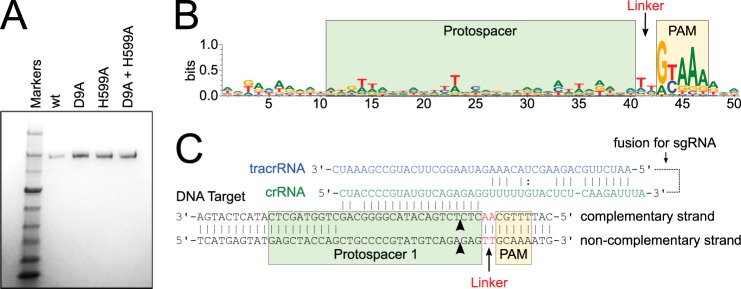
The type II CRISPR-Cas system of S. thermophilus LMG18311. A, Coomassie Blue-stained SDS-polyacrylamide gel of Cas9, Cas9 D9A, Cas9 D599A, and Cas9 D9A,D599A. B, logo plot revealing the PAM for LMG18311 Cas9. The positions of the protospacer, PAM, and linker are indicated. C, schematic representation of the crRNA (green), tracrRNA (blue), and DNA target (black). The positions of the protospacer, PAM, and linker are indicated. The site at which the crRNA and tracrRNA are fused to generate the sgRNA is indicated with a dotted line.
RNA Preparation
RNAs were generated by in vitro transcription using T7 RNA polymerase. Plasmid templates were linearized overnight with EcoRI and then purified by phenol:chloroform extraction and ethanol precipitation. 0.5 μg of linear plasmid template was incubated with 0.1 mg/ml T7 RNA polymerase and 5 mm each of CTP, GTP, ATP, and UTP in reaction buffer (25 mm Tris-HCl, pH 8.0, 1.5 mm MgCl2, 2 mm spermidine, 40 mm DTT) at 37 °C for 3 h. RNA transcripts were then gel-purified.
In Vivo Transformation Assay
The recipient cells were prepared by co-transforming E. coli BL21 (DE3) with plasmids encoding Cas9 (pMAT) and sgRNA (pRSFDuet-1) or empty vectors. All plasmids, including the targets, had unique selection markers and origins of replication. The transformation assay was performed using the CaCl2 heat-shock procedure described in Ref. 33 with minor changes. The recipient cells were transformed with 5 ng of plasmid DNA and recovered in LB medium containing 0.2 mm IPTG at 37 °C for 1 h and plated on LB agar containing appropriate antibiotics and 0.2 mm IPTG. Reported transformation efficiencies are the average of at least three biological replicates. All target plasmids used in this study transformed into control recipient cells with the same efficiency (∼200 colony-forming units per 5 ng of DNA).
Plasmid Cleavage Assay
Cas9 (25 nm), tracrRNA (25 nm), and crRNA (25 nm) were incubated in a cleavage buffer (20 mm HEPES, pH 7.5, 150 mm KCl, 10 mm MgCl2) at 37 °C for 30 min. The reactions were initiated by adding plasmid targets (4 nm), incubated at 37 °C for 30 min, and quenched with phenol. The aqueous layer was extracted and separated on a 0.8% agarose gel. Gels were stained by soaking in 1× Tris-Acetate-EDTA buffer supplemented with 5 μg/μl ethidium bromide for 1 h and then for a further hour in 1× Tris-Acetate-EDTA buffer. Bands were visualized using an FLA-7000 (Fuji) and quantified with ImageGauge (Fuji). To account for the different binding affinity of ethidium bromide to linear and supercoiled DNA, control samples with equal amounts of DNA in both forms were loaded on the same gel. The ratios of the fluorescence intensities of linear and supercoiled bands were measured and used to calculate a correlation coefficient K (34),
 |
where ILin and Isc are the intensities of the linear and supercoiled bands, respectively. In our case, K was determined to be 0.4 ± 0.05 and did not vary significantly between experiments. The percentage of linear product was then calculated as follows (34).
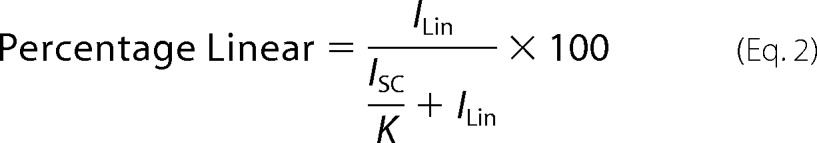 |
Electrophoresis Mobility Shift Assay
DNA oligonucleotides were purified on 10% denaturing polyacrylamide gels. dsDNA targets (Table 1) were made by annealing each strand and purified on 12% native polyacrylamide gels containing 1× Tris-borate-EDTA. dsDNA were 5′ end-labeled with [γ-32P]ATP using T4 polynucleotide kinase (New England Biolabs). A fixed concentration (10–100 pm) of labeled dsDNA targets was mixed with an increasing concentration of premixed Cas9D9A,H599A-sgRNA complex. Binding assays, performed in buffer (20 mm HEPES, pH 7.5, 150 mm KCl, 10 mm MgCl2, 0.1 mg/ml BSA, and 10% glycerol), were incubated at 37 °C for 30 min followed by separation on 5% native polyacrylamide gels. Gels were visualized by phosphorimaging (Fuji) and quantified with ImageGauge (Fuji). Fraction of DNA bound was plotted versus concentration of Cas9, and data were fit to a one-site binding isotherm using GraphPad Prism software. Reported Kd values are the average of at least three replicates.
RESULTS
Identifying the PAM for LMG18311 Cas9
The genome of S. thermophilus LMG18311 contains two CRISPR-Cas systems, of type II-A and III-A, each associated with a CRISPR loci: CRISPR-1 and CRISPR-2, respectively. The first study of PAM sequences identified a putative PAM for S. thermophilus as RYAAA (where R is a purine and Y is a pyrimidine) (19). This sequence was found in natural target sequences matching 41 spacers collected from 13 different S. thermophilus strains, including LMG18311. Subsequent studies showed that PAM sequences vary greatly, even between different strains (reviewed in Ref. 26). Therefore to confirm the PAM sequence for LMG18311 Cas9, we performed BLAST searches to identify potential protospacers in viral and plasmid genomes that matched any of the 33 spacer sequences from CRISPR-1. This search generated 41 unique target sequences, from the genomes of bacteriophage known to infect S. thermophilus. We then aligned 50-nucleotide segments from the identified target genomes, inclusive of the 30-nucleotide protospacer and 10-nucleotide flanking regions (Fig. 1B). In agreement with the previous study (19), inspection of this alignment clearly identified a 5-bp PAM with a consensus sequence, GYAAA, invariantly located 2 bp downstream of the protospacer (Fig. 1, B and C). The most commonly observed PAM sequence, found in 7 of the 41 target sequences, was GCAAA.
To confirm that the identified PAM was functional, we used a previously described transformation assay in which E. coli cells containing an exogenous type II CRISPR-Cas system are resistant to plasmid transformation, whereas cells lacking the system are competent for transformation (33, 35) (Fig. 2A). To generate cells containing the type II CRISPR-Cas system (CRISPR+ cells), compatible vectors encoding either LMG18311 Cas9 or its cognate sgRNA, engineered to contain a 20-nucleotide sequence derived from the first spacer of CRISPR-1 (Fig. 1C), were co-transformed into E. coli BL21(DE3). In this overexpression system, the Cas9 and sgRNA genes are under the control of an IPTG-inducible T7 promoter. Control cells lacking the CRISPR-Cas system (CRISPR− cells) were generated by co-transforming compatible empty vectors into E. coli BL21(DE3). We constructed a target and two control plasmids. The target plasmid contained protospacer-1 (whose sequence was identical to the first spacer of CRISPR-1), a 2-bp linker, and the identified PAM (GCAAA) (Fig. 1C). The first control plasmid contained only protospacer-1, whereas the second control plasmid lacked both protospacer-1 and PAM. The target and control plasmids were then tested for CRISPR-Cas silencing by transformation into the CRISPR+ and CRISPR− strains in the presence of IPTG and the appropriate antibiotics (Fig. 2A). The control plasmids transformed into both strains with similar efficiency (Fig. 2B). The target plasmid failed to transform into the CRISPR+ cells but transformed into the CRISPR− cells with an efficiency comparable with that of the control plasmids (Fig. 2B). All of the transformation efficiencies were comparable with those previously reported (35). These results indicate that the identified PAM is functional in vivo and that the type II CRISPR-Cas system of S. thermophilus LMG18311 protects E. coli cells from transformation by plasmid DNA.
FIGURE 2.

LMG18311 Cas9 and cognate sgRNA can provide resistance to plasmid transformation in E. coli. A, schematic representation of transformation assay. B, interference of plasmid transformation by LMG18311 Cas9 and sgRNA in E. coli cells. Transformation efficiency is expressed as cfu per 5 ng of plasmid DNA. Average values from at least three biological replicates are shown, with error bars representing 1 S.D. C, effect of mutation in the PAM sequence on plasmid transformation efficiency. D, effect of linker length on plasmid transformation efficiency. PS denotes the protospacer sequence.
Both the PAM Sequence and the Linker Length Are Important for Plasmid Interference
To investigate the PAM sequence requirements for LMG18311 Cas9, we transformed a series of plasmid targets harboring single-nucleotide mutations throughout the PAM sequence in the CRISPR+ strain (Fig. 2C). Only the plasmid containing a mutation at the position 1 guanosine (that is, the PAM nucleotide closest to the protospacer) was transformed, albeit with a reduced (∼66%) transformation efficiency as compared with the intact PAM sequence (Fig. 2C). Plasmids containing single mutations to any of the other four positions were resistant to transformation (Fig. 2C). These results indicate that the guanosine at position 1 is important for PAM function but individually the four other positions have little effect on PAM function.
A 2-bp linker separates the protospacer from the PAM for LMG18311 Cas9 (Fig. 1, B and C). To investigate how linker length affects Cas9 activity, we generated plasmid targets with linkers ranging from 0 to 5 bp in length (Fig. 2D). We then determined the transformation efficiency for these plasmids into the CRISPR+ cells. The CRISPR+ cells were equally resistant to transformation by a plasmid target with a linker length of either 2 bp or 3 bp (Fig. 2D). Plasmids with other linker lengths transformed with efficiencies more similar to the control plasmid (Fig. 2D), suggesting that plasmids with these linkers were able to escape CRISPR-Cas silencing.
In Vitro Reconstitution Recapitulates in Vivo Activity
To further investigate the requirements of PAM sequence and linker length, we reconstituted the activity of LMG18311 Cas9 in vitro. LMG18311 Cas9 was expressed and purified from E. coli (Fig. 1A). A 42-nucleotide tracrRNA mimicking the processed tracrRNA and a 42-nucleotide crRNA containing the sequence derived from first spacer of CRISPR-1 (Fig. 1C) were chemically synthesized. Plasmid targets were incubated with Cas9, tracrRNA, and crRNA and then analyzed by electrophoresis through agarose gels and ethidium bromide staining. As observed for other Cas9 orthologs, cleavage of the plasmid target occurred in the presence of Cas9, tracrRNA, crRNA, and Mg2+ (Fig. 3A). Cleavage also occurred when an sgRNA was substituted for the tracrRNA and crRNA (Fig. 3B). As expected, cleavage was dictated by the sequence of the sgRNA (Fig. 3C). Cas9 variants with active site mutations in either the RuvC-like domain (D9A) or the HNH domain (H599A) nicked the plasmid targets, whereas a variant with a double mutation (D9A,H599A) displayed no activity (Fig. 3D). Cleavage assays using short oligonucleotide substrates confirmed that the HNH domain cleaves the strand complementary to the guide RNA, whereas the RuvC-like domain cleaves the noncomplementary strand (Fig. 3E). Mapping the location of the cut sites revealed that, as seen with other Cas9 orthologs (23, 24, 36, 37), cleavage of both strands occurs within the protospacer, 3 bp from its PAM proximal end, producing a blunt-end dsDNA break (Fig. 3E).
FIGURE 3.

DNA cleavage by LMG18311 Cas9 in vitro. A, RNA-guided cleavage by Cas9. Reaction mixtures containing 5 nm target plasmid, 25 nm Cas9, 25 nm crRNA, 25 nm tracrRNA, and 10 mm Mg2+ were incubated for 30 min at 37 °C. B, a cognate sgRNA can substitute for crRNA and tracrRNA. C, cleavage of a plasmid target is dictated by the sgRNA sequence. D, cleavage of a plasmid target by active site mutants of Cas9. E, cleavage of a synthetic dsDNA by active site mutants of Cas9. The dsDNA was radiolabeled at the 5′ end of the complementary strand (left) or the noncomplementary strand (right). Reactions were performed as in A, and products were separated by 10% denaturing PAGE. The cleavage sites are indicated with arrows in the schematic diagram (bottom). 50 nt, 50 nucleotides; 37 nt, 37 nucleotides. F, cleavage of plasmid targets containing mutations in the PAM sequence. G, cleavage of plasmid targets containing the indicated linker lengths. Average values from at least three biological replicates are shown, with error bars representing 1 S.D. In A–C and E–F, the positions of negatively supercoiled (nSC), linear (L) and nicked or open circle (OC) plasmid are indicated. The linear control is a digestion of the plasmid target with the restriction enzyme AgeI.
We next wished to confirm that either mutations in the PAM or changes in linker length had the same effect on DNA interference in vitro as they did in vivo. Therefore we monitored cleavage of these variant plasmids by recombinant LMG18311 Cas9. The fraction plasmid cleaved was calculated using the procedure detailed under “Experimental Procedures,” which accounts for the different binding affinity of ethidium bromide to linear and supercoiled DNA. Consistent with the in vivo results, mutation of the guanosine at position 1 had the greatest effect, and individual mutations to the other four positions of the PAM had only a modest effect on plasmid cleavage (Fig. 3F). Cleavage of plasmid targets with different linker lengths was optimal at 2 or 3 bp and then decreased steadily with increasing or decreasing lengths (Fig. 3G).
Metal Dependence of DNA Cleavage by Cas9
To evaluate whether other divalent cations besides Mg2+ can activate DNA cleavage by Cas9, we performed plasmid cleavage assays in the presence of one of the following divalent cations: Ca2+, Mn2+, Co2+, Ni2+, and Cu2+. Reactions containing Ca2+ yielded nicked, instead of linear plasmid (Fig. 4A), suggesting that Ca2+ activates only one of the Cas9 nuclease domains. To identify which domain was activated, we assayed the single active site mutants of Cas9 (D9A or H599A) in a reaction buffer containing Ca2+. We observed little cleavage with the HNH mutant (H599A) but robust cleavage with the RuvC-like mutant (D9A) (Fig. 4B), suggesting that the HNH but not the RuvC-like domain was activated by Ca2+. None of the other divalent cations tested activated either nuclease domain of Cas9 (Fig. 4A).
FIGURE 4.
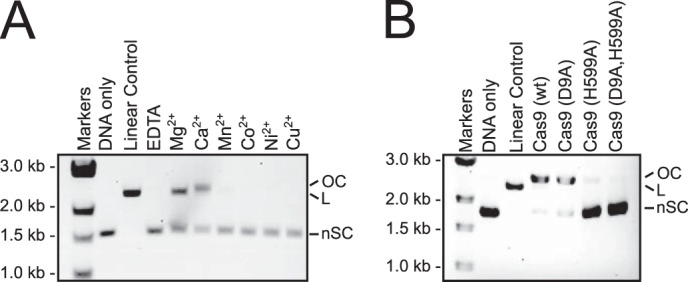
Metal dependence of DNA cleavage by Cas9. A, cleavage of a target plasmid by Cas9 with either no metal or 1 mm of the indicated metal ions. All reactions were treated with 0.5 mm EDTA prior to metal addition. B, cleavage of a target plasmid by active site mutants of Cas9 in the presence of 10 mm Ca2+. In both panels, the positions of negatively supercoiled (nSC), linear (L), and nicked or open circle (OC) plasmid are indicated. The linear control is a digestion of the plasmid target with the restriction enzyme AgeI.
Both the PAM Sequence and the Linker Length Are Important for Target Binding
Previous studies indicate that mutations within the PAM impair DNA cleavage by Cas9 due to weakened binding (23, 24, 38). To determine the effect of PAM sequence and linker length on binding of LMG18311 Cas9 to DNA targets, we determined the binding affinity (Kd) of the Cas9-sgRNA complex to 5′ end-labeled dsDNA targets using native gel electrophoresis (Fig. 5A). Binding experiments were conducted with the nuclease-deficient mutant of Cas9 (D9A,H599A) in the presence of Mg2+. Fixed concentrations of the dsDNA targets were incubated with increasing concentrations of the Cas9-sgRNA complex (Fig. 5A). A target containing a complementary protospacer, a 2-bp linker, and a functional PAM bound to Cas9-sgRNA with an affinity of 0.94 ± 0.27 nm (Fig. 5B). We were unable to detect binding to a target containing a noncomplementary protospacer or to a target that lacked a PAM. Mutation of the guanosine at position 1 of the PAM resulted in an ∼100-fold increase in Kd (Fig. 5B), whereas mutations at positions 2 through 5 did not significantly alter the affinity (all within ∼4-fold on the consensus PAM) (Fig. 5B). Changes in linker length had a larger effect on binding affinity (Fig. 5C). Under the conditions tested, we failed to detect binding to plasmid targets containing linker lengths of 0, 4, or 5 bp (Kd > 1000 nm), whereas linkers of 1 and 3 bp reduced the affinity by ∼400- and ∼20-fold, respectively (Fig. 5C).
FIGURE 5.

DNA target binding by Cas9. A, a representative gel shift assay for Cas9-sgRNA and the binding curve measured from the assay. B and C, bar graph plotting Kd values for DNA targets with PAM mutations (labeled red) (B) or DNA targets with different linker lengths (labeled red) (C). Average values from at least three replicates are shown, with error bars representing 1 S.D. Targets where binding was not observed are shown with Kd values at the lower limit (> 1000 nm). PS denotes the protospacer sequence.
HNH and RuvC-like Domains Determine the Location of Their Cut Sites Using Different Mechanisms
Previous studies reported that Cas9 cleaves both DNA strands within the protospacer, 3 bp from its PAM proximal end, producing a predominantly blunt-end dsDNA break (23, 24, 36, 37). To determine whether linker length has any effect on where the Cas9 nuclease domains cut, we mapped the location of the cut sites in plasmids containing protospacer-1 and different lengths of linker. Following cleavage by Cas9 (programmed with an sgRNA complementary to protospacer-1), the linear plasmid products were purified by agarose gel electrophoresis and sequenced. Sequencing data revealed that the position of the cleavage site on the noncomplementary strand, but not on the complementary strand, depended on linker length (Fig. 6A). Cleavage of the complementary strand always occurred 3 nucleotides from the 5′ end of the protospacer sequence, independent of the linker length (Fig. 6A). In contrast, cleavage of the noncomplementary strand occurred predominantly 5 nucleotides from the 3′ end of the PAM with linker lengths of 2 or more bp or at 4 and 5 nucleotides from the 3′ end of the PAM with a linker length of 1 bp (Fig. 6A). The site of cleavage on both strands of the DNA target was also found to be independent of spacer sequence. The location of Cas9 cut sites in plasmids containing protospacer-2 was found to be identical to plasmids containing protospacer-1 for all linker lengths investigated (Fig. 6B). We were unable to generate enough cleaved DNA from the plasmid target with a linker length of zero for sequencing.
FIGURE 6.

Mapping the Cas9 cleavage sites in plasmid targets with different linker lengths. A and B, direct sequencing electropherograms for plasmid-sp1 (A) and plasmid-sp2 (B) from complementary strand primer (bottom left) and noncomplementary strand primer (bottom right) are shown. Termination of primer extension in the sequencing reactions reveals the positions of the cleavage site (red lines). The positions of the protospacer, PAM, and linker are indicated. The 3′-terminal A addition, indicated by asterisk, is an artifact of the sequencing reaction.
DISCUSSION
Cas9, the RNA-guided endonuclease from the type II CRISPR-Cas system, has the potential to revolutionize our ability to manipulate the genomes of a wide variety of organisms (1–18). Targeting Cas9 to specific genomic sites relies on the presence of a PAM and complementarity between the sequence of its crRNA and the protospacer. A remarkably diverse set of PAM sequences is recognized by Cas9 orthologs (30). To date, PAM recognition and DNA cleavage have been experimentally studied in only a handful of Cas9 orthologs (23, 24, 28, 30). Characterization of additional orthologs is expected to improve our mechanistic understanding of Cas9 and likely expand our engineering capabilities. Here we present characterization of the Cas9 protein from S. thermophilus LMG18311.
We demonstrate LMG18311 Cas9 is active in vivo through transformation assays (Fig. 2) and in vitro by monitoring plasmid cleavage (Fig. 3). We also confirm that the PAM for LMG18311 Cas9 identified by sequence alignments is functional (Fig. 1B). As observed for other Cas9 orthologs, LMG18311 Cas9 activity requires tracrRNA, crRNA, and Mg2+ (Fig. 3). Metal ion substitution studies also reveal that Ca2+ likely activates the HNH but not the RuvC-like domain of LMG18311 Cas9 (Fig. 4B). Here however, we cannot rule out the possibility that the observed activation of the HNH domain may be due to trace Mg2+ contamination in the Ca2+ solution. Neither nuclease domain of S. pyogenes Cas9 is activated by Ca2+ (23).
Cas9 orthologs have been reported to cleave target DNA with a wide range of mutations in the PAM sequences (30). However, in natural targets, PAM sequences are highly conserved. This apparent discrepancy may arise from the dual function of the PAM (26, 30). The stringency on the PAM sequence is greater for spacer acquisition than for DNA cleavage by Cas9. Consistent with this, our results show that although the PAM for LMG18311 Cas9 is conserved (Fig. 1B), the nuclease activity of LMG18311 Cas9 tolerates a broad range of mutations in the PAM of the target DNA. Mutations to the guanosine at position 1 impair Cas9 activity, whereas individual mutations at positions 2 through 5 have little effect. The PAM for N. meningitides Cas9 also contains a single guanosine important for Cas9 activity. In addition, two recent in vivo studies show that an AG sequence can partially replace the consensus PAM, GG, for S. pyogenes Cas9 (13, 39). Thus, despite the varying sequence of PAM, Cas9 proteins from LMG18311, S. pyogenes, and N. meningitides all contain a guanosine that appears essential for DNA silencing in vivo.
A previously unexplored aspect of target binding and cleavage by Cas9 is the length of the linker between the PAM and protospacer. The 41 natural targets of LMG18311 Cas9 we identified in our sequence searches all contain a 2-bp linker. However, we found that DNA containing a 3-bp linker was silenced with the same efficiency as that with a 2-bp linker (Figs. 2D and 3F). Further lengthening or shortening of the linker eliminates CRISPR-Cas silencing and inhibits plasmid cleavage (Figs. 2D and 3F). Thus, our results on the type II system of S. thermophilus LMG18311 suggest that the requirements for the length of the linker appear to be less stringent for DNA silencing than for spacer acquisition, a pattern similar to that observed for requirements on PAM sequence.
Recognition of target DNA by either Cas9 or effector complexes from the type I CRISPR-Cas systems is thought to be a multistep process (23, 24, 38, 40, 41). First, cellular DNA is scanned for PAM sequences. Once a PAM is identified, the adjacent DNA duplex is destabilized, enabling Cas9 to probe sequence complementarity on the target strand. Target recognition is completed if this adjacent sequence contains a protospacer that can base-pair with the crRNA, stabilizing the complex. If this sequence lacks a protospacer, then the crRNA-DNA heteroduplex fails to form and Cas9 dissociates. We found the affinity of LMG18311 Cas9-sgRNA for its target sequence is ∼1.0 nm (Fig. 5), which is similar to the Kd of ∼0.5 nm reported for S. pyogenes Cas9 (38) and comparable with the affinity of the type I effector complexes for their DNA targets (42–44). Targets lacking a PAM had no detectable affinity for Cas9. As expected (23, 24), the impaired nuclease activity of LMG18311 Cas9 observed when PAM sequences are mutated arises from the weakened binding affinity between Cas9 and target DNA (Fig. 5B). Further analysis also revealed that the inhibition of cleavage of targets with different linker lengths was also due to weakened affinity (Fig. 5C). Although both PAM and linker mutations result in reduced target affinity, they likely affect different steps in binding. PAM mutations inhibit the initial recognition of a target sequence, whereas altering linker length likely impairs the efficiency of base-pairing between crRNA and the protospacer, thus destabilizing the complex.
The length of the linker between the PAM and protospacer affects both the efficiency of DNA target cleavage and the position of the cleavage sites. This suggests that the two nuclease domains of Cas9 select their cleavage sites by different mechanisms. The HNH domain cleaves the complementary strand at a fixed position, whereas the RuvC-like domain, employing a ruler mechanism, cleaves the noncomplementary strand at a position measured from the PAM (Fig. 6). These observations suggest that the relative positions of the Cas9 nuclease domains are highly flexible.
While this manuscript was in preparation, crystal structures of Cas9 from S. pyogenes and Actinomyces naeslundii (45) and Cas9 from S. pyogenes in complex with sgRNA and its ssDNA target (46) were reported. The domain organization observed in these structures is consistent with our data showing that the two nuclease domains of Cas9 select their cleavage sites by different mechanisms. These structures reveal that Cas9 adopts a bilobed architecture composed of target recognition and nuclease lobes. The target recognition lobe is essential for binding the sgRNA and the complementary strand of the DNA target. The nuclease lobe contains a C-terminal domain implicated in PAM binding (45, 46) as well as the HNH and RuvC-like nuclease domains. The position of the RuvC-like domain is fixed relative to the position of the PAM binding domain, supporting our observation that cleavage of the noncomplementary strand by the RuvC-like domain occurs at a fixed distance from the PAM (Fig. 7). In contrast, the position of the HNH domain is variable among the current structures (45, 46). In the structure of Cas9-sgRNA bound to ssDNA, which is in an inactive conformation because of the lack of a PAM sequence, the HNH domain is positioned away from the location of its cleave site (46). Therefore Cas9 must undergo a conformational change that repositions the HNH domain to engage the complementary strand before cleavage. Because the target recognition lobe holds the complementary strand, the HNH domain must dock with this lobe to engage its target (Fig. 7). This docking likely determines the cleavage site of the HNH domain in the complementary strand consistent with our observation that the HNH domain cleaves at a fixed position independent of linker length. The flexibility of the HNH domain and the flexibility between the two lobes of Cas9 (45, 46) likely accommodate the varying lengths of the linker DNA while maintaining the cleavage site of the HNH domain on the complementary strand (Fig. 7).
FIGURE 7.
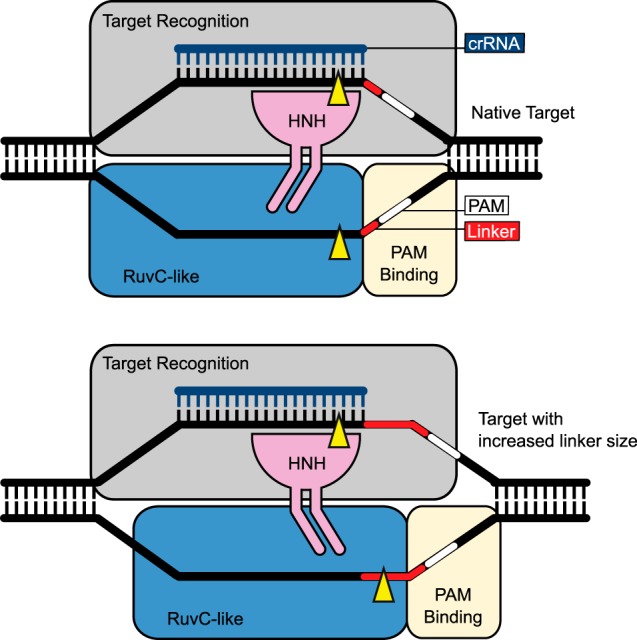
Schematic representation of cut site selection by the HNH and RuvC-like domains of Cas9. Top, a schematic with a native DNA target; bottom, a schematic with a DNA target containing a longer length linker. Cleavage sites on the complementary and noncomplementary strands are indicated by yellow arrows. For clarity, the tracrRNA is not shown.
In summary, we have characterized the substrate requirements of LMG18311 Cas9 both in vivo and in vitro. Our results enable wider target selection for genome manipulation through the use of a distinct PAM. They also reiterate the importance of considering which Cas9 ortholog to use in genome manipulation as those with longer PAM sequences are not necessarily more stringent in DNA cleavage. We also reveal the requirements for linker length in DNA cleavage by a Cas9 ortholog and, by varying the linker length, reveal that the two nuclease domains of Cas9 select their cut sites by different mechanisms.
Acknowledgment
We thank Jennifer M. Kavran for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM097330 (to S. B.).
- CRISPR
- clustered regularly interspaced short palindromic repeats
- Cas
- CRISPR-associated
- crRNA
- CRISPR-RNA
- sgRNA
- single guide RNA
- tracrRNA
- trans-activating RNA
- PAM
- protospacer adjacent motif
- MBP
- maltose-binding protein
- IPTG
- iso-propyl-β-d-thiogalactopyranoside.
REFERENCES
- 1. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. (2013) RNA-programmed genome editing in human cells. Elife 2, e00471–e00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cho S. W., Kim S., Kim J. M., Kim J.-S. (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232 [DOI] [PubMed] [Google Scholar]
- 5. Shen B., Zhang J., Wu H., Wang J., Ma K., Li Z., Zhang X., Zhang P., Huang X. (2013) Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 23, 720–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., Jaenisch R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J.-F., Norville J. E., Aach J., McCormack M., Zhang D., Bush J., Church G. M., Sheen J. (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31, 688–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nekrasov V., Staskawicz B., Weigel D., Jones J. D. G., Kamoun S. (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 691–693 [DOI] [PubMed] [Google Scholar]
- 9. Hwang W. Y., Fu Y., Reyon D., Maeder M. L., Tsai S. Q., Sander J. D., Peterson R. T., Yeh J.-R. J., Joung J. K. (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gratz S. J., Cummings A. M., Nguyen J. N., Hamm D. C., Donohue L. K., Harrison M. M., Wildonger J., O'Connor-Giles K. M. (2013) Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194, 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedland A. E., Tzur Y. B., Esvelt K. M., Colaiácovo M. P., Church G. M., Calarco J. A. (2013) Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10, 741–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DiCarlo J. E., Norville J. E., Mali P., Rios X., Aach J., Church G. M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang W., Bikard D., Cox D., Zhang F., Marraffini L. A. (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakayama T., Fish M. B., Fisher M., Oomen-Hajagos J., Thomsen G. H., Grainger R. M. (2013) Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis 51, 835–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qi L. S., Larson M. H., Gilbert L. A., Doudna J. A., Weissman J. S., Arkin A. P., Lim W. A. (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L. A. (2013) Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maeder M. L., Linder S. J., Cascio V. M., Fu Y., Ho Q. H., Joung J. K. (2013) CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilbert L. A., Larson M. H., Morsut L., Liu Z., Brar G. A., Torres S. E., Stern-Ginossar N., Brandman O., Whitehead E. H., Doudna J. A., Lim W. A., Weissman J. S., Qi L. S. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bolotin A., Quinquis B., Sorokin A., Ehrlich S. D. (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561 [DOI] [PubMed] [Google Scholar]
- 20. Pourcel C., Salvignol G., Vergnaud G. (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663 [DOI] [PubMed] [Google Scholar]
- 21. Mojica F. J. M., Díez-Villaseñor C., García-Martínez J., Soria E. (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182 [DOI] [PubMed] [Google Scholar]
- 22. Makarova K. S., Haft D. H., Barrangou R., Brouns S. J. J., Charpentier E., Horvath P., Moineau S., Mojica F. J. M., Wolf Y. I., Yakunin A. F., van der Oost J., Koonin E. V. (2011) Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gasiunas G., Barrangou R., Horvath P., Siksnys V. (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, E2579–E2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deltcheva E., Chylinski K., Sharma C. M., Gonzales K., Chao Y., Pirzada Z. A., Eckert M. R., Vogel J., Charpentier E. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shah S. A., Erdmann S., Mojica F. J. M., Garrett R. A. (2013) Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 10, 891–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deveau H., Barrangou R., Garneau J. E., Labonté J., Fremaux C., Boyaval P., Romero D. A., Horvath P., Moineau S. (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190, 1390–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y., Heidrich N., Ampattu B. J., Gunderson C. W., Seifert H. S., Schoen C., Vogel J., Sontheimer E. J. (2013) Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol. Cell 50, 488–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mali P., Esvelt K. M., Church G. M. (2013) Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esvelt K. M., Mali P., Braff J. L., Moosburner M., Yaung S. J., Church G. M. (2013) Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 10, 1116–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peränen J., Rikkonen M., Hyvönen M., Kääriäinen L. (1996) T7 vectors with modified T7lac promoter for expression of proteins in Escherichia coli. Anal. Biochem. 236, 371–373 [DOI] [PubMed] [Google Scholar]
- 32. Mulepati S., Bailey S. (2013) In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J. Biol. Chem. 288, 22184–22192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sapranauskas R., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V. (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 39, 9275–9282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panyutin I. V., Luu A. N., Panyutin I. G., Neumann R. D. (2001) Strand breaks in whole plasmid DNA produced by the decay of 125I in a triplex-forming oligonucleotide. Radiat. Res. 156, 158–166 [DOI] [PubMed] [Google Scholar]
- 35. Karvelis T., Gasiunas G., Miksys A., Barrangou R., Horvath P., Siksnys V. (2013) crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 10, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Magadán A. H., Dupuis M.-È., Villion M., Moineau S. (2012) Cleavage of phage DNA by the Streptococcus thermophilus CRISPR3-Cas system. PLoS ONE 7, e40913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garneau J. E., Dupuis M.-È., Villion M., Romero D. A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A. H., Moineau S. (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71 [DOI] [PubMed] [Google Scholar]
- 38. Sternberg S. H., Redding S., Jinek M., Greene E. C., Doudna J. A. (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Semenova E., Jore M. M., Datsenko K. A., Semenova A., Westra E. R., Wanner B., van der Oost J., Brouns S. J. J., Severinov K. (2011) Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 108, 10098–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sashital D. G., Wiedenheft B., Doudna J. A. (2012) Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell 46, 606–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mulepati S., Orr A., Bailey S. (2012) Crystal structure of the largest subunit of a bacterial RNA-guided immune complex and its role in DNA target binding. J. Biol. Chem. 287, 22445–22449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sashital D. G., Jinek M., Doudna J. A. (2011) An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat. Struct. Mol. Biol. 18, 680–687 [DOI] [PubMed] [Google Scholar]
- 44. Wiedenheft B., van Duijn E., Bultema J. B., Waghmare S. P., Zhou K., Barendregt A. J., Westphal W., Heck A. J. R., Boekema E. J., Dickman M. J., Doudna J. A. (2011) RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U.S.A. 108, 10092–10097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jinek M., Jiang F., Taylor D. W., Sternberg S. H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S., Kaplan M., Iavarone A. T., Charpentier E., Nogales E., Doudna J. A. (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nishimasu H., Ran F. A., Hsu P. D., Konermann S., Shehata S. I., Dohmae N., Ishitani R., Zhang F., Nureki O. (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]