

Background: The sodium chloride cotransporter NCC mediates NaCl reabsorption in the kidney distal convoluted tubule.
Results: NCC internalization from the plasma membrane is clathrin-mediated and regulated by NCC phosphorylation and ubiquitylation.
Conclusion: Phosphorylation of NCC can regulate NCC internalization and ubiquitylation.
Significance: Impaired NCC endocytosis could be implicated in salt-sensitive hypertension in vivo.
Keywords: Endocytosis, Epithelium, Phosphorylation, Plasma Membrane, Trafficking, Ubiquitylation
Abstract
The thiazide-sensitive sodium chloride cotransporter, NCC, is the major NaCl transport protein in the distal convoluted tubule (DCT). The transport activity of NCC can be regulated by phosphorylation, but knowledge of modulation of NCC trafficking by phosphorylation is limited. In this study, we generated novel tetracycline-inducible Madin-Darby canine kidney type I (MDCKI) cell lines expressing NCC to examine the role of NCC phosphorylation and ubiquitylation on NCC endocytosis. In MDCKI-NCC cells, NCC was highly glycosylated at molecular weights consistent with NCC monomers and dimers. NCC constitutively cycles to the apical plasma membrane of MDCKI-NCC cells, with 20–30% of the membrane pool of NCC internalized within 30 min. The use of dynasore, PitStop2, methyl-β-cyclodextrin, nystatin, and filipin (specific inhibitors of either clathrin-dependent or -independent endocytosis) demonstrated that NCC is internalized via a clathrin-mediated pathway. Reduction of endocytosis resulted in greater levels of NCC in the plasma membrane. Immunogold electron microscopy confirmed the association of NCC with the clathrin-mediated internalization pathway in rat DCT cells. Compared with controls, inducing phosphorylation of NCC via low chloride treatment or mimicking phosphorylation by replacing Thr-53, Thr-58, and Ser-71 residues with Asp resulted in increased membrane abundance and reduced rates of NCC internalization. NCC ubiquitylation was lowest in the conditions with greatest NCC phosphorylation, thus providing a mechanism for the reduced endocytosis. In conclusion, our data support a model where NCC is constitutively cycled to the plasma membrane, and upon stimulation, it can be phosphorylated to both increase NCC activity and decrease NCC endocytosis, together increasing NaCl transport in the DCT.
Introduction
The distal convoluted tubule (DCT)2 reabsorbs 5–10% of the filtered NaCl and is critical for fine-tuning the final urinary excretion of NaCl and thus homeostatic Na+ balance. The major transport protein responsible for reabsorbing NaCl in the DCT is the thiazide-sensitive NaCl cotransporter designated NCC or TSC (reviewed in Ref. 1). NCC is a member of the superfamily of cation-chloride cotransporters, including the sodium-potassium-chloride cotransporters, NKCC1 and NKCC2. The transporter is encoded by the solute carrier family 12 member 3 (SLC12A3) gene. NCC is expressed in the apical membrane and in subapical vesicles of the DCT (2, 3). In the plasma membrane NCC is functional as a glycosylated homodimer (4, 5). Inactivating mutations of the SLC12A3 gene results in the autosomal recessive disease Gitelman syndrome, characterized by hypokalemia, hypomagnesemia, metabolic alkalosis, and hypocalciuria (6–9), thus emphasizing the important role of NCC in cardiovascular and renal physiology and pathophysiology.
Knowledge of the molecular mechanisms that modulate trafficking events of NCC and thus regulate the apical membrane abundance of NCC is slowly emerging. A WNK4-induced decrease in NCC membrane abundance has been shown to occur via alteration of NCC forward trafficking (exocytosis) (10, 11). Ko et al. (12, 13) demonstrated that treatment of DCT cells with the phorbol ester 12-O-tetradecanoyl-phorbol-13-acetate reduced NCC membrane abundance via a Ras-GRP1 (Ras-guanyl nucleotide-releasing protein 1)-dependent ERK1/2-MAPK pathway (12) that increased ubiquitylation and dynamin-dependent internalization of NCC (13). However, the exact mechanisms behind the regulated endocytosis of NCC and whether, similarly to the water channel aquaporin 2 (AQP2) (14) and the sodium potassium chloride cotransporter NKCC2 (15), constitutive endocytosis of NCC plays a role in steady-state surface expression of NCC are not known.
Phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 is an essential mediator of NCC function (16, 17). Phosphorylation is likely to have a direct effect on the transport activity of NCC, as demonstrated in oocytes (16, 17). However, whether other regulatory mechanisms of phosphorylation exist that alter NCC function is a matter of debate. Phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 occurs directly in the DCT plasma membrane and thus is unlikely to play a major role in NCC forward trafficking (exocytosis) (18, 19). However, phosphorylation has also been demonstrated to play a regulatory role in endocytosis of membrane transport proteins, e.g. ENaC is directly phosphorylated by ERK2 and casein kinase 2 (CK2) to promote Nedd4-2-mediated ENaC internalization (20, 21) and phosphorylation of AQP2 regulates AQP2 endocytosis and recycling (22, 23).
In this study, we examined the internalization pathways of NCC and whether constitutive endocytosis of NCC is an important modulator of steady-state NCC membrane abundance. Furthermore, we examined whether phosphorylation of NCC plays a regulatory role in the ubiquitylation of NCC and thus the rate of NCC internalization. Our studies indicate that NCC undergoes constitutive trafficking to/from the apical plasma membrane via a clathrin-dependent mechanism that can modulate overall NCC cell surface expression. Furthermore, we demonstrate that phosphorylation of NCC can regulate the apical membrane abundance of NCC via reducing NCC ubiquitylation and subsequent endocytosis.
EXPERIMENTAL PROCEDURES
Antibodies
The primary antibodies used in this study are as follows: polyclonal rabbit antibodies against total NCC (SPC-402D, StressMarq, originally characterized in Ref. 24), proteasome 20 S (ab3325, Abcam), ZO-1 (40-2300, Zymed Laboratories Inc.), and FLAG (F7425, Sigma); mouse monoclonal antibodies against FLAG (MAB8183, Abnova), ubiquitin (P4D1, Cell Signaling), gp135 (105582, Abcam); a rabbit anti-human transferrin antibody (DAKO A0061), and a previously characterized rabbit polyclonal antibody against phosphorylated NCC (Thr(P)-58NCC) (18).
Immunoblotting
The preparation of samples and immunoblotting were as described previously (25). A horseradish peroxidase-conjugated secondary antibody (DAKO P448, goat anti-rabbit IgG, or DAKO P447, goat anti-mouse IgG) was used at 1:5,000, and antibody-antigen reactions were visualized using SuperSignal West Femto chemiluminescent substrate (Thermo Scientific, Denmark). Semi-quantitative data were obtained by analysis of band densities and calculated as relative abundance ratios for each individual sample for each time point or stimulant. All reported values are means ± S.E.
Immunofluorescence Confocal Microscopy
MDCKI-NCC cells were grown on 6-well filter plates (Costar, 0.4 μm) until confluent and induced. Cells were fixed in 4% paraformaldehyde/PBS for 15 min at room temperature, before permeabilization in 0.3% SDS in PBS for 5 min. Labeling was performed as in Ref. 22, but 0.05% Triton X-100 was substituted for saponin. A Leica TCS SL confocal microscope with an HCX PL Apo 63× oil objective lens (numerical aperture, 1.40) was used for obtaining image stacks with a z-distance of 0.1 μm between images.
Generation of an Inducible NCC-expressing MDCKI Cell Line
A FLAG tag (GACTACAAGGACGATGATGACAAG; amino acids DYKDDDDK) was introduced in a pcDNA5/FRT/TO/TOPO-rNCC vector (26) by PCR, creating pcDNA5/FRT/TO/TOPO-N-term-FLAG-rNCC. Phosphorylation-deficient and -mimicking mutations were introduced at the Thr-53, Thr-58, and Ser-71 sites (T53A/T58A/S71A or T53D/T58D/S71D) in the pcDNA5/FRT/TO/TOPO-N-term-FLAG-rNCC vector using site-directed mutagenesis (Stratagene) and standard methodologies. An MDCK type I cell line containing a single FRT site in its genome was used as the host cell line (27). Stable inducible cell lines were made by initially stably transfecting MDCKI-FRT cells with the pcDNA6/TR6 (Invitrogen) vector to promote tetracycline inducibility. The pcDNA6/TR6-positive clones were selected using 5 μg/ml blasticidin-HCl. These cells were subsequently cotransfected with the pcDNA5/FRT/TO/TOPO-N-term-FLAG-rNCC WT or mutant constructs and pOG44 (encoding flp recombinase) using Lipofectamine 2000 (Invitrogen). Positive clones were selected using 150–500 μg/ml hygromycin B. After clonal selection, cells were grown in DMEM GlutaMAX supplemented with 10% donor bovine serum, 5 μg/ml blasticidin-HCl, and 150 μg/ml hygromycin B. PCR on cell genomic DNA was used to verify the incorporation of rNCC into the genome. To induce NCC protein expression in the MDCKI-FRT-TO-N-term-FLAG-rNCC cells (termed MDCKI-NCC), 10 μg/ml tetracycline was added for a minimum of 16 h prior to performing an experiment.
Reverse Transcriptase-PCR (RT-PCR)
RNA was purified from the MDCKI-NCC WT and mutant cell lines using the RiboPureTM kit (Ambion) following the manufacturer's protocol. 2 μg of RNA was used for RT-PCR. Possible DNA contamination was removed by incubating RNA with DNase I Amp Grade 1 in DNase Reaction buffer (20 mm Tris-HCl, pH 8.4, 2 mm MgCl2, 50 mm KCl) (both Invitrogen) for 15 min at room temperature. 1.1 mm EDTA was added, and the sample was heated to 65 °C for 10 min to stop the DNase reaction. cDNA was produced following the protocol from SuperScriptTM II reverse transcriptase (Invitrogen). PCR with primers against actin were used to verify cDNA generation (Table 1).
TABLE 1.
RT-PCR primers
| Target | Forward primer | Reverse primer | Expected band size |
|---|---|---|---|
| bp | |||
| NCC | GCCAGGCGATGCTCTGTGCA | CATGGCCACACCCACGGCAT | 666 |
| OSR1 | AAAAAGGTTCGGAGAGTACC | AGTACTAGGCTGATTGGGAT | 368 |
| SPAK | GAGAGGCTATGACTTCAAGG | GACTGTATTTGCTCGGGTAT | 543 |
| WNK1 | CGTTTACGCTTTTGGAATGT | TTTGCCTTATGCATCCTTCA | 176 |
| WNK3 | ATCTCGACTTCCTAGTCCAA | CTGCTGAAAATTAGGGGACT | 847 |
| WNK4 | ACGAGGAAAAGTACGATGAG | TCAGCTTCACAGACTAAACC | 463 |
| V2R | CATGTATGCCTCCTCCTACA | ACGTAGACGATCACAATGAC | 477 |
Immunogold Electron Microscopy
The preparation of tissue and associated techniques for cryo-electron microscopy have been described previously in detail (18). Labeling with total NCC antibody was visualized with goat anti-rabbit IgG conjugated to 10 nm colloidal gold particles.
Deglycosylation Assay
Cells were lysed in ice-cold lysis buffer (20 mm Tris, 135 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, 5 μg/ml leupeptin, 100 μg/ml Pefabloc) for 40 min on ice followed by sonication. Samples were centrifuged for 10 min at 10,000 × g at 4 °C, and NCC was immunoprecipitated (see below) before elution in 1% SDS at 65 °C. Samples were diluted to 0.1% SDS and denatured in 1× denaturing buffer (New England Biolabs) for 10 min at 60 °C or 30 min at 37 °C, before deglycosylation using PNGase F (New England Biolabs) and standard protocols. 4× Laemmli gel sample buffer was added, and samples were heated for 10 min at 65 °C before SDS-PAGE.
Human Transferrin (hTf), Lactosylceramide (LacCer), and Dextran Endocytosis Assay
To characterize the inhibitor efficacy on different pathways of plasma membrane protein endocytosis in the inducible NCC MDCKI cell line, various internalization assays were performed using uptake of fluorescent transferrin, LacCer, or dextran, reported to enter cells via the clathrin-mediated pathway, a clathrin-independent mechanism, or by fluid phase endocytosis, respectively. These assays were supported by studies using immunoblotting where applicable (transferrin uptake). Cells were grown on 12-well plastic plates (Costar) to confluency for several days. On the day of the experiment, the cells were serum-starved for 2 h and then pretreated with inhibitors or vehicle in DMEM GlutaMAX with 0.2% BSA for 30–60 min at 37 °C. The cells were cooled on ice for 30–60 min. To study the various endocytic pathways, hTf-Alexa488 final concentration of 30 μg/ml (transferrin from human serum, AlexaFluor® 488 conjugate, Invitrogen), 0.3 μm LacCer (BODIPY® FL C5-lactosylceramide complexed to BSA, Invitrogen), or 25 μg/ml Dextran Oregon Green® 488; 10,000 MW (Invitrogen) in room temperature DMEM GlutaMAX with 0.2% BSA with inhibitors or DMSO were added, and the cells were returned to the incubator at 37 °C for up to 60 min. A 0-min control sample was left on ice. To remove the hTf nonspecifically bound to the cell surface or hTf complexed with transferrin receptor on the cell surface, the cells were washed twice in ice-cold 0.2 m acetic acid (in PBS) for 10 min and then twice in PBS, pH 7.4, for 10 min. The cells were lysed in lysis buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris-HCl, pH 7.5, 1% Triton X-100 with 5 μg/ml leupeptin and 100 μg/ml Pefabloc) and sonicated. The samples were spun at 10,000 × g for 5 min at 4 °C, and fluorescence from duplicate samples was recorded on a multimode plate reader (Enspire Multimode Plate Reader, PerkinElmer Life Sciences) equipped with a 490-nm bandpass filter for excitation and a 520-nm filter for emission.
Cell Surface Biotinylation Assays
Cells were grown in complete DMEM (DMEM GlutaMAX, 10% donor bovine serum) on filter plates to confluency for several days. When stimulated, cells were washed three times in pure media (without serum) before adding the stimulant (25 μm forskolin, 50 μm (Sp)-cAMP (Sigma), or hypotonic chloride condition (67.5 mm sodium gluconate, 2.5 mm potassium glucanate, 0.5 mm CaCl2, 0.5 mm MgCl2, 1 mm Na2HPO4, 1 mm Na2SO4, 7.5 mm sodium HEPES, pH 7.4)) and incubated for 30 min at 37 °C, as indicated. Otherwise, cells were washed in ice-cold PBS/CM (PBS, 1 mm CaCl2, 0.1 mm MgCl2, pH 7.5) and incubated with mild agitation for 45 min at 4 °C in ice-cold biotinylation buffer (10 mm triethanolamine, 2 mm CaCl2, 125 mm NaCl, pH 8.9) containing a 1.5 mg/ml final concentration of sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (EZ-link Sulfo-NHS-SS-biotin, Pierce) added to the apical compartment. Cells were washed in ice-cold quenching buffer (PBS/CM, 50 mm Tris-HCl, pH 8) followed by two washes of PBS/CM. Cells were lysed in lysis buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris-HCl, pH 7.5, 1% Triton X-100) with 5 μg/ml leupeptin, 100 μg/ml Pefabloc (both from Roche Applied Science), 10 μg/ml phosphatase inhibitor mixture (Sigma), and sonicated. A fraction of the sample was retained for total NCC protein estimates before the samples were spun at 10,000 × g for 5 min at 4 °C. The supernatant was transferred to spin columns containing NeutrAvidin gel slurry (Pierce) and incubated for 60 min at room temperature with end-over-end mixing. After extensive washing, biotinylated proteins were eluted in 1× Laemmli sample buffer. Total protein samples were added to 4× Laemmli sample buffer, and all samples were heated for 15 min at 60 °C. To verify that no biotinylation of cytosolic proteins occurred, immunoblots from each biotinylation experiment were probed with the intracellular marker protein proteasome 20 S. Additionally, to visualize and confirm side-specific biotinylation, cells were grown on filters as described and biotinylated from the apical or basolateral surface, fixed in 4% paraformaldehyde/PBS, permeabilized with 0.2% saponin, and labeled with fluorophore-conjugated streptavidin (STREP-488, Invitrogen). Filters were mounted on glass slides, and the biotinylated proteins were analyzed by confocal microscopy (Fig. 1).
FIGURE 1.

Characterization of a novel inducible MDCKI-NCC cell line. A, representative immunoblot of total protein samples extracted from MDCKI-NCC cells grown until confluent on semi-permeable supports and treated for the last 16 h with various concentrations of tetracycline. NCC is predominantly detected as a diffuse protein band between 100 and 150 kDa using an NCC-specific antibody. Higher molecular weight protein bands (possibly NCC dimers) can be detected above 250 kDa. i.b., immunoblot. B, representative immunoblot of total protein samples extracted from MDCKI-NCC cells grown until confluent on semi-permeable supports and treated with 10 μg/ml tetracycline for various time points. C, confocal laser micrographs (top, xy plane; bottom, xz plane) of MDCKI-NCC cells grown on semi-permeable supports labeled with mouse anti-FLAG (NCC, red) and anti-ZO-1 (blue). NCC labeling is observed intracellularly but also at the level of the tight junction complex. D, left, following immunoprecipitation (IP) of NCC (500 μg of lysate) using a rabbit FLAG antibody, the higher molecular mass bands above 250 kDa are more apparent. Control is noninduced MDCKI-NCC cells. Right, following PNGase treatment, both the higher and lower molecular weight protein bands are reduced in size, confirming that NCC exists as a complex glycosylated protein in MDCKI-NCC cells. Temperatures refer to those used for denaturation of the protein sample prior to PNGase treatment. E, expression of NCC and various genes involved in NCC regulation in MDCKI-NCC cells grown on semi-permeable supports. Conventional RT-PCR analysis of RNA extracted from MDCKI-NCC cells with primers specific for NCC, OSR1, SPAK, WNK1, WNK4, WNK3, and V2R.
Biotin Internalization Assay
Cells were grown in complete DMEM on filter plates to confluency for several days. The cells were pretreated with inhibitors or vehicle for 20 min at 37 °C. Cells were biotinylated from the apical side. After quenching of the biotinylation reaction, surface expression controls were made by following the standard biotinylation protocol. The remaining cells were incubated in pure media with vehicle or inhibitors (160 μm dynasore (Sigma D7693), 30 μm PitStop2 (Abcam Biochemicals ab120687), 20 mm methyl-β-cyclodextrin (MβCD) (Sigma C4555), 0.4 mg/ml filipin complex (Sigma F9765), or 25 μg/ml nystatin (Sigma N4014)) for 0, 5, 15, 30, or 60 min at 37 °C to allow plasma membrane proteins to internalize. The endocytosis was stopped by rapidly cooling the cells using ice-cold PBS/CM. To strip biotin from proteins not internalized and therefore still on the cell surface, the cells were treated three times for 20 min with the membrane-impermeable reducing agent sodium-MES (MesNa, 200 mm) in (100 mm NaCl, 1 mm EDTA, 50 mm Tris, pH 8.6, 0.2% BSA). Excess MesNa was washed away with PBS/CM, and the reaction was quenched with 120 mm iodoacetic acid in PBS/CM. The cells were washed in PBS/CM and lysed in lysis buffer, and biotinylated proteins were isolated according to the surface biotinylation protocol. Biotinylated and total protein fractions were analyzed for NCC abundance by Western blotting and densitometry. The biotinylated samples were initially normalized to the total protein fraction. The stripped sample signal density at the 0-min internalization time point was then utilized as background signal for all other time points.
Immunoprecipitation (IP) Assay
Cells were grown in complete DMEM on filter plates to confluency for several days. Cells were lysed in 20 mm Tris, 150 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, 20 mm N-ethylmaleimide, pH 7.4, containing protease inhibitors leupeptin and Pefabloc and phosphatase inhibitor mixture tablets (PhosSTOP, Roche Diagnostics). Following extensive sonication, samples were centrifuged at 10,000 × g for 10 min at 4 °C. IP of the lysates was performed at 4 °C overnight in a total volume of 500 μl using 2 μg of rabbit FLAG antibody preincubated for 2 h in room temperature with 20 μl of protein A-agarose (Santa Cruz Biotechnology), washed three times with lysis buffer, and eluted with 200 μg/ml FLAG peptide solution (Sigma) in TBS (10 mm Tris-HCl, 150 mm NaCl, pH 7.4). In some experiments, cells were biotinylated from the apical surface, purified using NeutrAvidin gel slurry (Pierce), and eluted using 50 mm DTT at room temperature for 1 h. These samples were subsequently utilized in IP experiments.
Statistics
One-way and two-way analyses of variance or Student's unpaired t tests were used for statistical comparisons where appropriate. Values were considered statistically significant at p < 0.05.
RESULTS
Generation and Characterization of an MDCK Cell Line with Tetracycline-inducible NCC Expression
We initially generated a suitable polarized kidney cell line model to study trafficking of NCC and the role of various mutations in NCC for modulation of NCC function. We generated various MDCKI cell lines with tetracycline-inducible NCC expression and characterized them based on cell morphology, the degree of NCC expression and glycosylation, and polarized trafficking of NCC to the apical plasma membrane. From these lines, an individual clonal line MDCKI-FRT-TO-N-term-FLAG-rNCC (termed MDCKI-NCC from this point) was utilized for the remainder of the studies. Time- and dose-response assays of NCC induction demonstrated robust NCC expression after 16 h (Fig. 1, A and B). In the MDCKI-NCC cells, NCC in total cell extracts was detected as various protein moieties as follows: a protein smear centered at 140 kDa, a single protein entity at ∼100 kDa, and an equivalent banding pattern at higher molecular weights. NCC labeling was observed both intracellularly and at the apical pole of MDCKI-NCC cells using immunofluorescence and confocal microscopy (Fig. 1C). Immunoprecipitation of NCC using FLAG antibodies followed by immunoblotting emphasizes the molecular weights of NCC in the MDCKI-NCC cells (Fig. 1D). PNGase F treatment of protein homogenates shifted the observed molecular weights, demonstrating that in our MDCKI-NCC cell, similar to what is observed for NCC in vivo (18), NCC exists as a highly glycosylated protein and an unglycosylated form (Fig. 1D). RT-PCR of the MDCKI-NCC cells demonstrated the presence of several components of signaling cascades involved in NCC regulation in the DCT (Fig. 1E). Cell surface biotinylation of confluent MDCKI-NCC cell monolayers showed NCC to be abundantly expressed in the apical plasma membrane (Fig. 2A). In the biotinylated fraction, NCC was also detected as higher molecular weight moieties (>200 kDa, data not shown), indicative of NCC as a functional dimer or as a larger protein complex.
FIGURE 2.

NCC is constitutively internalized from the apical plasma membrane. A, representative Western blots showing NCC internalization in MDCKI-NCC cells grown on semi-permeable supports. Following biotinylation, MDCKI-NCC cells were incubated for 5 min (7th to 9th lanes), 15 min (10th to 12th lanes), 30 min (13th to 15th lanes), or 60 min (16th to 18th lanes) at 37 °C to allow internalization before treatment with the reducing agent MesNa (stripping biotin). Top blot, 1st to 3rd lanes show expression of steady-state surface NCC. The 4th to 6th lanes show surface NCC after treatment with the reducing agent MesNa. Internalized NCC was isolated, detected by Western blot, and quantified by densitometry. Note: all biotinylated samples are run on the same SDS-polyacrylamide gel, and all total samples are run on the same SDS-polyacrylamide gel, so a direct comparison between the biotinylated and total protein pool to estimate relative abundances is not possible. This applies to all subsequent figures. Bottom blot, lanes show total NCC abundance from corresponding samples. B, semi-quantitative assessment of the percentage of internalized NCC (steady-state surface levels equals 100%), showing a significant increase in NCC endocytosis over time. Data are means ± S.E. (n = 3). * represents significant change compared with 0-min time point. C, confocal laser micrographs (top, xy plane; bottom, xz plane) of MDCKI-NCC cells grown on semi-permeable supports and biotinylated from the apical or basolateral surface and labeled with fluorophore-conjugated streptavidin demonstrating side-specific membrane biotin labeling. D, immunoblotting of apically biotinylated MDCKI-NCC cells detects proteasome 20 S, a marker of intracellular proteins, only in the total protein pool. The Gp135/podocalyxin, an apical plasma membrane marker, is enriched in the biotinylated protein pool but is also detected in lower quantities in the total protein pool.
NCC Undergoes Constitutive Endocytosis in MDCKI Cells
Surface NCC levels at steady-state regulate basal NaCl reabsorption in the DCT, yet whether NCC can be transported to the plasma membrane via a constitutive trafficking pathway alongside a regulated pathway remains unknown. Therefore, we investigated whether NCC is present in the apical cell surface without any acute stimulation and whether this pool of NCC is constitutively internalized from the plasma membrane. A time course using biotin-based internalization assays on polarized MDCKI-NCC cells (biotinylation from the apical side) demonstrated that NCC was constitutively endocytosed (Fig. 2, A and B). A significant increase in the internalized pool of NCC was observed after 15 min, which reached a maximum after 30 min. The reduced quantity of NCC in the biotinylated fraction after 60 min (relative to 30 min) suggests that either biotinylated NCC is being degraded at later time points or that NCC is recycled back to the apical plasma membrane. No significant differences were observed in the total pool of NCC at any time point examined (Fig. 2A). Specificity of the site-specific biotinylation of MDCKI-NCC cells was performed using fluorescent streptavidin (Fig. 2C). Furthermore, no proteasome 20 S was observed in the biotinylated samples, demonstrating specificity of the assay for plasma membrane proteins (Fig. 2D).
Characterization of Constitutive NCC Endocytosis Pathways Using Chemical Inhibition
In our MDCKI-NCC cell line, various proteins implicated in clathrin-mediated or caveolae-mediated endocytotic pathways were detected, including clathrin heavy chain, caveolin-1, flotillin-1, and flotillin-2 (data not shown). These proteins were also detected in purified mouse DCT cells (data not shown). Thus, it was plausible that NCC could be internalized via clathrin-mediated or caveolae-mediated pathways. To identify the pathway of constitutive NCC internalization, we utilized various inhibitors of dynamin-dependent, clathrin-dependent, and caveolae-mediated internalization pathways. To characterize inhibitor efficacy and to determine a suitable concentration for use in our MDCKI-NCC cell line, uptakes of fluorescent transferrin (clathrin-mediated pathway), LacCer (clathrin-independent pathway), or dextran (fluid phase endocytosis) were examined (Figs. 3 and 4). Transferrin, dextran, and LacCer uptakes into MDCKI-NCC cells increased in a time-dependent manner under control conditions (Figs. 3 and 4). Compared with controls, dynasore, an inhibitor of dynamin GTPase and thus dynamin-dependent endocytosis, significantly decreased transferrin uptake within 15 min (Fig. 3A). Dynasore had no significant effect on dextran uptake (Fig. 3B). PitStop2, a molecule developed to specifically inhibit clathrin-mediated endocytosis but also capable of decreasing clathrin-independent pathways (28), significantly decreased transferrin uptake but had no significant effect on dextran uptake (Fig. 3). Pitstop2 had no significant effect on LacCer uptake (Fig. 4). MβCD, which selectively removes cholesterol from membranes and disrupts formation of clathrin-coated pits and caveolae invaginations, significantly decreased transferrin uptake after 30 min and had no significant effect on dextran internalization (Fig. 3). MβCD had a profound effect on LacCer, significantly decreasing uptake at both 15- and 30-min time points (Fig. 4). Transferrin and dextran uptakes were not affected by nystatin or filipin (Fig. 3), cholesterol-sequestering compounds that inhibit caveolin-mediated internalization. However, nystatin decreased the internalization of LacCer after 15 or 30 min incubation (Fig. 4). Fluorescent transferrin uptake assays were supported by immunoblotting (Fig. 5).
FIGURE 3.
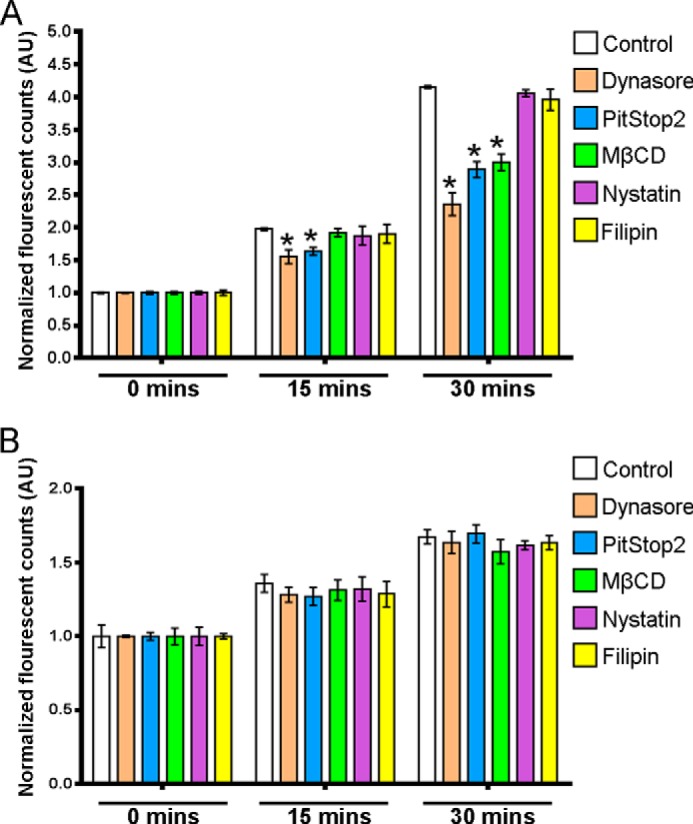
Characterization of clathrin-dependent and fluid phase endocytosis inhibitors in MDCKI-NCC cells. Internalization assays of fluorescent transferrin (marker for clathrin-mediated endocytosis) or dextran (marker of fluid phase internalization) in MDCKI-NCC cells were grown on plastic. A, internalization of transferrin after a 15- and 30-min incubation time is significantly decreased by 160 μm dynasore and 30 μm PitStop2. 20 mm MβCD significantly reduces transferrin endocytosis after 30 min. 0.4 mg/ml filipin or 25 μg/ml nystatin have no significant inhibitory effect on transferrin uptake. B, 160 μm dynasore, 30 μm PitStop2, 20 mm MβCD, 0.4 mg/ml filipin, or 25 μg/ml nystatin have no significant effect on dextran endocytosis. Data are means ± S.E. (n = 3). * represents significant change compared with control treatment at same time point.
FIGURE 4.
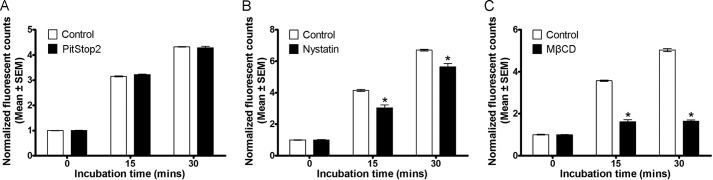
Characterization of clathrin-independent endocytosis inhibitors in MDCKI-NCC cells. Internalization assay of LacCer (BODIPY® FL C5-lactosylceramide complexed to BSA) (marker for clathrin-independent endocytosis) in MDCKI-NCC cells grown on plastic. A, 30 μm PitStop2 has no significant effect on the internalization of LacCer. B, internalization of LacCer after 15 and 30 min is significantly reduced following 25 μg/ml nystatin treatment. C, LacCer endocytosis is significantly inhibited at all time points examined with 20 mm MβCD. Data are means ± S.E. (n = 3). * represents significant change compared with control treatment at same time point.
FIGURE 5.

Dynasore and PitStop2, but not filipin and nystatin, inhibit internalization of human transferrin in MDCKI-NCC cells. Internalization assays of fluorescent transferrin (marker for clathrin-mediated endocytosis) in MDCKI-NCC cells grown on plastic. hTf was added to the cells and subjected to internalization by incubation at 37 °C for 0, 15, or 30 min. Unspecifically bound or nonendocytosed transferrin was removed by acid wash. Representative immunoblots and quantitative assessment of duplicate samples are shown. 160 μm dynasore and 30 μm PitStop2 but not 0.4 mg/ml filipin or 25 μg/ml nystatin decreased internalization of transferrin after 30 min. Data are means ± S.E. * represents significant change compared with control treatment at same time point.
To investigate whether the chemical inhibitors increased accumulation of NCC at the apical surface in the MDCKI-NCC cells, apical surface biotinylation of cells pretreated with the inhibitors was performed. PitStop2 (30 μm), dynasore (160 μm), and MβCD (20 mm) significantly increased steady-state surface expression of NCC by 41, 60, and 75%, respectively (Fig. 6, A–F). 10 mm MβCD also had a small but significant cumulative effect on surface abundance of NCC of 9% (data not shown). In contrast, nystatin (25 μg/ml) and filipin (0.4 mg/ml) had no significant effect on the apical plasma membrane abundance of NCC (Fig. 6, G–J). These data indicate that in the absence of acute stimulation, inhibition of clathrin-mediated endocytosis increases NCC plasma membrane abundance.
FIGURE 6.

PitStop2, dynasore, and MβCD, but not nystatin or filipin, increase steady-state surface abundance of NCC. A, C, E, G, and I, representative Western blots showing steady-state levels of NCC in the apical plasma membrane of MDCKI-NCC cells grown on semi-permeable supports and treated with vehicle or 30 μm PitStop2 (A), 160 μm dynasore (C), 20 mm MβCD (E), 25 μg/ml nystatin (G), or 0.4 mg/ml filipin (I). Top blots, apical plasma membrane NCC abundance (biotinylated pool). Bottom blots, total NCC abundance. Blots are cropped to fit the figure, but samples are run on the same SDS-PAGE. B, D, F, H, and J, quantitative assessment of steady-state levels of NCC at the apical membrane. 30 μm PitStop2 (B), 160 μm dynasore (D), or 20 mm MβCD (F) significantly increase steady-state apical membrane levels of NCC. 25 μg/ml nystatin (H) or 0.4 mg/ml filipin (J) have no significant effect on the steady-state levels of NCC in the apical membrane. Data are means ± S.E. (n = 6). * represents significant change compared with control treatment.
To confirm that the increased accumulation of NCC on the cell surface was due to decreased endocytosis, biotin-based internalization assays were performed in the presence of inhibitors. In all experiments, as shown previously (Fig. 2, A and B), NCC undergoes constitutive endocytosis over time, with ∼20% of the initial membrane pool of NCC internalized within 30 min. PitStop2 (30 μm) significantly inhibited NCC internalization by 90% after 5 min, 72% after 15 min, and 64% after 30 min (Fig. 7, A and B). Dynasore (160 μm) significantly decreased the internalized pool of NCC at the time points examined (Fig. 7, C and D). MβCD (20 mm) significantly decreased NCC internalization by 83% after 15 min and 82% after 30 min (Fig. 7, E and F). 10 mm MβCD also significantly decreased NCC internalization at the same time points but to a lesser degree (60 and 67% after 15 and 30 min, respectively) (data not shown). In line with the apical surface biotinylation assays, inhibition of the caveolin-mediated pathway with filipin complex or nystatin did not have any significant effect on NCC internalization (Fig. 8). The intracellular protein proteasome 20 S was not detected in any of the biotinylated fractions (data not shown).
FIGURE 7.
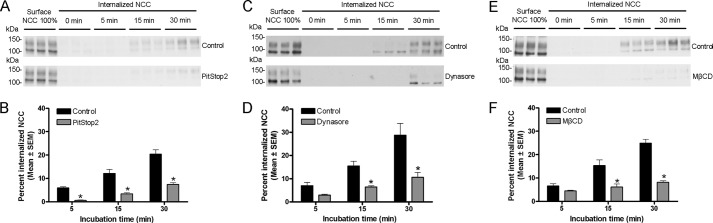
PitStop2, dynasore, and MβCD inhibit NCC internalization. A, C, and E, representative Western blots showing NCC internalization in MDCKI-NCC cells grown on semi-permeable supports and treated with vehicle or 30 μm PitStop2 (A), 160 μm dynasore (C), or 20 mm MβCD (E). Following biotinylation, MDCKI-NCC cells were incubated for 5 min (7th to 9th lanes), 15 min (10th to 12th lanes), or 30 min (13th to 15th lanes) at 37 °C to allow internalization before treatment with the reducing agent MesNa (stripping biotin). Top blots, 1st to 3rd lanes show expression of steady-state surface NCC. 4th to 6th lanes show surface NCC after treatment with the reducing agent MesNa. Internalized NCC was isolated, detected by Western blot, and quantified by densitometry. Bottom blots, similar setup but following PitStop2, dynasore, or MβCD treatment. B, D, and F, semi-quantitative assessment of the percentage of internalized NCC (steady-state surface levels equals 100%) with vehicle or 30 μm PitStop2 (B), 160 μm dynasore (D), or 20 mm MβCD (F) treatment. PitStop2, dynasore, and MβCD significantly reduce internalization of NCC. Data are means ± S.E. (n = 6). * represents significant change compared with control treatment at same time point.
FIGURE 8.
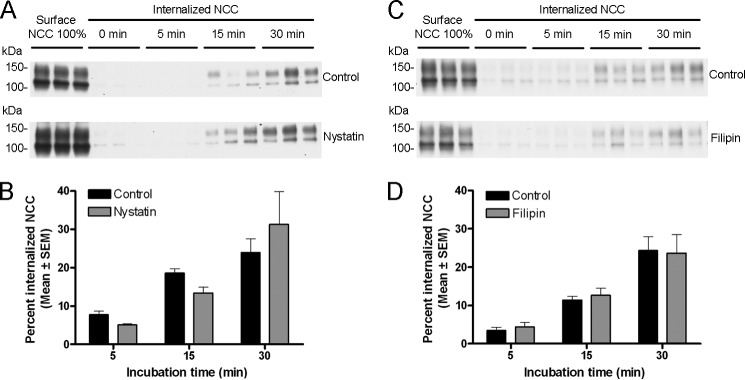
Nystatin and filipin do not significantly change internalization of NCC. A and C, representative Western blots showing NCC internalization in MDCKI-NCC cells grown on semi-permeable supports and treated with vehicle or 25 μg/ml nystatin (A) or 0.4 mg/ml filipin (C). Following biotinylation, MDCKI-NCC cells were incubated for 5 min (7th to 9th lanes), 15 min (10th to 12th lanes), or 30 min (13th to 15th lanes) at 37 °C to allow internalization before treatment with the reducing agent MesNa (stripping biotin). Top blots, 1st to 3rd lanes show expression of steady-state surface NCC. 4th to 6th lanes show surface NCC after treatment with the reducing agent MesNa. Internalized NCC was isolated, detected by Western blot, and quantified by densitometry. Bottom blots, similar setup but following nystatin or filipin treatment. B and D, semi-quantitative assessment of the percentage of internalized NCC (steady-state surface levels equals 100%) with vehicle or 25 μg/ml nystatin (B) or 0.4 mg/ml filipin (D). Nystatin and filipin have no significantly effect on internalization of NCC. Data are means ± S.E. (n = 6).
NCC Localizes within Clathrin-coated Pits and Clathrin-coated Vesicles in Rat DCT
The results from the inhibitor studies (Figs. 6–8) indicate that NCC is internalized from the apical plasma membrane of MDCKI cells via a clathrin-mediated endocytic pathway. To find evidence of a similar pathway in vivo, we examined the localization of NCC in rat kidney DCT cells using immunogold electron microscopy. In DCT cells, gold particles representing NCC were observed in structures morphologically resembling clathrin-coated endocytic invaginations (coated pits) and clathrin-coated vesicles (Fig. 9), suggesting that our observations in MDCKI-NCC cells closely match the in vivo situation. No caveolae were observed in the apical domain of rat DCT cells.
FIGURE 9.
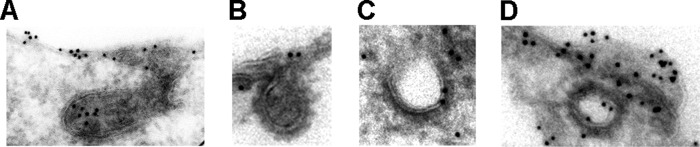
NCC localizes to clathrin-coated pits and vesicles in rat DCT cells. Immunogold electron micrographs demonstrating NCC in structures resembling clathrin-coated pits (A and B) and clathrin-coated vesicles (C and D) in the apical domain of rat DCT cells. Gold particle diameter, 10 nm.
Phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 Is Important for NCC Constitutive Internalization
Phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 is the essential mediator of NCC function (16, 17). Recent studies have implied that plasma membrane abundance of NCC can be regulated by phosphorylation of NCC at these sites (29), but it has not been demonstrated whether this effect of phosphorylation is due to increased NCC exocytosis or decreased endocytosis. As NCC phosphorylation in vivo occurs directly in the plasma membrane (18), it is likely that phosphorylation does not play a role in forward trafficking (exocytosis) of NCC. We have previously shown that poly-phosphorylation of AQP2 results in accumulation of AQP2 at the cell surface by reducing the rate of AQP2 internalization from the plasma membrane (22). To examine whether phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 plays a similar role, we generated multiple inducible phospho-deficient (Ala) and phospho-mimicking (Asp) NCC mutant cell lines and examined constitutive NCC membrane accumulation via biotinylation. Pilot studies showed each cell line to be comparable in terms of NCC expression and NCC surface abundance; thereafter, individual clonal lines were utilized. Compared with WT-NCC, no significant difference was observed in the membrane abundance of T53A/T58A/S71A-NCC (1.00 ± 0.024 versus 1.076 ± 0.068). In contrast, compared with WT-NCC, there was a significantly greater cell surface abundance of T53D/T58D/S71D-NCC (1.00 ± 0.024 versus 1.36 ± 0.125, p < 0.05).
To assess whether increased accumulation of NCC on the cell surface was due to decreased endocytosis, biotin-based internalization assays were performed. In MDCKI-T53A/T58A/S71A-NCC cells, the rate of NCC internalization was similar to that observed previously for WT-NCC, with ∼20% of the membrane pool of NCC internalized after 30 min (Fig. 10). In contrast, mutant T53D/T58D/S71D-NCC was internalized significantly less after 15 and 30 min, reaching a maximum of ∼8% at the latter time point (Fig. 10B).
FIGURE 10.

Mimicking phosphorylation at Thr-53, Thr-58, and Ser-71 decreases the internalization of NCC from the apical plasma membrane of MDCKI-NCC cells grown on semi-permeable supports. A, representative Western blots showing NCC internalization in MDCKI-NCC cells expressing phospho-deficient (T53A/T58A/S71A) or phospho-mimicking (T53D/T58D/S71D) NCC mutants. Following biotinylation, MDCKI-NCC cells were incubated for 5 min (7th to 9th lanes), 15 min (10th to 12th lanes), or 30 min (13th to 15th lanes) at 37 °C to allow internalization before treatment with the reducing agent MesNa (stripping biotin). Top blot, 1st to 3rd lanes show expression of steady-state surface T53A/T58A/S71A-NCC. 4th to 6th lanes show surface NCC after treatment with the reducing agent MesNa. Internalized NCC was isolated, detected by Western blot, and quantified by densitometry. Bottom blot, similar setup for T53D/T58D/S71D-NCC. B, semi-quantitative assessments of the percentage of internalized T53A/T58A/S71A-NCC or T53D/T58D/S71D-NCC (steady-state surface levels equals 100%). T53D/T58D/S71D-NCC displays reduced NCC abundance in the internalized pool at 15 and 30 min compared with T53A/T58A/S71A-NCC. Data are means ± S.E. (n = 3). * represents significant change in T53D/T58D/S71D-NCC compared with T53A/T58A/S71A-NCC at same time point.
Hypotonic Low Chloride Increases Apical Membrane Abundance of NCC and Decreases NCC Internalization
The results from the mutational studies suggest that phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 is important for regulating the plasma membrane abundance of NCC by reducing NCC internalization. To confirm this regulatory role of phosphorylation without the use of NCC mutants, endogenous phosphorylation of NCC in MDCKI cells was induced using hypotonic low chloride (16, 17) followed by analysis of apical membrane abundance using biotin. In addition, increased phosphorylation of NCC at Thr-53, Thr-58 (18), and Ser-71 (30) has been demonstrated with acute arginine vasopressin stimulation in vivo. As arginine vasopressin predominantly signals through cAMP-mediated pathways, we also stimulated cells with either cAMP or forskolin (an activator of adenylyl cyclase) to address whether these stimuli would increase NCC phosphorylation. As demonstrated in Fig. 11, a significant increase in NCC phosphorylation at Thr-58 (both in the total pool and biotinylated pool) was observed after treatment with hypotonic low chloride stimulation, which was accompanied by a significantly greater apical membrane abundance of NCC. In contrast, cAMP or forskolin treatment did not increase NCC phosphorylation in the MDCKI-NCC cells, and neither had a significant effect on NCC membrane abundance (Fig. 11). Internalization assays demonstrated that the rate of NCC internalization was significantly less with low chloride stimulation after 15 and 30 min compared with control conditions (Fig. 12). Immunoblotting of the same samples with a Thr(P)-58NCC antibody confirmed the increase in phosphorylated NCC following hypotonic low chloride stimulation (Fig. 12C). However, no signal for Thr(P)-58NCC was detectable in the internalized fraction (even following long exposure), suggesting that the phosphorylated form of NCC was not internalized from the plasma membrane or is rapidly dephosphorylated during endocytosis.
FIGURE 11.
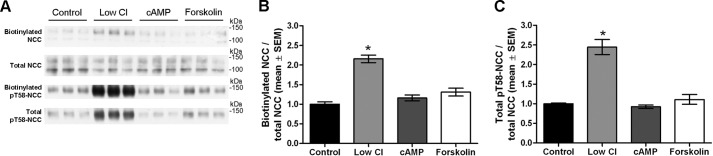
Hypotonic low chloride treatment of MDCKI-NCC cells increases NCC membrane abundance and Thr-58 phosphorylation. A, representative Western blot of samples from an apical surface biotinylation experiment of MDCKI-NCC cells grown on semi-permeable supports and treated with hypotonic low chloride, 25 μm forskolin, 50 μm (Sp)-cAMP, or control conditions. Biotinylated and total pools were blotted for total NCC and Thr(P)-58NCC. B, semi-quantitative assessment of apical membrane NCC abundance. Hypotonic low chloride conditions significantly increased surface levels of NCC. C, semi-quantitative assessment of total Thr(P)-58NCC levels. Thr(P)-58NCC abundance is significantly increased with hypotonic low chloride. Data are means ± S.E. (n = 6). * represents significant change compared with control treatment.
FIGURE 12.
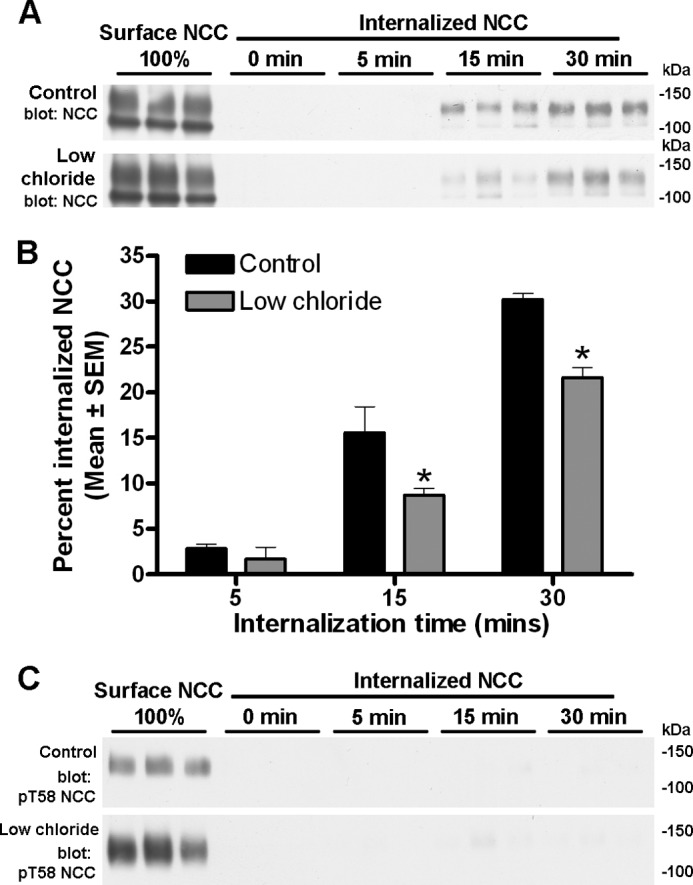
Hypotonic low chloride decreases NCC endocytosis in MDCKI-NCC cells grown on semi-permeable supports. A, representative western blots showing NCC internalization in MDCKI-NCC cells treated with vehicle or low chloride. Following biotinylation, MDCKI-NCC cells were incubated for 5 min (7th to 9th lanes), 15 min (10th to 12th lanes), or 30 min (13th to 15th lanes) at 37 °C to allow internalization before treatment with the reducing agent MesNa (stripping biotin). Top blot, 1st to 3rd lanes show expression of steady-state surface NCC. 4th to 6th lanes show surface NCC after treatment with MesNa. Internalized NCC was isolated, detected by Western blot, and quantified by densitometry. Bottom blot, similar set-up but following low chloride treatment. B, semi-quantitative assessments of the percentage of internalized NCC (steady-state surface levels equals 100%) with hypotonic low chloride or control conditions. Cells stimulated with hypotonic low chloride have reduced abundance of NCC in the internalized pool at 15 and 30 min compared with control conditions. Data are means ± S.E. (n = 6). * represents significant change compared with control treatment at same time point. C, representative Western blots of the same samples demonstrating no Thr(P)-58NCC in the internalized pool.
Phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 Decreases NCC Ubiquitylation at the Cell Surface
A recent study in COS7 cells demonstrated an inverse relationship between the levels of phosphorylated NCC at the cell surface and NCC ubiquitylation (29). To examine whether a similar mechanism occurs in our polarized NCC cell system and can account for the altered endocytosis rates observed under different conditions, MDCKI cells expressing WT-NCC or phospho-mutant NCC were apically biotinylated and the surface pool of proteins was utilized for NCC immunoprecipitation and subsequent examination of NCC ubiquitylation levels. Correlating with the decreased rates of NCC internalization (Figs. 10 and 12), the pool of ubiquitylated NCC isolated from MDCKI-NCC cells treated with low chloride or T53D/T58D/S71D-NCC cells was significantly lower compared with MDCKI-NCC cells under control conditions or T53A/T58A/S71A-NCC cells (Fig. 13). As before, the low chloride treatment resulted in increased NCC phosphorylation and greater steady-state abundance of NCC at the apical plasma membrane (Fig. 13B). Together, these data indicate that NCC phosphorylation regulates the levels of NCC ubiquitylation and in doing so can modulate the rate of NCC endocytosis.
FIGURE 13.
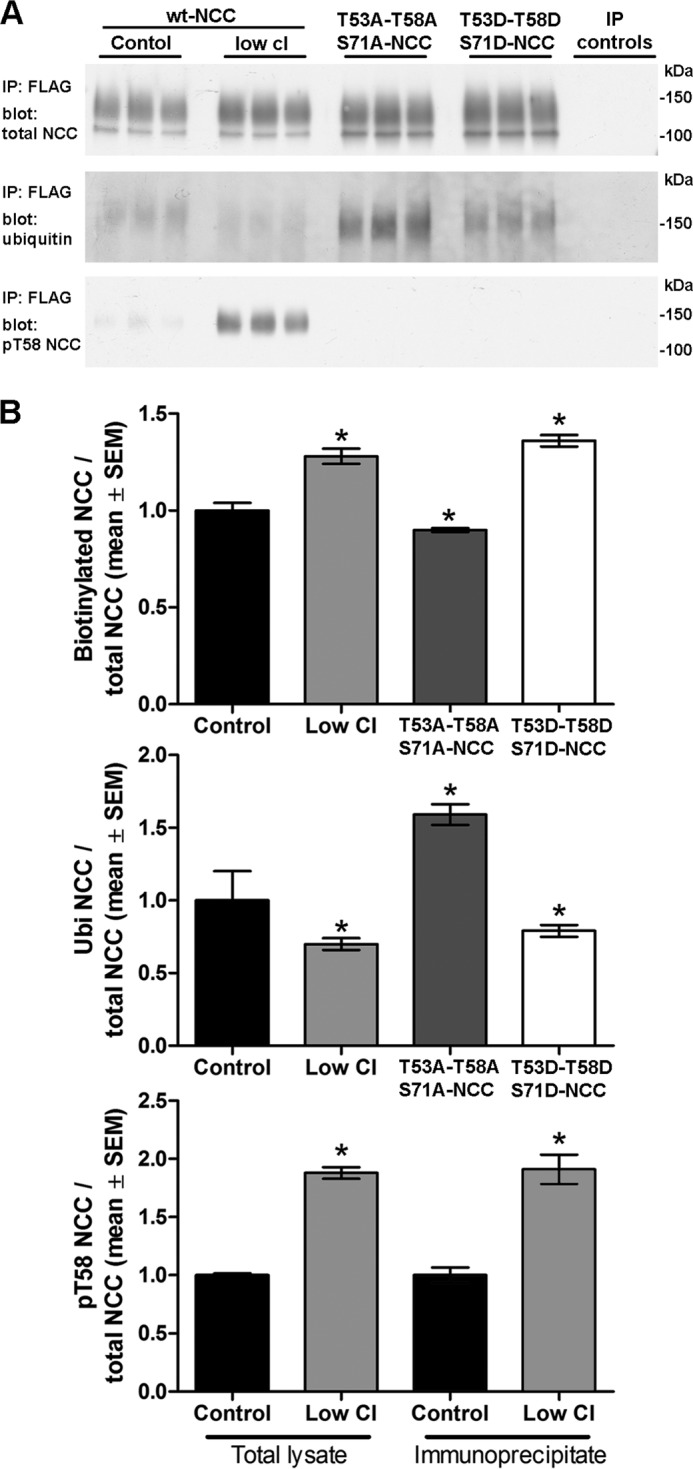
Phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 decreases NCC ubiquitylation at the cell surface. MDCKI-NCC cells grown on semi-permeable supports under control or after treatment with low chloride, T53D/T58D/S71D-NCC cells, and T53A/T58A/S71A-NCC cells were apically biotinylated, and the surface pool of proteins was utilized for NCC immunoprecipitation using FLAG antibodies. A, representative immunoblots of the levels of total NCC, ubiquitylated NCC, and Thr(P)-58NCC. The pool of ubiquitylated NCC isolated from MDCKI-NCC cells treated with low chloride or T53D/T58D/S71D-NCC cells was lower compared with MDCKI-NCC cells under control conditions or T53A/T58A/S71A-NCC cells. Control IPs are FLAG antibody without lysate, no antibody without lysate, and anti-AQP1 antibody with lysate. B, semi-quantitative assessments of the levels of surface (biotinylated) NCC, ubiquitylated NCC, and Thr(P)-58NCC under the various conditions (n = 6). Data are means ± S.E. * represents significant change compared with MDCKI-NCC control cells.
DISCUSSION
Modulation of NCC function is an essential mechanism for maintaining body sodium balance. Multisite phosphorylation of NCC has been demonstrated to be a key regulator of NCC transport activity (16, 17). The role of phosphorylation has focused predominantly on the direct activation of the cotransporter, with increased phosphorylation resulting in greater NaCl transport activity (16, 17). However, under certain conditions, NCC abundance has been shown to be increased or decreased in the apical plasma membrane of DCT cells in vivo (31, 32), which presumably also modulates the total NaCl transport capacity of this nephron segment. However, very little is known about whether phosphorylation plays a role in modulating this plasma membrane targeting of NCC and whether both forward trafficking (exocytosis), retrograde transport (endocytosis), or both mechanisms are involved in maintaining the surface pool of NCC. Furthermore, whether NCC can cycle to/from the plasma membrane in the absence of hormonal stimulation, similar to the water channel AQP2 (14), has not been examined. Thus, in this study we attempted to resolve the role of constitutive NCC trafficking for maintaining NCC surface expression, the role of NCC endocytosis and the internalization pathways of NCC, and the possible involvement of NCC phosphorylation and ubiquitylation in modulating these trafficking events.
Previous studies of NCC trafficking have been performed using immortalized DCT cell lines (33–36), stably transfected NCC-expressing MDCKII cell lines (26, 37), or transiently transfected cells such as COS and HEK (10, 11, 38). However, these cell lines are not ideal for studying trafficking events of NCC due to, for example, a lack of polarized delivery of NCC, absence of NCC complex glycosylation, and in the case of immortalized DCT cells the inability to assess the roles of site-directed mutations for NCC activity. Therefore, we generated novel polarized epithelial cell lines with inducible NCC expression based on tetracycline-inducible MDCKI cells containing FRT sites (27). The use of FRT sites ensures the same genomic insertion of a single copy of wild type or mutant NCC in the MDCKI cell, making a direct comparison between different NCC variants possible. As demonstrated in Fig. 1, these MDCKI-NCC cells demonstrated expression of a highly glycosylated form of NCC following tetracycline induction at low and high molecular weights (representing monomers and dimers, respectively), with abundant NCC expression in the apical plasma membrane. As NCC is associated with apical plasma membrane domains in DCT cells and exists as a highly glycosylated protein moiety in vivo that is important for both NCC activity and NCC trafficking to the plasma membrane (5, 39, 40), we consider our MDCKI-NCC cells an excellent model to study NCC trafficking events.
Under basal conditions, a pool of NCC was constitutively targeted to and accumulated in the apical plasma membrane. Despite repeated attempts (data not shown), we were unsuccessful in developing a satisfactory exocytosis assay to assess the role of forward trafficking events in this constitutive trafficking of NCC. However, we determined that in the absence of a hormonal stimulation, NCC was constitutively internalized from the apical plasma membrane. We consistently observed that 20–30% of the membrane pool of NCC was internalized after 30 min, which contrasts to a previous study that reported negligible constitutive NCC internalization after 20 min in mouse DCT cells (13). The reasons for this discrepancy are unknown, but the rates of constitutive endocytosis of NCC in our model are similar to those of NKCC2 in native TAL cells, where ∼25% of the surface NKCC2 was internalized after 30 min (41). In contrast, both the water channel AQP2 and the membrane K+ channel ROMK display a much more rapid internalization rate than NCC, with 40% of ROMK internalized within 7.5 min (42) and 40% of AQP2 being endocytosed within 30 min (22, 23). Taking into consideration technical differences between experiments and the use of different cell lines, it is still likely that the rates of constitutive endocytosis of renal membrane proteins are highly variable, suggesting that a variety of alternative regulatory mechanisms is involved and highlighting the different modes of major regulation namely trafficking versus transport activity.
A variety of pathways for membrane protein internalization has been characterized, including clathrin- or caveolae-mediated endocytosis, macropinocytosis, and clathrin- and caveolae-independent endocytosis (43, 44). Although a variety of proteins implicated in these pathways were detected in the MDCKI-NCC cells, the use of various pharmacological inhibitors indicated that constitutive NCC endocytosis in these cells occurs via a clathrin-mediated mechanism. In contrast, eliminating caveolae-dependent endocytosis reduced NKCC2 internalization in TAL segments (15). Although caveolae have been observed in mouse DCT cells in vivo (45) and various caveola-associated proteins were detected in isolated mouse DCT cells, NCC was only observed in clathrin-coated pits and vesicles in rat DCT cells using immunogold electron microscopy, thus supporting the conclusions from our MDCKI-NCC cell model and in accordance with the internalization pathways of various other renal apical membrane-associated sodium transport proteins (15, 46–49).
Several conserved phosphorylation sites in the cytoplasmic amino-terminal tail of NCC play vital roles in NCC regulation (16, 50, 51). Although the involvement of these sites for moderating NCC activity is well documented (16, 50, 51), little was known regarding their role in NCC membrane targeting. Expression of a T53D/T58D/S71D NCC mutant (mimicking constitutive phosphorylation at these residues) resulted in significantly increased levels of NCC in the apical membrane compared with expression of either WT-NCC or a T53A/T58A/S71A NCC mutant. These results were corroborated using hypotonic low chloride (16, 17) to induce endogenous phosphorylation of NCC. Our results verify recent findings reported for NCC phospho-mutants expressed in COS7 cells (29) and indicate that phosphorylation can modulate plasma membrane abundance of NCC. As phosphorylated NCC has been demonstrated to be only detectable in the apical plasma membrane of DCT cells in vivo (18, 19), we postulated that our results were due to a role of phosphorylation in reducing the internalization rate of NCC, rather than an effect on NCC forward trafficking. Indeed, inducing phosphorylation of NCC via low chloride treatment or mimicking phosphorylation using a T53D/T58D/S71D NCC mutant resulted in reduced rates of NCC internalization. The reduced internalization rate of phosphorylated NCC coincided with decreased levels of ubiquitylated NCC at the cell surface, indicating that NCC phosphorylation, either directly or indirectly, modulates NCC ubiquitylation. NCC is highly ubiquitylated, with up to 11 ubiquitylated residues (52). As two of these sites, Lys-79 and Lys-126, lie close to the known NCC phosphorylation sites, it would be informative to determine the effects of phosphorylation on the ubiquitin levels at these residues.
Our previous studies have demonstrated that NCC phosphorylated at Thr-53 or Thr-58 is only detected in the apical plasma membrane of DCT cells (18). In line with these observations, no phosphorylated NCC was detectable in the internalized pool of proteins either under basal conditions or following hypotonic low chloride stimulation. These results indicate a rapid dephosphorylation of NCC prior to or during the early stages of NCC endocytosis. It would be informative to assess whether the protein phosphatases PP1 or PP4, both of which have been implicated in NCC dephosphorylation (53, 54), interact with NCC directly in the apical plasma membrane of DCT cells.
In summary we have generated various novel polarized epithelial cell lines to study regulation of NCC. We demonstrate that NCC undergoes constitutive internalization from the apical plasma membrane via a clathrin-dependent pathway, which can modulate steady-state membrane abundance of NCC. Furthermore, phosphorylation of NCC at Thr-53, Thr-58, and Ser-71 plays a role in modulation of NCC surface abundance via regulation of NCC ubiquitylation and endocytosis.
Acknowledgments
We thank Christian Westberg, Tina Drejer, Inger-Merete Paulsen, Else-Merete Locke, and Helle Hoeyer for technical assistance.
This work was supported by the Danish Medical Research Council, the Lundbeck Foundation, the Novo Nordisk Foundation, the Carlsberg Foundation, and the Aarhus University Research Foundation.
- DCT
- distal convoluted tubule
- PNGase
- peptide:N-glycosidase
- MβCD
- methyl-β-cyclodextrin
- hTf
- human transferrin
- LacCer
- lactosylceramide
- IP
- immunoprecipitation
- MDCK
- Madin-Darby canine kidney cell
- MDCKI
- MDCK type I
- FRT
- Flp recombinase target.
REFERENCES
- 1. Gamba G. (2005) Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493 [DOI] [PubMed] [Google Scholar]
- 2. Plotkin M. D., Kaplan M. R., Verlander J. W., Lee W. S., Brown D., Poch E., Gullans S. R., Hebert S. C. (1996) Localization of the thiazide sensitive Na-Cl cotransporter, rTSC1 in the rat kidney. Kidney Int. 50, 174–183 [DOI] [PubMed] [Google Scholar]
- 3. Gamba G., Miyanoshita A., Lombardi M., Lytton J., Lee W. S., Hediger M. A., Hebert S. C. (1994) Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 269, 17713–17722 [PubMed] [Google Scholar]
- 4. de Jong J. C., Willems P. H., Mooren F. J., van den Heuvel L. P., Knoers N. V., Bindels R. J. (2003) The structural unit of the thiazide-sensitive NaCl cotransporter is a homodimer. J. Biol. Chem. 278, 24302–24307 [DOI] [PubMed] [Google Scholar]
- 5. Hoover R. S., Poch E., Monroy A., Vázquez N., Nishio T., Gamba G., Hebert S. C. (2003) N-Glycosylation at two sites critically alters thiazide binding and activity of the rat thiazide-sensitive Na(+):Cl(−) cotransporter. J. Am. Soc. Nephrol. 14, 271–282 [DOI] [PubMed] [Google Scholar]
- 6. Simon D. B., Nelson-Williams C., Bia M. J., Ellison D., Karet F. E., Molina A. M., Vaara I., Iwata F., Cushner H. M., Koolen M., Gainza F. J., Gitleman H. J., Lifton R. P. (1996) Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 12, 24–30 [DOI] [PubMed] [Google Scholar]
- 7. Mastroianni N., Bettinelli A., Bianchetti M., Colussi G., De Fusco M., Sereni F., Ballabio A., Casari G. (1996) Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am. J. Hum. Genet. 59, 1019–1026 [PMC free article] [PubMed] [Google Scholar]
- 8. Lemmink H. H., Knoers N. V., Károlyi L., van Dijk H., Niaudet P., Antignac C., Guay-Woodford L. M., Goodyer P. R., Carel J. C., Hermes A., Seyberth H. W., Monnens L. A., van den Heuvel L. P. (1998) Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. 54, 720–730 [DOI] [PubMed] [Google Scholar]
- 9. Monkawa T., Kurihara I., Kobayashi K., Hayashi M., Saruta T. (2000) Novel mutations in thiazide-sensitive Na-Cl cotransporter gene of patients with Gitelman's syndrome. J. Am. Soc. Nephrol. 11, 65–70 [DOI] [PubMed] [Google Scholar]
- 10. Subramanya A. R., Liu J., Ellison D. H., Wade J. B., Welling P. A. (2009) WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J. Biol. Chem. 284, 18471–18480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai H., Cebotaru V., Wang Y. H., Zhang X. M., Cebotaru L., Guggino S. E., Guggino W. B. (2006) WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 69, 2162–2170 [DOI] [PubMed] [Google Scholar]
- 12. Ko B., Joshi L. M., Cooke L. L., Vazquez N., Musch M. W., Hebert S. C., Gamba G., Hoover R. S. (2007) Phorbol ester stimulation of RasGRP1 regulates the sodium-chloride cotransporter by a PKC-independent pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 20120–20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ko B., Kamsteeg E. J., Cooke L. L., Moddes L. N., Deen P. M., Hoover R. S. (2010) RasGRP1 stimulation enhances ubiquitination and endocytosis of the sodium-chloride cotransporter. Am. J. Physiol. Renal Physiol. 299, F300–F309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu H., Sun T. X., Bouley R., Blackburn K., McLaughlin M., Brown D. (2004) Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am. J. Physiol. Renal Physiol. 286, F233–F243 [DOI] [PubMed] [Google Scholar]
- 15. Ares G. R., Ortiz P. A. (2012) Dynamin2, clathrin, and lipid rafts mediate endocytosis of the apical Na/K/2Cl cotransporter NKCC2 in thick ascending limbs. J. Biol. Chem. 287, 37824–37834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richardson C., Rafiqi F. H., Karlsson H. K., Moleleki N., Vandewalle A., Campbell D. G., Morrice N. A., Alessi D. R. (2008) Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 121, 675–684 [DOI] [PubMed] [Google Scholar]
- 17. Pacheco-Alvarez D., Cristóbal P. S., Meade P., Moreno E., Vazquez N., Muñoz E., Díaz A., Juárez M. E., Giménez I., Gamba G. (2006) The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 281, 28755–28763 [DOI] [PubMed] [Google Scholar]
- 18. Pedersen N. B., Hofmeister M. V., Rosenbaek L. L., Nielsen J., Fenton R. A. (2010) Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 78, 160–169 [DOI] [PubMed] [Google Scholar]
- 19. McCormick J. A., Mutig K., Nelson J. H., Saritas T., Hoorn E. J., Yang C. L., Rogers S., Curry J., Delpire E., Bachmann S., Ellison D. H. (2011) A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 14, 352–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi H., Asher C., Chigaev A., Yung Y., Reuveny E., Seger R., Garty H. (2002) Interactions of β and γ ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J. Biol. Chem. 277, 13539–13547 [DOI] [PubMed] [Google Scholar]
- 21. Shi H., Asher C., Yung Y., Kligman L., Reuveny E., Seger R., Garty H. (2002) Casein kinase 2 specifically binds to and phosphorylates the carboxy termini of ENaC subunits. Eur. J. Biochem. 269, 4551–4558 [DOI] [PubMed] [Google Scholar]
- 22. Moeller H. B., Praetorius J., Rützler M. R., Fenton R. A. (2010) Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 107, 424–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rice W. L., Zhang Y., Chen Y., Matsuzaki T., Brown D., Lu H. A. (2012) Differential, phosphorylation-dependent trafficking of AQP2 in LLC-PK1 cells. PLoS One 7, e32843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim G. H., Masilamani S., Turner R., Mitchell C., Wade J. B., Knepper M. A. (1998) The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc. Natl. Acad. Sci. U.S.A. 95, 14552–14557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fenton R. A., Brønd L., Nielsen S., Praetorius J. (2007) Cellular and subcellular distribution of the type-2 vasopressin receptor in the kidney. Am. J. Physiol. Renal Physiol. 293, F748–F760 [DOI] [PubMed] [Google Scholar]
- 26. Rosenbaek L. L., Assentoft M., Pedersen N. B., MacAulay N., Fenton R. A. (2012) Characterization of a novel phosphorylation site in the sodium-chloride cotransporter, NCC. J. Physiol. 590, 6121–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fröhlich O., Klein J. D., Smith P. M., Sands J. M., Gunn R. B. (2004) Urea transport in MDCK cells that are stably transfected with UT-A1. Am. J. Physiol. Cell Physiol. 286, C1264–C1270 [DOI] [PubMed] [Google Scholar]
- 28. Dutta D., Williamson C. D., Cole N. B., Donaldson J. G. (2012) Pitstop 2 is a potent inhibitor of clathrin-independent endocytosis. PLoS One 7, e45799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hossain Khan M. Z., Sohara E., Ohta A., Chiga M., Inoue Y., Isobe K., Wakabayashi M., Oi K., Rai T., Sasaki S., Uchida S. (2012) Phosphorylation of Na-Cl cotransporter by OSR1 and SPAK kinases regulates its ubiquitination. Biochem. Biophys. Res. Commun. 425, 456–461 [DOI] [PubMed] [Google Scholar]
- 30. Mutig K., Saritas T., Uchida S., Kahl T., Borowski T., Paliege A., Böhlick A., Bleich M., Shan Q., Bachmann S. (2010) Short-term stimulation of the thiazide-sensitive Na+-Cl− cotransporter by vasopressin involves phosphorylation and membrane translocation. Am. J. Physiol. Renal Physiol. 298, F502–F509 [DOI] [PubMed] [Google Scholar]
- 31. Sandberg M. B., Riquier A. D., Pihakaski-Maunsbach K., McDonough A. A., Maunsbach A. B. (2007) ANG II provokes acute trafficking of distal tubule Na+-Cl− cotransporter to apical membrane. Am. J. Physiol. Renal Physiol. 293, F662–F669 [DOI] [PubMed] [Google Scholar]
- 32. Sandberg M. B., Maunsbach A. B., McDonough A. A. (2006) Redistribution of distal tubule Na+-Cl− cotransporter (NCC) in response to a high-salt diet. Am. J. Physiol. Renal Physiol. 291, F503–F508 [DOI] [PubMed] [Google Scholar]
- 33. Ko B., Mistry A. C., Hanson L., Mallick R., Cooke L. L., Hack B. K., Cunningham P., Hoover R. S. (2012) A new model of the distal convoluted tubule. Am. J. Physiol. Renal Physiol. 303, F700–F710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gesek F. A., Friedman P. A. (1992) Mechanism of calcium transport stimulated by chlorothiazide in mouse distal convoluted tubule cells. J. Clin. Invest. 90, 429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peng K. C., Cluzeaud F., Bens M., Duong Van Huyen J. P., Wioland M. A., Lacave R., Vandewalle A. (1999) Tissue and cell distribution of the multidrug resistance-associated protein (MRP) in mouse intestine and kidney. J. Histochem. Cytochem. 47, 757–768 [DOI] [PubMed] [Google Scholar]
- 36. Markadieu N., San-Cristobal P., Nair A. V., Verkaart S., Lenssen E., Tudpor K., van Zeeland F., Loffing J., Bindels R. J., Hoenderop J. G. (2012) A primary culture of distal convoluted tubules expressing functional thiazide-sensitive NaCl transport. Am. J. Physiol. Renal Physiol. 303, F886–F892 [DOI] [PubMed] [Google Scholar]
- 37. Yang S. S., Yamauchi K., Rai T., Hiyama A., Sohara E., Suzuki T., Itoh T., Suda S., Sasaki S., Uchida S. (2005) Regulation of apical localization of the thiazide-sensitive NaCl cotransporter by WNK4 in polarized epithelial cells. Biochem. Biophys. Res. Commun. 330, 410–414 [DOI] [PubMed] [Google Scholar]
- 38. Zhou B., Zhuang J., Gu D., Wang H., Cebotaru L., Guggino W. B., Cai H. (2010) WNK4 enhances the degradation of NCC through a sortilin-mediated lysosomal pathway. J. Am. Soc. Nephrol. 21, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Jong J. C., Van Der Vliet W. A., Van Den Heuvel L. P., Willems P. H., Knoers N. V., Bindels R. J. (2002) Functional expression of mutations in the human NaCl cotransporter: evidence for impaired routing mechanisms in Gitelman's syndrome. J. Am. Soc. Nephrol. 13, 1442–1448 [DOI] [PubMed] [Google Scholar]
- 40. Kunchaparty S., Palcso M., Berkman J., Velázquez H., Desir G. V., Bernstein P., Reilly R. F., Ellison D. H. (1999) Defective processing and expression of thiazide-sensitive Na-Cl cotransporter as a cause of Gitelman's syndrome. Am. J. Physiol. 277, F643–F649 [DOI] [PubMed] [Google Scholar]
- 41. Ares G. R., Ortiz P. A. (2010) Constitutive endocytosis and recycling of NKCC2 in rat thick ascending limbs. Am. J. Physiol. Renal Physiol. 299, F1193–F1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fang L., Garuti R., Kim B. Y., Wade J. B., Welling P. A. (2009) The ARH adaptor protein regulates endocytosis of the ROMK potassium secretory channel in mouse kidney. J. Clin. Invest. 119, 3278–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mayor S., Pagano R. E. (2007) Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8, 603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Traub L. M. (2009) Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 10, 583–596 [DOI] [PubMed] [Google Scholar]
- 45. Fujimoto T. (1993) Calcium pump of the plasma membrane is localized in caveolae. J. Cell Biol. 120, 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shimkets R. A., Lifton R. P., Canessa C. M. (1997) The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J. Biol. Chem. 272, 25537–25541 [DOI] [PubMed] [Google Scholar]
- 47. Sun T. X., Van Hoek A., Huang Y., Bouley R., McLaughlin M., Brown D. (2002) Aquaporin-2 localization in clathrin-coated pits: inhibition of endocytosis by dominant-negative dynamin. Am. J. Physiol. Renal Physiol. 282, F998–F1011 [DOI] [PubMed] [Google Scholar]
- 48. Chow C. W., Khurana S., Woodside M., Grinstein S., Orlowski J. (1999) The epithelial Na+/H+ exchanger, NHE3, is internalized through a clathrin-mediated pathway. J. Biol. Chem. 274, 37551–37558 [DOI] [PubMed] [Google Scholar]
- 49. Huang H., Feng X., Zhuang J., Fröhlich O., Klein J. D., Cai H., Sands J. M., Chen G. (2010) Internalization of UT-A1 urea transporter is dynamin dependent and mediated by both caveolae- and clathrin-coated pit pathways. Am. J. Physiol. Renal Physiol. 299, F1389–F1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chiga M., Rai T., Yang S. S., Ohta A., Takizawa T., Sasaki S., Uchida S. (2008) Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int. 74, 1403–1409 [DOI] [PubMed] [Google Scholar]
- 51. Vallon V., Schroth J., Lang F., Kuhl D., Uchida S. (2009) Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiol. Renal Physiol. 297, F704–F712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wagner S. A., Beli P., Weinert B. T., Schölz C., Kelstrup C. D., Young C., Nielsen M. L., Olsen J. V., Brakebusch C., Choudhary C. (2012) Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell. Proteomics 11, 1578–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Glover M., Mercier Zuber A., Figg N., O'Shaughnessy K. M. (2010) The activity of the thiazide-sensitive Na+-Cl− cotransporter is regulated by protein phosphatase PP4. Can. J. Physiol. Pharmacol. 88, 986–995 [DOI] [PubMed] [Google Scholar]
- 54. Sorensen M. V., Grossmann S., Roesinger M., Gresko N., Todkar A. P., Barmettler G., Ziegler U., Odermatt A., Loffing-Cueni D., Loffing J. (2013) Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 83, 811–824 [DOI] [PubMed] [Google Scholar]