

Background: Sulfotransferases with distinct specificities act in sequence in the heparan sulfate biosynthetic pathway.
Results: The crystal structure of 2-O-sulfotransferase with bound substrate reveals its requirements for substrate recognition.
Conclusion: The 2-O-sulfotransferase recognizes N-sulfate but excludes 6-O-sulfate on substrates.
Significance: The results advance the understanding of cellular control for the biosynthesis of heparan sulfate.
Keywords: Heparan Sulfate, Heparin, Heparin-binding Protein, Oligosaccharide, Sulfotransferase
Abstract
Heparan sulfate (HS) is an abundant polysaccharide in the animal kingdom with essential physiological functions. HS is composed of sulfated saccharides that are biosynthesized through a complex pathway involving multiple enzymes. In vivo regulation of this process remains unclear. HS 2-O-sulfotransferase (2OST) is a key enzyme in this pathway. Here, we report the crystal structure of the ternary complex of 2OST, 3′-phosphoadenosine 5′-phosphate, and a heptasaccharide substrate. Utilizing site-directed mutagenesis and specific oligosaccharide substrate sequences, we probed the molecular basis of specificity and 2OST position in the ordered HS biosynthesis pathway. These studies revealed that Arg-80, Lys-350, and Arg-190 of 2OST interact with the N-sulfo groups near the modification site, consistent with the dependence of 2OST on N-sulfation. In contrast, 6-O-sulfo groups on HS are likely excluded by steric and electrostatic repulsion within the active site supporting the hypothesis that 2-O-sulfation occurs prior to 6-O-sulfation. Our results provide the structural evidence for understanding the sequence of enzymatic events in this pathway.
Introduction
Heparan sulfate (HS)3 is a highly sulfated polysaccharide that is widely present on the mammalian cell surfaces and in the extracellular matrix. HS participates in a wide range of physiological and pathophysiological functions, including embryonic development, inflammatory responses, blood coagulation, and assisting in viral/bacterial infections (1). HS consists of a disaccharide repeating unit with glucosamine and either glucuronic acid (GlcA) or iduronic acid (IdoA), each of which is capable of carrying sulfo groups. The position of the sulfo groups and the distributions of IdoA and GlcA units dictate the function of HS (2). A specialized biosynthetic pathway, involving multiple HS sulfotransferases and an epimerase, is required for the preparation of functional HS with elaborate sulfation patterns (3). The heparan sulfate 2-O-sulfotransferase (2OST) is a key enzyme in the HS biosynthetic pathway.
2OST is responsible for the transfers of a sulfo group to the 2-OH position of preferentially IdoA, and to a limited extent GlcA, within HS polysaccharides to form 2-O-sulfated iduronic acid (IdoA2S) or 2-O-sulfated glucuronic acid (GlcA2S). The IdoA2S unit is a common structural motif found in cellular HS and is required for binding several growth factors and is essential for triggering growth factor-mediated signal transduction pathways (4, 5). The critical physiological functions of IdoA2S residues have been revealed through numerous studies. For example, the 2OST-knock-out mice exhibit renal agenesis and die in the neonatal period. These animals also display retardation of eye development and skeletal over mineralization, demonstrating the essential role of 2OST in regulating embryonic development (6). Furthermore, decreased expression of 2OST leads to the reduction of neutrophil infiltration during acute inflammation in mice, implicating the function of IdoA2S motif in inflammatory responses (7). In Caenorhabditis elegans, 2OST has been implicated in axon migration/guidance and nervous system development (8, 9). Dependence on 2-O-sulfation is notably less critical in Drosophila. The 2OST−/− Drosophila meanogaster synthesizes HS with higher levels of 6-O-sulfation, which may compensate for the loss of 2-O-sulfation (10).
Understanding the biosynthesis of HS plays a significant role in developing a new generation of heparin, a commonly used anticoagulant drug (11). Heparin is a HS-like polysaccharide that is isolated from porcine intestine or bovine lung through a poorly regulated supply chain. The worldwide distribution of contaminated heparin in 2007 raised the concerns over the safety and reliability of animal-sourced heparins (12). A cost-effective method for producing pharmaceutical grade synthetic heparin is highly desirable. A chemoenzymatic approach to synthesize heparin and HS oligosaccharides is becoming a very promising approach to accomplish this goal (13–16). The new method has dramatically improved the synthesis efficiency and product purity. HS is also being investigated for possible use in the treatment of inflammation-related diseases and cancer (17). Better understanding of the enzymes used in the chemoenzymatic approach should aid in the production of heparin-based therapeutics tailored for specific disease targets.
The cellular mechanism regulating HS biosynthesis is largely unknown. Understanding the substrate specificities and mechanisms of HS biosynthetic enzymes is crucial for dissecting this fundamental process. In this article, we report a crystal structure of a ternary complex of 2OST with 3′-phosphoadenosine 5′-phosphate (PAP) and a heptasaccharide acceptor substrate. Numerous critical interactions between the enzyme and the heptasaccharide were identified, providing structural evidence for the substrate specificity of this enzyme. A series of structurally defined oligosaccharides were synthesized to examine the contribution of oligosaccharide size and functional groups to 2OST modification. Our results suggest that 2OST utilizes multiple structural elements to distinguish sulfation patterns of the substrate, controlling the order and extent of the 2-O-sulfation in the HS biosynthetic process.
EXPERIMENTAL PROCEDURES
Crystallization and Data Collection
Maltose-binding protein (MBP)-2OST was expressed and purified as described previously (18). Crystals of MBP-2OST were grown using the vapor diffusion sitting drop method by mixing 400 nl of MBP-2OST at 20 mg/ml in 25 mm Tris, pH 7.5, 75 mm NaCl, 5 mm maltose, 1 mm DTT, 4 mm PAP, and 10 mm heptasaccharide with 400 nl of the reservoir solution consisting of 17.9% PEG 4000, 60 mm sodium citrate, pH 5.6, 120 mm ammonium acetate, 10.5% glycerol, and 10 mm hexamine cobalt chloride. Prior to mounting the reservoir solution was switched to 25.5% PEG 4000, 15% glycerol, 85 mm sodium citrate, and 170 mm ammonium acetate. After 5.5 h of equilibration the crystal was harvested and frozen directly into liquid nitrogen. Data were collected to 3.45 Å on the SER-CAT bending magnet beamline at APS with a wavelength of 1 Å. Molecular replacement was carried out in PHENIX using the 2OST biological trimer and the individual MBP coordinates from Protein Data Bank ID code 3F5F as the search models (18, 19). The model was refined using multiple cycles of manual model building in Coot followed by refinement in PHENIX using minimization, individual ADP B-factor refinement, and torsion angle simulated annealing (Table 1) (19, 20). TLS (Translation/Libration/Screw) refinement was also used in later rounds of refinement. Noncrystallographic symmetry restraints were utilized between the 2OST molecules, saccharides E and F of the heptasaccharides bound and between the MBP molecules excluding loops that had different conformations in the density. The final model consisted of two trimers of the MBP-2OST fusion monomers labeled A–F. There is electron density for all six 2OST molecules, with each containing PAP. All monomers but monomer C contain density for the heptasaccharide substrate. There is strong density for three monomers of MBP with weak density for molecules B and F and no density for molecule A. The model was refined to a Rwork of 17.9% and Rfree 22.4% (Protein Data Bank ID code 4NDZ). The quality of the structure was analyzed with MolProbity (Table 1) (21).
TABLE 1.
Crystallographic data
| Parameters | |
| Unit cell | a = 155.72 Å, b = 170.69 Å, c = 183.96 Å; α = β = γ = 90° |
| Space group | P212121 |
| Resolution (Å) | 50.0-3.45 |
| No. of observations | 386,263 |
| Unique reflections | 64,322 |
| Rsym (%) (last shell)a | 10.7 (63.8) |
| I/σI (last shell) | 8.8 (2.3) |
| Mosaicity range | 0.39–0.56 |
| Completeness (%) (last shell) | 99.1 (93.8) |
| Refinement statistics | |
| Rwork (%)b | 17.6 |
| Rfree (%)c | 22.8 |
| Overall Wilson B value (Å) | 94.3 |
| B-factors (for molecule A) | |
| Protein | 88.3 |
| PAP | 72.1 |
| Heptasaccharide | 114.3 |
| Trisaccharides (D-E-F) | 83.9 |
| Root mean square deviation from ideal values | |
| Bond length (Å) | 0.005 |
| Bond angle (°) | 0.84 |
| Dihedral angle (°) | 11.2 |
| Ramachandran statisticsd | |
| Favored regions (98%) | 95.0 |
| Allowed regions (>99.8%) | 99.5 |
a Rsym = Σ (|Ii − <I>|)/Σ(Ii) where Ii is the intensity of the ith observation and <I> is the mean intensity of the reflection.
b Rwork = Σ‖Fo| − | Fc‖/Σ| Fo| calculated from working data set.
c Rfree was calculated from 5% of data randomly chosen not to be included in refinement.
d Ramachandran results were determined by MolProbity.
2-O-Sulfation Assay
Oligosaccharides (0.05 mg/ml) were incubated at 37 °C for 2 h with 2OST (0.2 mg/ml) and 35S-PAPS in 200 μl of MES buffer system. The reaction mixture was analyzed using a silica-based polyamine HPLC column. Determinations were carried out in duplicate.
Disaccharide Analysis
To determine the size of the 35S sulfation, the sample was subjected to nitrous acid degradation at pH 1.5 to produce 35S-labeled disaccharide as described by Shively and Conrad (22). The resultant 35S-labeled disaccharides were resolved using a C18 reverse-phase column (0.46 × 25 cm; Vydac) under reverse-phase ion-pairing HPLC conditions (23). The identities of the disaccharides were determined by co-elution with the appropriate 35S-labeled disaccharide standards.
Quantum Mechanics and Molecular Dynamics Calculations
Gaussian optimization calculations were carried out for the trisaccharide unit, GlcNS-IdoA-GlcNS, with the central IdoA unit in the 4C1, 1C4, and 2S0 conformations, as well as replaced by a GlcA in the 4C1 conformation. All optimizations were done at the ωB97-XD level with the 6-31+g(d,p) basis set. We also included the polarization continuum model with water as the solvent in all calculations. The four optimized structures yield energies within a range of 6.8 kcal/mol for sulfated systems in which 4C1 and 1C4 displayed similar energies whereas 2S0 was less favored by 6.8 kcal/mol. In the nonsulfated cases, 4C1 and 2S0 have similar energies and 1C4 is less favored by only 3.3 kcal/mol. In both cases when GlcA is substituted for IdoA, the trisaccharides tend to become less favorable in energy (by 4.2 kcal/mol compared with 4C1 in the sulfated case and by 9.7 kcal/mol 4C1 in the nonsulfated case).
Using molecular dynamics, solution structures of the HS heptasaccharide fragment in its free and 2OST bound forms were generated. The initial structure of the HS bound to the 2OST was based on the x-ray crystal structure. Necessary conformation changes (from 4C1 to 1C4 and 2S0) were introduced to the acceptor IdoA as well as substitution with a GlcA in a 4C1 conformation. The models were first energy-minimized in a vacuum using the program Amber.12 (24). Next, each optimized configuration was solvated in a box of water (8,113 water molecules in 2OST-free systems and 24,121 water molecules in 2OST-bound systems). Prior to equilibration, all systems were subjected to 1) 1-ns belly dynamics runs with fixed HS (or HS/2OST), 2) minimization, 3) low temperature constant pressure dynamics at fixed HS (or HS/2OST) to assure a reasonable starting density, 4) minimization, 5) stepwise heating molecular dynamics at constant volume, and 6) constant volume molecular dynamics for 1 ns. All final unconstrained trajectories were calculated at 300 K under constant volume (25 ns, time step 1 fs) using PMEMD (Amber.12) to accommodate long range interactions (24). The protein parameters were taken from the FF12 force field (24). Charges for the sugars were obtained using the Gaussian (25) calculations of appropriate fragments at the ωB97-XD level with the higher extended 6-311++g(d,p) basis set (with the polarization continuum model with water as the solvent) (supplemental Fig. S1).
Preparation of Oligosaccharide Substrates
Preparation of the substrates followed essentially the same procedures described in our previous work (16, 26). All oligosaccharides were elongated from p-nitrophenol (PNP) glucuronide (GlcA-PNP), which was purchased from Sigma. Briefly, GlcA-PNP (0.20 mg/ml) was incubated with UDP-GlcNTFA (0.50 mg/ml) or UDP-GlcNAc (0.46 mg/ml) and KfiA (N-acetyl glucosaminyl transferase of Escherichia coli K5 strain, 0.05 mg/ml) in buffer containing 25 mm Tris-HCl, pH 7.2, and 10 mm MnCl2 for 24 h at room temperature. The reaction was monitored using a polyamine-based HPLC column. Upon the complete consumption of UDP-GlcNTFA/GlcNAc, UDP-GlcA (0.44 mg/ml), and PmHS2 (Pasteurella multocida heparosan synthase 2, 0.05 mg/ml) were added, and the reaction was incubated for another 24 h at room temperature. The reaction was purified using a C18 column (0.75 × 20 cm; Biotage), and product eluted with a linear gradient of 0–100% methanol in H2O and 0.1% TFA over 60 min at a flow rate of 2.0 ml/min. The trisaccharide product was determined by exploiting the UV absorbance of the PNP tag at 310 nm, and the identity of the product was confirmed by electrospray ionization-MS. Further elongation to the desired size of oligosaccharides was achieved by an additional reaction with the incubation of UDP donors, KfiA, or PmHS2 following the same procedure as described above.
Oligosaccharide backbones (1∼2 mg) were dried and resuspended in 0.1 m LiOH solution (20 ml). The reaction was incubated on ice for 2 h and then stopped by adjusting the pH to 7.0. N-Sulfation of oligosaccharides was carried out by incubating the de-NTFA oligosaccharide substrates with N-sulfotransferase and PAPS. The reaction mixture typically contained 1∼2 mg of de-NTFA oligosaccharide, 500 μm PAPS, 50 mm MES, pH 7.0, and 1 mg of N-sulfotransferase in a total volume of 15 ml. The reaction mixture was incubated overnight at 37 °C.
The N-sulfated oligosaccharides were incubated overnight at 37 °C with C5-epimerase in 50 mm MES buffer, pH 7.0, containing 2 mm CaCl2. To get the IdoA2S saccharide, the oligosaccharides were incubated overnight at 37 °C with C5-epimerase, 2OST, and PAPS in a buffer containing 50 mm MES, pH 7.0, and 2 mm CaCl2. For 6-O-sulfation, the reaction mixture contained 6-O-sulfotransferases (6OST-1 and 6OST-3) and PAPS. Each of the reactions was incubated overnight at 37 °C and then purified by Q-Sepharose FF column (GE Healthcare).
Site-directed Mutagenesis
The 2OST mutants were prepared using MBP-2OST plasmid as the template and a method from the Stratagene QuikChange mutagenesis protocol. The mutagenesis primers were synthesized by Invitrogen. The resultant constructs were sequenced to confirm the expected mutation (University of North Carolina at Chapel Hill Genome Sequencing Facility). The expression of mutants was carried out in Origami B cells (Novagen) using a protocol described previously (18). The mutant proteins were purified by an Amylose-agarose column (New England Biolabs). All mutants described in this study, including Y94A, H106A, R184A, R189A, and K354A, have an expression level very comparable with that of wild type protein, as determined by SDS-PAGE analysis.
Activity Measurement of 2OST Mutants Using Substrates 3 and 4
The procedures for the analysis of 2OST mutants were essentially identical to those described previously (18), with the exception of using two structurally defined oligosaccharide substrates, compounds 3 and 4. To use compound 3 as a substrate, the substrate (0.1 mg/ml) was incubated with the mutant protein (0.1 mg/ml) and 35S-PAPS (1 × 106 cpm) in 25 mm MES buffer, pH 7.0. The reaction was incubated at 37 °C for 30 min and was terminated by heating at 100 °C for 2 min. The reaction was then analyzed by silica-based polyamine HPLC. To use compound 4 as a substrate, the substrate (0.1 mg/ml) was incubated with the mutant protein (0.1 mg/ml) and 35S-PAPS (1 × 106 cpm) in 25 mm MES buffer, pH 7.0. The reaction was incubated at 37 °C for 5 min and was terminated by heating at 100 °C for 2 min.
Mass Spectrometry Analysis
MS analysis was performed on a Thermo LCQ-Deca. The oligosaccharides were dissolved in H2O and injected via direct infusion (40 μl/min) into the instrument. The experiments were performed utilizing a negative ionization mode with a spray voltage of 3 kV and a capillary temperature of 160 °C. The MS data were acquired and processed using Xcalibur 1.3 software.
NMR Analysis of Heptasaccharide Substrate
The structure of oligosaccharides constructs and intermediates were analyzed by NMR experiments, including one-dimensional (1H and 13C) and two-dimensional (1H-1H COSY, TOCSY, and 1H-13C HSQC) NMR. NMR experiments were performed at 298 K with a Varian Inova 500 MHz spectrometer equipped with 5-mm triple resonance XYZ or broadband PFG probe and processed by VnmrJ 2.2D software. Samples (2 mg) were each dissolved in 0.5 ml of D2O (99.994%; Sigma) and lyophilized three times to remove the exchangeable protons. The samples were redissolved in 0.5 ml of D2O and transferred to NMR microtubes (OD 5 mm; Norrell). One-dimensional 1H NMR experiments were performed with 256 scans and an acquisition time of 768 ms. One-dimensional 13C NMR experiments were performed with 40,000 scans, 1.0-s relaxation delay, and an acquisition time of 1000 ms. Two-dimensional (1H-1H COSY, TOCSY, and 1H-13C HSQC) spectra were recorded with carbon decoupling, 1.5-s relaxation delay, during 204-ms acquisition time with 500 increments for 48 scans.
Taking advantage of one-dimensional and two-dimensional NMR experiments, we were able to assign anomeric protons and chemical shifts of saccharide E with GlcA or IdoA in the heptasaccharides. At low field region of compound 11 spectrum, the anomeric protons are D1 (5.63 ppm), F1 (5.60 ppm), B1 (5.37 ppm), and G1 (5.28 ppm). Comparing the spectra of compound 11 and 12, we found another new signal 5.34 ppm was represented only in compound 12. However, the GlcA proton chemical shifts at saccharide E of compound 11 are 4.52, 3.84, 3.83, 3.78, and 3.36 ppm; and as expected, another series of IdoA proton signals, 5.34, 4.94, 4.09, 4.04, 3.99 ppm, showed up for compound 12. Comparing the HSQC spectra of compound 11 and 12 also confirmed the presence of IdoA residue in compound 12. In the 1H NMR spectrum of compound 12, the proton peaks of IdoA also showed as smaller coupling constants or broad shape, not typically larger axial-axial coupling constants in GlcA, supporting the existence of IdoA and fast conformational equilibrium among chair (4C1 and 1C4) and skew-boat (2S0) conformers. In addition, based on the integration value of the proton spectra, the epimerization product in compound 12 contained approximately 40% IdoA and 60% GlcA, which was also in agreement with the HPLC analysis.
RESULTS
Crystal Structure of 2OST with Bound Heptasaccharide
To crystalize 2OST, a MBP fusion with 2OST from chicken (92% identity to human) was utilized (18, 27). The structure of the ternary complex of MBP-2OST·PAP·heptasaccharide was solved at 3.45 Å resolution (Table 1 and Fig. 1, B and C). The crystal structure contains two symmetrical MBP-2OST trimers in the asymmetric unit. These trimers are similar to those of the MBP-2OST·PAP binary complex and are believed to represent the functional oligomeric state of the enzyme (18). Intermolecular interactions within the trimer are formed by extension of the C-terminal tail of one monomer across the active site of a neighboring monomer, forming an antiparallel β-strand on the edge of the central parallel β-sheet of the neighboring monomer (Fig. 1, B and C) (18). This arrangement results in 24% buried surface area for each monomer. In the first trimer (monomers A–C), there is no electron density for the MBP portion of monomer A, and the MBP portion of monomer B is substantially disordered. As well, there is no interpretable electron density for the heptasaccharide binding to monomer C. In the second trimer (monomers D–F), all monomers contain heptasaccharide bound, but the MBP portion of monomer F shows substantial disorder. All MBP molecules in the asymmetric unit are found in different orientations relative to the fused 2OST, suggesting flexibility in the linker between MBP and 2OST. Such flexibility in the linker region was not observed in the MBP-2OST·PAP binary complex. Attempts to shorten the linker to decrease flexibility failed to improve crystal quality.
FIGURE 1.

Crystal structure of the 2OST trimer. A, chemical structure of the heptasaccharide substrate used in the ternary complex. B, crystal structure of the 2OST trimer with each monomer (colored dark blue, slate, and light blue) bound to a molecule of PAP (pink) and heptasaccharide substrate (green) (monomers D, E, and F). The C terminus of each subunit (circled in magenta) extends into the active site of the neighboring molecule. C, B rotated 90° with respect to the horizontal axis in the plane of the paper.
The substrate preparation is a mixture of two heptasaccharides: one contains the IdoA at position E (structure shown in Fig. 1A) and the other a GlcA residue at position E. HPLC analysis revealed that the ratio of IdoA to GlcA at position E is 1.0:1.2 (Fig. 2A). Because 2OST preferentially sulfates the IdoA-containing heptasaccharide (as demonstrated in Fig. 2, B–F), we used the epimeric mixture for this study because of the difficulty in purifying large quantities of the specific IdoA epimer for crystallography.
FIGURE 2.

Anion-exchange HPLC chromatograms of heptasaccharide modified by 2OST. A, starting mixture of heptasaccharide substrate with no 2OST present. B–F, results after mixing heptasaccharide substrates in the presence of 2OST and PAPS at different concentrations (2.5, 12.5, 50, 100, and 400 μm, respectively). The mixture of heptasaccharide substrate yielded a very similar ratio of GlcA2S-containing product (1) and IdoA2S-containing product (2) under different concentrations of substrate, suggesting that 2OST preferentially sulfates IdoA-containing substrate in a wide range of substrate concentrations. The ratio of the areas under peak 2 and peak 1 for each analysis is shown.
The heptasaccharide substrates are bound in a surface cleft on the outside of each monomer at a trimer interface and are separated by ∼50 Å (Figs. 1, B and C, and 3, A–D). The cleft is formed by positively charged residues in the active site and C-terminal residues from the neighboring monomer. The orientation of the heptasaccharides in the active sites was determined by the clear electron density for the N-sulfo groups and the number or residues that could fit into the density from the acceptor saccharide (Fig. 3A). The heptasaccharide binds with the reducing end at the top of the trimer and the nonreducing end nearest the N-terminal side of the trimer. All five of the heptasaccharide molecules in the crystal structure bind in a similar manner with the greatest uniformity at the acceptor saccharide (saccharide E) and the adjacent GlcNS saccharides (Fig. 3B). The greatest differences exist at the ends of the heptasaccharide away from the acceptor site.
FIGURE 3.
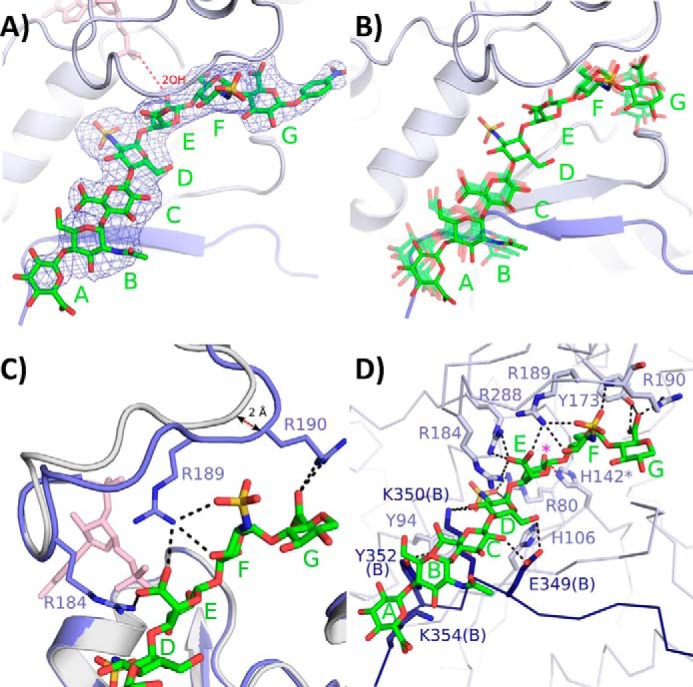
Heptasaccharide binding. A, electron density from a simulated annealing omit Fo − Fc map of the heptasaccharide contoured at 2.5σ (from monomer A). The PAP is shown in pink with the distance between the leaving group oxygen of the PAP and the acceptor hydroxyl 2-OH of the IdoA (saccharide E) displayed in a red dashed line (4.9 Å). B, superposition of the substrates from molecules B, D, E, and F (semitransparent green) into the active site of monomer A (substrate solid green). As the distance increases from the acceptor IdoA there is more variation in conformation. C, binding of the heptasaccharide resulting in a >2 Å movement of the loop containing residues Arg-184 through Lys-196 toward the hepatsaccahride. Residues Arg-184 and Arg-190, which are disordered in the substrate-free binary structure, become ordered in the bound ternary structure. D, residues involved in putative heptasaccharide-substrate interactions from monomer A shown in light blue. The residues from the C-terminal tail of neighboring monomer B are shown in dark blue. The magenta asterisk denotes the acceptor 2-OH position. Putative hydrogen bonds are displayed in black dashed lines.
To determine the identity and conformation of the acceptor saccharide crystallographic refinement was carried out with this sugar modeled as GlcA in the 4C1 conformation and as IdoA in 4C1, 1C4, and 2S0 conformations. Based on the best fit to the electron density, the acceptor saccharide was modeled as an IdoA in the 4C1 conformation (Fig. 4, A–D). Because the resolution (3.45 Å) does not allow for unambiguous identification of the sugar or ring conformation at the acceptor site, molecular dynamic simulations were carried out on all four sugar conformations for 25 ns. The conformations 1C4 and 2S0 have been previously reported as preferred conformations for HS structures (28, 29). Among the three possible conformations for IdoA, the 4C1 conformation displayed the strongest binding energy (ΔE) (Table 2), supporting the crystallographic assignment of saccharide E as an IdoA in the 4C1 conformation while binding 2OST. The GlcA showed weaker interaction energy than the IdoA in the 4C1 conformation(ΔE) (Table 2), consistent with experimental results demonstrating a 5-fold higher affinity for IdoA-containing versus GlcA-containing substrates (30). However, given the relatively small range for the interaction energies of the IdoA conformations, coupled with the inherent errors in the calculation of interaction energy for this system, it is difficult to rule out any of the three IdoA conformations. The 4C1 conformation, nevertheless, provides the best fit to the electron density. The decision to rule out a GlcA residue is also based on IdoA being the preferred substrate by a >10:1 ratio when a mixture of this heptasaccharide substrate is provided to 2OST (Fig. 2, B–F). Crystallographic refinement of the acceptor as an IdoA in the 4C1 conformation also positions the acceptor 2-OH 4.9 Å from the leaving group oxygen of PAP, consistent with a catalytically relevant binding position (Fig. 3A).
FIGURE 4.
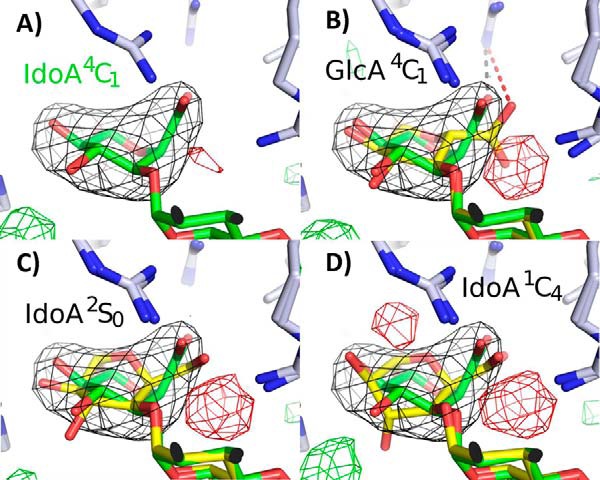
Refinement of different conformations of the acceptor saccharide. A, refinement of saccharide E as IdoA in the 4C1 conformation (green). B–D, refinement of saccharide E as either GlcA in the 4C1 conformation, IdoA 2S0, or IdoA 1C4, respectively (yellow) superimposed with the IdoA 4C1 structure (green). The simulated annealing omit Fo − Fc map contoured at 4.0σ is displayed in black. Positive Fo − Fc density from each respective refinement is shown in green (3.0σ), and negative density (3.0σ) is shown in red. In B the black dashed line represents a salt bridge (2.9 Å) between the IdoA and Arg-189. For GlcA this distance is 4.0 Å (red dashed line) and thus likely does not form a salt bridge.
TABLE 2.
Molecular dynamics interaction energies of HS with 2OST
Only the five saccharides (C-G)' that are required for activity and are well ordered in the crystal structure were used in the energy calculation of the heptasaccharide substrate with different conformations at position E (the last 20 ns of the 25-ns simulations were used for analysis).
| Acceptor saccharide E and conformation | Total interaction energy of HS with 2OST in presence of water | Total interaction energy of HS with water | Interaction energy (ΔE) | N(His)-O2 (saccharide E) distance |
|---|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | ||
| 1C4, IdoA | −1847.8±144.0 | −974.8±100.1 | −873.0 | 2.91±0.19 |
| 4C1, IdoA | −1893.0±135.6 | −966.8±105.5 | −926.2 | 2.84±0.18 |
| 2S0, IdoA | −1857.2±143.2 | −966.3±110.1 | −890.9 | 3.10±0.24 |
| 4C1, GlcA | −1859.7±145.6 | −978.0±111.3 | −881.7 | 3.10±0.30 |
| 4C1, IdoA 2OST(R189A) | −1836.3±150.2 | −966.8±105.5 | −869.5 | 3.31±0.36 |
Upon binding substrate, a number of residues have become ordered or are found in a different conformation due to interactions with the substrate. Most notably, the loop containing residues 185–196 shifts more than 2.0 Å closing down on the heptasaccharide compared with the substrate-free binary structure (Fig. 3C).
A number of specific interactions exist between the protein and heptasaccharide (Fig. 3D). The reducing end GlcA (saccharide G) is positioned between Asn-112 and Arg-190. Arg-190 has become ordered upon substrate binding and along with Tyr-173 is in position to form hydrogen bonds with the carboxylate group of this saccharide. For saccharide F (GlcNS) the N-sulfate is positioned within hydrogen bonding distance of the backbone amide of Arg-190 as well as the side chain from Arg-189. A number of putative interactions exist with the acceptor IdoA (saccharide E). Residues Arg-184, Arg-189, and Arg-288 are in position to interact with the carboxylate group of the acceptor IdoA residue. Residue His-142, the proposed catalytic base, has assumed a single conformation within hydrogen bonding distance to the acceptor 2-OH. This differs from the binary structure where His-142 is observed in multiple conformations (18). The N-sulfate of saccharide D lies within hydrogen bonding distance to Arg-80. Amino acid residues from the C-terminal tail of the neighboring monomer also interact with this GlcNS because Glu-349(B) is within hydrogen bonding distance to the 6-OH and Lys-350(B) is near the N-sulfate and the 3-OH. Tyr-352(B) from the C-terminal tail assumes a different orientation from that of the binary structure and is located within hydrogen bonding distance to the carboxylate group of saccharide C (GlcA). In addition, Glu-349(B) may potentially interact with this sugar as it is near the 2-OH group. Neither saccharide B (GlcNAc) nor saccharide A (GlcA) appears to form specific hydrogen bonding interactions with the protein; however, in the ternary complex the side chain of Lys-354 becomes ordered and stacks with saccharide A. The lack of specific interactions with saccharides A and B is consistent with their increased disorder compared with saccharides C–F (Fig. 3, A and B).
Substrate Specificity of 2OST
To eliminate the confounding effects from structurally heterogeneous polysaccharide substrates, such as completely de-O-sulfated N-sulfated heparin or N-sulfoheparosan (18), we investigated the substrate specificity of 2OST using a series of structurally defined oligosaccharides. A total of 14 compounds with different lengths and sulfation content was synthesized using the chemoenzymatic synthesis approach (Tables 3 and 4) (16, 31). The synthesis of IdoA-containing substrates is somewhat limited by the substrate specificity of C5-epimerase (32, 33); therefore, only a few selective IdoA-containing substrates were prepared. Because 2OST is capable of sulfating either IdoA or GlcA, the majority of the study focused on substrates containing GlcA acceptor sites avoiding the C5-epimerase step. The oligosaccharide substrates were carefully analyzed by using HPLC and MS to determine the purity (Fig. 5 and 6) and extent of sulfation (Table 3). The results from this study indicate that a pentasaccharide is the minimum size required for sulfation by 2OST based on its ability to sulfate the pentasaccharide compound 2 but not the tetrasaccharide compound 1. Within the required pentasaccharide 2OST recognizes a trisaccharide motif with the structure of -GlcNS-GlcA-GlcNS- or -GlcNS-IdoA-GlcNS-. The acceptor IdoA or GlcA residue must be flanked by two N-sulfoglucosamine residues. Replacing one of the two flanking GlcNS residues with either GlcNAc (as the case for compounds 6 and 7) or GlcNH2 (as the case for compound 5) resulted in the loss of 2OST modification. 6-O-Sulfation on the flanking GlcNS residues of the acceptor site abolishes the reactivity to 2OST modification as demonstrated for the compounds 8, 9, and 10. The presence of an IdoA2S residue on the reducing end of the trisaccharide domain has no effect on the reactivity to 2OST modification, as observed for compounds 13 and 14.
TABLE 3.
Reactivity of oligosaccharides to 2OST modification
| Substrate | Size | Abbreviated structure of oligosaccharide substratesa | 2-O-Sulfated productb |
|---|---|---|---|
| pmol | |||
| 1 | Tetrasaccharide | GlcNS-GlcA-GlcNS-GlcA-PNP | 0.47 ± 0.08 |
| 2 | Pentasaccharide | GlcA-GlcNS-GlcA-GlcNS-GlcA-PNP | 4.17 ± 0.03 |
| 3 | Hexasaccharide | GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-PNP | 7.20 ± 0.38 |
| 4 | Hexasaccharide | GlcNAc-GlcA-GlcNS-IdoA-GlcNS-GlcA-PNP | 10.71 ± 0.63 |
| 5 | Hexasaccharide | GlcNAc-GlcA-GlcNH2-GlcA-GlcNS-GlcA-PNP | <0.08 |
| 6 | Hexasaccharide | GlcNAc-GlcA-GlcNHAc-GlcA-GlcNS-GlcA-PNP | <0.08 |
| 7 | Hexasaccharide | GlcNAc-GlcA-GlcNS-GlcA-GlcNHAc-GlcA-PNP | <0.08 |
| 8 | Hexasaccharide | GlcNAc6S-GlcA-GlcNS6S-GlcA-GlcNS6S-GlcA-PNP | <0.08 |
| 9 | Hexasaccharide | GlcNAc-GlcA-GlcNS-GlcA-GlcNS6S-GlcA-PNP | <0.08 |
| 10 | Hexasaccharide | GlcNAc-GlcA-GlcNS-GlcA-GlcNAc6S-GlcA-PNP | <0.08 |
| 13 | Octasaccharide | GlcNAc-GlcA-GlcNS-GlcA-GlcNS-IdoA2S-GlcNS-GlcA-PNP | 8.22 ± 0.40 |
| 14 | Octasaccharide | GlcNAc-GlcA-GlcNS-IdoA-GlcNS-IdoA2S-GlcNS-GlcA-PNP | 14.08 ± 0.68 |
a The boxed trisaccharide domain, -GlcNS-GlcA-GlcNS- or–GlcNS-IdoA-GlcNS-, is designated the recognition site by 2OST, where PNP represents the p-nitrophenol group.
b The susceptibility of the oligosaccharide substrates to 2OST modification was determined by the analysis using anion exchange-HPLC. The presented value is the average of two determinations ± range.
TABLE 4.
Summary of the HPLC and MS analysis of oligosaccharide substrates
| Compound | Abbreviated structures | Purity | Amount prepared | Molecular weight |
|---|---|---|---|---|
| % | mg | |||
| 1 | GlcNS-GlcA-GlcNS-GlcA-PNP | 98 | 1.5 | 973.6 ± 0.0 (973.8) |
| 2 | GlcA-GlcNS-GlcA-GlcNS-GlcA-PNP | 97 | 2.0 | 1149.8 ± 0.4 (1149.9) |
| 3 | GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-PNP | 98 | 12.5 | 1353.4 ± 0.2 (1353.1) |
| 4 | GlcNAc-GlcA-GlcNS-IdoA-GlcNS-GlcA-PNP | 96 | 0.7 | 1353.0 ± 0.4 (1353.1) |
| 5 | GlcNAc-GlcA-GlcNH2-GlcA-GlcNS-GlcA-PNP | 99 | 2.5 | 1272.9 ± 0.2 (1273.0) |
| 6 | GlcNAc-GlcA-GlcNAc-GlcA-GlcNS-GlcA-PNP | 99 | 8.5 | 1315.0 ± 0.4 (1315.1) |
| 7 | GlcNAc-GlcA-GlcNS-GlcA-GlcNAc-GlcA-PNP | 99 | 10 | 1315.0 ± 0.5 (1315.1) |
| 8 | GlcNAc6S-GlcA-GlcNS6S-GlcA-GlcNS6S-GlcA-PNP | 96 | 0.3 | 1593.1 ± 0.3 (1593.3) |
| 9 | GlcNAc-GlcA-GlcNS-GlcA-GlcNS6S-GlcA-PNP | 97 | 5.0 | 1433.2 ± 0.3 (1433.2) |
| 10 | GlcNAc-GlcA-GlcNS-GlcA-GlcNAc6S-GlcA-PNP | 98 | 6.0 | 1395.3 ± 0.6 (1395.2) |
| 11 | GlcA-GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-PNP | 98 | 25 | 1529.4 ± 0.6 (1529.3) |
| 12a | GlcA-GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-PNP and GlcA-GlcNAc-GlcA-GlcNS-IdoA-GlcNS-GlcA-PNP | 52 and 48 | 60 | 1529.4 ± 0.9 (1529.3) |
| 13 | GlcNAc-GlcA-GlcNS-GlcA-GlcNS-IdoA2S-GlcNS-GlcA-PNP | 99 | 5.0 | 1850.0 ± 0.1 (1850.5) |
| 14 | GlcNAc-GlcA-GlcNS-IdoA-GlcNS-IdoA2S-GlcNS-GlcA-PNP | 98 | 0.5 | 1850.4 ± 0.5 (1850.5) |
a Compound 12 was used to obtain the co-crystal structure with 2-OST.
FIGURE 5.

Anion-exchange HPLC chromatograms of synthetic oligosaccharide substrates. Each synthesized oligosaccharide substrate was analyzed by anion-exchange HPLC to determine the purity of the compound.
FIGURE 6.

MS spectra of synthetic oligosaccharide substrates. Each synthesized oligosaccharide substrate was analyzed by electrospray ionization-MS to determine the molecular mass.
Site-directed Mutagenesis
The contribution of specific amino acid residues in sulfation preference of IdoA versus GlcA was investigated using site-directed mutants with two hexasaccharide substrates, compounds 3 (GlcA) and 4 (IdoA). Based on the ternary complex crystal structure we generated five 2OST mutants Y94A, H106A, R184A, R189A, and K354A for this analysis. Our previous studies using polysaccharide substrates demonstrated that mutants Y94A and H106A prefer IdoA-containing substrates, whereas R189A prefers a GlcA-containing substrate (18, 34). Arg-184 and Lys-354, which were not predicted from the binary structure, were chosen for mutagenesis because the ternary structure revealed interactions between these residues and the heptasaccharide. The mutant proteins were incubated with compounds 3 and 4 in the presence of 35S-labeled PAPS, and the reaction mixtures were analyzed by anion-exchange HPLC to identify formation of the 35S-labeled hexasaccharide product (Fig. 7). It should be noted that the experiment was intended to compare the reactivity of different 2OST mutants toward GlcA-containing and IdoA-containing substrates. Here, only 35S-labeled products were detected. Because the substrates (compounds 3 and 4) have no radioactive tag, they were invisible in this analysis. It is likely that only a small portion of compounds 3 and 4 participated in the reaction. Thus, the analysis was not used to assess the completion of the 2-O-sulfation for each substrate.
FIGURE 7.

Anion-exchange HPLC chromatograms of oligosaccharide substrates modified by 2OST wild type and mutant proteins. Two oligosaccharide substrates, compound 3 and 4, were incubated with 2OST proteins in the presence of 35S-labeled PAPS followed by anion-exchange HPLC analysis. A–C represent chromatograms of the GlcA-containing substrate incubated with wild type 2OST (red tracing), 2OST R189A, and 2OST R354A, respectively. D–F represent chromatograms of the IdoA-containing substrate incubated with wild type 2OST, 2OST R189A, and 2OST R354A, respectively. Control reactions have no enzyme (blue tracing in both A and D). For the left column (A–C), the 35S-labeled peak eluted at 32–33 min contains the desired product. The 35S-labeled peak eluted at 38 min is PAPS. * indicates an unidentified peak. For the right column (D–F), the35S-labeled peak eluted at 35–36 min is the desired product.
Measuring the relative reactivity of the mutants toward compounds 3 and 4 permitted us to assess the contribution of the amino acid residues to recognize a GlcA-containing substrate versus an IdoA-containing substrate (Table 5). Indeed, R189A remains reactive to compound 3 (GlcA) but loses its reactivity to compound 4 (IdoA) (Fig. 7, B and E, and Table 5). This result is consistent with our previous findings using polysaccharide substrates (18). Mutant K354A exhibits comparable reactivity of the wild type protein to compounds 3 and 4, suggesting that Lys-354 is not important for substrate recognition or enzymatic activity (Fig. 7, C and F, and Table 5). However, 2OST R184A loses reactivity toward both substrates, suggesting that Arg-184 is crucial for activity (Table 5). Mutants 2OST Y94A and H106A display a modest reduction in reactivity toward both compounds 3 and 4 (Table 5). The data demonstrate that Tyr-94 and His-106 residues are not directly involved in distinguishing between GlcA and IdoA within the context of compounds 3 and 4 but may be involved in recognizing alternative sequence patterns present in the previously utilized heterogeneous polysaccharide substrates. Indeed, no direct interactions between Tyr-94 and His-106 with the hexasaccharide substrate are observed in the ternary crystal structure.
TABLE 5.
Summary of mutant analysis using both GlcA-containing and IdoA-containing substrates
| Name of mutants | Reactivity to compound 3 (GlcA-containing substrate) | Reactivity to compound 4 (IdoA-containing substrate) |
|---|---|---|
| % | % | |
| 2OST WT | 100 | 100 |
| 2OST Y94A | 23.0 | 35.7 |
| 2OST H106A | 16.8 | 27.7 |
| 2OST R184A | 10.1 | 8.3 |
| 2OST R189A | 81.7 | 2.4 |
| 2OST K354A | 82.0 | 101.2 |
DISCUSSION
The biosynthesis of HS involves the actions of multiple sulfotransferases and an epimerase. 2OST plays an essential role in assembling HS with desired structures for a multitude of biological functions. Although HS biosynthesis is not a template-driven process, the overall structure of HS from a single cell type remains largely unchanged for generations (35). Determining the substrate specificity via crystallography and structurally defined oligosaccharide substrates helps define the regulatory role of 2OST in HS biosynthesis.
The results from this study reveal several central conclusions. First, the acceptor IdoA residue in the heptasaccharide appears to be in the 4C1 conformation. This conformation is distinct from previous data that suggests IdoA assumes either the 1C4 or 2S0 conformation within the context of HS (36). Because GlcA is only observed in the 4C1 conformation, one possible advantage for the IdoA being in the 4C1 conformation is that the active site of 2OST needs only to bind one sugar conformation to be able to accommodate and sulfate both IdoA and GlcA. Residue Arg-189 may function to stabilize the 4C1 conformation for the IdoA substrate as the R189A mutant loses activity for IdoA containing substrates while still maintaining significant activity for GlcA-containing substrates. This notion is supported by the fact that in the crystal structure Arg-189 forms a putative salt bridge with the carboxylate of the IdoA, but based on the crystallographic refinement, would not be in position to interact with the carboxylate of GlcA (Fig. 4B). In addition, the molecular dynamics studies show a decrease in the interaction energy (ΔE) between the IdoA-containing 4C1 substrate and the R189A mutant (Table 2), potentially destabilizing binding by the mutant compared with the wild type protein. In the present study, a heptasaccharide substrate was used for the crystal study. Because the heptasaccharide only represents a small portion of a polysaccharide substrate, our model does not rule out that additional interaction may exist between the enzyme and substrate outside the heptasaccharide.
Second, 2OST requires at least a pentasaccharide for displaying its full activity. This is consistent with the crystal structure which suggests saccharides C–G of the heptasaccharide form all of the observed specific interactions with the protein. The tetrasaccharide domain D–G represents the structure of compound 1, whereas the pentasaccharide domain C–G represents the structure of compound 2. Removal of the nonreducing end GlcA from compound 2 would result in potentially two fewer hydrogen bonds with the substrate at residue Glu-349(B) and Tyr-352(B) (Fig. 3D), supporting the dependence of this saccharide.
Third, 2OST has evolved in a way that enables it to discriminate based on sulfation patterns, ensuring that a programmed sulfation sequence is followed. The substrate specificity study demonstrates that a 2OST acceptor site must be flanked by two GlcNS residues, consistent with the fact that the N-sulfo groups interact with numerous points of 2OST. In the crystal structure, both flanking GlcNS saccharides show strong electron density for their 2S-sulfates and are very well ordered (Fig. 3A). The N-sulfo group from saccharide F(GlcNS) on the reducing side forms putative hydrogen bonds with the backbone amide from Arg-190 and possibly side chain interactions with Arg-189. Arg-189 also appears to interact with the carboxylate on the acceptor IdoA (saccharide E). Although R189A loses substantial activity for compound 4 (IdoA), it maintains substantial activity for compound 3 (GlcA), suggesting that its interaction with the N-sulfo group is not the predominant role. The N-sulfo groups from saccharide D (GlcNS) on the nonreducing side are within hydrogen bonding distance to Arg-80 and Lys-350(B) (Fig. 3D). The 2OST R80A mutant was previously shown to have <2% of wild type activity, whereas 2OST K350A displayed <25%, suggesting that these interactions are important for substrate binding (18). Taken together, these observations imply that N-sulfation is a prerequisite for 2-O-sulfation.
Finally, unlike the requirement for N-sulfation near the acceptor site, the 6-O-sulfation has precisely the opposite effect on 2OST activity. The interactions between proteins and HS generally involve charge-charge interactions (37); namely, an increase in the binding affinity would be expected when the total number of sulfo groups is increased. On the contrary, 6-O-sulfation on the reducing side of the acceptor saccharide (compound 9) prevents modification by 2OST. Based on the crystal structure, 6-O-sulfation on saccharide F, on the reducing side of the acceptor saccharide, would create steric clashes with Pro-82 and/or Tyr-173, interfering with its ability to bind within the substrate binding cleft (Fig. 8A). 6-O-Sulfation is also likely selected against on the nonreducing adjacent glucosamine due to charge repulsion with Glu-349(B) from the neighboring monomer (Fig. 8B). These results suggest that 2-O-sulfation must occur prior to 6-O-sulfation to create a domain containing -IdoA2S-GlcNS6S-, a common disaccharide repeating unit found in HS (38). Our conclusion is consistent with the current sequential model for the HS biosynthetic pathway with N-sulfation occurring first, followed by C5-epimerization/2-O-sulfation, 6-O-sulfation, and 3-O-sulfation, respectively.
FIGURE 8.
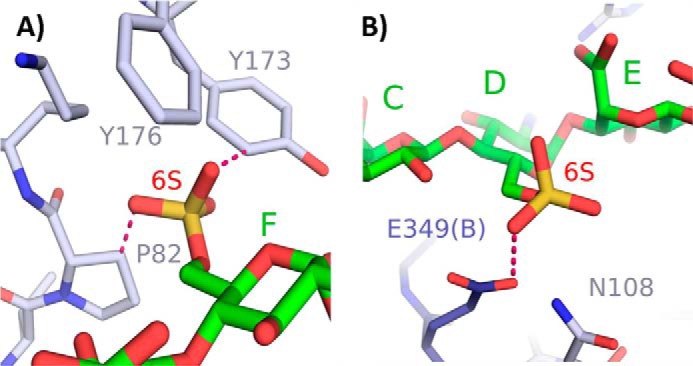
Modeling of 6-O-sulfates on the substrate heptasaccharide. A, a 6-O-sulfate on saccharide F would result in steric clashes with Tyr-173 (2.2 Å) and Pro-82 (2.4 Å). B, 6-O-sulfate on saccharide D would likely create electrostatic repulsion with Glu-349(B) (3.0 Å).
Sulfation patterns of HS determine the binding specificity to protein targets. The crystal structure of HS oligosaccharide bound to 2OST provides an example of the mechanism used by a protein to recognize a specific sulfation pattern. Decoding the recognition mechanism between sulfated saccharides and proteins will advance our understanding of the fundamental functions of HS. Furthermore, understanding the substrate recognition by 2OST and other HS biosynthetic enzymes will likely improve the capability of the chemoenzymatic synthesis technology to prepare targeted heparin-based therapeutics.
Supplementary Material
Acknowledgments
We thank Dr. L. G. Pedersen and A. F. Moon for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GM102137 and HL094463 and by the Intramural Research Program of the National Institutes of Health, NIEHS Project ZIAES102645 and ZICES043010-28 (to L. C. P.).

This article contains supplemental Fig. 1.
The atomic coordinates and structure factors (code 4NDZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HS
- heparan sulfate
- GlcA2S
- 2-O-sulfated glucuronic acid
- IdoA2S
- 2-O-sulfated iduronic acid
- MBP
- maltose-binding protein
- 2OST
- 2-O-sulfotransferase
- PAP
- 3′-phosphoadenosine 5′-phosphate
- GlcNTFA
- N-trifluoroacetyl glucosamine
- PNP
- p-nitrophenol.
REFERENCES
- 1. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 2. Liu J., Moon A. F., Sheng J., Pedersen L. C. (2012) Understanding the substrate specificity of the heparan sulfate sulfotransferases by a combined synthetic crystallographic approach. Curr. Opin. Struct. Biol. 22, 550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Esko J. D., Lindahl U. (2001) Molecular diversity of heparan sulfate. J. Clin. Invest. 108, 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kreuger J., Salmivirta M., Sturiale L., Giménez-Gallego G., Lindahl U. (2001) Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J. Biol. Chem. 276, 30744–30752 [DOI] [PubMed] [Google Scholar]
- 5. Witt R. M., Hecht M. L., Pazyra-Murphy M. F., Cohen S. M., Noti C., van Kuppevelt T. H., Fuller M., Chan J. A., Hopwood J. J., Seeberger P. H., Segal R. A. (2013) Heparan sulfate proteoglycans containing a glycpican 5 core and 2-O-sulfoiduronic acid function as sonic hedgehog co-receptors to promote proliferation. J. Biol. Chem. 288, 26275–26288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bullock S. L., Fletcher J. M., Beddington R. S., Wilson V. A. (1998) Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 12, 1894–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Axelsson J., Xu D., Kang B. N., Nussbacher J. K., Handel T. M., Ley K., Sriramarao P., Esko J. D. (2012) Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood 120, 1742–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bülow H. E., Hobert O. (2004) Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron 41, 723–736 [DOI] [PubMed] [Google Scholar]
- 9. Kinnunen T., Huang Z., Townsend J., Gatdula M. M., Brown J. R., Esko J. D., Turnbull J. E. (2005) Heparan 2-O-sulfotransferase, hst-2, is essential for normal cell migration in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 102, 1507–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamimura K., Koyama T., Habuchi H., Ueda R., Masu M., Kimata K., Nakato H. (2006) Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling. J. Cell Biol. 174, 773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Linhardt R. J., Liu J. (2012) Synthetic heparin. Curr. Opin. Pharmacol. 12, 217–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu H., Zhang Z., Linhardt R. J. (2009) Lessons learned from the contamination of heparin. Nat. Prod. Rep. 26, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J., Avci F. Y., Muñoz E. M., McDowell L. M., Chen M., Pedersen L. C., Zhang L., Linhardt R. J., Liu J. (2005) Enzymatically redesigning of biologically active heparan sulfate. J. Biol. Chem. 280, 42817–42825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J., Jones C. L., Liu J. (2007) Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures. Chem. Biol. 14, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Z., McCallum S. A., Xie J., Nieto L., Corzana F., Jiménez-Barbero J., Chen M., Liu J., Linhardt R. J. (2008) Solution structure of chemoenzymatically synthesized heparin and its precursors. J. Am. Chem. Soc. 130, 12998–13007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu Y., Masuko S., Takieddin M., Xu H., Liu R., Jing J., Mousa S. A., Linhardt R. J., Liu J. (2011) Chemoenzymatic synthesis of homogeneous ultra-low molecular weight heparin. Science 334, 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shriver Z., Raguram S., Sasisekharan R. (2004) Glycomics: a pathway to a class of new and improved therapeutics. Nat. Rev. Drug Discov. 3, 863–873 [DOI] [PubMed] [Google Scholar]
- 18. Bethea H. N., Xu D., Liu J., Pedersen L. C. (2008) Redirecting the substrate specificity of heparan sulfate 2-O-sulfotransferase by structurally guided mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 18724–18729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol Crystallogr 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 21. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shively J. E., Conrad H. E. (1976) Formation of anhydrosugars in the chemical depolymerization of heparin. Biochemistry 15, 3932–3942 [DOI] [PubMed] [Google Scholar]
- 23. Liu J., Shworak N. W., Sinaÿ P., Schwartz J. J., Zhang L., Fritze L. M., Rosenberg R. D. (1999) Expression of heparan sulfate d-glucosaminyl 3-O-sulfotransferase isoforms reveals novel substrate specificities. J. Biol. Chem. 274, 5185–5192 [DOI] [PubMed] [Google Scholar]
- 24. Case D. A., Darden T. A., Cheatham T. E., 3rd, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2012) AMBER 12, University of California, San Francisco [Google Scholar]
- 25. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J. (2009) Gaussian 09, Revision C, 1, Gaussian, Inc., Wallingford CT [Google Scholar]
- 26. Peterson S., Liu J. (2012) Deciphering mode of action heparanase using structurally defined oligosaccharides. J. Biol. Chem. 287, 34836–34843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moon A. F., Mueller G. A., Zhong X., Pedersen L. C. (2010) A synergistic approach to protein crystallization: combination of a fixed-arm carrier with surface entropy reduction. Protein Sci. 19, 901–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moon A. F., Edavettal S. C., Krahn J. M., Munoz E. M., Negishi M., Linhardt R. J., Liu J., Pedersen L. C. (2004) Structural analysis of the sulfotransferase (3-OST-3) involved in the biosynthesis of an entry receptor of herpes simplex virus 1. J. Biol. Chem. 279, 45185–45193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moon A. F., Xu Y., Woody S. M., Krahn J. M., Linhardt R. J., Liu J., Pedersen L. C. (2012) Dissecting substrate recognition mode of heparan sulfate 3-O-sulfotransferases for the biosynthesis of anticoagulant heparin. Proc. Natl. Acad. Sci. U.S.A. 109, 5265–5270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rong J., Habuchi H., Kimata K., Lindahl U., Kusche-Gullberg M. (2001) Substrate specificity of the heparan sulfate hexuronic acid 2-O-sulfotransferase. Biochemistry 40, 5548–5555 [DOI] [PubMed] [Google Scholar]
- 31. Liu R., Xu Y., Chen M., Weïwer M., Zhou X., Bridges A. S., DeAngelis P. L., Zhang Q., Linhardt R. J., Liu J. (2010) Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem. 285, 34240–34249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sheng J., Xu Y., Dulaney S. B., Huang X., Liu J. (2012) Uncovering biphasic catalytic mode of C5-epimerase in heparan sulfate biosynthesis. J. Biol. Chem. 287, 20996–21002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J. P., Gong F., El Darwish K., Jalkanen M., Lindahl U. (2001) Characterization of the d-glucuronyl C5-epimerase involved in the biosynthesis of heparin and heparan sulfate. J. Biol. Chem. 276, 20069–20077 [DOI] [PubMed] [Google Scholar]
- 34. Li K., Bethea H. N., Liu J. (2010) Determining the activity of C5-epimerase using an engineered heparan sulfate 2-O-sulfotransferase. J. Biol. Chem. 285, 11106–11113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kreuger J., Spillmann D., Li J. P., Lindahl U. (2006) Interactions between heparan sulfate and proteins: the concept of specificity J. Cell Biol. 174, 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mulloy B., Forster M. (2000) Conformation and dynamics of heparin and heparan sulfate. Glycobiology 10, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 37. Linhardt R. J. (2003) 2003 Claude S. Hudson Award address in carbohydrate chemistry. Heparin: structure and activity. J. Med. Chem. 46, 2551–2564 [DOI] [PubMed] [Google Scholar]
- 38. Lindahl U., Kusche-Gullberg M., Kjellén L. (1998) Regulated diversity of heparan sulfate. J. Biol. Chem. 273, 24979–24982 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.