

Background: The TGFβ and Hippo pathways are dysregulated in metastatic breast cancers.
Results: TGFβ-induced cues and nuclear TAZ/YAP converge at the transcriptional level to control gene expression important for tumorigenesis.
Conclusion: TAZ/YAP are required to promote TGFβ-induced tumorigenic phenotypes in breast cancer cells.
Significance: Our study reveals novel cross-talk between the TGFβ pathway and TAZ/YAP in late-stage breast cancers.
Keywords: Breast Cancer, Cell Migration, Cell Signaling, Coregulator Transcription, Signal Transduction, Transforming Growth Factor β (TGFβ), Hippo Pathway, Signaling Cross-talk, YAP/TAZ
Abstract
Uncontrolled transforming growth factor-β (TGFβ) signaling promotes aggressive metastatic properties in late-stage breast cancers. However, how TGFβ-mediated cues are directed to induce tumorigenic events is poorly understood, particularly given that TGFβ has clear tumor suppressing activity in other contexts. Here, we demonstrate that the transcriptional regulators TAZ and YAP (TAZ/YAP), key effectors of the Hippo pathway, are necessary to promote and maintain TGFβ-induced tumorigenic phenotypes in breast cancer cells. Interactions between TAZ/YAP, TGFβ-activated SMAD2/3, and TEAD transcription factors reveal convergent roles for these factors in the nucleus. Genome-wide expression analyses indicate that TAZ/YAP, TEADs, and TGFβ-induced signals coordinate a specific pro-tumorigenic transcriptional program. Importantly, genes cooperatively regulated by TAZ/YAP, TEAD, and TGFβ, such as the novel targets NEGR1 and UCA1, are necessary for maintaining tumorigenic activity in metastatic breast cancer cells. Nuclear TAZ/YAP also cooperate with TGFβ signaling to promote phenotypic and transcriptional changes in nontumorigenic cells to overcome TGFβ-repressive effects. Our work thus identifies cross-talk between nuclear TAZ/YAP and TGFβ signaling in breast cancer cells, revealing novel insight into late-stage disease-driving mechanisms.
Introduction
Elevated nuclear levels of the transcriptional regulators TAZ and YAP (TAZ/YAP) are associated with a broad range of aggressive cancers (1). For instance, the extent of nuclear TAZ or YAP levels corresponds with breast cancer tumor grade (2–4). In breast cancer cells, enhanced nuclear TAZ and YAP levels promote oncogenic transformation and endow cells with tumorigenic properties, including the ability to proliferate, subvert apoptotic cues, migrate, invade, and grow under anchorage-independent conditions (5–9). Moreover, high nuclear TAZ levels induce cancer stem cell-like activity (2, 10) and promote evasion of certain breast cancer drug therapies (2, 11). Thus, understanding the roles of TAZ/YAP is critical for directing efficient breast cancer therapies.
The tumor initiating activity of TAZ/YAP relies on their binding to the TEAD family of transcription factors (TEAD1–4) (10, 12, 13), indicating that together these factors direct a tumorigenic transcriptional program. Supporting this premise, TAZ/YAP·TEAD complexes directly promote the expression of genes encoding oncogenic factors, such as CTGF (also known as CCN2) and CYR61 (also known as CCN1) (12, 13), which contribute to human breast cancer progression (14). Nuclear TAZ/YAP activity is highly regulated and governed in large part by the Hippo pathway-regulated LATS1 and LATS2 kinases (15). LATS1/2 kinases phosphorylate TAZ/YAP on conserved serine residues, which promote 14-3-3 binding and subsequent sequestration in the cytoplasm (16, 17), and also prime TAZ/YAP for further phosphorylation by CK1ϵ/δ kinases that evoke TAZ/YAP degradation via proteasome-dependent mechanisms (18, 19). Additional phosphorylation events destabilize TAZ, including those regulated by Wnt, phosphatidylinositol 3-kinase, and GSK3β (20, 21). Thus, dysregulation of multiple upstream signals likely contributes to the aberrant nuclear TAZ/YAP activity that is observed in cancers.
TAZ/YAP modify the activity of other transcription factors besides TEADs, including the transforming growth factor-β (TGFβ)-activated SMAD complexes (22). TGFβ is the prototypic member of a family of secreted factors that regulates numerous developmental and homeostatic processes (23). SMAD2 and SMAD3 (SMAD2/3) are the primary mediators of TGFβ-induced transcription. SMAD2/3 are phosphorylated by TGFβ-bound membrane receptors, which induce binding to SMAD4 (24, 25), forming active transcriptional complexes that accumulate in the nucleus upon binding to TAZ/YAP (26). In cancer, the role of TGFβ is complex, as it can suppress early oncogenic events but also promote aggressive late-stage metastatic phenotypes (27, 28). What mechanistically distinguishes the different TGFβ-dependent responses is poorly understood.
Several lines of evidence indicate that TGFβ, like TAZ/YAP, promotes aggressive tumorigenic properties in late-stage breast carcinomas (29, 30). Given that TAZ/YAP bind to SMAD transcription factors and direct TGFβ signaling in other contexts (26, 31, 32), we sought to characterize whether TAZ/YAP define TGFβ-mediated tumorigenic cues in breast cancer cells. Our observations indicate that TGFβ-induced tumorigenic events, such as increased cell migration, invasion, and anchorage-independent growth, require TAZ/YAP. Our data also indicate that, like TAZ/YAP, the TEAD transcription factors interact with TGFβ-induced SMAD2/3 in the nucleus, suggesting that TAZ/YAP·TEAD·SMAD2/3 complexes coordinate transcriptional events in a concerted manner. Genome-wide microarray analysis of gene expression changes that occur upon knockdown of TAZ/YAP or TEADs, or inhibition of TGFβ signaling, revealed that TAZ/YAP, TEAD, and TGFβ regulate overlapping target genes. Interestingly, the direct gene targets NEGR1 and UCA1, which are synergistically regulated by TAZ/YAP, TEAD, and TGFβ, are necessary for maintaining tumorigenic activity in metastatic breast cancer cells, suggesting that the convergence of TAZ/YAP·TEAD-TGFβ signals is critical for driving late-stage breast cancer phenotypes. Supporting this premise, expression of nuclear-localized TAZ or YAP mutants direct transcriptional events that sensitize untransformed breast cancer cells to adopt tumorigenic phenotypes in response to TGFβ, while also suppressing TGFβ-induced cytostasis. These findings reveal novel cross-talk between TGFβ and Hippo signaling that we propose is important for late stage tumorigenic events in breast cancer.
EXPERIMENTAL PROCEDURES
Cell Culture, Plasmids, and Transfections
MCF10A, MCF-12A, HMLE, and MCF7 cells were cultured using DMEM/F-12 media (1:1) supplemented with 5% horse serum, 20 ng/ml epithelial growth factor (EGF; PeproTech), 0.5 μg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma), 10 μg/ml insulin (Sigma). MDA-MB-231 (MDA-231) and MDA-MB-231-LM2-4 (LM2-4) cells were cultured using RPMI media supplemented with 10% FBS. SUM-149 cells were cultured using Ham's F-12 media supplemented with 5% FBS, 10 μg/ml insulin (Sigma), 0.5 μg/ml hydrocortisone (Sigma). BT20, HS578T, SKBR3, and HEK293T cells were cultured using DMEM supplemented with 10% FBS. HEK293T cells were transfected using TurboFect (Thermo Scientific) according to the manufacturer's protocol. MCF10A doxycycline-inducible stable cell lines were generated using the lentiviral Tet-On system (Clontech). 3×FLAG-tagged mutants of TAZ (4SA: S66A, S89A, S117A, and S311A) or YAP (5SA: S61A, S109A, S127A, S164A, and S397A) were generated by site-directed mutagenesis and cloned into the pLVX-Tight-Puro plasmid (catalog no. 632162, Clontech). Tet-On cells were selected with 1 mg/ml G-418 sulfate (Gold Biotechnology) and 1 μg/ml puromycin (American Bioanalytical). RNA interference was performed by transfecting siRNA using Dharmafect 1 (Thermo Scientific) according to manufacturer's protocol. Sequences for the siRNAs used are outlined in supplemental Table S1.
Immunofluorescence and Proximity Ligation Assay (PLA)
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100/PBS, blocked in 2% BSA/PBS, and probed with primary and secondary antibodies outlined in supplemental Table S2. LM2-4 cells were treated with or without TGFβ1 (500 pm, R&D Systems) or SB-431542 (5 μm, Sigma) for 24 h before fixing. For the PLA,2 LM2-4 cells were plated on 96-well microplates (Falcon) and treated with or without TGFβ1 for 24 h. Cells were fixed and permeabilized as described, blocked according to the manufacturer's protocol (Duolink), and probed with the primary antibodies outlined in supplemental Table S2. Anti-mouse MINUS and anti-rabbit PLUS PLA probes (Duolink) were used. Nuclei were stained with Hoechst. All immunofluorescence was visualized by confocal microscopy (LSM 700), and images were processed using Volocity software (PerkinElmer Life Sciences). Images were quantitated using ImageJ software.
Mammospheres
LM2-4 cells were transfected with siRNA, dissociated 24 h later, and resuspended in Mammary Epithelium Growth Medium (MEGM; Lonza) supplemented with B27 (Invitrogen), 20 ng/ml epidermal growth factor (EGF; PeproTech), 20 ng/ml basic fibroblast growth factor (bFGF; PeproTech). Single cells were seeded at 5 × 103 cells/ml in 6-well ultra-low attachment plates (Corning Glass) and treated with or without TGFβ1 or SB-431542. Primary spheres were photographed after 7 days and either lysed for RNA (Quick-RNA MiniPrep, Zymo Research) to examine knockdown or dissociated in 0.05% trypsin for 10 min and resuspended as single cells in MEGM for passage. Secondary spheres were photographed after an additional 14 days. Images were analyzed using ImageJ software, and statistics were calculated using Prism software (GraphPad) using a two-tailed unpaired Student's t test.
Immunoprecipitation and Immunoblots
LM2-4 cells examined for endogenous protein expression were treated with or without TGFβ1 or SB-431542 for 2 h and were lysed and examined by immunoblotting. Transfected HEK293T cells expressing the indicated proteins were lysed, subjected to immunoprecipitation using anti-FLAG-conjugated protein-G beads (Sigma), and analyzed by immunoblotting. MCF10A doxycycline-inducible cells were treated with or without doxycycline (0.1 to 100 ng/ml) or TGFβ1 for 24 h and were lysed and examined by immunoblotting. Antibodies are outlined in supplemental Table S2.
Cell Morphology Analysis, Wound Healing, and Transwell Migration
Low density MCF10A doxycycline-inducible cells were pretreated with doxycycline (100 ng/ml, Clontech) for 24 h and then treated with or without TGFβ1 for an additional 24 h. For the wound-healing scratch assays, LM2-4 cells were transfected with siRNA and 24 h later were treated with or without TGFβ1 or SB-431542 for an additional 24 h. MCF10A doxycycline-inducible cells were treated with or without doxycycline or TGFβ1 for 24 h. Monolayers were wounded and photographed after an additional 24 h (LM2-4) or 12 h (MCF10A). Images were analyzed using ImageJ software, and statistics were calculated using Prism software (GraphPad) using a two-tailed unpaired Student's t test. Cells used in the transwell assay were transfected with siRNA, trypsinized 24 h later, and resuspended in low serum media (0.25% FBS). Cells were plated at 105 cells/ml on 0.4-μm transwell filters (BD Biosciences) pretreated for 24 h with 1 μg/ml fibronectin (Millipore). Media + 10% FBS were used in the bottom chamber. Cells were allowed to migrate for 24 h in the presence of TGFβ1 and were subsequently stained with 0.5% crystal violet.
Three-dimensional Invasion
Stable knockdown of TAZ and YAP in LM2-4 cells was accomplished by lentivirus-mediated transduction of shRNA using the pLKO1-puro and pLKO1-neo vectors and subsequent selection with 2 μg/ml puromycin and 1.5 mg/ml G418. The shRNA sequences used are listed in supplemental Table S1. Single cells were plated on 100% growth factor-reduced Matrigel (BD Biosciences) using the overlay method (33). Assay media contained 2% Matrigel added to supplemented MEGM, and cells were cultured with puromycin and G418 with medium changes every 3 days. TGFβ1 and SB-431542 were added after 9 days and then cultured for an additional 3 days before being photographed.
Microarrays
LM2-4 cells were transfected with control siRNA or siRNAs targeting TAZ/YAP or all four TEADs (outlined in supplemental Table S1) and were treated 24 h later with TGFβ1 or SB-431542 for an additional 24 h. Total RNA was isolated and purified by Quick-RNA MiniPrep (Zymo Research). Twelve microarrays in total were performed, with each condition carried out three times on separate days. The Boston University Microarray Core generated the data using the Affymetrix Human Gene 1.0 St Array, which covers 27,300 probe sets. The data were filtered using a moderated p value of less than 0.01, and the average fold change in expression of each gene, for each condition, relative to the siCTL + TGFβ sample was calculated. Fold expression changes relative to siCTL + TGFβ-treated cells were calculated, and statistical significance was assessed using a moderated t test and p values. Hierarchical gene clustering was performed on overlapping genes displaying a p value of <0.01 with the open source program Cluster 3.0 (34).
Quantitative Real Time PCR (qPCR)
LM2-4 cells were transfected with siRNA and were treated 24 h later with or without TGFβ1 or SB-431542 for an additional 24 h. MCF10A doxycycline-inducible cells were treated with or without doxycycline (0.1 to 100 ng/ml) or TGFβ1 for 24 h. Total RNA was purified using Quick-RNA MiniPrep kit, and cDNA synthesis was performed using 1 μg RNA and iScript cDNA synthesis kit (Bio-Rad) according to manufacturer's protocol. qPCR was performed using Fast SYBR Green enzyme (Applied Biosystems) and measured on ViiA 7 real time PCR system (Applied Biosystems). Transcript levels were analyzed using the ΔΔCt method and normalized to GAPDH. Primer sequences are indicated in supplemental Table S3.
Chromatin Immunoprecipitation (ChIP)
LM2-4 cells were fixed with 1 mm EGS (Thermo Scientific) for 30 min, 1% formaldehyde for 10 min, and quenched in 0.125 m glycine in PBS. Cells were collected and lysed in Cell Lysis buffer (10 mm KOH/HEPES, pH 7.8, 85 mm KCl, 1 mm EDTA, pH 8.0, 1% Nonidet P-40) with a protease inhibitor mixture. Nuclei were lysed in Nuclear Lysis buffer (50 mm Tris/HCl, pH 7.4, 1% SDS, 10 mm EDTA, pH 8.0) with protease inhibitors, and genomic DNA was fragmented to <400 bp using Bioruptor bath sonicator (Diagenode). Immunoprecipitations were performed using antibodies outlined in supplemental Table S2 (note: anti-TEAD4 also recognizes TEAD1 and -3 (35)) followed by incubation with protein-G Dynabeads (Invitrogen), and then washing sequentially in buffer A (20 mm Tris/HCl, pH 7.6, 140 mm NaCl, 1 mm EDTA, pH 8.0, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100), buffer B (20 mm Tris/HCl, pH 7.6, 500 mm NaCl, 1 mm EDTA, pH 8.0, 0.5% sodium deoxycholate, 1% Triton X-100), buffer C (20 mm Tris/HCl, pH 7.6, 1 mm EDTA, pH 8.0, 0.5% sodium deoxycholate, 1% Triton X-100, 250 mm LiCl), and TBS. Samples were eluted in Elution buffer (50 mm NaHCO3, 50 mm Tris/HCl, pH 8.0, 2 mm EDTA, pH 8.0, 1% SDS). Cross-links were reversed overnight at 65 °C in 0.2 m NaCl in Elution buffer, and DNA was purified using QIAquick PCR purification columns (Qiagen). Samples were then analyzed by qPCR using the primers outlined in supplemental Table S3.
Cell Proliferation and Cell Cycle Analysis
MCF10A doxycycline-inducible cells were plated (5 × 104 cells) and treated with doxycycline with or without TGFβ1 (day 0). Cells were counted each day for 6 consecutive days (day 1–6). For cell cycle analysis, MCF10A doxycycline-inducible cells were treated with doxycycline with or without TGFβ1 for 48 h. 1 × 106 cells were fixed overnight in 100% ethanol and stained using 50 μg/ml propidium iodide (Sigma) and 100 μg/ml RNase A (Sigma). Samples were acquired on the FACScan (BD Biosciences), collecting 1 × 104 events, and analyzed using FlowJo software (Tree Star). Statistical analysis was conducted using a two-tailed unpaired Student's t test.
RESULTS
Nuclear TAZ/YAP Are Required to Promote TGFβ-induced Tumorigenic Phenotypes in Breast Cancer Cells
In cancer, the role of TGFβ is complex, as it can suppress early oncogenic events, such as cell cycle progression, but can also promote late-stage metastatic phenotypes (27, 28). What distinguishes these different TGFβ-dependent responses is poorly understood. Several lines of evidence indicate that nuclear TAZ/YAP, like TGFβ, induce tumorigenic properties in late-stage breast carcinomas (29, 30). In untransformed mammary epithelium, TAZ/YAP localization is restricted to the cytoplasm by cell compaction/polarity-regulated cues (9, 32). Dysregulation of cell polarity cues, which is a hallmark of cancer progression (36), induces nuclear TAZ/YAP localizations. Given our prior work showing that TAZ/YAP bind to and regulate the localization and activity of TGFβ-activated SMAD transcription factors (26, 32), we sought to test whether TAZ and/or YAP promote TGFβ-induced tumorigenic events. We began our analysis by examining the relationship between TAZ and YAP localizations and the TGFβ-induced cytostatic response in a panel of mammary epithelial and breast cancer cell lines. Based on published data, we divided the panel into cells that are responsive to TGFβ-induced cytostasis (MCF10A, BT20, HMLE, HS578T, MCF7, and MCF-12) and cells in which TGFβ induces pro-tumorigenic signals but not growth arrest (MDA-MB-231, MDA-MB-231-LM2-4, SKRB3, and SUM149) (37–44). Interestingly, we observed that cells displaying high levels of nuclear TAZ/YAP correlate with those in which TGFβ induces tumorigenic cues (Fig. 1A).
FIGURE 1.

TAZ/YAP are required for TGFβ-induced tumorigenic events. A, panel of breast cancer cell lines was divided by TGFβ-induced tumor suppression and promoting responses and examined by immunofluorescence for endogenous TAZ and YAP localization. B, LM2-4 cells were transiently transfected with control siRNA (siCTL) or siRNA targeting TAZ (siTAZ), YAP (siYAP), or TAZ and YAP (siTAZ/YAP). Cells were left untreated, treated with TGFβ or SB-431542 + TGFβ, and grown in anchorage-independent conditions. Primary mammospheres were examined for knockdown or were passaged into secondary spheres. Secondary mammospheres following SB-431542 (SB) treatment, or transfection with siTAZ, siYAP, or siTAZ/YAP, were unable to be determined due to low numbers. Representative images are shown, and three independent experiments from each condition were quantitated, measuring the number of colonies formed and the size of each colony. Black error bars represent the average + S.E., and red error bars represent the average ± S.E., *, p < 0.025; **, p < 0.005; ***, p < 0.0001 (t test). C, LM2-4 cell lysates were immunoblotted to examine endogenous levels of the indicated proteins upon TGFβ or SB-431542 treatment compared with GAPDH (loading control). D, LM2-4 cells were transiently transfected with siCTL or siTAZ/YAP. Cells were left untreated, treated with TGFβ, or SB-431542 + TGFβ. Monolayers were wounded and analyzed for cell migration. E, LM2-4 cells stably expressing control shRNA (shCTL) or shRNA targeting TAZ and YAP (shTAZ/YAP) were treated with TGFβ or SB-431542 + TGFβ and incubated in three-dimensional Matrigel culture conditions. Representative images from three independent experiments are shown.
To further investigate this relationship, we sought to determine the roles of nuclear TAZ/YAP in the human MDA-MB-231-LM2-4 (herein referred to as LM2-4) metastatic breast cancer cell line (45), a highly aggressive derivative of triple-negative basal subtype MDA-MB-231 cells (46). A fraction of LM2-4 cells in culture are capable of generating clonal mammospheres under anchorage-independent conditions (Fig. 1B), which is often used as a measure of the self-renewing potential of tumorigenic cells in vitro (47). TGFβ treatment of LM2-4 cells led to dramatic increases in the number and size of mammospheres observed (Fig. 1B), similar to that observed with TGFβ treatment of other mammary cells (30). The self-renewing properties of the cells within the mammospheres were assessed by their ability to form secondary clonal spheres (47), and we found that TGFβ also promoted secondary mammosphere formation. Co-treatment of the cells with the TGFβ receptor antagonist SB-431542 abolished the formation of primary mammospheres, validating that the observed effects are indeed generated via canonical TGFβ receptor-mediated signals (Fig. 1B) (48, 49). As expected, SB-431542 treatment eliminated the TGFβ-induced phosphorylation of SMAD2 and SMAD3 in these cells (Fig. 1C). Individual TAZ or YAP knockdown also repressed the number and size of TGFβ-induced mammospheres (Fig. 1B). However, simultaneous knockdown of both TAZ and YAP dramatically reduced mammosphere formation (Fig. 1B), indicating redundant roles for TAZ and YAP in transducing TGFβ-mediated cues required for anchorage-independent growth.
We further investigated other hallmark tumorigenic properties that may be mediated by TGFβ and TAZ/YAP in metastatic breast cancers, including cell migration and invasion (36). We found that treatment of LM2-4 cells with TGFβ led to increases in cell migration in an in vitro wound-healing scratch assay (Fig. 1D), similar to previous work (50). As expected, co-treatment with the TGFβ receptor antagonist SB-431542 blocked TGFβ-induced cell migration (Fig. 1D). Simultaneous knockdown of TAZ/YAP using siRNA also abolished TGFβ-induced LM2-4 cell migration (Fig. 1D). Similarly, SB-431542 treatment or shRNA-mediated TAZ/YAP knockdown abolished the ability of three-dimensional colonies of LM2-4 cells to invade into the surrounding Matrigel matrix in the presence of TGFβ (Fig. 1E). Taken together, our observations indicate that TAZ/YAP are critical mediators of TGFβ-induced tumorigenic events, including mammosphere formation, cell migration, and invasion.
TAZ/YAP, TEADs, and SMADs Converge to Regulate a TGFβ-induced Transcriptional Program in Breast Cancer Cells
Studies indicate that TAZ/YAP-induced cell transformation relies on the recruitment of TAZ/YAP to DNA by the TEAD family of transcription factors (TEAD1–4) (12, 13). TAZ and YAP also bind TGFβ-activated SMAD complexes to control SMAD localization and activity in a variety of cell types, including mammary epithelial cells (26, 32). Recent work has shown that TAZ/YAP·TEAD·SMAD2/3 complexes control transcriptional events important for maintaining human embryonic stem cell pluripotency (35). Thus, we hypothesized that similar complexes may also be present in late stage breast cancers such that TEAD and SMAD transcription factors can cooperatively facilitate TAZ/YAP-mediated tumorigenic activity. We found that TEAD2 and TEAD4 associate with SMAD3, as well as YAP (Fig. 2A), and these interactions were unaffected by stimulation with a constitutively active TGFβ receptor (TGFβR1-T240D (51)). Given that TAZ/YAP exhibit a predominantly nuclear localization in LM2-4 cells (Fig. 2B), we speculated that TAZ/YAP·TEAD might be interacting with TGFβ-activated SMAD2/3 to specify pro-tumorigenic transcriptional events. To acquire both protein interaction and localization information, we performed in situ PLA. PLA is a sensitive technique used to visualize the localization and association of endogenous protein complexes (proteins localized within 40 nm of each other) by microscopy (52). Using PLA, we observed TAZ/YAP·SMAD2/3 interactions in both the nucleus and cytoplasm of untreated LM2-4 cells (Fig. 2C). Upon TGFβ treatment, nuclear TAZ/YAP-SMAD2/3 binding became much more apparent in the nucleus (Fig. 2C), consistent with nuclear TAZ/YAP·SMAD2/3 complexes directing transcriptional events (26, 32). We also detected endogenous TAZ/TEAD1 interactions in the nucleus of LM2-4 cells with or without TGFβ stimulation (Fig. 2D), which were increased slightly upon TGFβ treatment (Fig. 2D). TEAD1·SMAD2/3 interactions were readily detected in the nucleus of LM2-4 cells, particularly after TGFβ treatment (Fig. 2E), suggesting these complexes stabilize upon nuclear accumulation of SMADs. Taken together, our observations indicate that TAZ/YAP, TEAD, and SMAD interact in TGFβ-stimulated metastatic breast cancer cells and suggest that they may form transcriptional complexes that function together in the nucleus.
FIGURE 2.

TAZ/YAP, TEADs, and SMAD2/3 interact endogenously. A, HEK293T cells expressing the indicated proteins were lysed and subjected to immunoprecipitation (IP) with a FLAG antibody followed by immunoblotting with the indicated antibodies. B, LM2-4 cells were left untreated or treated with SB-431542 (SB) or TGFβ and examined by immunofluorescence for endogenous TAZ or YAP localization. C and D, LM2-4 cells left untreated or treated with TGFβ were probed with primary antibodies recognizing TAZ/YAP and SMAD2/3 (C), TEAD1 and TAZ (D), or TEAD1 and SMAD2/3 (E). In situ PLA followed by confocal microscopy were performed using mouse and rabbit secondary probes. Red dots indicate endogenous interactions, and nuclei were visualized with Hoechst stain. Representative images are shown, and three fields from each condition were quantitated, measuring the nuclear-cytoplasmic localization of the interactions and the number of interactions per nucleus. Black error bars either represent the average + S.E. or the average ± S.E.
To explore the possible overlap in transcriptional activity by TAZ/YAP, TEAD, and SMAD complexes, we used microarrays to compare the global expression profiles of LM2-4 cells treated as follows (complete data available in supplemental Table S4): 1) transfected with control siRNA (siCTL) and treated with TGFβ; 2) transfected with siRNA targeting both TAZ/YAP (siTAZ/YAP) and treated with TGFβ; 3) transfected with siRNA targeting all four TEAD (TEAD1–4) family members (siTEAD) and treated with TGFβ; and 4) transfected with control siRNA (siCTL) and treated simultaneously with TGFβ and SB-431542. In terms of significant gene expression differences (p value < 0.01) relative to siCTL + TGFβ treatment, 461 genes overlapped between siTAZ/YAP and siTEAD conditions (Fig. 3A). This gene set displayed a high degree of correlation in expression (R = 0.86). The expression of 594 genes changed following SB-431542 treatment, and of these, 176 genes overlapped with siTAZ/YAP conditions. Of these 176 genes, 80 were also altered following TEAD knockdown (Fig. 3A).
FIGURE 3.

TAZ/YAP, TEADs, and TGFβ direct different and overlapping transcriptional events. A, LM2-4 cells were transfected with control siRNA (siCTL), siRNA targeting TAZ and YAP (siTAZ/YAP), or siRNA targeting all four TEADs (siTEAD1–4), and then treated with TGFβ or SB-431542 + TGFβ. RNA from cell lysates was harvested, and global gene expression profiles were examined using Affymetrix microarrays. The Venn diagram highlights the number of genes with significant expression changes occurring for the indicated condition relative to the siCTL + TGFβ sample. Hierarchical clustering was performed on the significantly changing genes, which revealed four major clusters as indicated. Top significantly changing genes of interest are highlighted in each of the four clustered groups. B–F, LM2-4 cells were transiently transfected with siCTL, siTAZ, siYAP, siTAZ/YAP, or siTEADs and treated with or without TGFβ or SB-431542 + TGFβ. Relative expression of genes indicated in the microarray analysis was determined by qPCR. All data are shown as the average of three independent experiments + S.E. B, confirmation of knockdown. C, group 1, genes repressed by siTAZ/YAP, siTEAD1–4, and SB-431542 treatment. D, group 2, genes repressed by siTAZ/YAP and siTEAD1–4 but induced by SB-431542 treatment. E, group 3, genes induced by siTAZ/YAP, siTEAD1–4, and SB-431542 treatment. F, group 4, genes induced by siTAZ/YAP and siTEAD1–4 but repressed by SB-431542 treatment.
Interestingly, genes for which expression was altered among all three experimental conditions exhibited distinct expression correlations. Unbiased clustering segregated TAZ/YAP·TEAD-TGFβ-regulated genes into four different groups as follows: group 1, repressed following siTAZ/YAP, siTEADs, or TGFβ inhibition (therefore normally induced by the presence of these factors); group 2, repressed following siTAZ/YAP or siTEAD treatment but induced by TGFβ inhibition; group 3, induced following siTAZ/YAP, siTEADs, or TGFβ inhibition (therefore normally repressed by the presence of these factors); and group 4, induced by siTAZ/YAP and siTEADs but repressed by TGFβ inhibition. The top five genes with altered expression in each group are listed in Fig. 3A. Quantitative PCR analysis confirmed the respective knockdown of TAZ/YAP and TEADs knockdown in each sample (Fig. 3B), as well as the expression changes observed from our microarray results for each group (Fig. 3, C–F). Notable genes for group 1 included the following: neuronal growth regulator 1 (NEGR1), urothelial cancer associated 1 (UCA1), and CTGF. Elevated expression of the group 1 genes NEGR1, UCA1, and CTGF relied on the presence of TAZ/YAP, TEADs, and active TGFβ signaling (Fig. 3C), suggesting that TAZ/YAP·TEAD-TGFβ synergize to promote the expression of these genes. In agreement with our observations, CTGF has recently been confirmed as an important transcriptional target of YAP·TEAD·SMAD complexes that promotes tumorigenesis in human malignant mesothelioma (31). NEGR1, UCA1, and CTGF expression was abolished following TAZ/YAP or TEAD knockdown in the absence of TGFβ (Fig. 3C), suggesting that although specific TGFβ signals rely on TAZ/YAP·TEAD, the basal level of TAZ/YAP·TEAD activity does not require TGFβ, and therefore TAZ/YAP·TEAD complexes may function dominantly to TGFβ signals.
The group 2 genes we confirmed by qPCR included the following: Occludin (OCLN) and cytoplasmic FMR1-interacting protein 2 (CYFIP2) (Fig. 3D). The group 3 genes confirmed include the following: killer cell lectin-like receptor subfamily C protein (KLRC3) and serine palmitoyltransferase long chain base subunit 3 (SPTLC3) (Fig. 3E). The group 4 genes we confirmed include the following: limb bud and heart development (LBH) and prostate transmembrane protein androgen-induced 1 (PMEPA1) (Fig. 3F). Notably, many genes were found to be differentially regulated by TAZ/YAP·TEADs and TGFβ, suggesting that although TAZ/YAP·TEAD complexes synergize with some TGFβ-mediated signals (group 1 and 3 targets), they repress others (group 2 and 4 targets).
NEGR1 and UCA1 Are Direct Targets of TEADs and Are Necessary to Maintain Tumorigenic Breast Cancer Phenotypes
Our analysis of LM2-4 cells indicates that TAZ/YAP, TEAD, and TGFβ co-regulate the expression of a distinct subset of genes. To examine the importance of these genes in tumorigenesis, we focused our attention on group 1 genes, as these are synergistically induced by TAZ/YAP, TEAD, and TGFβ and include CTGF, a defined mediator of TAZ/YAP-induced tumorigenesis and cancer stem cell-like phenotypes (2, 31). The top two genes synergistically induced by TAZ/YAP·TEAD and TGFβ identified in our analysis were NEGR1 and UCA1. NEGR1 encodes a cell adhesion molecule that plays a role in neuronal growth and development (53–59). UCA1 encodes a long noncoding RNA that is expressed in development, is turned off in homeostatic tissues, and has been found to be highly expressed in bladder carcinomas (60). To determine whether these are direct transcriptional targets of TAZ/YAP, TEAD, and SMAD2/3, we performed chromatin immunoprecipitation (ChIP). Examination of the promoter regions of NEGR1, UCA1, and CTGF revealed consensus TEAD binding (61) and SMAD-binding motifs (62). ChIP of TAZ/YAP, TEAD, and SMAD2/3 from LM2-4 cell lysates revealed enrichment of these factors at the NEGR1, UCA1, and CTGF promoters, with SMAD2/3 recruitment only apparent after TGFβ treatment (Fig. 4, A–C).
FIGURE 4.

NEGR1 and UCA1 are direct transcriptional targets of TAZ/YAP, TEADs, and SMADs. LM2-4 cells treated with TGFβ or SB-431542 (SB) were subjected to ChIP analysis using control rabbit IgG, TAZ/YAP, TEAD4, or SMAD2/3 antibodies. Samples were analyzed by qPCR using primers recognizing the indicated regions in the promoter of NEGR1 (A), UCA1 (B), or CTGF (C). Normalized values are shown as the average of three independent experiments + S.E.
To further investigate the role of NEGR1 and UCA1 in TGFβ-mediated tumorigenesis, we examined the consequences of reducing their expression following siRNA-mediated knockdown. Knockdown of NEGR1 or UCA1 repressed the migration of LM2-4 cells treated with TGFβ in wound-healing scratch assays (Fig. 5A) and in transwell migration assays (Fig. 5B). Knockdown of either NEGR1 or UCA1 also suppressed the ability of LM2-4 cells to form large mammosphere colonies in the presence of TGFβ (Fig. 5C), consistent with pro-tumorigenic roles for NEGR1 and UCA1. The results of these experiments reflect our observations with TGFβ inhibition (SB-431542 treatment) or TAZ/YAP knockdown, suggesting that cooperative regulation of NEGR1 and UCA1 expression by TAZ/YAP·TEAD·SMAD complexes is necessary to promote tumorigenic phenotypes.
FIGURE 5.

NEGR1 and UCA1 are necessary for TGFβ-induced tumorigenic events. A, LM2-4 cells were transiently transfected with control siRNA (siCTL) or siRNA targeting NEGR1 (siNEGR1) or UCA1 (siUCA1) and treated with TGFβ. Monolayers were wounded and analyzed for cell migration. Representative images of three independent experiments are shown. B, LM2-4 cells transfected with siCTL, siNEGR1, or siUCA1 were plated on transwell filters to assess cell migration. Migrated cells are shown as the average number in 10 random fields over two independent experiments + S.E. C, LM2-4 cells were transfected with siCTL, siNEGR1, or siUCA1 and then grown under anchorage-independent conditions in the presence of TGFβ to examine primary mammosphere formation. Representative images are shown, and three independent experiments from each condition were quantitated, measuring the number of colonies formed and the size of each colony. Black error bars represent the average + S.E., and red error bars represent the average ± S.E., **, p < 0.005; ***, p < 0.0001 (t test).
Nuclear TAZ and YAP Cooperate with TGFβ to Promote Phenotypic and Transcriptional Changes in Nontumorigenic Cells
Based on the results uncovered from our gene expression studies, we decided to test whether ectopic expression of nuclear TAZ/YAP in nontumorigenic human mammary MCF10A cells would lead to the induction of TGFβ-dependent transcriptional events similar to those we characterized in the malignant LM2-4 cells. Stable expression of nuclear TAZ or YAP mutants can transform epithelial cells (2, 5, 7, 8), but this occurs following weeks of stable selection. Similarly, treatment of cells with TGFβ for several days to weeks is required to observe tumorigenic events in mammary epithelial cells (30, 63). To prevent confounding issues with long term culture conditions, we generated MCF10A cells that express a nuclear-localized and stable TAZ mutant (TAZ(4SA)) (7) or YAP mutant (YAP(5SA)) (9) in a doxycycline-inducible manner. These TAZ/YAP mutants have the LATS kinase-induced phosphorylation sites substituted to alanines, preventing their cytoplasmic sequestration and proteasomal degradation (7, 9). Titration of increasing amounts of doxycycline evoked subtle to high expression of TAZ(4SA) or YAP(5SA) in these cells (Fig. 6A). High levels of TAZ(4SA) or YAP(5SA) expression for short time frames (24 h) had minimal effects on the morphology of these cells (Fig. 6B). Short treatments of TGFβ led to flattening of cells (Fig. 6B), a morphology indicative of cells undergoing cell cycle arrest, as has been described for MCF10A cells post-TGFβ treatment (64). Strikingly, simultaneous doxycycline and TGFβ treatment led to rapid cell morphology changes that differed from either condition alone, with the cells becoming more spindle-like and elongated (Fig. 6B). Furthermore, TAZ(4SA)- or YAP(5SA)-expressing cells treated with TGFβ displayed much more rapid cell migration in a wound-healing scratch assay, as compared with either condition alone (Fig. 6C), indicating that nuclear TAZ/YAP synergize with TGFβ to promote cell morphology and cell migration changes.
FIGURE 6.

TAZ and YAP synergize with TGFβ to promote distinct morphological changes and gene transcription. A, doxycycline (Dox)-inducible MCF10A cells expressing 3×FLAG-TAZ(4SA) or 3×FLAG-YAP(5SA) were treated with increasing levels of doxycycline with or without TGFβ. Expression of TAZ or YAP was determined by immunoblotting along with GAPDH (loading control). B, doxycycline-inducible MCF10A control cells or cells expressing 3×FLAG-TAZ(4SA) or 3×FLAG-YAP(5SA) were treated with doxycycline with or without TGFβ and examined for cell morphology. Representative images of three independent experiments are shown. C, doxycycline-inducible MCF10A cells expressing 3×FLAG-TAZ(4SA) or 3×FLAG-YAP(5SA) were treated with or without doxycycline and/or TGFβ. Monolayers were wounded and analyzed for cell migration. Representative images are shown, and three independent experiments were quantitated. Error bars represent the average + S.E., *, p < 0.05; **, p < 0.01; ***, p < 0.0005 (t test). D–F, doxycycline-inducible MCF10A cells expressing 3×FLAG-TAZ(4SA) or 3×FLAG-YAP(5SA) were treated with increasing levels of doxycycline with or without TGFβ. Relative expression of group 1 genes (D), group 2 genes (E), and group 4 genes (F) was analyzed by qPCR and is shown as the average of three independent experiments + S.E.
In accordance with our expression analysis of LM2-4 cells, we found that nuclear TAZ or YAP function in concert with TGFβ to control transcriptional events in MCF10A cells. For example, TAZ or YAP synergized with TGFβ to promote the transcription of group 1 genes in an inducible fashion, including the expression of NEGR1, UCA1, and CTGF (Fig. 6D). Increased TAZ(4SA) or YAP(5SA) levels also induced the expression of group 2 genes (e.g. OCLN and CYFIP2), whereas TGFβ repressed this group of genes (Fig. 6E). Conversely, group 4 genes, such as LBH and PMEPA1, were induced by TGFβ but repressed in an inducible fashion by nuclear TAZ or YAP (Fig. 6F). Intriguingly, group 3 genes were undetectable in MCF10A cells, which may reflect the more differentiated state of these cells compared with LM2-4 cells. Together, our data indicate that the relationship between TAZ/YAP and TGFβ is conserved in mammary-derived cells, and our observations support the idea that dysregulated TAZ/YAP and TGFβ work in concert to control transcriptional events.
Nuclear TAZ and YAP Overcome TGFβ-induced Cytostasis in Nontumorigenic Cells
A hallmark trait of TGFβ is its ability to suppress tumorigenesis in normal epithelium and early stage cancers, particularly through cell cycle inhibition. However, TGFβ signals lose their ability to induce cytostasis in late stage cancers via poorly understood mechanisms (27, 28). TGFβ-induced cell cycle arrest has been previously described in MCF10A cells (64), so we sought to explore the relationship between TGFβ, nuclear TAZ/YAP, and cell cycle progression. We performed proliferation assays using control MCF10A cells or cells with doxycycline-inducible nuclear TAZ(4SA) or YAP(5SA) expression. TGFβ-induced cytostasis was evident in control MCF10A cells (Fig. 7A). Strikingly, we found that expression of TAZ(4SA) or YAP(5SA) overcomes TGFβ growth arrest, as cells treated simultaneously with doxycycline and TGFβ proliferated similarly to control cells (Fig. 7A). To investigate whether the proliferative differences were due to cell cycle alterations, we used fluorescence-activated cell sorting analysis (FACS) to examine the DNA content of these cells. We found that TGFβ treatment arrests cells in the G1 phase of the cell cycle, and that TAZ(4SA) or YAP(5SA) expression reverses the G1 phase arrest (Fig. 7, B and C). Our data therefore suggest that nuclear TAZ/YAP are responsible for the switch in TGFβ activity from tumor-suppressive to tumorigenic in later stage breast cancers by converging to direct a distinct transcriptional program (see model in Fig. 8).
FIGURE 7.
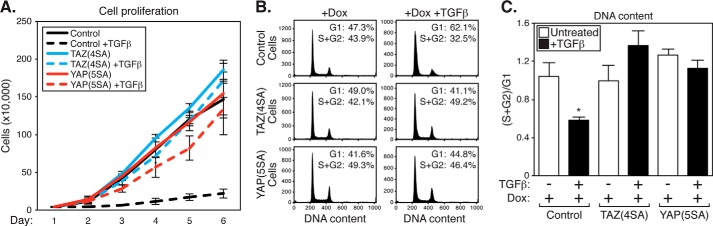
Nuclear TAZ and YAP overcome TGFβ-induced cell cycle arrest. A, doxycycline-inducible MCF10A control cells or cells expressing 3×FLAG-TAZ(4SA) or 3×FLAG-YAP(5SA) were treated with doxycycline (Dox) with or without TGFβ. Cells were counted over 6 days and graphed to determine their rate of proliferation. Cell number counts are shown as the average of three independent experiments ± S.E. B, doxycycline-inducible MCF10A control cells or cells expressing 3×FLAG-TAZ(4SA) or 3×FLAG-YAP(5SA) were treated with doxycycline with or without TGFβ. Cells were subject to propidium iodide staining and flow cytometry analysis to determine DNA content. Data from a representative experiment are shown. C, cell cycle phase quantitation from the data in B is represented as the ratio of cells in S + G2 to cells in G1. The average of three independent experiments + S.E. is shown, *, p < 0.015 (t test).
FIGURE 8.
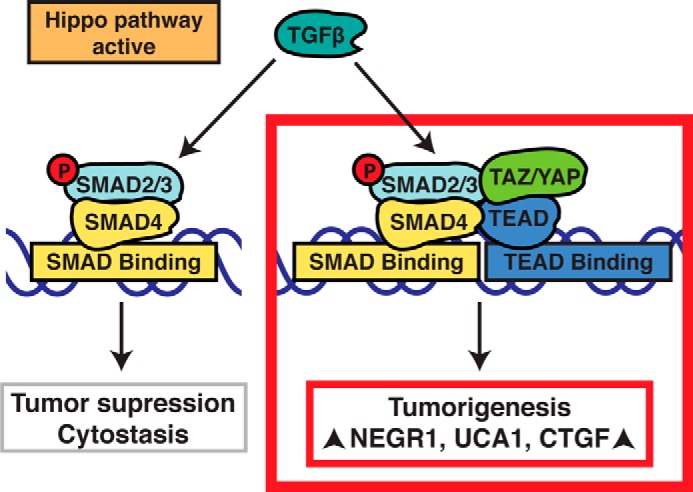
Model for how TAZ/YAP direct TGFβ-induced tumorigenic events. We propose that increased nuclear TAZ/YAP, resulting from defects in upstream Hippo pathway signals, overcome TGFβ-mediated tumor suppressive functions (e.g. cytostasis) and concomitantly drive tumorigenic transcriptional events by promoting the activity of TEAD·SMAD complexes.
DISCUSSION
We have found TAZ/YAP to be necessary for transduction of TGFβ-induced tumorigenic phenotypes in metastatic breast cancer cells, such as clonal anchorage-independent growth, cell migration, and invasion. Interactions between endogenous TAZ/YAP, TEAD, and SMAD2/3 in the nucleus suggest that these complexes coordinate their activities at the transcriptional level. Through genome-wide expression analysis, we show that TAZ/YAP, TEAD, and TGFβ regulate individual and common gene targets both positively and negatively, implying a complex level of transcriptional regulation and cross-talk between these factors. Of those gene targets we identified, many have yet to be characterized in breast cancer, and therefore our work may highlight previously unrecognized factors contributing to tumorigenesis. Of note, epithelial-mesenchymal transition-related genes were not enriched among the overlapping TAZ/YAP·TEAD-TGFβ-regulated subset, indicating that the TAZ/YAP·TEAD·SMAD2/3 complex drives aggressive behaviors of metastatic breast cancer cells downstream from the loss of epithelial cell polarity. Our transcriptional signature may thus reveal insight into the TAZ/YAP-mediated tumorigenic program occurring in late-stage cancers, as MDA-MB-231 cells, and their LM2-4 derivatives possess mesenchymal properties. Indeed, the two genes that we characterized, NEGR1 and UCA1, proved to be necessary for the anchorage-independent growth and migratory properties of LM2-4 cells. TAZ/YAP and TGFβ synergistically induce the expression of NEGR1 and UCA1 (group 1 genes), and given that TAZ/YAP, TEADs, and SMAD2/3 are enriched at the promoters of these genes, direct transcriptional synergy between TAZ/YAP·TEAD·SMAD complexes likely promotes their expression in breast cancer.
Out of the 80 genes co-regulated by TAZ/YAP, TEAD, and TGFβ, 21 of them encode membrane proteins, several of which function as cell surface receptors, and 13 of them encode secreted proteins. The enrichment of such genes may reflect important non-cell autonomous alterations that are regulated by TAZ/YAP·TEAD and TGFβ signals. Such signals are important for the pro-tumorigenic activity of TAZ and YAP (65, 66), and thus we propose that cross-talk between TAZ/YAP·TEAD and TGFβ signals demarcate a distinct local cellular environment that may promote a tumor-initiating niche. The well documented TAZ/YAP·TEAD target CTGF best highlights a secreted factor that is cooperatively induced by TGFβ. CTGF is a well established target of TGFβ-activated SMAD2/3 transcription factors (67) but is also an important driver of TAZ/YAP-induced tumorigenic events (2, 13). We observe that CTGF expression relies on the presence of TAZ/YAP, TEADs, and TGFβ signaling, and nuclear TAZ or YAP mutants synergize with TGFβ to strongly induce CTGF expression. Therefore, as in malignant mesotheliomas (31), the synergistic regulation of the CTGF promoter likely promotes aggressive breast cancer phenotypes.
We have additionally identified genes that are activated by both TAZ and YAP but repressed by TGFβ signaling (group 2 genes) and, reciprocally, genes repressed by TAZ/YAP but induced by TGFβ (group 4 genes). These groups of genes were somewhat surprising as they indicate that TAZ/YAP and TGFβ direct opposing transcriptional events, and therefore suggest that a subset of TGFβ-activated SMAD activity does not rely on TAZ/YAP and vice versa. Based on the products encoded by several of these genes, we speculate that nuclear TAZ/YAP may override tumor-suppressive or negative feedback mechanisms initiated by TGFβ. For example, PMEPA1, which we found is induced by TGFβ and inhibited by TAZ/YAP (group 4 gene), encodes a transmembrane protein that sequesters SMAD complexes in the cytoplasm (68). Thus, nuclear TAZ/YAP may function to overcome the induced expression of this gene to sustain pro-tumorigenic TGFβ signals.
Historically, TAZ and YAP have been considered to be activators of gene transcription. However, our data indicate that TAZ/YAP play repressive roles as well (group 3 and 4 genes). We hypothesize TAZ/YAP·TEAD complexes execute this repressive function by various means. Recent work has shown that TAZ/YAP recruit the nucleosome remodeling and deacetylation (NuRD) complex to repress gene expression (35). Yorkie (Yki), the homolog of TAZ/YAP in Drosophila melanogaster, is also known to associate with chromatin-modifying proteins (69, 70). Thus, TAZ/YAP·TEAD complexes likely function directly to inhibit transcription in breast cancers through similar recruitment of repressive factors to control local chromatin remodeling at promoters. However, TAZ/YAP·TEAD complexes may also function in an indirect manner, particularly in conjunction with TGFβ signaling, by binding, and re-localizing SMAD complexes (26, 32). SMAD redistribution by TAZ/YAP may explain why nuclear TAZ or YAP affects the expression of certain target genes (group 2 and 4) more dramatically in MCF10A cells in the presence of TGFβ. Moreover, TAZ/YAP binding to SMADs is evident in the nucleus and in the cytoplasm (Fig. 2C), suggesting that interactions between these proteins in different localizations may direct distinct events.
Of interest, nuclear TAZ or YAP is capable of overcoming TGFβ-induced cytostasis (Fig. 7), which is a major mechanism by which TGFβ functions as a tumor suppressor in early stage cancers (27). Consistent with this, we find that constitutively nuclear TAZ/YAP is evident in breast cancer cell lines where TGFβ has lost its ability to induce cytostatic signals (Fig. 1A). TAZ/YAP drive the expression of cell cycle regulators (6), which may account for the ability of these factors to overcome cell cycle arrest. Indeed, our gene expression analysis in LM2-4 cells identified several cell cycle regulators as TAZ/YAP-regulated genes (e.g. CDKL1, CCNA1, CCNB1, and CCND3). However, given that TAZ/YAP bind SMAD complexes, we also speculate that TAZ/YAP may be capable of redirecting TGFβ-induced SMADs away from their cell cycle-repressive transcriptional roles toward those that promote tumorigenesis.
Our phenotypic and transcriptional analysis revealed redundant functions for TAZ and YAP. For example, TAZ and YAP have redundant roles in mediating TGFβ-induced mammosphere formation. Additionally, TAZ and YAP redundantly regulate the expression of group 1 genes NEGR1 and UCA1 (Fig. 3C). Interestingly, TAZ knockdown alone led to increases in UCA1 expression, which may reflect compensatory YAP hyperactivity in this context. A redundant role for these factors is further implied on account of similar effects resulting from nuclear TAZ or YAP mutant expression in MCF10A cells. Such redundancy is consistent with the overlapping roles of TAZ/YAP in early development (71). However, we also present evidence for divergent transcriptional activity, based on specific gene expression reliance on either TAZ or YAP exclusively. For example, the expression of CTGF was repressed by TAZ or TAZ/YAP knockdown in LM2-4 cells but not by YAP knockdown alone (Fig. 3C). Thus, TAZ appears to have a dominant role in regulating CTGF expression in LM2-4 cells. Interestingly, recent work has revealed that YAP, in cooperation with TGFβ, has critical roles in controlling the expression of CTGF in malignant mesotheliomas (31). Thus, it appears that context defines dominance of TAZ or YAP.
Effective treatments of late-stage breast cancers are lacking, and our current understanding of the important signals driving and maintaining proliferation and metastasis is unclear. Our work has revealed critical intersections between TAZ/YAP, TEAD, and TGFβ signaling in directing pro-tumorigenic phenotypes in breast cancer, and provides novel mechanisms by which the TGFβ program may be directed toward aggressive tumorigenic phenotypes. Given the well documented roles of TGFβ in late-stage cancers, recent efforts have been focused on optimizing new TGFβ signaling inhibitors, which are currently in pre-clinical and clinical trials (72). Although advancement with such treatments is logical, our work suggests that enhanced efficacy may be achieved by treatment or co-treatment with current (73) or future TAZ/YAP·TEAD inhibitors.
Supplementary Material
Acknowledgments
We thank the Boston University Clinical and Translational Science Institute for funds to perform the microarray analysis (CTSA Grant UL1-TR000157) and Adam Gower and the Boston University Microarray Core for help with microarray data analysis. We thank Alicia Viloria-Petit (University of Guelph, Canada) for the LM2-4 cells; Kathrin Kirsch (Boston University) for the BT20, HS578T, MCF7, and SKBR3 cells; and David Sherr (Boston University) for the SUM149 cells.
This work was supported in part by funds from the Concern Cancer Foundation, the Karin Grunebaum Cancer Research Foundation, and Research Grant 5-FY11-578 from the March of Dimes Foundation.
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1–S4.
- PLA
- proximity ligation assay
- qPCR
- quantitative real time PCR
- OCLN
- occludin.
REFERENCES
- 1. Harvey K. F., Zhang X., Thomas D. M. (2013) The Hippo pathway and human cancer. Nat. Rev. Cancer 13, 246–257 [DOI] [PubMed] [Google Scholar]
- 2. Cordenonsi M., Zanconato F., Azzolin L., Forcato M., Rosato A., Frasson C., Inui M., Montagner M., Parenti A. R., Poletti A., Daidone M. G., Dupont S., Basso G., Bicciato S., Piccolo S. (2011) The hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772 [DOI] [PubMed] [Google Scholar]
- 3. Vlug E. J., van de Ven R. A., Vermeulen J. F., Bult P., van Diest P. J., Derksen P. W. (2013) Nuclear localization of the transcriptional coactivator YAP is associated with invasive lobular breast cancer. Cell. Oncol. 36, 375–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang X., Su L., Ou Q. (2012) Yes-associated protein promotes tumour development in luminal epithelial derived breast cancer. Eur. J. Cancer 48, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 5. Chan S. W., Lim C. J., Guo K., Ng C. P., Lee I., Hunziker W., Zeng Q., Hong W. (2008) A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 68, 2592–2598 [DOI] [PubMed] [Google Scholar]
- 6. Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S. A., Gayyed M. F., Anders R. A., Maitra A., Pan D. (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lei Q. Y., Zhang H., Zhao B., Zha Z. Y., Bai F., Pei X. H., Zhao S., Xiong Y., Guan K. L. (2008) TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 28, 2426–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Overholtzer M., Zhang J., Smolen G. A., Muir B., Li W., Sgroi D. C., Deng C. X., Brugge J. S., Haber D. A. (2006) Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. U.S.A. 103, 12405–12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao B., Wei X., Li W., Udan R. S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L., Zheng P., Ye K., Chinnaiyan A., Halder G., Lai Z. C., Guan K. L. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lamar J. M., Stern P., Liu H., Schindler J. W., Jiang Z. G., Hynes R. O. (2012) The hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. U.S.A. 109, E2441–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lai D., Ho K. C., Hao Y., Yang X. (2011) Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 71, 2728–2738 [DOI] [PubMed] [Google Scholar]
- 12. Zhang H., Liu C. Y., Zha Z. Y., Zhao B., Yao J., Zhao S., Xiong Y., Lei Q. Y., Guan K. L. (2009) TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 284, 13355–13362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C. Y., Chinnaiyan A. M., Lai Z. C., Guan K. L. (2008) TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xie D., Nakachi K., Wang H., Elashoff R., Koeffler H. P. (2001) Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers associated with more advanced features. Cancer Res. 61, 8917–8923 [PubMed] [Google Scholar]
- 15. Pan D. (2010) The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanai F., Marignani P. A., Sarbassova D., Yagi R., Hall R. A., Donowitz M., Hisaminato A., Fujiwara T., Ito Y., Cantley L. C., Yaffe M. B. (2000) TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Basu S., Totty N. F., Irwin M. S., Sudol M., Downward J. (2003) Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 11, 11–23 [DOI] [PubMed] [Google Scholar]
- 18. Liu C. Y., Zha Z. Y., Zhou X., Zhang H., Huang W., Zhao D., Li T., Chan S. W., Lim C. J., Hong W., Zhao S., Xiong Y., Lei Q. Y., Guan K. L. (2010) The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J. Biol. Chem. 285, 37159–37169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao B., Li L., Tumaneng K., Wang C. Y., Guan K. L. (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev. 24, 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Azzolin L., Zanconato F., Bresolin S., Forcato M., Basso G., Bicciato S., Cordenonsi M., Piccolo S. (2012) Role of TAZ as mediator of Wnt signaling. Cell 151, 1443–1456 [DOI] [PubMed] [Google Scholar]
- 21. Huang W., Lv X., Liu C., Zha Z., Zhang H., Jiang Y., Xiong Y., Lei Q. Y., Guan K. L. (2012) The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J. Biol. Chem. 287, 26245–26253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mauviel A., Nallet-Staub F., Varelas X. (2012) Integrating developmental signals: a hippo in the (path)way. Oncogene 31, 1743–1756 [DOI] [PubMed] [Google Scholar]
- 23. Wu M. Y., Hill C. S. (2009) Tgf-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 16, 329–343 [DOI] [PubMed] [Google Scholar]
- 24. Abdollah S., Macías-Silva M., Tsukazaki T., Hayashi H., Attisano L., Wrana J. L. (1997) TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 272, 27678–27685 [DOI] [PubMed] [Google Scholar]
- 25. Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C. H., Miyazono K., ten Dijke P. (1997) TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 16, 5353–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varelas X., Sakuma R., Samavarchi-Tehrani P., Peerani R., Rao B. M., Dembowy J., Yaffe M. B., Zandstra P. W., Wrana J. L. (2008) TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 10, 837–848 [DOI] [PubMed] [Google Scholar]
- 27. Bierie B., Moses H. L. (2006) Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520 [DOI] [PubMed] [Google Scholar]
- 28. Massagué J. (2008) TGFβ in Cancer. Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shipitsin M., Campbell L. L., Argani P., Weremowicz S., Bloushtain-Qimron N., Yao J., Nikolskaya T., Serebryiskaya T., Beroukhim R., Hu M., Halushka M. K., Sukumar S., Parker L. M., Anderson K. S., Harris L. N., Garber J. E., Richardson A. L., Schnitt S. J., Nikolsky Y., Gelman R. S., Polyak K. (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell 11, 259–273 [DOI] [PubMed] [Google Scholar]
- 30. Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Brooks M., Reinhard F., Zhang C. C., Shipitsin M., Campbell L. L., Polyak K., Brisken C., Yang J., Weinberg R. A. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujii M., Toyoda T., Nakanishi H., Yatabe Y., Sato A., Matsudaira Y., Ito H., Murakami H., Kondo Y., Kondo E., Hida T., Tsujimura T., Osada H., Sekido Y. (2012) TGF-β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med. 209, 479–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Varelas X., Samavarchi-Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B. G., Rossant J., Wrana J. L. (2010) The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 19, 831–844 [DOI] [PubMed] [Google Scholar]
- 33. Aranda V., Haire T., Nolan M. E., Calarco J. P., Rosenberg A. Z., Fawcett J. P., Pawson T., Muthuswamy S. K. (2006) Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat. Cell Biol. 8, 1235–1245 [DOI] [PubMed] [Google Scholar]
- 34. de Hoon M. J., Imoto S., Nolan J., Miyano S. (2004) Open source clustering software. Bioinformatics 20, 1453–1454 [DOI] [PubMed] [Google Scholar]
- 35. Beyer T. A., Weiss A., Khomchuk Y., Huang K., Ogunjimi A. A., Varelas X., Wrana J. L. (2013) Switch enhancers interpret TGF-β and hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 5, 1611–1624 [DOI] [PubMed] [Google Scholar]
- 36. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 37. Wu J., Mukherjee A., Lebman D. A., Fang X. (2011) Lysophosphatidic acid-induced p21Waf1 expression mediates the cytostatic response of breast and ovarian cancer cells to TGFβ. Mol. Cancer Res. 9, 1562–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choki I., Sourla A., Reyes-Moreno C., Koutsilieris M. (1998) Osteoblast-derived growth factors enhance adriamycin-cytostasis of MCF-7 human breast cancer cells. Anticancer Res. 18, 4213–4224 [PubMed] [Google Scholar]
- 39. Dai M., Al-Odaini A. A., Arakelian A., Rabbani S. A., Ali S., Lebrun J. J. (2012) A novel function for p21Cip1 and acetyltransferase p/CAF as critical transcriptional regulators of TGFβ-mediated breast cancer cell migration and invasion. Breast Cancer Res. 14, R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frey R. S., Mulder K. M. (1997) TGFβ regulation of mitogen-activated protein kinases in human breast cancer cells. Cancer Lett. 117, 41–50 [DOI] [PubMed] [Google Scholar]
- 41. Bouquet F., Pal A., Pilones K. A., Demaria S., Hann B., Akhurst R. J., Babb J. S., Lonning S. M., DeWyngaert J. K., Formenti S. C., Barcellos-Hoff M. H. (2011) TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 17, 6754–6765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen C. R., Kang Y., Massagué J. (2001) Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor β growth arrest program. Proc. Natl. Acad. Sci. U.S.A. 98, 992–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohmori T., Yang J. L., Price J. O., Arteaga C. L. (1998) Blockade of tumor cell transforming growth factor-βs enhances cell cycle progression and sensitizes human breast carcinoma cells to cytotoxic chemotherapy. Exp. Cell Res. 245, 350–359 [DOI] [PubMed] [Google Scholar]
- 44. Kang Y., He W., Tulley S., Gupta G. P., Serganova I., Chen C. R., Manova-Todorova K., Blasberg R., Gerald W. L., Massagué J. (2005) Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 13909–13914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Munoz R., Man S., Shaked Y., Lee C. R., Wong J., Francia G., Kerbel R. S. (2006) Highly efficacious nontoxic preclinical treatment for advanced metastatic breast cancer using combination oral UFT-cyclophosphamide metronomic chemotherapy. Cancer Res. 66, 3386–3391 [DOI] [PubMed] [Google Scholar]
- 46. Cailleau R., Olivé M., Cruciger Q. V. (1978) Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro 14, 911–915 [DOI] [PubMed] [Google Scholar]
- 47. Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., Wicha M. S. (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laping N. J., Grygielko E., Mathur A., Butter S., Bomberger J., Tweed C., Martin W., Fornwald J., Lehr R., Harling J., Gaster L., Callahan J. F., Olson B. A. (2002) Inhibition of transforming growth factor (TGF)- β1-induced extracellular matrix with a novel inhibitor of the TGF-β type I receptor kinase activity: SB-431542. Mol. Pharmacol. 62, 58–64 [DOI] [PubMed] [Google Scholar]
- 49. Inman G. J., Nicolás F. J., Callahan J. F., Harling J. D., Gaster L. M., Reith A. D., Laping N. J., Hill C. S. (2002) SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74 [DOI] [PubMed] [Google Scholar]
- 50. Bakin A. V., Rinehart C., Tomlinson A. K., Arteaga C. L. (2002) p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115, 3193–3206 [DOI] [PubMed] [Google Scholar]
- 51. Wieser R., Wrana J. L., Massagué J. (1995) GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 14, 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koos B., Andersson L., Clausson C. M., Grannas K., Klaesson A., Cane G., Soderberg O. (2013) Analysis of Protein Interactions in situ by Proximity Ligation Assays. Curr. Top. Microbiol. Immunol. 10.1007/82_2013_334 [DOI] [PubMed] [Google Scholar]
- 53. Funatsu N., Miyata S., Kumanogoh H., Shigeta M., Hamada K., Endo Y., Sokawa Y., Maekawa S. (1999) Characterization of a novel rat brain glycosylphosphatidylinositol-anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J. Biol. Chem. 274, 8224–8230 [DOI] [PubMed] [Google Scholar]
- 54. Hashimoto T., Maekawa S., Miyata S. (2009) IgLON cell adhesion molecules regulate synaptogenesis in hippocampal neurons. Cell Biochem. Funct. 27, 496–498 [DOI] [PubMed] [Google Scholar]
- 55. Levitt P. (1984) A monoclonal antibody to limbic system neurons. Science 223, 299–301 [DOI] [PubMed] [Google Scholar]
- 56. Marg A., Sirim P., Spaltmann F., Plagge A., Kauselmann G., Buck F., Rathjen F. G., Brümmendorf T. (1999) Neurotractin, a novel neurite outgrowth-promoting Ig-like protein that interacts with CEPU-1 and LAMP. J. Cell Biol. 145, 865–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pimenta A. F., Zhukareva V., Barbe M. F., Reinoso B. S., Grimley C., Henzel W., Fischer I., Levitt P. (1995) The limbic system-associated membrane protein is an Ig superfamily member that mediates selective neuronal growth and axon targeting. Neuron 15, 287–297 [DOI] [PubMed] [Google Scholar]
- 58. Spaltmann F., Brümmendorf T. (1996) CEPU-1, a novel immunoglobulin superfamily molecule, is expressed by developing cerebellar Purkinje cells. J. Neurosci. 16, 1770–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Struyk A. F., Canoll P. D., Wolfgang M. J., Rosen C. L., D'Eustachio P., Salzer J. L. (1995) Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J. Neurosci. 15, 2141–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang F., Li X., Xie X., Zhao L., Chen W. (2008) UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma and embryo, influencing cell growth and promoting invasion. FEBS Lett. 582, 1919–1927 [DOI] [PubMed] [Google Scholar]
- 61. Lian I., Kim J., Okazawa H., Zhao J., Zhao B., Yu J., Chinnaiyan A., Israel M. A., Goldstein L. S., Abujarour R., Ding S., Guan K. L. (2010) The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 24, 1106–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 63. Brown K. A., Aakre M. E., Gorska A. E., Price J. O., Eltom S. E., Pietenpol J. A., Moses H. L. (2004) Induction by transforming growth factor-β1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 6, R215–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iavarone A., Massagué J. (1997) Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature 387, 417–422 [DOI] [PubMed] [Google Scholar]
- 65. Yang N., Morrison C. D., Liu P., Miecznikowski J., Bshara W., Han S., Zhu Q., Omilian A. R., Li X., Zhang J. (2012) TAZ induces growth factor-independent proliferation through activation of EGFR ligand amphiregulin. Cell Cycle 11, 2922–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang J., Ji J. Y., Yu M., Overholtzer M., Smolen G. A., Wang R., Brugge J. S., Dyson N. J., Haber D. A. (2009) YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the hippo pathway. Nat. Cell Biol. 11, 1444–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leask A., Sa S., Holmes A., Shiwen X., Black C. M., Abraham D. J. (2001) The control of ccn2 (ctgf) gene expression in normal and scleroderma fibroblasts. Mol. Pathol. 54, 180–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Watanabe Y., Itoh S., Goto T., Ohnishi E., Inamitsu M., Itoh F., Satoh K., Wiercinska E., Yang W., Shi L., Tanaka A., Nakano N., Mommaas A. M., Shibuya H., Ten Dijke P., Kato M. (2010) TMEPAI, a transmembrane TGF-β-inducible protein, sequesters Smad proteins from active participation in TGF-β signaling. Mol. Cell 37, 123–134 [DOI] [PubMed] [Google Scholar]
- 69. Bayarmagnai B., Nicolay B. N., Islam A. B., Lopez-Bigas N., Frolov M. V. (2012) Drosophila GAGA factor is required for full activation of the dE2f1-Yki/Sd transcriptional program. Cell Cycle 11, 4191–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oh H., Slattery M., Ma L., Crofts A., White K. P., Mann R. S., Irvine K. D. (2013) Genome-wide association of Yorkie with chromatin and chromatin-remodeling complexes. Cell Rep. 3, 309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nishioka N., Inoue K., Adachi K., Kiyonari H., Ota M., Ralston A., Yabuta N., Hirahara S., Stephenson R. O., Ogonuki N., Makita R., Kurihara H., Morin-Kensicki E. M., Nojima H., Rossant J., Nakao K., Niwa H., Sasaki H. (2009) The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev. Cell 16, 398–410 [DOI] [PubMed] [Google Scholar]
- 72. Connolly E. C., Freimuth J., Akhurst R. J. (2012) Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 8, 964–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu-Chittenden Y., Huang B., Shim J. S., Chen Q., Lee S. J., Anders R. A., Liu J. O., Pan D. (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.