

Background: FcRn controls the long serum half-life of albumin.
Results: A single amino acid substitution of albumin considerably improved binding to FcRn and extended serum half-life in mice and rhesus monkeys.
Conclusion: Serum half-life of albumin may be tailored by engineering the FcRn-albumin interaction.
Significance: This study reports on engineered albumin that may be attractive for improving the serum half-life of biopharmaceuticals.
Keywords: Albumin, Animal Models, Antibody Engineering, Biodegradation, Bioengineering, FC Receptors, pH Regulation, Pharmacokinetics
Abstract
A major challenge for the therapeutic use of many peptides and proteins is their short circulatory half-life. Albumin has an extended serum half-life of 3 weeks because of its size and FcRn-mediated recycling that prevents intracellular degradation, properties shared with IgG antibodies. Engineering the strictly pH-dependent IgG-FcRn interaction is known to extend IgG half-life. However, this principle has not been extensively explored for albumin. We have engineered human albumin by introducing single point mutations in the C-terminal end that generated a panel of variants with greatly improved affinities for FcRn. One variant (K573P) with 12-fold improved affinity showed extended serum half-life in normal mice, mice transgenic for human FcRn, and cynomolgus monkeys. Importantly, favorable binding to FcRn was maintained when a single-chain fragment variable antibody was genetically fused to either the N- or the C-terminal end. The engineered albumin variants may be attractive for improving the serum half-life of biopharmaceuticals.
Introduction
The two most abundant soluble proteins in our body, IgG and albumin, account for more than 80% of the total plasma protein pool and amount to an impressive 12 and 40 mg/ml, respectively, in mouse and man. Although albumin acts as a versatile transporter of an array of small molecules, supports colloidal osmotic pressure, and buffers the pH of the blood, IgG is the major antibody class that protects against invading pathogens. However, IgG and albumin also share two remarkable features. First, they both have prolonged half-lives of 19 days compared with few days or less for other circulating proteins. Second, they are unique among the plasma proteins in that there is a direct relationship between their blood concentrations and their fractional catabolic rates. It has now become apparent that their homeostatic regulation is controlled by a cellular receptor, named the neonatal Fc receptor (FcRn),4 which rescues both from intracellular degradation and thus is responsible for their prolonged half-lives (1, 2).
FcRn is a major histocompatibility class I-related molecule that is built up of a unique transmembrane heavy chain (HC), which noncovalently associates with the common β2-microglobulin (β2m) (3, 4). The extracellular part of the HC consists of three domains (α1, α2, and α3) where the N-terminal α1-α2 domains form eight antiparallel β-pleated strands topped by two long α-helices followed by the membrane proximal α3-domain (5, 6). The β2m subunit is bound to the HC via both the α1-α2 platform and the α3-domain.
IgG and albumin bind FcRn simultaneously in a noncooperative and strictly pH-dependent manner, with strong binding at pH 6.0 that becomes progressively weaker approaching neutral pH (2, 7, 8). These pH-sensitive interactions are regulated by ionic networks at the interfaces and internally in each protein (5, 9–11). FcRn rescues the proteins from degradation in hematopoietic cells and endothelial cells lining the vascular space by binding IgG and albumin within intracellular endosomal compartments, which then results in transport of the ternary complex to the cell membrane for release of ligands back into the circulation as a consequence of exposure to the neutral pH of the blood (1, 12, 13).
Full-length antibodies of the IgG type and IgG Fc fusions are the fastest growing classes of biopharmaceuticals (14). Their remarkably long serum half-life provided by FcRn has surely contributed to their clinical success. Moreover, the central role of FcRn in controlling the pharmacokinetics of IgG has inspired the development of novel Fc-engineered IgG molecules with improved FcRn binding that result in extended half-lives and improved therapeutic efficacy (15–18). The lesson learned from such studies is that the major engineering challenge is to increase binding affinity at pH 6.0 while at the same time retaining low affinity at nearly neutral pH, so as to allow efficient FcRn-mediated recycling.
Although the molecular interaction between FcRn and IgG has been studied for decades, the characterization of the albumin-FcRn interaction is just starting (7, 8, 10, 11, 19). Still, the knowledge of its prolonged half-life has been utilized for some time to improve the in vivo efficacy of therapeutics. The approaches taken have been to associate or covalently couple a protein of interest by genetic fusion or chemical conjugation to albumin leading to increase in vivo half-life of the fusion compared with the original molecule (20, 21). The great advantage of extended serum persistence is a more even serum concentration of the drug, lower dosing frequency, and the fact that the doses given may be decreased without compromising pharmacological efficacy. This may well translate into less toxicity and side effects, as well as improved compliance. Using albumin to increase half-life has been shown to be useful for a wide range of different proteins that includes coagulation factors, antibody fragments, interferon 2b, and glucagon-like peptide-1, as reviewed elsewhere (20–23). Thus, we aimed to develop novel engineered human serum albumin (HSA) variants with improved pH-dependent binding to human FcRn (hFcRn) and enhanced serum half-life that can be used as improved carriers for diagnostics and therapeutics.
Here, we report on a panel of engineered single HSA variants with substitutions at position 573 within the C-terminal helix of domain III (DIII). All show considerably improved binding to hFcRn. Surprisingly, replacement of Lys-573 with any amino acid resulted in enhanced binding to hFcRn at acidic pH. In particular, K573P, where Lys-573 was replaced by a proline, had more than 12-fold improved affinity toward hFcRn, resulting in extended serum half-life in WT mice, mice transgenic (Tg) for hFcRn and cynomolgus monkeys. Importantly, fusion of an antibody derived single-chain variable fragment (scFv) to the K573P variant did not negatively affect the favorable binding. Thus, the K573P variant may be an ideal carrier for half-life extension of both conjugated and genetically fused diagnostics and therapeutics.
EXPERIMENTAL PROCEDURES
Construction and Production of Recombinant FcRn
The construction and production of recombinant soluble mouse, cynomolgus monkey, and human forms of FcRn have been described previously (22, 24, 25).
Construction and Production of HSA Variants
Construction of WT HSA, mouse serum albumin (MSA), cynomolgus monkey serum albumin (CSA), and variants was done essentially by following the procedures previously described (10, 22). The albumin variants were produced in Saccharomyces cerevisiae and monomeric fractions were purified using the AlbuPureTM matrix (ProMetic BioSciences) followed by chromatography as described (10). Construction and production of WT HSA, K500A, and K573P with N- or C-terminal fusions of a scFv fragment with specificity for fluorescein isothiocyanate were done in accordance with previously described procedures (22, 26). WT HSA and K573P with N-terminal fusions of a c-Myc tag (EQKLISEEDL) without a linker sequence were constructed essentially as previously described (10), followed by purification using AlbuPureTM, diethylaminoethyl weak anion exchange Sepharose Fast Flow (GE Healthcare), and Sephacryl S200 high resolution gel filtration (GE Healthcare) as to reduce the level of a +2058-Da miscleaved leader to below 5% (w/v).
Determination of FcRn Binding by Surface Plasmon Resonance (SPR)
A Biacore 3000 instrument (GE Healthcare) was used with CM5 sensor chips coupled with mFcRn, cynomolgus monkey FcRn, or hFcRn (1,000 resonance units) using amine coupling chemistry as described by the manufacturer. The coupling was performed by injecting 5–10 μg/ml of each protein into 10 mm sodium acetate, pH 5.0 (GE Healthcare). For all experiments, phosphate buffer (67 mm phosphate buffer, 0.15 m NaCl, 0.005% Tween 20) with pH 5.5, 6.0, or 7.4 was used as running buffer and dilution buffer. Regeneration of the surfaces were achieved using injections of HBS-EP buffer (0.01 m HEPES, 0.15 m NaCl, 3 mm EDTA, 0.005% surfactant P20) at pH 7.4 (GE Healthcare). Kinetic measurements were performed by injecting serial dilutions of 100–1.4 μm or 10–0.03 μm of albumin variants with a flow rate of 40 μl/min at 25 °C. For all sensorgrams, data were zero adjusted, and the reference cell value was subtracted. Binding kinetic constants were calculated using the predefined binding models provided by the BIAevaluation 4.1 software.
ELISA
Ovalbumin conjugated to 4-hydroxy-3-iodo-5-nitrophenylacetic acid (1 μg/ml) was coated in Microtiter wells (Nunc) overnight at 4 °C and then blocked with 4% skimmed milk (Acumedia) in PBS for 1 h at room temperature. Subsequently, the wells were washed with PBS, 0.005% Tween 20 (PBS/T), pH 6.0, before titrated amounts of an anti-4-hydroxy-3-iodo-5-nitrophenylacetic acid human IgG1 antibody (10 nm-0.07 nm) were added for 1 h. After washing as above, GST-tagged hFcRn (0.5 μg/ml) was added alone or in the presence of 1000 nm of WT HSA, WT MSA, K500A, or K573P followed by incubated for 1 h and washing as above. Bound receptor was detected using a horseradish peroxidase-conjugated goat anti-GST antibody (GE Healthcare) that was added followed incubation and washing. Visualization was done by adding tetramethylbenzidine substrate (Calbiochem). Quantification of total mouse IgG was done using a mouse IgG quantification kit (Bethyl Laboratories Inc.).
Circular Dichroism Spectroscopy
Circular dichroism spectra were recorded using a Jasco J-810 spectropolarimeter (Jasco International Co., Ltd., Tokyo, Japan) calibrated with ammonium d-camphor-10-sulfonate (Icatayama Chemicals). All measurements were performed with a HSA concentration of 2 mg/ml in 10 mm phosphate-buffered saline (pH 6.0) without NaCl added, at 23 °C using a quartz cuvette (Starna) with a path length of 0.1 cm. Each sample was scanned seven times at 20 nm/min with a bandwidth of 1 nm at a response time of 1 s, and the wavelength range was set to 190–260 nm. The collected data were averaged, and the spectrum of a sample-free control was subtracted. The content of secondary structural elements was calculated after smoothing (means-movement, convolution width 5) from ellipticity data, using the neural network program CDNN version 2.1 and the supplied neural network based on the 33-member basis set (27).
In Vivo Mouse and Cynomolgus Monkey Studies
NMRI mice (female, aged 8–10 weeks, weight between 20 and 30 g, 18 mice/group, 6 mice/time point) received 10 mg/kg intravenous injections of WT HSA, K500A, and K573P (dose volume, 5 ml/kg) in 1× PBS. Blood samples (30 μl) were collected from the tail vessel in time intervals of (a) predose, 0.08, 6, 48, 120, 192, and 264 h for mouse numbers 1–6; (b) predose, 1, 12, 72, 144, 216, and 288 h for mouse numbers 7–12; and (c) predose, 2, 24, 96, 168, 240, and 312 h for numbers 13–18. Mice Tg for hFcRn (Tg32) on a C57BL/6J genetic background were obtained from the Jackson Laboratory (Bar Harbor, ME) (28). Tg32 Tg mouse strain, isogenic on a B6 background, harbors a knock-out allele of the FcRn HC (Fcgrttm1Dcr) and expresses an hFcRn HC (FCGRT) transgene under control of the hFcRn promoter. The Tg32 mice (male, aged 6–10 weeks, weight > 15 g, 10 mice/group, 5 mice/time point) received a 10 mg/kg intravenous injections of WT HSA, K500A, and K573P (dose volume 5 ml/kg). Blood samples (30 μl) were collected from the tail vessel in time intervals of (a) predose, 2, 12, 72, 120, 192, and 264 h for numbers 1–5 and (b) predose, 6, 48, 96, 168, 240, and 312 h for numbers 6–10.
Female cynomolgus monkeys (Macaca fascicularis) (weight between 2.5 and 3.5 kg, 2 animals/group) were injected with c-Myc tagged WT HSA and K575P in 1× PBS (dose volume, 5 ml/kg). Prior to injections, a predose blood sample was collected, whereas postdosing of blood (0.8 ml) was drawn at 0.25, 2, 8, 24, 48, 72, 96, 120, 144, 192, 240, 288, 336, 384, 432, 480, 528, 576, 624, 720, 816, 912, 1008, 1104, and 1200 h. Plasma samples were prepared from blood collected onto a 0.13 m sodium citrate as anticoagulant, mixed thoroughly by inversion, and centrifuged at 2000 × g for 10 min at 4 °C.
All animal experiments and procedures were reviewed and approved by the institutional animal care and use committee prior to procedure initiation. All studies were conducted according to the Animal Welfare Act 1986 and conducted in a manner to avoid or minimize distress or pain to animals.
Analysis of Pharmacokinetics
All serum samples were stored at −80 °C until analysis by AlphaLISA using a HSA kit (PerkinElmer Life Sciences) (mouse) or a c-Myc ELISA (primate). The latter was performed with EIA Maxisorb plates (Nunc) coated overnight with an anti-c-Myc antibody (Abcam) at 1.25 μg/ml in 1× PBS before the plates were washed three times with 1× PBS containing 0.05% Tween 20 (PBST) (Sigma-Aldrich), pH 7.4. Then the plates were blocked for 2 h with PBS containing 5% skimmed milk powder, 1% Tween 20, and 10% rat serum, pH 7.4. Plasma samples were diluted 1:10 in 1× PBS and mixed with 10% female cynomologus monkey sodium citrate plasma (SeraLab) in PBST at final dilutions of 1:10, 1:20, 1:40, and 1:80. A standard curve was included on each plate with purified c-Myc HSA diluted in 10% cynomologus monkey plasma in PBST. Plates were incubated for 1 h before washed as above. Next 100 μl of an biotinylated anti-HSA antibody (Abcam) at 1.5 μg/ml in PBST was added to the plates followed incubation for 30 min. Plates were washed as above, and 100 μl of 1.25 μg/ml HRP-conjugated streptavidin (Sigma-Aldrich) in PBST was added to the plates and incubated for 30 min. Subsequently, the plates were washed as above, and the signal was developed with 100 μl of 3,3′,5,5′-tetramethylbenzidine-Ultra substrate (Pierce) for 5 min before the reactions were stopped with 0.2 m sulfuric acid. The signals were measured on an EnSpire Multimode plate reader (PerkinElmer Life Sciences) as 450–550 nm. The standard curve on each plate was fitted to a four-parameter nonlinear regression model using an in-house Excel Macro, and the plasma HSA concentration was calculated at each time point using the dilutions that fell within the linear range of the standard curve.
Group mean serum or plasma concentration profiles were subjected to noncompartmental pharmacokinetic analysis using WinNonLin version 5.3 (mouse) (Pharsight Corporation) or WinNonLin version 6.3 (primate). Nominal time points and doses were used, and all data points were equally weighted in the analysis. Mean serum concentrations versus time profiles for each of the HSA variants were fit with a two-compartment model to generate the curve fit shown in the figures. The following pharmacokinetic parameters were assessed: Cmax, AUC, Vz, CL, and T½.
Sequence and Structural Analysis
The ClustalW software was used for amino acid sequence alignments. The NCBI accession numbers of the albumin sequences used were: NP_001129491 (human), AAT90502 (elephant), AF375971.1 (tuatara), NP_001127106 (orangutan), AAA30988 (pig), XP_001103956 (rhesus monkey), NP_001075972 (horse), NP_001009961 (cat), NP_001081244 (frog), NP_001117164 (salmon), AAV28861 (donkey), AAA51411 (cow), NP_001009376 (sheep), ACF10391 (goat), CAA76841 (dog), AAB58347 (rabbit), NP_001166576 (guinea pig), NP_001268578 (hamster), AAH85359 (rat), and AAH49971 (mouse).
The coordinates of the crystal structures of HSA (Protein Data Bank code 1bm0) (29) and hFcRn in complex with a HSA variant (HSA13) containing four mutations (Protein Data Bank code 4k71) (11) were retrieved from the protein database. The structures were inspected using PyMOL (Schrodinger Inc.).
RESULTS
Identification of HSA Variants with Altered Binding
We have previously obtained mechanistic knowledge about the FcRn-albumin interaction where DIII of HSA was identified as crucial for pH-dependent binding to the receptor (10, 19). DIII is divided into two subdomains, DIIIa and DIIIb, which consist of α-helices connected by a long flexible linker (Fig. 1A). Both subdomains are important for FcRn binding, and removal of the whole DIII or only DIIIb eliminates binding (8, 10).
FIGURE 1.

Amino acid substitutions in the last C-terminal end of HSA DIII modulate binding to hFcRn. A, an illustration of the crystal structure of full-length HSA with the three domains DI (pink), DII (orange), and DIII (cyan/blue) highlighted. The DIII is split into subdomains DIIIa (cyan) and DIIIb (blue). B, a close-up of DIIIb and the last α-helix in the C-terminal end with the amino acid residues Lys-573, Lys-574, and Gln-580 indicated. The figures were made using the software PyMOL, using coordinates from Protein Data Bank entry 1bm0. C–E, representative SPR sensorgrams showing binding of 1 μm of WT HSA, K574A, and Q580A (C); WT HSA and K573A (D); and WT HSA and HSA K573X (E) to immobilized hFcRn at pH 6.0. Injections were performed at 25 °C, and the flow rate was set to 40 μl/min. The kinetic rate constants were obtained using a simple first order (1:1) bimolecular interaction model (Langmuir) or a steady state affinity model supplied by the BIAevaluation 4.1 software. The kinetic values represent the average of triplicates.
Interestingly, inspection of the crystal structures of HSA reveals that the electron density corresponding to the C-terminal part is not clearly defined, probably as a result of conformational flexibility (29, 30). This led to the speculation that binding of hFcRn may stabilize this part of HSA. To address the impact of the C-terminal end, three charged amino acid residues (Lys-573, Lys-574, and Gln-580) in the last α-helix of HSA (Fig. 1B) were targeted by mutating each to an alanine. The HSA variants were subsequently expressed in S. cerevisiae, and binding to hFcRn was measured using SPR. Equal amounts of each variant were injected over immobilized receptor at pH 6.0. Compared with WT HSA, the K574A and Q580A variants showed reversible binding to hFcRn at pH 6.0 but with reduced capacity to interact (Fig. 1C). In stark contrast, K573A showed considerably stronger binding than the WT (Fig. 1D). Kinetic calculations revealed that K573A had 2.5-fold improved binding affinity. The importance of the stretch of amino acids corresponding to positions 573–585 was further confirmed by introducing a stop codon at position 573 (HSA K573X), which resulted in a more than 18-fold reduction in binding affinity (Fig. 1E).
Substitution of Lys-573 with a Proline Improves Binding Considerably
To investigate the evolutionary conservation of amino acids in the C-terminal end of albumin, we aligned the sequences from 20 different species and found that position 573 was found to be one of the most conserved, harboring a proline residue in 18 of 20 species, whereas human and orangutan albumins have a lysine (Fig. 2A). This prompted us to investigate how a proline at this position in HSA influences binding to hFcRn. Using SPR, we demonstrated that introduction of the single substitution K573P did not disrupt pH-dependent binding but instead resulted in a considerable improvement in binding to hFcRn without a concomitant increase in binding at neutral pH (Fig. 2B). Kinetic measurements demonstrated a more than 12-fold improvement compared with the WT, almost exclusively because of the slower off rate (kD).
FIGURE 2.

Considerably improved pH-dependent binding to hFcRn of a HSA variant containing K573P. A, a ClustalW amino acid sequence alignment of the last C-terminal α-helix of albumin derived from 20 species. The amino acid residues at positions 573, 574, and 580 are indicated. A lysine or a proline at position 573 is highlighted in blue or red, respectively. B, a representative SPR sensorgrams showing binding of 1 μm of K573P to immobilized hFcRn at pH 6.0 and 7.4. Injections were performed at 25 °C, and the flow rate was 40 μl/min. The kinetic rate constants were obtained using a Langmuir binding model supplied by the BIAevaluation 4.1 software. The kinetic values represent the averages of triplicates.
A Panel of HSA Single Point Variants with Improved FcRn Binding
To further investigate the potential of modulating position 573, we designed a library consisting of all 20 naturally occurring amino acids at this specific site. The variants were well expressed in S. cerevisiae and purified as monomers (Fig. 3A). Using SPR as above, we screened the panel of 573 variants for binding to hFcRn at pH 6.0 (Fig. 3, B–S). Strikingly, all 573 variants showed improved binding to the receptor with distinct differences in binding kinetics, which generated a hierarchy of novel HSA variants with superior binding to hFcRn. Although 16 of 19 variants bound less well than K573P, the largest improvement in binding strength was obtained when Lys-573 was swapped to a hydrophobic tryptophan or a tyrosine, which both showed 2-fold better affinity than K573P, and consequently more than 30-fold stronger binding at acidic pH compared with WT HSA. However, these variants also showed a somewhat improved binding at neutral pH compared with K573P (data not shown).
FIGURE 3.

A panel of single point HSA variants with improved binding to hFcRn. A, WT HSA and HSA variants were produced in S. cerevisiae and subsequently purified on an albumin affinity matrix before analysis by 4–12% (w/v) SDS-PAGE. B–S, representative sensorgrams showing binding of titrated amounts of WT HSA and 573 variants to immobilized hFcRn at pH 5.5. Injections were performed at 25 °C, and the flow rate was 40 μl/min. The kinetic rate constants were obtained using a Langmuir binding model supplied by the BIAevaluation 4.1 software. The kinetic values represent the averages of triplicates.
HSA K573P Shows Extended Half-life in Mice and Cynomolgus Monkeys
Based on the facts that proline is the most common amino acid at position 573 (conserved in 18 of 20 species) and that the K573P variant solely bound strongly to FcRn at acidic pH with no sign of binding at neutral pH, we chose this variant for in vivo studies.
Importantly, preclinical pharmacokinetic evaluations of HSA and HSA fusion molecules are routinely performed in rodents. However, we recently showed that such half-life evaluations are somewhat limited by the fact that FcRn from these species bind poorly to HSA compared with albumin from the animals themselves (22, 24). In line with this, recombinant WT HSA bound with 25-fold weaker affinity than MSA to mouse FcRn (mFcRn) (Fig. 4, A and B). Consequently, HSA injected into mice competes for binding to mFcRn with roughly 40 mg/ml endogenous MSA, resulting in a shorter half-life of HSA than that of endogenous MSA (24).
FIGURE 4.

HSA K573P shows improved binding to mFcRn. A–C, representative sensorgrams showing binding of titrated amounts of WT MSA (A, 10–0.03 μm), WT HSA (B, 100–1.4 μm), and HSA K573P (C, 10–0.03 μm) to immobilized mFcRn at pH 6.0. D–F, representative sensorgrams showing binding of titrated amounts of WT CSA (D, 10–0.03 μm), WT HSA (E, 10–0.03 μm), and HSA K573P (F, 10–0.03 μm) to immobilized cynomolgus monkey FcRn (cmFcRn) at pH 6.0. All injections were performed at 25 °C, and the flow rate was 40 μl/min. The kinetic rate constants were obtained using a Langmuir binding model or a steady state affinity model supplied by the BIAevaluation 4.1 software. The kinetic values represent the averages of triplicates.
Although mFcRn binds 25-fold less well to HSA than MSA, introduction of the single point mutation (K573P) greatly affected binding, because the affinity improved almost 15-fold (Fig. 4C). Thus, K573P binds almost as strongly as MSA. Hence, we aimed for pharmacokinetic experiments in WT NMRI mice. WT HSA and K573P were compared with a HSA variant, which contains a point mutation (K500A), located within an extended loop connecting the two DIII subdomains, that reduces binding to hFcRn by more than 30-fold (10). Calculation of their half-life from serum concentration measurements showed that K573P gave rise to a 1.5-fold enhancement in half-life relative to that of WT HSA (Fig. 5A and Table 1). Notably, and in agreement with poor binding of WT HSA to mFcRn, the half-life of WT HSA was only slightly longer than that of K500A (Fig. 5A and Table 1).
FIGURE 5.
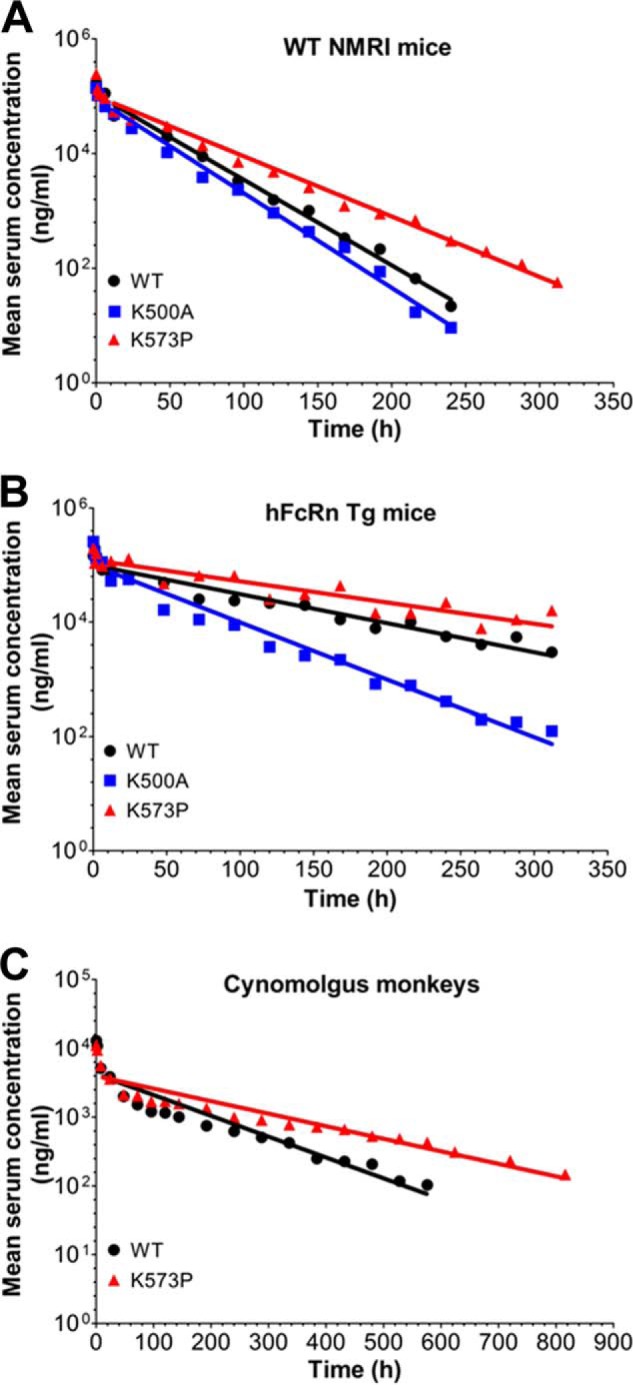
HSA K573P shows extended serum half-life in mice and cynomolgus monkeys. A and B, log-linear changes in serum concentrations of WT HSA, K500A, and K573P in WT NMRI mice (A, 18 mice/group, 6 mice/time point) and FcRn−/− hFcRn Tg32 mice (B, 10 mice/group, 5 mice/time point). HSA variants were administered by intravenous infusions at 10 mg/kg, followed by collecting serum samples from the tail vein before the serum HSA concentrations were determined using an AlphaLISA immunoassay. C, log-linear changes in serum concentrations of WT HSA and K573P in cynomolgus monkeys (2 monkeys/group). HSA variants were administered by intravenous infusions at 1 mg/kg, followed by collecting serum samples before the HSA concentrations were determined using an anti-c-Myc ELISA. The results are means ± standard errors.
TABLE 1.
Pharmacokinetic evaluations of HSA variants in mice
Cmax indicates the maximum concentration reached after administration. T½ indicates the elimination phase half-life. AUC indicates the area under the curve, a time integral of the blood uptake. Vz indicates the volume of distribution. CL indicates clearance, the volume of plasma in the vascular compartment cleared of the drug per unit time by the processes of metabolism and excretion. i.v. indicates intravenous injection.
| HSA | Dose (i.v.) | Cmax | AUC | Vz | CL | T½ |
|---|---|---|---|---|---|---|
| mg/kg | μg/ml | h × μg × ml | mg/kg | ml/h/kg | h | |
| WT NMRI mice | ||||||
| WT | 10 | 172 | 2,816 | 108.0 | 3.55 | 21.0 |
| K500A | 10 | 139 | 2,156 | 128.0 | 4.64 | 19.1 |
| K573P | 10 | 244 | 2,157 | 120.0 | 2.72 | 30.6 |
| FcRn−/− hFcRn Tg32 mice | ||||||
| WT | 10 | 106 | 7,527 | 128.0 | 1.33 | 67.0 |
| K500A | 10 | 156 | 4,490 | 128.0 | 2.23 | 31.3 |
| K573P | 10 | 128 | 12,506 | 110.0 | 0.80 | 95.2 |
| Rhesus monkeys | ||||||
| WT | 1 | 12.9 | 523.3 | 351.7 | 1.85 | 131.7 |
| K573P | 1 | 11.0 | 803.2 | 358.5 | 1.17 | 210.7 |
The fact that MSA binds more than 5-fold more strongly to hFcRn than HSA (24) may impact pharmacokinetic evaluations in mice Tg for hFcRn. Nevertheless, K573P binds more than 2-fold stronger than MSA. Thus, we compared the HSA variants in a Tg mouse strain lacking the expression of mFcRn and overexpressing hFcRn under the control of the human promoter (FcRn−/− hFcRn Tg mice) (28). The pharmacokinetics revealed that K573P gave rise to almost 1.5-fold longer serum half-life than WT HSA and 3-fold longer persistence than the K500A variant (Fig. 5B and Table 1). Notably, a comparison of the two mouse models revealed that the serum half-life of WT and K573P increased 3-fold in hFcRn Tg mice compared with NMRI mice (Table 1).
Next, these encouraging data prompted us to perform a pharmacokinetic study in cynomolgus monkeys to evaluate the capacity of K573P to improve serum half-life. Although HSA shows high amino acid sequence similarities with that of CSA, three amino acids differ in the C-terminal α-helix, including position 573, which is a proline (Fig. 2A). Nevertheless, WT HSA was shown to bind cynomolgus monkey FcRn slightly weaker than CSA, whereas K573P bound with almost 11-fold improved affinity compared with CSA, similar to the improvement measured toward hFcRn (Fig. 4, D–F).
To quantify the amounts of the HSA variants in blood samples from cynomolgus monkeys, a short c-Myc tag was added to the N-terminal end to distinguish the variants from endogenous CSA (data not shown). The pharmacokinetics demonstrated that introduction of a single point mutation in HSA prolonged the serum half-life from 5.4 to 8.8 days (Fig. 5C and Table 2).
TABLE 2.
Pharmacokinetic evaluations of HSA variants in cynomolgus monkeys
Cmax indicates the maximum concentration reached after administration. T½ indicates the elimination phase half-life. AUC indicates the area under the curve, a time integral of the blood uptake. Vz indicates the volume of distribution. CL indicates clearance, the volume of plasma in the vascular compartment cleared of the drug per unit time by the processes of metabolism and excretion. i.v. indicates intravenous injection.
| HSA | Dose (i.v.) | Cmax | AUC | Vz | CL | T½ |
|---|---|---|---|---|---|---|
| mg/kg | μg/ml | h × μg × ml | mg/kg | ml/h/kg | h | |
| WT | 1 | 12.9 ± 2.0 | 542.5 ± 43.9 | 351.7 ± 34.3 | 1.85 ± 0.20 | 131.7 ± 2.2 |
| K573P | 1 | 11.0 ± 1.0 | 848.3 ± 25.0 | 358.5 ± 5.5 | 1.17 ± 0.04 | 210.8 ± 3.0 |
Engineering of HSA Does Not Affect Binding to IgG
To investigate whether engineering of HSA for improved pH-dependent binding to hFcRn affected IgG binding, we performed an ELISA where hFcRn was added to titrated amounts of human IgG1 in the absence or presence of 100-fold excess amounts of HSA, MSA, K500A, or K573P. The results showed that hFcRn bound human IgG1 equally well under all conditions (Fig. 6A). Furthermore, we quantified the total serum levels of mouse IgG in blood of WT NMRI mice and hFcRn Tg mice before and after injections of WT HSA, K500A, and K573P, which showed that the levels were similar preinjection and 24 and 72 h postinjection in both strains (Fig. 6, B and C). Notably, hFcRn Tg mice showed very low levels of mouse IgG, which were more than 10-fold lower than that of WT mice. This is explained by the fact that hFcRn does not bind strongly to mouse IgG subclasses (2, 24, 31). Our data are in agreement with previous findings showing that the ligands bind to separated binding sites on FcRn (8, 10, 11, 24, 32).
FIGURE 6.
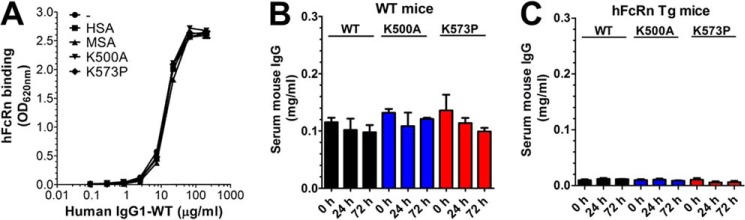
Engineering of HSA does not affect binding to IgG. A, ELISA showing binding of hFcRn-GST to titrated amounts of human IgG1 (10–0.07 nm) in the absence or presence of 1000 nm of HSA, MSA, K500A, or K573P. The ELISA was performed at pH 6.0. The numbers given represent the mean of triplicates. B and C, ELISA quantification of the serum concentrations of total mouse IgG WT mice (B) and hFcRn Tg mice (C) before and after injections (0, 24, and 72 h) of WT HSA, K500A, and K573P.
Fusion of scFv to HSA K573P Does Not Negatively Affect Binding
We have recently shown that genetic fusion of a peptide or an antibody-derived scFv fragment to the N-terminal end of WT HSA has no major impact on binding to hFcRn, whereas fusion to the C-terminal end slightly affects binding negatively (22). To utilize K573P as a carrier of therapeutics, it is important that favorable binding is retained postfusion. To address this, we compared the binding integrity of the WT with that of K573P after fusion of a scFv fragment to the N-terminal or the C-terminal end. The resulting SPR sensorgrams showed that the K573P fusions bound strongly to hFcRn postfusion at pH 6.0 compared with the WT, whereas fusions containing K500A bound poorly (Fig. 7, A and B). Importantly, the derived binding kinetics showed that scFv fusions harboring K573P have KD values 11–12-fold stronger than that of the WT formats, and a slight shift toward weaker affinity was observed for the C-terminal fusions.
FIGURE 7.

Binding of HSA scFv fusions to hFcRn and a structural overview of the C-terminal end of HSA in complex with hFcRn. A and B, representative sensorgrams showing binding of 1 μm of scFv-WT HSA, scFv-K500A, and scFv-K573P (A) and WT HSA-scFv, K500A-scFv, and K573P-scFv (B) to immobilized hFcRn at pH 6.0. Injections were performed at 25 °C, and the flow rate was 40 μl/min. C, an overview of the crystal structure complex between recombinant hFcRn and a HSA variant with four point substitutions (Protein Data Bank entry 4k71). The three HSA domains, DI, DII, and DIII, are shown in pink, orange, and cyan/blue, respectively. DIII is split into subdomains DIIIa (cyan) and DIIIb (blue). The HC of the hFcRn is shown in green, whereas the β2m subunit is shown in gray. The last α-helix of DIIIb is indicated by an arrow. D, a close-up of the structural area of the complex showing the last α-helix of DIIIb and the interaction of Lys-573 with Glu-69 and Ser-20 of the β2m unit. The neighboring hydrophobic Phe-22 is also indicated. The figures were made using the software PyMOL.
A Structural Explanation for Improved Binding
Introduction of K573P on the global structure was investigated and compared with that of the WT and K500A variants using circular dichroism. The obtained spectra at pH 6.0 were very similar for the HSA variants (data not shown), and calculation of their secondary structure elements revealed that the mutations did not have detectable impact on global folding (Table 3).
TABLE 3.
Secondary structural elements determined by circular dichroism
| HSA | Structural elements (pH 6.0) |
||||
|---|---|---|---|---|---|
| Helix | Antiparallel | Parallel | Beta turn | Random coil | |
| WT | 61.4 | 0.1 | 2.5 | 12.8 | 23.4 |
| K500A | 61.7 | 0.1 | 2.2 | 12.5 | 23.6 |
| K573P | 57.5 | 0.3 | 3.5 | 13.5 | 25.2 |
Furthermore, to understand why the panel of position 573 variants gave rise to a hierarchy of improved binders, we scrutinized a recently published co-crystal structure of hFcRn in complex with a HSA variant (HSA13) containing four amino acid substitutions (V418M/T420A/E505G/V547A) within DIII (11). The co-crystal structure shows that Lys-573 of HSA13 makes contact with Glu-69 and Ser-20 of the β2m, where Lys-573 forms a salt bridge with Glu-69 (Fig. 7, C and D). Our SPR binding studies show that a lysine at this position is the least favorable residue, because replacement with any other amino acid results in improved binding, a finding that is not easily explained by inspection of the interaction interface. Potentially, loss of the salt bridge between Lys-573 and Glu-69 could permit DIIIb to adopt a position promoting formation of the hydrophobic interface with hFcRn. Interestingly, the HSA13-hFcRn crystal structure contains two independent copies, one of which lacks the salt bridge between Lys-573 and Glu-69. Additionally, it is tempting to speculate that one or more of the mutations introduced in HSA13 may have structurally altered this region of HSA. A candidate in this regard is V547A, in DIIIb, which potentially facilitates reorientation of and/or altered contacts between the last two C-terminal helices (11).
DISCUSSION
In this report we used rational design to engineer a panel of HSA variants with altered hFcRn binding affinities. One candidate, K573P, showed extended serum half-life in normal mice and mice Tg for the human receptor, as well as in cynomolgus monkeys. Importantly, the enhanced affinity of the K573P mutation was retained postfusion of a scFv fragment to either the N- or C-terminal end. This variant is one of a panel of variants where all have increased binding to hFcRn at acidic pH relative to WT HSA. This work thus paves the way for a new generation of HSA fusion therapeutics utilizing the novel engineered HSA scaffolds as drug carriers. Our results indicate that serum half-life can be tailored using the HSA variants developed, which ultimately may lead to superior efficacy of biopharmaceuticals and greater convenience for patients.
We have previously reported that MSA binds much more strongly to mFcRn than does HSA (22, 24). This must be considered when mice are utilized as preclinical models to determine the pharmacokinetics of HSA variants and fusions. When small amounts of HSA fusions are injected into mice, they compete for mFcRn binding with large amounts of endogenous MSA (40 mg/ml). Biopharmaceutical fusion to HSA has resulted in improved pharmacokinetics, likely as the result of increased molecular weight above the renal clearance threshold; however, the serum half-lives did not reach that of endogenous albumin, underscoring the importance of efficient FcRn-mediated recycling and the route of clearance of HSA fusions. The nature of the fused protein may also potentially affect receptor binding, which needs to be addressed for each unique fusion. Thus, prior to in vivo evaluations in rodents, the FcRn binding kinetics should be determined.
Here we demonstrate that single point mutations in HSA have a large effect on the binding kinetics toward FcRn from mouse, cynomolgus monkey, and human. Despite the shortcomings of existing mouse models, we provide evidence that such engineering translates into altered half-life, where K573P and K500A showed increased and decreased half-life, respectively. Notably, whereas WT HSA showed almost a similar half-life as K500A in WT mice, it demonstrated 2-fold longer half-life in the hFcRn Tg mice. This is likely to be due to the fact that this mouse strain heavily overexpresses the receptor, which results in more available FcRn that can engage in binding of injected HSA in the presence of competing MSA. Nevertheless, the K573P variant demonstrated nearly 50% increase in serum persistence, and these encouraging results prompted us to address the half-life of K573P in cynomolgus monkeys. Here it was revealed that the single substitution extended the half-life by 3.4 days (5.4–8.8 days). Thus, it is expected that the effect will be at least as great in humans.
We have previously shown that swapping of DIII derived from MSA onto DI-DII of HSA gave rise to a hybrid albumin with considerably improved binding toward mFcRn, whereas swapping of DIII of HSA onto DI-DII of MSA reduced binding (22). Further, we swapped the stretch of amino acids corresponding to the C-terminal α-helix of MSA onto HSA (HSA-Cm), which resulted in a 4-fold improved binding compared with WT HSA (22). To our knowledge, this was the first HSA variant reported to have improved pH-dependent binding toward hFcRn. HSA-Cm harbors eight amino acid changes compared with the WT HSA, one of which is K573P. By introducing only K573P into HSA, the binding affinity to mFcRn was improved by more than 20-fold, demonstrating the importance of a proline at this position for optimal binding to mFcRn, and it partly explains why WT HSA binds the mouse receptor poorly.
Furthermore, we showed that a lysine at position 573, as found in WT HSA, is the residue that gives the lowest affinity toward hFcRn, because replacement of this charged residue with any other amino acid improves binding at pH 6.0. Some variants also showed low, but detectable binding at pH 7.4. As of now, one cannot easily predict how this will affect the half-life of variants with very strong binding at acidic pH. Moreover, the fact that all species have a proline at position 573, except human and orangutan, is an interesting observation. A proline at this position in the context of the endogenous HSA would result in extended half-life beyond 19–20 days. This would not necessarily have been favorable, because albumin serves as a carrier, not only for nutrients, but also toxins and waste products such as heme and bilirubin (33, 34); however, in a therapeutic context, extended half-life is desirable.
Interestingly, K573E, which showed 2.4-fold improved binding to hFcRn, also exists as a very rare mutation in humans, first identified in a homozygote male (35). It is expected that this variant has a longer serum half-life in such individuals. However, no reports exist of pathogenic manifestations.
HSA is increasingly utilized as a versatile carrier for therapeutic and diagnostic agents (20, 21, 36). This technology allows for genetic fusion to HSA by fusion to the N- or C-terminal end of a protein of interest (Albufuse® technology), or alternatively, by chemical conjugation to a free cysteine residue within DI (Cys-34). These strategies have been broadly applied for half-life extension of small protein and chemical drugs (reviewed in Refs. 20, 21, and 36). Moreover, great benefits may be achieved by this technology for small proteins such as drugs for diabetes control, coagulation factors, hormones, or cytokines.
Importantly, we recently showed that fusion of a peptide or an scFv fragment to either ends of HSA has only a minor negative impact on binding to the receptor, where the most pronounced effect was detected for C-terminal fusions, although still minor (22). This may easily be compensated by introducing mutations, such as K573P, which will result in more than 12-fold better hFcRn binding at acidic pH than the WT counterpart that consequently will result in extended half-life. This may lead to superior pharmacokinetics tailored to disease state and improved patient compliance.
Acknowledgments
We are grateful to Sathiaruby Sivaganesh for excellent technical assistance and all our colleagues at the Novozymes Biopharma sites in UK and Denmark.
This work was supported in part by the Research Council of Norway through Centres of Excellence Funding Scheme Project Number 179573. This work was also supported by Norwegian Research Council Grant 179573/V40 and South-Eastern Norway Regional Health Authority Grant 39375 (to J. T. A.), Norwegian Research Council Grant 179573 (to K. S. G.), and South-Eastern Norway Regional Health Authority through Regional Technology Platform for Structural Biology and Bioinformatics Grants 2009100, 2011040, and 2012085 (to M. B. and B. D.). I. S., J. T. A., B. D., J. C., A. P., L. E., and D. S. are co-inventors of pending patent applications related to the data described in this paper.
- FcRn
- neonatal Fc receptor
- hFcRn
- human FcRn
- mFcRn
- mouse FcRn
- DIII
- domain III
- HC
- heavy chain
- HSA
- human serum albumin
- MSA
- mouse serum albumin
- scFv
- single-chain fragment variable
- SPR
- surface plasmon resonance
- Tg
- transgenic
- β2m
- β2-microglobulin
- CSA
- cynomolgus monkey serum albumin.
REFERENCES
- 1. Montoyo H. P., Vaccaro C., Hafner M., Ober R. J., Mueller W., Ward E. S. (2009) Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2788–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chaudhury C., Mehnaz S., Robinson J. M., Hayton W. L., Pearl D. K., Roopenian D. C., Anderson C. L. (2003) The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 197, 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simister N. E., Mostov K. E. (1989) Cloning and expression of the neonatal rat intestinal Fc receptor, a major histocompatibility complex class I antigen homolog. Cold Spring Harbor Symp. Quant. Biol. 54, 571–580 [DOI] [PubMed] [Google Scholar]
- 4. Story C. M., Mikulska J. E., Simister N. E. (1994) A major histocompatibility complex class I-like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J. Exp. Med. 180, 2377–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burmeister W. P., Huber A. H., Bjorkman P. J. (1994) Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 372, 379–383 [DOI] [PubMed] [Google Scholar]
- 6. West A. P., Jr., Bjorkman P. J. (2000) Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor. Biochemistry 39, 9698–9708 [DOI] [PubMed] [Google Scholar]
- 7. Andersen J. T., Dee Qian J., Sandlie I. (2006) The conserved histidine 166 residue of the human neonatal Fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur. J. Immunol. 36, 3044–3051 [DOI] [PubMed] [Google Scholar]
- 8. Chaudhury C., Brooks C. L., Carter D. C., Robinson J. M., Anderson C. L. (2006) Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry 45, 4983–4990 [DOI] [PubMed] [Google Scholar]
- 9. Kim J. K., Firan M., Radu C. G., Kim C. H., Ghetie V., Ward E. S. (1999) Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur. J. Immunol. 29, 2819–2825 [DOI] [PubMed] [Google Scholar]
- 10. Andersen J. T., Dalhus B., Cameron J., Daba M. B., Plumridge A., Evans L., Brennan S. O., Gunnarsen K. S., Bjørås M., Sleep D., Sandlie I. (2012) Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat. Commun. 3, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidt M. M., Townson S. A., Andreucci A. J., King B. M., Schirmer E. B., Murillo A. J., Dombrowski C., Tisdale A. W., Lowden P. A., Masci A. L., Kovalchin J. T., Erbe D. V., Wittrup K. D., Furfine E. S., Barnes T. M. (2013) Crystal structure of an HSA/FcRn complex reveals recycling by competitive mimicry of HSA ligands at a pH-dependent hydrophobic interface. Structure 21, 1966–1978 [DOI] [PubMed] [Google Scholar]
- 12. Kobayashi K., Qiao S. W., Yoshida M., Baker K., Lencer W. I., Blumberg R. S. (2009) An FcRn-dependent role for anti-flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology 137, 1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akilesh S., Christianson G. J., Roopenian D. C., Shaw A. S. (2007) Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J. Immunol. 179, 4580–4588 [DOI] [PubMed] [Google Scholar]
- 14. Chan A. C., Carter P. J. (2010) Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 10, 301–316 [DOI] [PubMed] [Google Scholar]
- 15. Ghetie V., Popov S., Borvak J., Radu C., Matesoi D., Medesan C., Ober R. J., Ward E. S. (1997) Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat. Biotechnol. 15, 637–640 [DOI] [PubMed] [Google Scholar]
- 16. Hinton P. R., Johlfs M. G., Xiong J. M., Hanestad K., Ong K. C., Bullock C., Keller S., Tang M. T., Tso J. Y., Vásquez M., Tsurushita N. (2004) Engineered human IgG antibodies with longer serum half-lives in primates. J. Biol. Chem. 279, 6213–6216 [DOI] [PubMed] [Google Scholar]
- 17. Vaccaro C., Zhou J., Ober R. J., Ward E. S. (2005) Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat. Biotechnol. 23, 1283–1288 [DOI] [PubMed] [Google Scholar]
- 18. Zalevsky J., Chamberlain A. K., Horton H. M., Karki S., Leung I. W., Sproule T. J., Lazar G. A., Roopenian D. C., Desjarlais J. R. (2010) Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 28, 157–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andersen J. T., Daba M. B., Sandlie I. (2010) FcRn binding properties of an abnormal truncated analbuminemic albumin variant. Clin. Biochem. 43, 367–372 [DOI] [PubMed] [Google Scholar]
- 20. Elsadek B., Kratz F. (2012) Impact of albumin on drug delivery: new applications on the horizon. J. Control. Release 157, 4–28 [DOI] [PubMed] [Google Scholar]
- 21. Sleep D., Cameron J., Evans L. R. (2013) Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 1830, 5526–5534 [DOI] [PubMed] [Google Scholar]
- 22. Andersen J. T., Cameron J., Plumridge A., Evans L., Sleep D., Sandlie I. (2013) Single-chain variable fragment albumin fusions bind the neonatal Fc receptor (FcRn) in a species-dependent manner: implications for in vivo half-life evaluation of albumin fusion therapeutics. J. Biol. Chem. 288, 24277–24285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santagostino E., Negrier C., Klamroth R., Tiede A., Pabinger-Fasching I., Voigt C., Jacobs I., Morfini M. (2012) Safety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood 120, 2405–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andersen J. T., Daba M. B., Berntzen G., Michaelsen T. E., Sandlie I. (2010) Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J. Biol. Chem. 285, 4826–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andersen J. T., Justesen S., Fleckenstein B., Michaelsen T. E., Berntzen G., Kenanova V. E., Daba M. B., Lauvrak V., Buus S., Sandlie I. (2008) Ligand binding and antigenic properties of a human neonatal Fc receptor with mutation of two unpaired cysteine residues. FEBS J. 275, 4097–4110 [DOI] [PubMed] [Google Scholar]
- 26. Evans L., Hughes M., Waters J., Cameron J., Dodsworth N., Tooth D., Greenfield A., Sleep D. (2010) The production, characterisation and enhanced pharmacokinetics of scFv-albumin fusions expressed in Saccharomyces cerevisiae. Protein Expr. Purif. 73, 113–124 [DOI] [PubMed] [Google Scholar]
- 27. Böhm G., Muhr R., Jaenicke R. (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5, 191–195 [DOI] [PubMed] [Google Scholar]
- 28. Petkova S. B., Akilesh S., Sproule T. J., Christianson G. J., Al Khabbaz H., Brown A. C., Presta L. G., Meng Y. G., Roopenian D. C. (2006) Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int. Immunol. 18, 1759–1769 [DOI] [PubMed] [Google Scholar]
- 29. Sugio S., Kashima A., Mochizuki S., Noda M., Kobayashi K. (1999) Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng. 12, 439–446 [DOI] [PubMed] [Google Scholar]
- 30. Curry S., Mandelkow H., Brick P., Franks N. (1998) Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 5, 827–835 [DOI] [PubMed] [Google Scholar]
- 31. Ober R. J., Radu C. G., Ghetie V., Ward E. S. (2001) Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int. Immunol. 13, 1551–1559 [DOI] [PubMed] [Google Scholar]
- 32. Oganesyan V., Damschroder M. M., Cook K. E., Li Q., Gao C., Wu H., Dall'acqua W. F. (2014) Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 289, 7812–7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zunszain P. A., Ghuman J., Komatsu T., Tsuchida E., Curry S. (2003) Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zunszain P. A., Ghuman J., McDonagh A. F., Curry S. (2008) Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXα. J. Mol. Biol. 381, 394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanzetti G., Porta F., Prencipe L., Scherini A., Fraccaro M. (1979) A homozygote for a serum albumin variant of the fast type. Hum. Genet. 46, 5–9 [DOI] [PubMed] [Google Scholar]
- 36. Andersen J. T., Sandlie I. (2009) The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab. Pharmacokinet. 24, 318–332 [DOI] [PubMed] [Google Scholar]